
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid access to Asp/Glu sidechain hydrazides as thioester precursors for peptide cyclization and glycosylation†
Natalie G.
Barnes
,
Kudakwashe
Nyandoro
,
Hanzhang
Jin
and
Derek
Macmillan
 *
*
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H0AJ, UK. E-mail: d.macmillan@ucl.ac.uk
First published on 5th January 2021
Abstract
Head-to-sidechain macrocylic peptides, and neoglycopeptides, were readily prepared by site-specific amidation of aspartic and glutamic acid sidechain hydrazides. Hydrazides, serving as latent thioesters, were introduced through regioselective opening of the corresponding Nα-Fmoc protected anhydride precursors.
The use of peptide Cα-thioesters as building blocks for synthetic proteins using Native Chemical Ligation (NCL) is well established.1 These thioesters have additionally functioned as precursors for NCL-mediated head-to-tail peptide cyclisation.2 Both C-terminal thioesters and hydrazides (which function as stable thioester precursors) can be readily prepared from synthetic or bacterially produced materials which, in part, overcomes the size limitations associated with traditional stepwise solid phase peptide synthesis (SPPS).3 Meanwhile, selective formation of thioesters at the sidechain carboxyl groups of aspartic (Asp) and glutamic (Glu) acids within fully unprotected peptides and proteins is much less common, yet valuable for the production of branched and cyclic peptides possessing novel architectures relevant to natural products, therapeutics, and new biomaterials. For example, bioactive lariat peptides contain a head-to-sidechain macrocycle connecting their N-terminal amino and sidechain carboxyl groups of specific Asp or Glu residues.4 Sidechain amidation has also served as a key macrocyclisation strategy for preparation of synthetic stapled peptides, invariably employing fully protected precursors.5 Sidechain thioesters are difficult to incorporate into synthetic peptides by Fmoc-based SPPS due to their instability in the presence of piperidine. Consequently, alternative building blocks possessing a latent thioester activated via an N–S(e) acyl transfer have been described.6 Sidechain acyl hydrazides are also potentially valuable here as air and moisture stable alternatives, but there are only few reports of their use as sidechain thioester precursors.7 Some protected Cβ/γ-hydrazide building blocks of Asp7a and Glu8 respectively, suitable for use in Fmoc-based SPPS, have previously been prepared albeit by disparate, and often protracted, synthetic pathways.
Here we show how various sidechain protected Asp and Glu hydrazides can be accessed rapidly in only two steps from the corresponding Nα-Fmoc protected amino acid, via regioselective opening of the cyclic anhydride.9 Protected analogues were introduced to model synthetic peptides to demonstrate their utility in head-to-sidechain macrocyclisation and NCL-like glycosylation (Fig. 1).
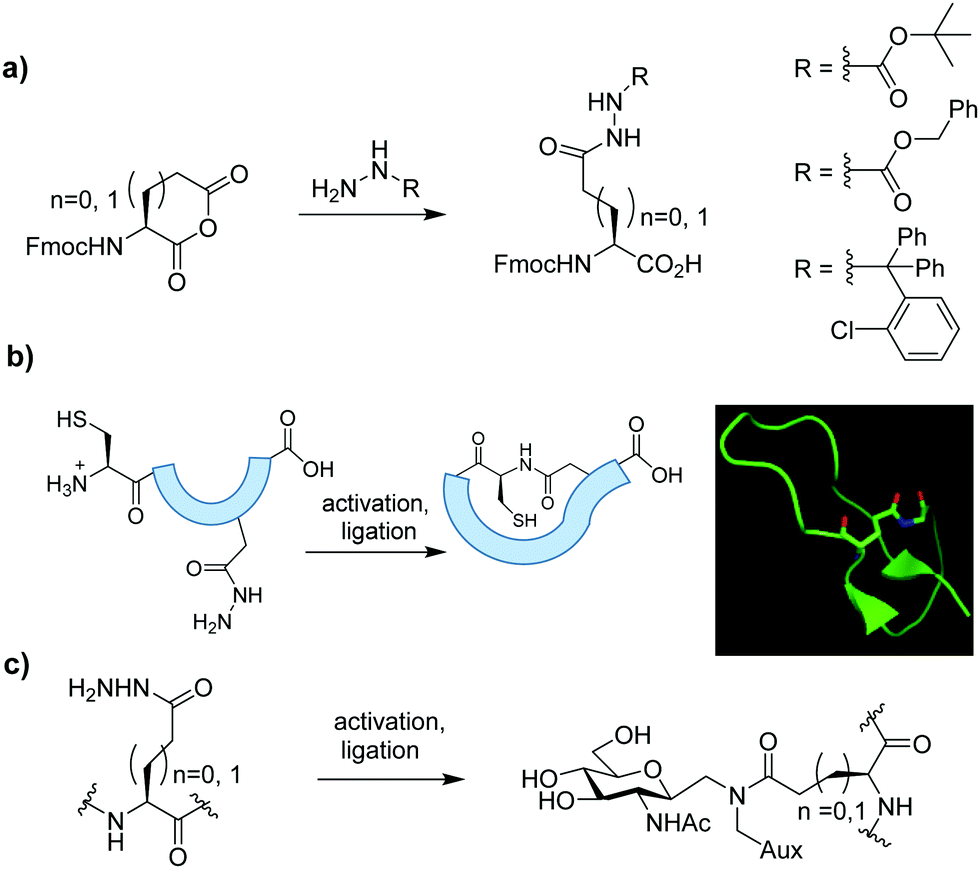 | ||
| Fig. 1 (a) Rapid synthesis of various sidechain protected Glu and Asp hydrazides by anhydride opening, by reaction with a suitable carbazate or hydrazine (b) Head-to sidechain cyclisation, typical of lariat peptides (inset) and (c) site-selective bioconjugation via NCL. | ||
Table 1 shows that several protected Asp/Glu building blocks were prepared via regioselective opening of the corresponding Fmoc-protected amino acid anhydride in as little as 1 h. Hydrazide building blocks were then introduced to short model peptides derived from microcin J25 (H-![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) GAGHVPEF-NH2, 7),10 and lassomycin (H-LRRLFADQ-NH2, 8).11 To assess their performance in NCL-type cyclisation, the N-terminal Gly residues were replaced with Cys, and the target sidechain carboxyl group (indicated in bold) was primed as the hydrazide derivative. 7 was prepared using building block 3 under standard (HBTU/HOBt) coupling, whilst Boc-protected 1 failed to couple under similar reaction conditions. Cyclisation at the Glu sidechain via acyl azide3c and acyl pyrazole12 intermediates was then examined (Fig. 2).
GAGHVPEF-NH2, 7),10 and lassomycin (H-LRRLFADQ-NH2, 8).11 To assess their performance in NCL-type cyclisation, the N-terminal Gly residues were replaced with Cys, and the target sidechain carboxyl group (indicated in bold) was primed as the hydrazide derivative. 7 was prepared using building block 3 under standard (HBTU/HOBt) coupling, whilst Boc-protected 1 failed to couple under similar reaction conditions. Cyclisation at the Glu sidechain via acyl azide3c and acyl pyrazole12 intermediates was then examined (Fig. 2).
|
|
|||||
|---|---|---|---|---|---|
| Hydrazide | n | Ra | hydrazine/carbazate equiv. | Time/h | Yieldb/% |
| a tert-Butyloxycarbonyl (Boc), Benzyloxycarbonyl (Cbz), and 2-Chlorotrityl (2-ClTrt). b Isolated yields following column chromatography, although crude amino acids were often sufficiently pure to be employed directly in peptide synthesis. | |||||
| 1 | 1 | Boc | 1.5 | 2 | 62 |
| 2 | 1 | Cbz | 1.5 | 2 | 88 |
| 3 | 1 | 2-ClTrt | 1.5 | 16 | 73 |
| 4 | 0 | Boc | 1.5 | 1 | 77 |
| 5 | 0 | Cbz | 1.5 | 1 | 63 |
| 6 | 0 | 2-ClTrt | 1.5 | 16 | 66 |
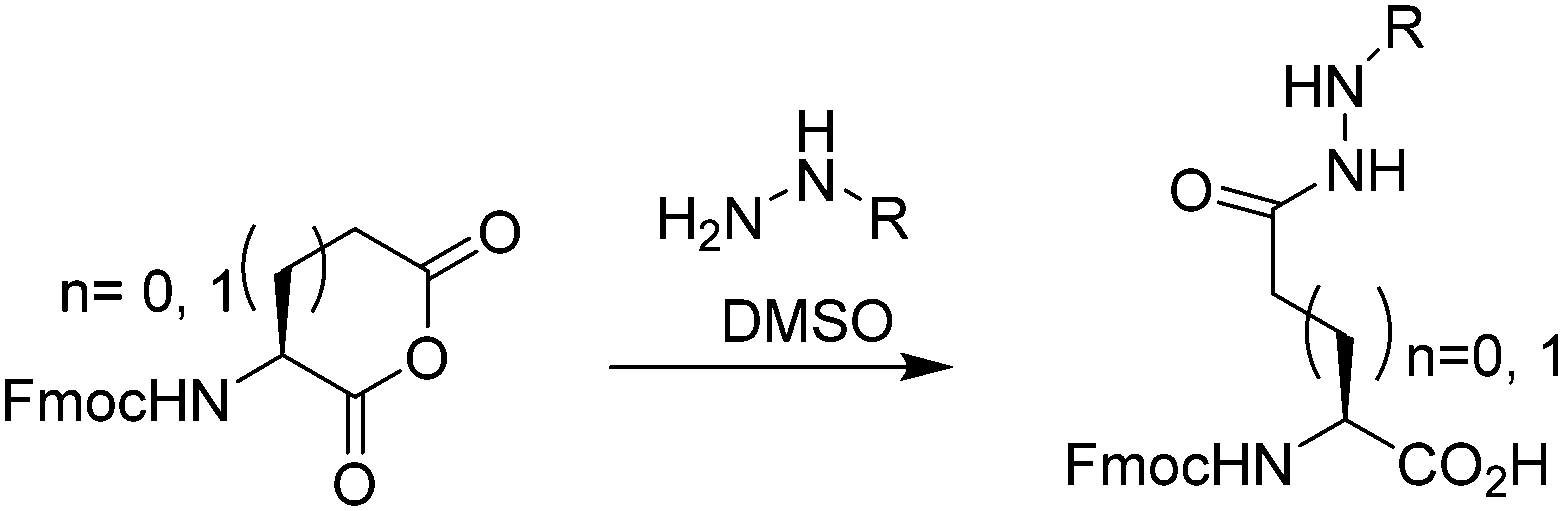
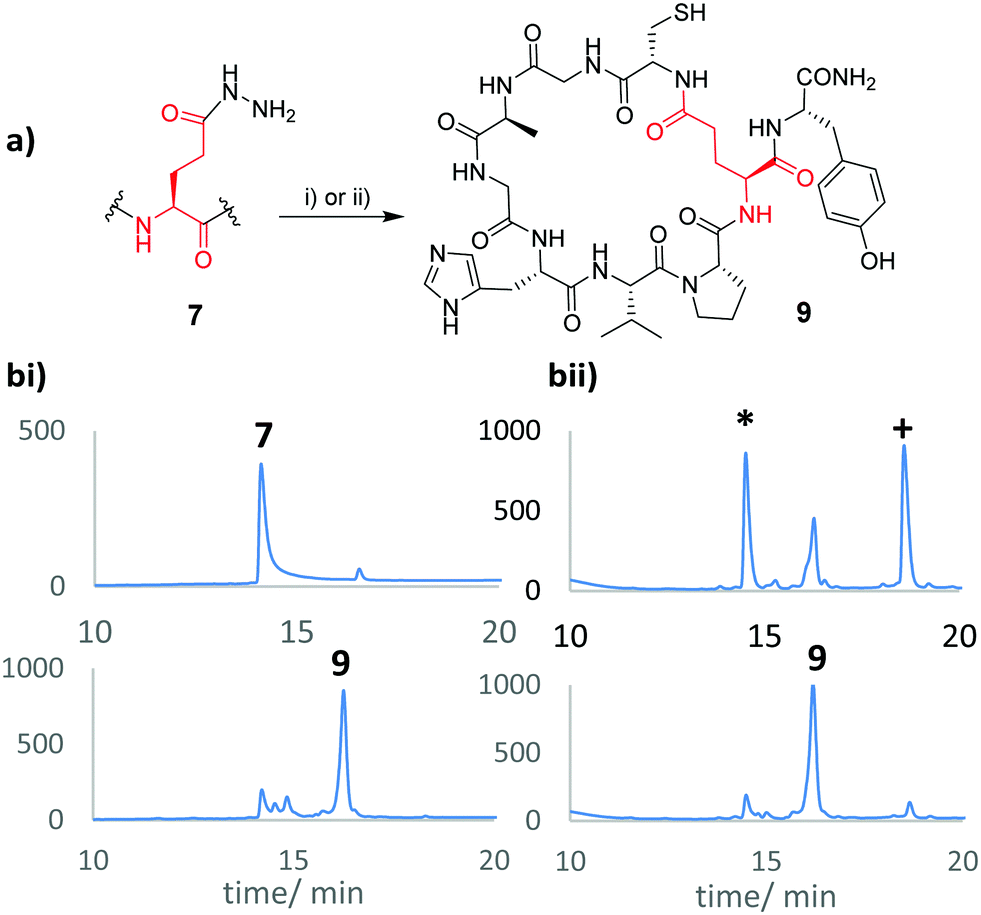 | ||
| Fig. 2 (a) Acyl hydrazide activation to form macrocycle 9, monitored by HPLC. (bi) Upper trace shows initial hydrazide 7 while the lower trace shows the reaction mixture after NaNO2 activation (20 min at pH3, 0 °C), followed by addition of sodium 2-mercaptoethane sulfonate (MESNa), 1 h, RT, pH 7.3c (bii) Activation of 7 with acetyl acetone (acac)/MESNa (pH 3–4, 2 h, upper trace) leads to a mix of MESNa thioester (*) and acyl pyrazole (+) which are both consumed upon adjusting to pH7 for 1 h (lower trace).12 | ||
In both reactions the desired macrocycle 9 was observed within 1–3 h. In the case of the acyl pyrazole, formed using acetyl acetone (acac) as activator, a mixture of 2-mercaptoethane sulfonic acid (MESNa) thioester and acyl pyrazole were initially observed, both of which were rapidly consumed upon adjusting the pH to 7. Studies were next extended to Asp derivatives and model peptide 8. Unfortunately all Asp derivatives 4–6 failed to couple efficiently under identical reaction conditions employed to prepare 7. These results underscored the utility of the Cβ-benzyl ester of Asp as a useful alternative hydrazide precursor.7b Coincidentally, the benzyl ester could also be prepared by regioselective opening of the Fmoc protected aspartic anhydride with benzyl alcohol (see ESI†).
Encouragingly coupling of 4 or 6via the symmetrical anhydride method13 allowed 8 to be isolated in 14% yield from a crude mixture comprising significant deletion by-product (H-![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) LRRLFAQ-NH2). Cyclisation was attempted following NaNO2 activation (Fig. 3). Mindful that aspartimide formation could potentially compete with NCL upon hydrazide activation, giving rise to a product of identical mass, the isolated product was treated with trypsin (0.01 mg ml−1 trypsin, pH 8, 30 °C, 1.5 h). Successful formation of 10 was confirmed by the appearance of two major species (see ESI†) with m/z = 1120.6, corresponding to amide bond hydrolysis at one of the scissile arginine (Arg) residues, and m/z = 964.5, corresponding to excision of one Arg residue. Had cyclisation been unsuccessful we would have expected to observe cleavage products resulting from loss of all 4 N-terminal residues. Overall the cyclisation product was isolated in 64% yield.
LRRLFAQ-NH2). Cyclisation was attempted following NaNO2 activation (Fig. 3). Mindful that aspartimide formation could potentially compete with NCL upon hydrazide activation, giving rise to a product of identical mass, the isolated product was treated with trypsin (0.01 mg ml−1 trypsin, pH 8, 30 °C, 1.5 h). Successful formation of 10 was confirmed by the appearance of two major species (see ESI†) with m/z = 1120.6, corresponding to amide bond hydrolysis at one of the scissile arginine (Arg) residues, and m/z = 964.5, corresponding to excision of one Arg residue. Had cyclisation been unsuccessful we would have expected to observe cleavage products resulting from loss of all 4 N-terminal residues. Overall the cyclisation product was isolated in 64% yield.
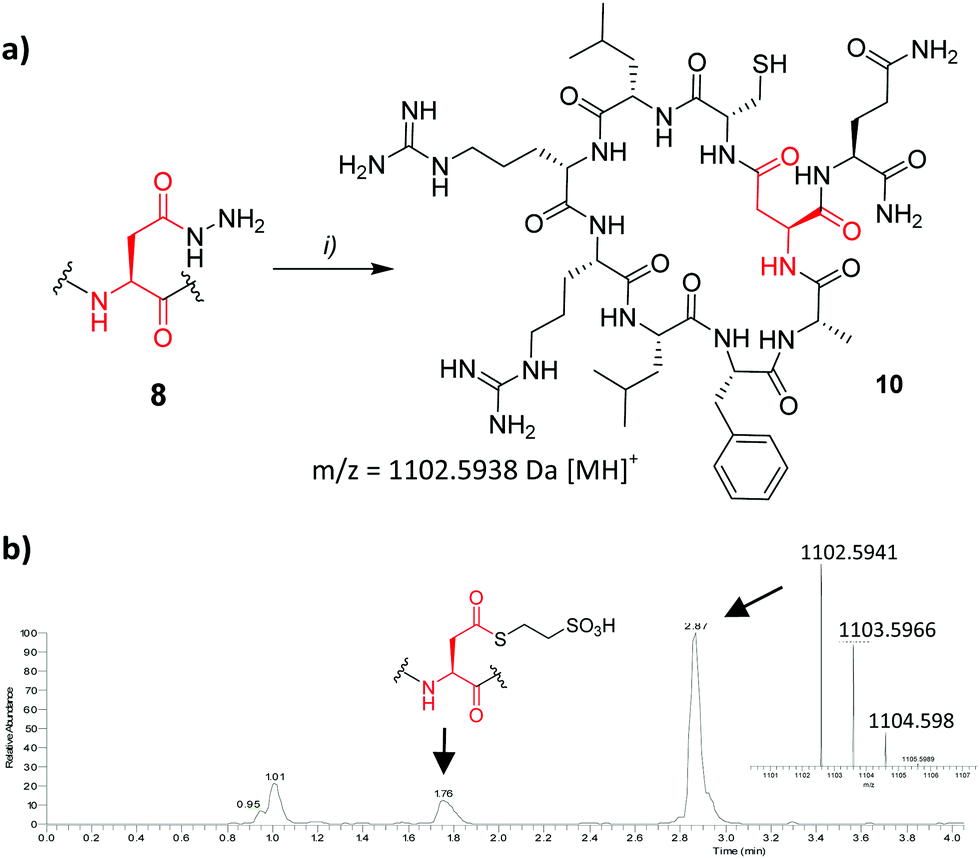 | ||
| Fig. 3 (a) Cyclisation of Lassomycin derivative 8. (i) NaNO2 (1.2 equiv. Na phosphate buffer; pH3, 0 °C, 20 min), followed by addition of MESNa (pH7, rt, 48 h). (b) LC-MS trace of the reaction mixture after 24 h, showing 10 as the major species. | ||
Sidechain hydrazide activation and NCL is not limited to intramolecular reactions. In initial attempts to extend this process to include convergent N-glycopeptide synthesis, we explored the reaction between sugar-linked auxiliary 1114,15 and peptides 12–13 (Fig. 4). Auxiliary-mediated ligation removes the requirement for cysteine to be incorporated at the ligation site.16 Peptides 12 and 13, corresponding to a glycosylated fragment of β-interferon, were prepared as their Glu and Asp sidechain hydrazides respectively, in a similar manner to 7 and 8. Alternatively 13 was prepared from the Asp sidechain benzyl ester (see ESI†). Whilst conversion from the benzyl ester to the hydrazide proceeded well (96% yield), in our hands much of the benzyl ester precursor itself (isolated in 19% yield) was lost due to competing aspartimide formation that occurred during peptide synthesis, cleavage and isolation. Consequently, syntheses employing the hydrazides 4 or 6, appeared comparable in terms of efficiency. 11 was employed in ligation reactions as a chemically robust alternative to the naturally occurring glycosylamine derived from N-acetyl glucosamine.16 The additional methylene spacer at the anomeric centre additionally serves to render eventual glycopeptide products resistant to enzymatic protein deglycosylation mediated by N-glycanases.17 Peptides 12 or 13 were treated with acac in sodium phosphate buffer at pH 3–4 for 2 h, followed by addition of 11, 4-mercaptophenyl acetic acid (MPAA) and tris-carboxyethylphosphine (TCEP) at pH 7, and monitored by LCMS. Ligation products were observed after 1 h and continued to accumulate over time, emerging as the major products after 48 h (Fig. 4b). Glycoconjugates 14 and 15 were isolated by preparative HPLC in 58 and 36% yields respectively.
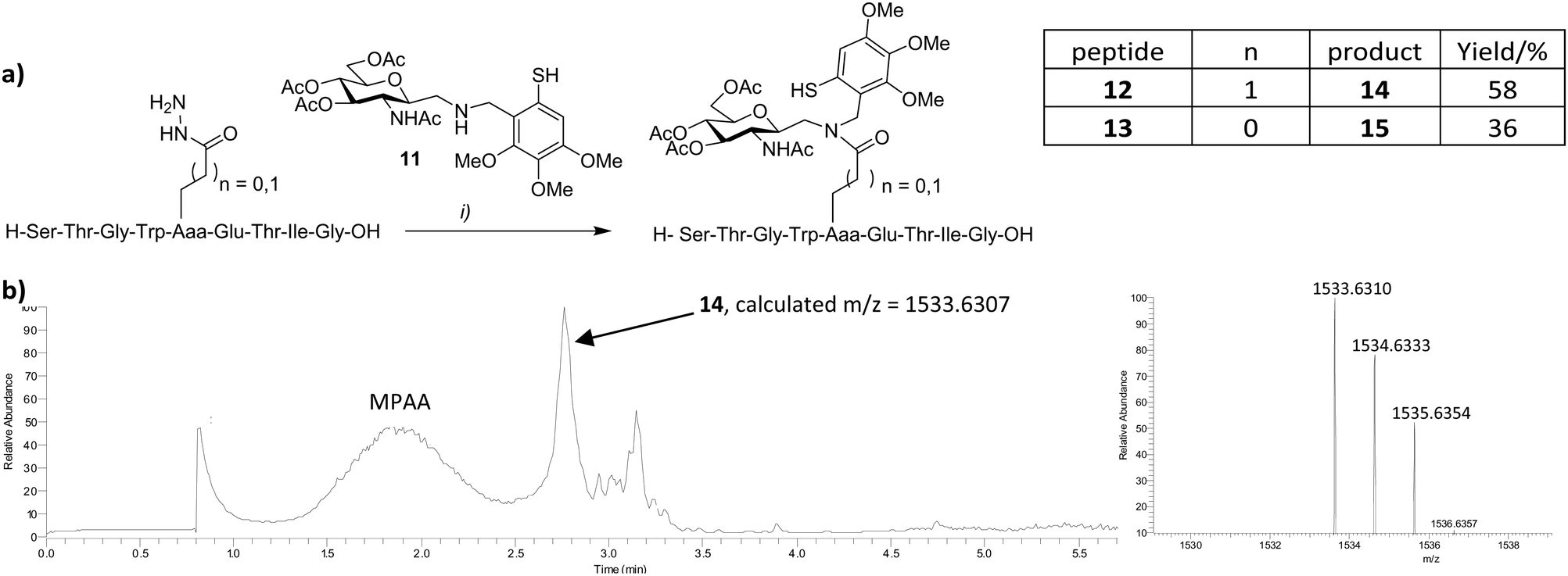 | ||
| Fig. 4 (a) Convergent N-neoglycopeptide synthesis: (i) acac (2 equiv.), 0.1 M MPAA, Na phosphate buffer pH 3, 2 h, then, 11 and TCEP (50 mM), pH 7, 48 h. (b) LC-MS analysis of the reaction mixture after 48 h showing predominance of glycoconjugate 14. | ||
In summary, we have shown that Asp and Glu hydrazide derivatives, compatible with Fmoc-based SPPS, can be simply and rapidly prepared from their amino acid anhydrides. As stable thioester precursors, they facilitate cyclisation at the amino acid sidechain upon activation with acac or NaNO2. Both activation methods are known for their broad amino acid tolerance and are applied widely in NCL processes.3f,12 Furthermore, we showed that glycopeptide analogues could be prepared using an auxiliary mediated NCL-type approach. Glycoaminoacid building blocks for use in Fmoc-based SPPS have previously been prepared using regioselective opening of Asp anhydrides.9a However these building blocks still require introduction into synthetic peptide thioester segments, to enable protein backbone assembly via NCL. With recombinant protein sidechain thioesters already available through an expanded genetic code,18 the convergent union of carbohydrates with fully recombinant proteins is edging closer.19 It is likely that genetically encoded hydrazide building blocks will also become available in the future, functioning as versatile intermediates on the path to modified proteins. We envisage that fully recombinant proteins modified with 11, or auxiliary-linked N-acetyl glucosamine, may provide a viable convergent route to glycoprotein therapeutics. Once installed, the monosaccharide can be extended enzymatically using endo-glucosidases,20 dramatically reducing the requirement for protein synthesis by multi-component NCL.
The authors acknowledge financial support from The Leverhulme Trust (RPG-2018-133) and UCL.
Conflicts of interest
There are no conflicts to declare.References
- (a) V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119(12), 7328–7443 CrossRef CAS; (b) A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC; (c) L. R. Malins and R. J. Payne, Curr. Opin. Chem. Biol., 2014, 22, 70–78 CrossRef CAS; (d) S. B. H. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- J. P. Tam and C. T. T. Wong, J. Biol. Chem., 2012, 287, 27020–27025 CrossRef CAS.
- (a) N. H. Shah and T. W. Muir, Chem. Sci., 2014, 5, 446–461 RSC; (b) A. L. Adams, B. Cowper, R. E. Morgan, B. Premdjee, S. Caddick and D. Macmillan, Angew. Chem., Int. Ed., 2013, 52, 13062–13066 CrossRef CAS; (c) J.-S. Zheng, S. Tang, Y. Guo, H.-N. Chang and L. Liu, ChemBioChem, 2012, 13, 542–546 CrossRef CAS; (d) Y.-M. Li, M.-Y. Yang, Y.-C. Huang, Y.-T. Li, P. R. Chen and L. Liu, ACS Chem. Biol., 2012, 7, 1015–1022 CrossRef CAS; (e) G.-M. Fang, Y.-M. Li, F. Shen, Y.-C. Huang, J.-B. Li, Y. Lin, H.-K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS; (f) Y.-C. Huang, G.-M. Fang and L. Liu, Nat. Sci. Rev., 2015, 3, 107–116 CrossRef.
- (a) H. Martin-Gómez and J. Tulla-Puche, Org. Biomol. Chem., 2018, 16, 5065–5080 RSC; (b) J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909–1919 CrossRef CAS.
- Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- (a) L. Raibaut, H. Drobecq and O. Melnyk, Org. Lett., 2015, 17, 3636–3639 CrossRef CAS; (b) D. J. Castillo-Pazos and D. Macmillan, Synlett, 2017, 1923–1928 CAS; (c) E. Boll, J. Dheur, H. Drobecq and O. Melnyk, Org. Lett., 2012, 14, 2222–2225 CrossRef CAS.
- (a) J. Lu, X.-B. Tian and W. Huang, Chin. Chem. Lett., 2015, 26, 946–950 CrossRef CAS; (b) X. Tian, P. Yu, Y. Tang, Z. Le and W. Huang, Synlett, 2017, 1966–1970 CAS.
- (a) K. Hofmann, A. Lindenmann, M. Z. Magee and N. H. Khan, J. Am. Chem. Soc., 1952, 74, 470–476 CrossRef CAS; (b) D. Gazis, J. Glass, I. L. Schwartz, G. Stavropoulos and D. Theodoropoulos, Int. J. Pept. Protein Res., 1989, 34, 353–357 CrossRef CAS; (c) C. M. Haney and W. S. Horne, J. Pept. Sci., 2014, 20, 108–114 CrossRef CAS.
- (a) F. M. Ibatullin and S. I. Selivanov, Tetrahedron Lett., 2009, 50, 6351–6354 CrossRef CAS; (b) X. Huang, X. Luo, Y. Roupioz and J. W. Keillor, J. Org. Chem., 1997, 62, 8821–8825 CrossRef CAS.
- M. J. Bayro, J. Mukhopadhyay, G. V. T. Swapna, J. Y. Huang, L.-C. Ma, E. Sineva, P. E. Dawson, G. T. Montelione and R. H. Ebright, J. Am. Chem. Soc., 2003, 125, 12382–12383 CrossRef CAS.
- S. Lear, T. Munshi, A. S. Hudson, C. Hatton, J. Clardy, J. A. Mosely, T. J. Bull, C. S. Sit and S. L. Cobb, Org. Biomol. Chem., 2016, 14, 4534–4541 RSC.
- D. T. Flood, J. C. J. Hintzen, M. J. Bird, P. A. Cistrone, J. S. Chen and P. E. Dawson, Angew. Chem., Int. Ed., 2018, 57, 11634–11639 CrossRef CAS.
- (a) J. Meienhofer, M. Waki, E. P. Heimre, T. J. Lambros, R. C. Makofske and C.-D. Chang, Int. J. Pept. Protein Res., 1979, 13, 35–42 CrossRef CAS; (b) M. Goodman, C. Toniolo, L. Moroder and A. L. Felix, Houben-Weyl Methods in Organic Chemistry, Volume E22, – Synthesis of Peptides and Peptidomimetics, Thieme Medical Publishers Inc, 2004, pp. 495–516 Search PubMed.
- D. Carrière, S. J. Meunier, F. D. Tropper, S. Cao and R. Roy, J. Mol. Catal. A: Chem., 2000, 154, 9–22 CrossRef.
- F. Burlina, A.-B. M. Abdel-Aal, R. Raz, I. Pinzuti, G. Papageorgiou, J. Li, R. Antrobus, S. R. Martin, S. Kunzelmann, B. Stieglitz and J. Offer, Commun. Chem., 2019, 2, 111 CrossRef.
- H. Chai, K. Le Mai Hoang, M. D. Vu, K. Pasunooti, C.-F. Liu and X.-W. Liu, Angew. Chem., Int. Ed., 2016, 55, 10363–10367 CrossRef CAS.
- L.-X. Wang, M. Tang, T. Suzuki, K. Kitajima, Y. Inoue, S. Inoue, J.-Q. Fan and Y. C. Lee, J. Am. Chem. Soc., 1997, 119, 11137–11146 CrossRef CAS.
- W. Xuan, D. Collins, M. Koh, S. Shao, A. Yao, H. Xiao, P. Garner and P. G. Schultz, ACS Chem. Biol., 2018, 13, 578–581 CrossRef CAS.
- N. Holloran, D. Collins, U. Rathnayake, B. Zhang, M. Koh, C. Kang and P. Garner, Bioconjugate Chem., 2020, 31, 2362–2366 CrossRef CAS.
- A. J. Fairbanks and Curr Opin., Chem. Biol., 2019, 53, 9–15 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of all synthetic procedures, and NMR spectra, HPLC traces and LC-MS analysis for selected compounds is available. See DOI: 10.1039/d0cc07404g |
| This journal is © The Royal Society of Chemistry 2021 |