
A new spin crossover FeII coordination environment in a two-fold interpenetrated 3-D Hofmann-type framework material†
Manan
Ahmed
 a,
Zixi
Xie
b,
Shannon
Thoonen
c,
Carol
Hua
c,
Cameron J.
Kepert
b,
Jason R.
Price
ad and
Suzanne M.
Neville
*a
a,
Zixi
Xie
b,
Shannon
Thoonen
c,
Carol
Hua
c,
Cameron J.
Kepert
b,
Jason R.
Price
ad and
Suzanne M.
Neville
*a
aSchool of Chemistry, The University of New South Wales, Sydney, 2052, Australia. E-mail: s.neville@unsw.edu.au
bThe School of Chemistry, The University of Sydney, Sydney, 2006, Australia
cSchool of Chemistry, The University of Melbourne, Parkville, Victoria 3010, Australia
dAustralian Synchrotron, ANSTO Clayton, Victoria 3800, Australia
First published on 27th November 2020
Abstract
A 3-D FeII Hofmann-type framework material has been prepared which contains a three-connecting pyridyl-donor ligand with amide functionality and [Au(CN)2]− metallo-ligands. The FeII sites display a rare FeII(py)3(N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C⋯)3 coordination environment, which we show for the first time to be conducive to spin crossover (SCO).
C⋯)3 coordination environment, which we show for the first time to be conducive to spin crossover (SCO).
Molecular materials which are responsive to external stimuli have gained considerable attention for utility in sophisticated technologies such as switches, sensors and memory devices.1,2 In this regard, materials which undergo the spin crossover (SCO) effect are an important class of molecular switch, whereby high spin (HS) and low spin (LS) electronic configurations can be accessed in response to external stimuli, such as temperature, light irradiation, pressure and molecular analytes.3–12 Importantly, this switching is reversible, detectable, and occurs in a controllable fashion, thereby providing ready accessibility to these bistable states. This, and the intrinsic link between structure and electronic state, continues to motivate the development of new SCO materials, and drives an enhanced understanding of structure and function through a combination of theory and experimental studies.13–18
The incorporation of electronically-active FeII SCO building blocks into coordination polymers has been shown to be an effective route for developing SCO materials with a range of impressive switching properties,19–25 such as hysteretic spin-state transitions, room temperature switching, multistep transitions and molecular guest sensitivity. This is particularly the case for Hofmann-type frameworks,21–25 where effective propagation of SCO throughout the lattice architecture is facilitated by the elasticity of metallocyanide and organic units which link the SCO sites. These systems are, in the majority of cases, constructed by the equatorial coordination of [MII(CN)4] (MII = Pt, Pd, Ni) or [MI(CN)2] (MI = Au, Ag, Cu, Hg) metallo-ligands and the axial coordination of aromatic N-donor ligands. The axial ligands may bridge the layers to form a 3-D framework or support the separation of layers in a 2-D framework. Here, we report a 3-D Hofmann-type framework material which contains a unique FeII coordination environment comprised of three pyridyl-donor groups from a tris-monodentate ligand with amide functionality (Fig. 1a),26,27 and three cyanido-donor groups from [Au(CN)2]− anions. This FeII(py)3(NC⋯)3 coordination environment (Fig. 1b) is rare28–30 and has not previously been observed to yield SCO behaviour.
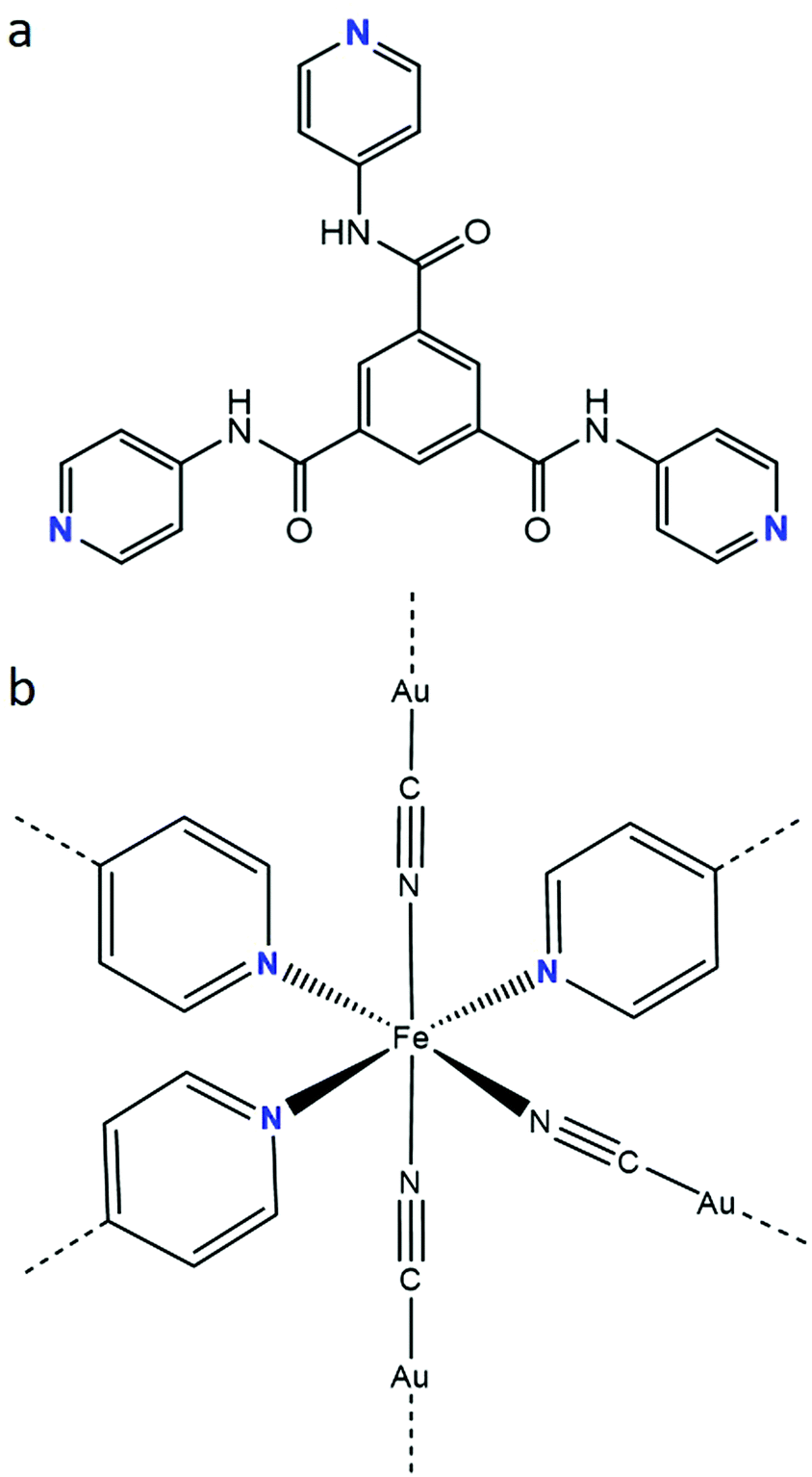 | ||
| Fig. 1 (a) N1,N3,N5-tri(pyridin-4-yl)benzene-1,3,5-tricarboxamide (Tbenpy) and (b) FeII(py)3(NC⋯)3 (mer) coordination environment. | ||
Yellow block-shaped crystals of [FeII(Tbenpy){Au(CN)2}2]·{2H2O,DMF} were formed by slow diffusion of Fe(ClO4)2·6H2O, Tbenpy (N1,N3,N5-tri(pyridin-4-yl)benzene-1,3,5-tricarboxamide)26,27 and K[Au(CN)2] in a mixture of ethanol and DMF. The identity and phase purity of the bulk sample was confirmed by CHN analysis, IR spectroscopy, thermogravimetric analysis (Fig. S1, ESI†) and powder X-ray diffraction (Fig. S2–S4, ESI†).
Single crystal structural analyses were performed at 10 K intervals over the range 100–250 K (Table S1, ESI†). At all temperatures, the asymmetric unit contains one FeII site, one Tbenpy ligand, two [Au(CN)2]− anions, two water molecules and one DMF molecule (Fig. S8–S10, ESI†). Each FeII centre shows a distorted [FeIIN6] octahedral connectivity comprised of three pyridine groups from Tbenpy ligands and three NC⋯ groups from [Au(CN)2]− anions in a mer-arrangement (Fig. 1b).
The 3-D Hofmann-type framework is a pillared-layered architecture, whereby 2-D layers are bridged by [Au(CN)2]− anions (Fig. 2). This is in contrast to the broad family of Hofmann-like frameworks which typically show a trans-[FeII(py)2(NC)4] coordination environment,21–25 in which the Hofmann-layers are bridged by the organic ligand. Here, in contrast, the 2-D layers contain the pyridyl-ligand, and the layer bridges are the metal-cyanide-containing component. More specifically, the 2-D layers here are formed by dimers of FeII sites bridged by two Tbenpy ligands, as depicted in Fig. 2a. Each dimer is then connected into a 2-D layer motif via the remaining pyridyl-group, such that all of the Tbenpy ligands are three-connecting and each FeII site shows a T-shaped connectivity (i.e., mer; Fig. 2b). The remaining equatorial FeII site is occupied by a terminal [Au(CN)2]− group (Fig. 2a) which is engaged in a hydrogen bonding interaction with a water molecule (Fig. 2a and Table S2, ESI† H2O⋯NC). The axial FeII sites are thus occupied by bridging [Au(CN)2]− ligands, thereby connecting the 2-D layers into a pillared layered 3-D network (Fig. 2c). The 3-D pillared-layered framework is two-fold interpenetrated (Fig. 2d), with Au⋯Au and C(H)⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C interactions linking the two-interpenetrated nets (Fig. S12 and Table S2, ESI†). In other cases where three-connecting organic ligands have been employed,31–33 novel framework topologies have emerged, such as a NbO-net; however, the ‘classical’ [FeII(py)2(NC)4] coordination environment is always retained.
C interactions linking the two-interpenetrated nets (Fig. S12 and Table S2, ESI†). In other cases where three-connecting organic ligands have been employed,31–33 novel framework topologies have emerged, such as a NbO-net; however, the ‘classical’ [FeII(py)2(NC)4] coordination environment is always retained.
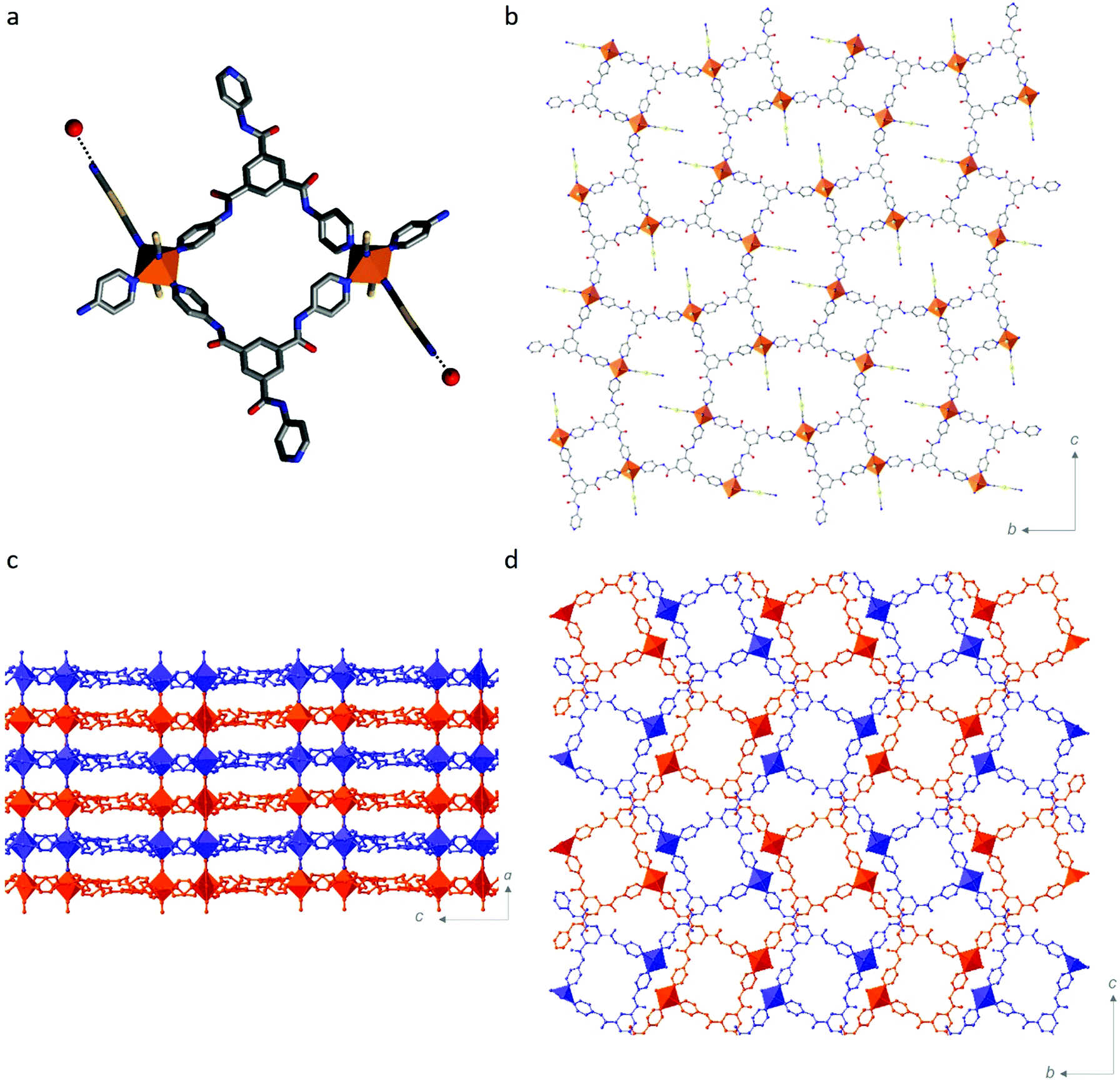 | ||
| Fig. 2 Structural representation (250 K) of [FeII(Tbenpy){Au(CN)2}2]·{2H2O,DMF} showing (a) one FeII dimer connected by two-Tbenpy ligands and illustrating the terminal [Au(CN)2]− group with hydrogen bond to water; (b) one 2-D layer (bc-plane) comprised of FeII with T-shaped equatorial coordination (mer) of Tbenpy ligands and one terminal [Au(CN)2]− ligand; View of the interpenetrated 3-D networks (red and blue) along (c) the b-axis and (d) the a-axis. | ||
Part of the motivation for utilising an organic ligand with amide functionality was to exploit its well-known propensity for hydrogen bonding26,27,34–39 to enhance lattice communication. There are three amide groups per Tbenpy ligand, with each of the N(H) groups participating in host–guest hydrogen bonding interactions (Fig. S13 and Table S2: N(H)⋯H2O & N(H)⋯DMF, ESI†). None of the OC groups form host–guest hydrogen-bonding interactions, only the one host–host interaction, as described above. This drive to form hydrogen-bonding interactions, alongside the considerable steric bulk of the Tbenpy ligand, may play a part in the formation of this unique FeII coordination environment and Hofmann framework topology.
Variable temperature magnetic susceptibility measurements were performed on a bulk crystalline sample (300–50–300 K; Fig. 3a), revealing a gradual, two-step SCO transition. Between 300 and ∼230 K, the χMT values (∼3.49 cm3 Kmol−1) are indicative of FeII in the HS state. Below ∼230 K, there is a relatively abrupt, then more gradual decrease in χMT values, with a minimum of ∼0.33 cm3 Kmol−1 achieved by 70 K indicating a near complete HS to LS transition of the FeII sites. There is negligible thermal hysteresis in the cooling and heating profiles. The derivative of the spin transition curve (Fig. 3a: inset) reveals two-steps with characteristic transition temperatures (T½) of 191 and 120 K and with the subtle undulation occurring at an approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 HS:LS ratio. This SCO behaviour is reproducible over consecutive thermal cycles and at a range of thermal scan rates (4, 2, 1 K min−1; Fig. S18, ESI†).
:1 HS:LS ratio. This SCO behaviour is reproducible over consecutive thermal cycles and at a range of thermal scan rates (4, 2, 1 K min−1; Fig. S18, ESI†).
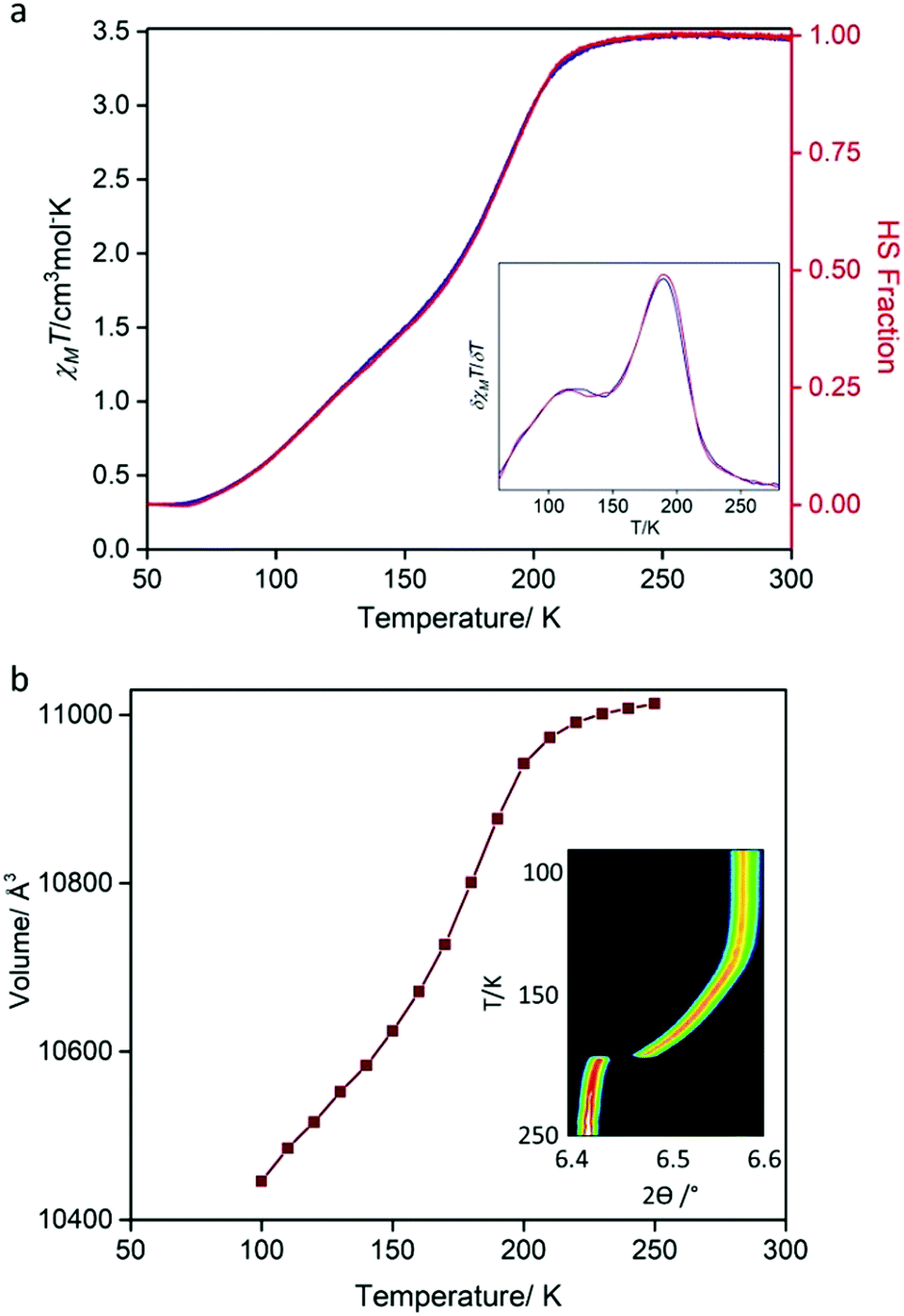 | ||
| Fig. 3 (a) Temperature-dependent magnetic susceptibility (χMT) for [FeII(Tbenpy){Au(CN)2}2]·{2H2O,DMF}. Data were recorded at a scan rate of 0.5 K min−1 over the range 300–50–300 K (continuous mode; blue = cooling, red = heating). Other scan rates (4, 1, 2 K min−1) are provided in the ESI.† Inset: Plot of δχMT/δT highlighting the two spin-state transition steps. (b) Variable temperature (250–90 K) synchrotron powder diffraction unit cell evolution derived from Le bail analysis. Inset: Contour peak evolution of the (400) reflection (λ = 0.589063 Å). | ||
Variable temperature powder (250–90 K) and single crystal (100–250 K) X-ray diffraction data, each using synchrotron radiation, were collected to monitor the structural evolution of the SCO transition. The variation in peak position (Fig. 3b: inset, ESI†) from powder diffraction matches the gradual two-step character observed by magnetic susceptibility, with a sharp initial transition followed by a more gradual transition at lower temperatures. Likewise, plots of the unit cell parameters versus temperature, derived from both powder (Fig. 3b and Fig. S5, ESI†) and single crystal diffraction (Fig. S14, ESI†), match that of the magnetic susceptibility. Most importantly, both techniques show a continuous transition with retention of C-centred orthorhombic symmetry over the entire two-step SCO transition. Therefore, over the entire HS to LS transition, there is one structurally unique FeII site. Inspection of the average Fe–N bond distance variation over the 250–100 K temperature range shows contraction of ∼0.2 Å consistent with a complete HS to LS transition (Table S2, ESI†). The retention of a single FeII site over the entire two-step SCO transition indicates that there is no 3-D long-range order of HS and LS sites at the intermediate step plateau region. Careful inspection of the single crystal precession images did not reveal any diffuse features indicative of short-range ordering or lower dimensional ordering at the intermediate temperature region (Fig. S15–S17, ESI†). Furthermore, variable temperature Raman spectra taken over the first step (i.e., the HS to HS:LS transition; 250–150 K; Fig. S19 and S20, ESI†) do not show any features that could be attributed to a locally ordered HS–LS state.
There are only three previous reports of the more general [FeII(py)3(NC⋯)3] coordination environment; two display a fac-[FeII(py)3(NCCH3)3] environment28 with LS FeII sites and the other shows a mer-[FeII(py)3(NC)3] environment29,30 with HS FeII sites. The SCO which emerges in [FeII(Tbenpy){Au(CN)2}2]·{2H2O,DMF} likely arises from the use of coordinating [Au(CN)2]− ions, which would provide an intermediate ligand field strength between the previously utilised NCCH3 and CN− units. Furthermore, the use of a mono-dentate pyridyl ligand, rather than rigid tridentate ligands (fac or mer), would likely provide more flexibility around the FeII core to facility the HS to LS transition. In comparison to other Hofmann-type frameworks containing [Au(CN)2]−, this overall [FeII(py)3(NC–Au⋯)3] coordination environment (cf. [FeII(py)2(NC–Au⋯)4]), would present a slightly weaker ligand field and therefore may account for the observed gradual spin-state transition rather than the more typical abrupt transitions characteristic of Hofmann-type materials.21–25
In conclusion, a two-fold interpenetrated 3-D FeII Hofmann framework has been constructed using a three-connecting organic ligand with amide functional groups (Tbenpy) and [Au(CN)2]− metallo-ligands. There are two important distinctions between this and other Hofmann-type frameworks that incorporate [Au(CN)2]− anions: firstly, one of the pyridyl donor groups is replaced with a [Au(CN)2]− group; and secondly, the cyanido- and pyridyl-group location is exchanged with respect to the Hofmann layers. Collectively, this has led to a new FeII Hofmann-type coordination environment, [FeII(py)3(NC⋯)3], and a new Hofmann-like framework topology, whereby the 2-D layers contain the organic ligands and layer bridging occurs via the metal–cyanide. We show that this new Hofmann-like coordination environment is conducive to SCO. Therefore, this study provides a new FeII coordination environment platform to explore in Hofmann-like frameworks and more broadly in molecular switching materials. Preliminary studies on this 3-D framework indicate robust porosity and an affinity for a wide range of liquid and gaseous molecules. This result opens up a variety of molecular sensing opportunities, in particular with respect to the presence of both amide and free cyanido-groups (from the terminal [Au(CN)2]− site) on the framework surface, both of which should show diverse analyte affinity.26,27,31–36
This work was supported by Fellowships and Discovery Project funding from the Australian Research Council. Access and use of the facilities of the Australian Synchrotron was supported by ANSTO, using the MX2 beamline and made use of the Australian Cancer Research Foundation (ACRF) detector.
Conflicts of interest
There are no conflicts to declare.References
- B. L. Feringa and W. R. Browne, Molecular Switches, Wiley-VCH Verlarg GmbH & Co., 2011 Search PubMed.
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS.
- J.-F. Létard, P. Guionneau and L. Goux-Capes, Top. Curr. Chem., 2004, 235, 221–249 CrossRef.
- A. Bousseksou, G. Molnar, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313–3335 RSC.
- M. A. Halcrow, Spin-Crossover Materials, John Wiley & Sons Ltd, Chichester, 2013 Search PubMed.
- P. Gütlich, Eur. J. Inorg. Chem., 2013, 581–591 CrossRef.
- G. Molnár, L. Salmon, W. Nicolazzi, F. Terki and A. Bousseksou, J. Mater. Chem. C, 2014, 2, 1360–1366 RSC.
- S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC.
- M. M. Khusniyarov, Chem. – Eur. J., 2016, 22, 15178–15191 CrossRef CAS.
- M. D. Manrique-Juárez, S. Rat, L. Salmon, G. Molnár, C. M. Quintero, L. Nicu, H. J. Shepherd and A. Bousseksou, Coord. Chem. Rev., 2016, 308, 395–408 CrossRef.
- K. Senthil Kumar and M. Ruben, Coord. Chem. Rev., 2017, 346, 176–205 CrossRef CAS.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- H. Spiering, Top. Curr. Chem., 2004, 235, 171–195 CrossRef CAS.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC.
- P. Gütlich, A. B. Gaspar and Y. Garcia, Beilstein J. Org. Chem., 2013, 9, 342–391 CrossRef.
- P. Guionneau, Dalton Trans., 2014, 43, 382 RSC.
- M. Paez-Espejo, M. Sy and K. Boukheddaden, J. Am. Chem. Soc., 2016, 138, 3202–3210 CrossRef CAS.
- R. W. Hogue, S. Singh and S. Brooker, Chem. Soc. Rev., 2018, 47, 7303 RSC.
- J. A. Real, E. Andres, M. C. Muñoz, M. Julve, T. Granier, A. Bousseksou and F. Varret, Science, 1995, 268, 265 CrossRef CAS.
- G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and J. D. Cashion, Science, 2002, 298, 1762–1765 CrossRef CAS.
- K. S. Murray and C. J. Kepert, Top. Curr. Chem., 2004, 233, 195–228 CrossRef CAS.
- M. C. Muñoz, A. B. Gaspar, A. Galet and J. A. Real, Inorg. Chem., 2007, 46, 8182 CrossRef.
- G. Agustí, M. C. Muñoz, A. B. Gaspar and J. A. Real, Inorg. Chem., 2008, 47, 2552 CrossRef.
- M. C. Muñoz and J. A. Real, Coord. Chem. Rev., 2011, 255, 2068 CrossRef.
- Z.-P. Ni, J.-L. Liu, N. Hoque, W. Liu, J.-Y. Li, Y.-C. Chen and M.-L. Tong, Coord. Chem. Rev., 2017, 335, 28–43 CrossRef CAS.
- S. Hong, Y. Zou, D. Moon and M. S. Lah, Chem. Commun., 2007, 1707–1709 RSC.
- S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607–2614 CrossRef CAS.
- H. Zheng, L. Zhao, T. Liu, P.-F. Zhuang, C.-Q. Jiao, J.-X. Hu, Y. Xu, C. He and C.-Y. Duan, Inorg. Chem. Commun., 2015, 57, 33–35 CrossRef CAS.
- R. Ramasubramanian, K. Anandababu, N. C. Mösch-Zanetti, F. Belaj and R. Mayilmurugan, Dalton Trans., 2019, 48, 14326–14336 RSC.
- K. Anandababu, R. Ramasubramanian, H. Wadepohl, P. Comba, N. Britto, M. Jaccob and R. Mayilmurugan, Chem. – Eur. J., 2019, 25, 9540–9547 CrossRef CAS.
- Z. Arcís-Castillo, M. C. Muñoz, G. Molnár, A. Bousseksou and J. A. Real, Chem. – Eur. J., 2013, 19, 6851–6861 CrossRef.
- F. J. Valverde-Muñoz, M. C. Muñoz, S. Ferrer, C. Bartual-Murgui and J. A. Real, Inorg. Chem., 2018, 57, 12195–12205 CrossRef.
- L. Piñeiro-López, Z. Arcís-Castillo, M. C. Muñoz and J. A. Real, Cryst. Growth Des., 2014, 14, 6311–6319 CrossRef.
- W. Lan, F. J. Valverde-Muñoz, Y. Dou, X. Hao, M. C. Muñoz, Z. Zhou, H. Liu, Q. Liu, J. A. Real and D. Zhang, Dalton Trans., 2019, 48, 17014–17021 RSC.
- Y. Gong, Y. Zhou, J. Li, R. Cao, J. Qin and J. Li, Dalton Trans., 2010, 39, 9923–9928 RSC.
- Y. Gong, J. Li, J. Qin, T. Wu, R. Cao and J. Li, Cryst. Growth Des., 2011, 11, 1662–1674 CrossRef CAS.
- K. Kim, S. Park, K.-M. Park and S. S. Lee, Cryst. Growth Des., 2011, 11, 4059–4067 CrossRef CAS.
- F. J. Valverde-Muñoz, C. Bartual-Murgui, L. Piñeiro-López, M. C. Muñoz and J. A. Real, Inorg. Chem., 2019, 58(15), 10038–10046 CrossRef.
- D. J. Mondal, S. Roy, J. Yadav, M. Zeller and S. Konar, Inorg. Chem., 2020, 59(18), 13024–13028 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2042801–2042816. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc07326a |
| This journal is © The Royal Society of Chemistry 2021 |