
Insight into the organocatalytic arylation of azonaphthalenes with α-chloroaldehydes: the general mechanism and origin of selectivities†
Xinghua
Wang
 a,
Yang
Wang
b,
Donghui
Wei
*a and
Yu
Lan
*a
a,
Yang
Wang
b,
Donghui
Wei
*a and
Yu
Lan
*a
aGreen Catalysis Center, and College of Chemistry, Zhengzhou University, Zhengzhou, Henan 450001, China. E-mail: donghuiwei@zzu.edu.cn; lanyu@cqu.edu.cn
bDepartment of Material and Chemical Engineering, Zhengzhou University of Light Industry, 136 Science Avenue, Zhengzhou, Henan Province 450002, P. R. China
First published on 26th November 2020
Abstract
A systemical computational study was performed to explore the mechanism and origin of selectivities on the organocatalytic arylation of azonaphthalenes with α-chloroaldehydes. The calculated results reveal that the nucleophilicity of active sites and the hydrogen bonds respectively regulate chemo- and stereoselectivities and, for the first time, N-heterocyclic carbene (NHC) can act as a multiple functional base (MFB) catalyst in one system.
Transition-metal-catalyzed inert aryl C–H activation/functionalization has been widely investigated as a powerful method for carbon–carbon (C–C) and carbon–heteroatom (C–X) bond formation. In transition-metal-catalyzed aryl C–H activation and transformation, aromatic rings generally act as formal electrophiles to react with different nucleophiles.1,2 However, nucleophilic aromatic substitution involving aryl C–H cleavage, especially organocatalytic arylation, has been rarely reported. Nicewicz and co-workers were the first to report aryl C–H amination and cyanation by direct C–H functionalization using an acridinium photoredox organocatalyst.3,4 Subsequently, owing to the advantages of high stereoselectivity and low toxicity, organocatalytic arylations have attracted increasing attention and become one of the greatest challenges in the organocatalysis field.5–8
Organocatalytic arylation of azonaphthalenes has been reported in a few experimental studies since 2017 (Scheme 1).9–12 In a pioneering study, Tan performed chiral phosphoric acid catalyzed enantioselective [3+2] annulation of azonaphthalenes with 2-substituted indoles to construct benzindole derivatives (Scheme 1A).9 Independently, Shi et al. reported chiral phosphoric acid catalyzed [3+2] annulation of azonaphthalenes with azlactones (Scheme 1B).10 In addition, Li et al. found that a secondary amine can also catalyze the [3+2] annulation of azonaphthalenes with enolizable carbonyl compounds (Scheme 1C).11 Note that Wang et al. reported unexpected [4+2] rather than [3+2] annulation of azonaphthalenes with α-chloroaldehydes catalyzed by N-heterocyclic carbene (NHC) (Scheme 1D).12 Although these excellent experimental reports greatly contributed to the determination of the possible mechanisms of these types of organocatalytic annulation reactions of azonaphthalenes, to the best of our knowledge, no systematic theoretical study of the possible mechanisms has been reported, and the origin of chemoselectivity and stereoselectivity remains unclear.
 | ||
| Scheme 1 Organocatalyst-mediated arylation reactions of azonaphthalenes. | ||
The above issue and our continuous interest in organocatalysis prompted us to perform a theoretical study,13–20 in which the NHC-catalyzed [4+2] annulation reaction of azonaphthalene with α-chloroaldehyde was selected as the computational model (Scheme 2). In the present study, we aim to answer the following questions: (1) what is the detailed mechanism of this reaction? Four possible [n + 2] (n = 3, 4) annulation pathways were considered for the chemoselective annulation step: two ring-closure (RC) first and two proton transfer (PT) first mechanisms. (2) What is the origin of the high stereoselectivity? The R- and S-configured pathways in the stereoselective C–C bond formation process were calculated. (3) What is the role of the NHC catalyst in the whole reaction? For the first time, we propose that NHC can act as a multiple functional base (MFB, i.e. Lewis base, Brønsted base (BB) and hydrogen bond donor/acceptor) to promote the rearomatization process, which is remarkably different from the traditional role of the NHC catalyst as a Lewis base (LB) in one system. Due to the reliability of density functional theory (DFT) on the mechanistic studies of organocatalytic21–26 and transition metal-catalyzed reactions,27–30 we performed DFT calculations at the M06-2X31,32/6-311++G(2df,2pd)/IEF-PCMTHF33,34//M06-2X/6-31G(d,p)/IEF-PCMTHF level to investigate the detailed mechanism, the role of NHC, and the origin of selectivities. More computational details and a benchmark study are provided in the ESI.†
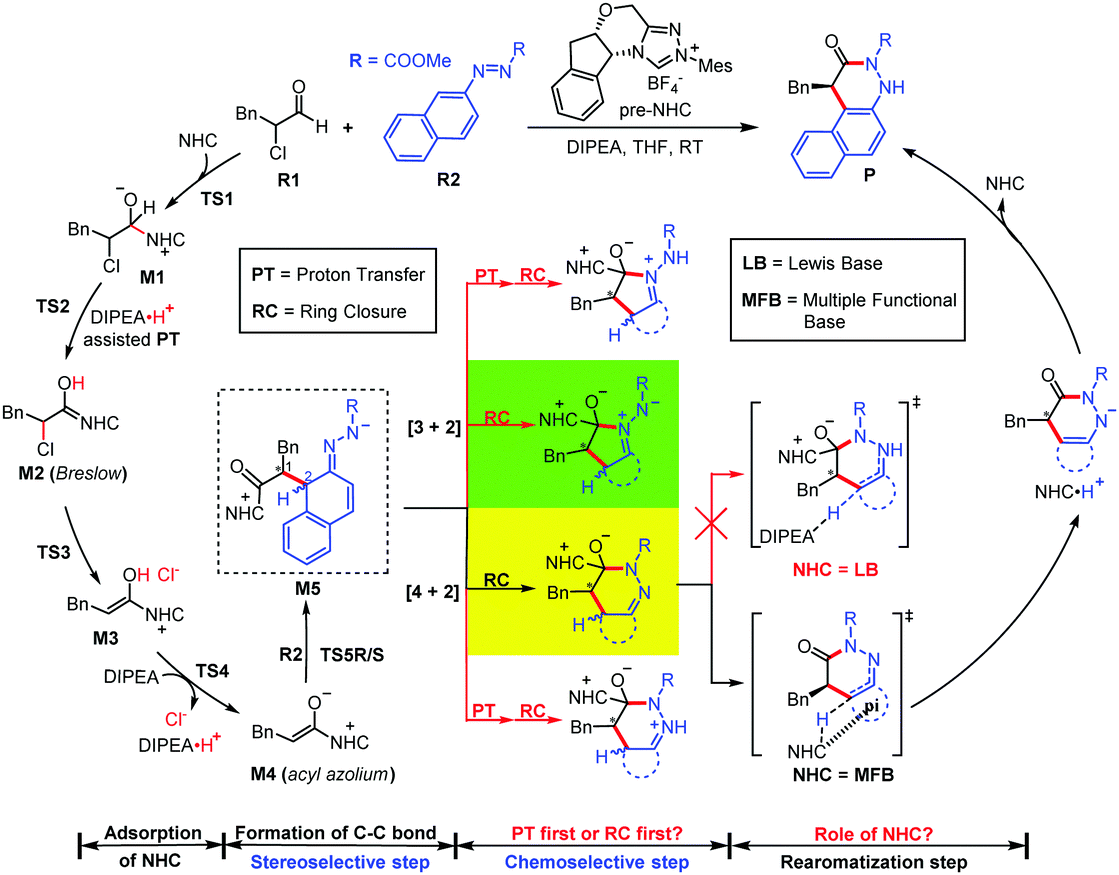 | ||
| Scheme 2 Selected NHC-catalyzed reaction model with the unresolved questions. | ||
As shown in Fig. 1, the whole reaction is initiated by nucleophilic addition to α-chloroaldehyde R1 by the NHC obtained by the deprotonation of pre-NHC to generate zwitterionic intermediate M1via the transition state TS1. With the aid of DIPEA·H+, intermediate M1 transforms to Breslow intermediate M2 through [1, 2]-proton transfer via transition state TS2. Owing to the attack direction, NHC can be divided into Re- and Si-faces, and both cases are discussed in more detail in the ESI.† The calculated results show that the energy barrier through transition state Re-TS1 (ΔG‡ = 13.4 kcal mol−1) is slightly higher than that through transition state Si-TS1 (ΔG‡ = 12.0 kcal mol−1), but the energy barrier of Re-TS2 (ΔG‡ = 7.6 kcal mol−1) is much lower than that of Si-TS2 (ΔG‡ = 24.5 kcal mol−1), so the pathway associated with Re-face attack on R1 by NHC is more energetically favorable. Subsequently, the C–Cl bond of Breslow intermediate M2 is cleaved to afford enolate intermediate M3via transition state TS3 (ΔG‡ = 4.9 kcal mol−1). The enolate intermediate M3 can then be easily deprotonated by DIPEA to form acyl azolium intermediate M4via transition state TS4 (ΔG‡ = 4.1 kcal mol−1).
 | ||
| Fig. 1 Relative Gibbs free energy profiles for the adsorption of NHC and the formation of the C–C bond. | ||
For the stereoselectivity-determining step, the nucleophilic addition of M4 to azonaphthalene R2 first undergoes C–C bond formation via four diastereoselective transition states TS5RR/RS/SR/SS to generate intermediates M5RR/RS/SR/SS, respectively. As shown in Scheme 2, the two letters after the names represent the molecular chirality associated with the chiral C1 and C2 atoms, respectively. Inspired by the previous studies, conformational searches of TS5RR/RS/SR/SS were performed to ensure the optimized diastereoselective transition states associated with the lowest energy configurations, and more discussions can be seen in Scheme S3 of the ESI.† The chiral C2 atom will become the achiral one after the following rearomatization process, so we only selected and discussed TS5RR/SS with the lowest energies to represent TS5R and TS5S (Fig. 1 and 2). The pathway associated with the R-configurational product is the energetically favorable pathway, and the energy barrier difference (ΔΔG‡) between ΔG‡TS5R and ΔG‡TS5S is 3.9 kcal mol−1, which corresponds to an enantiomeric excess (ee) value of greater than 99% according to the equations provided in the ESI,† which is close to the experimental observation (99% ee).12 Atom-in-molecule (AIM) quantitative analysis35 (Fig. 2 and Table S1, ESI†) indicates that a larger number of C–H⋯O and C–H⋯π interactions exist in TS5R than those in TS5S, and these interactions are stronger in the favorable TS5R, so it can be concluded that the C–H⋯O and C–H⋯π interactions are important in controlling the stereoselectivity. It should be mentioned that the energy barrier viaTS5R (20.4 kcal mol−1) can be overcome at room temperature, and the following transformations continue to happen to result in the final product P depicted in Fig. 3.
 | ||
| Fig. 2 Quantitative analysis and comparison of the non-covalent interactions involved in TS5R and TS5S (distances in Å and Laplacian of electron densities in a.u.). | ||
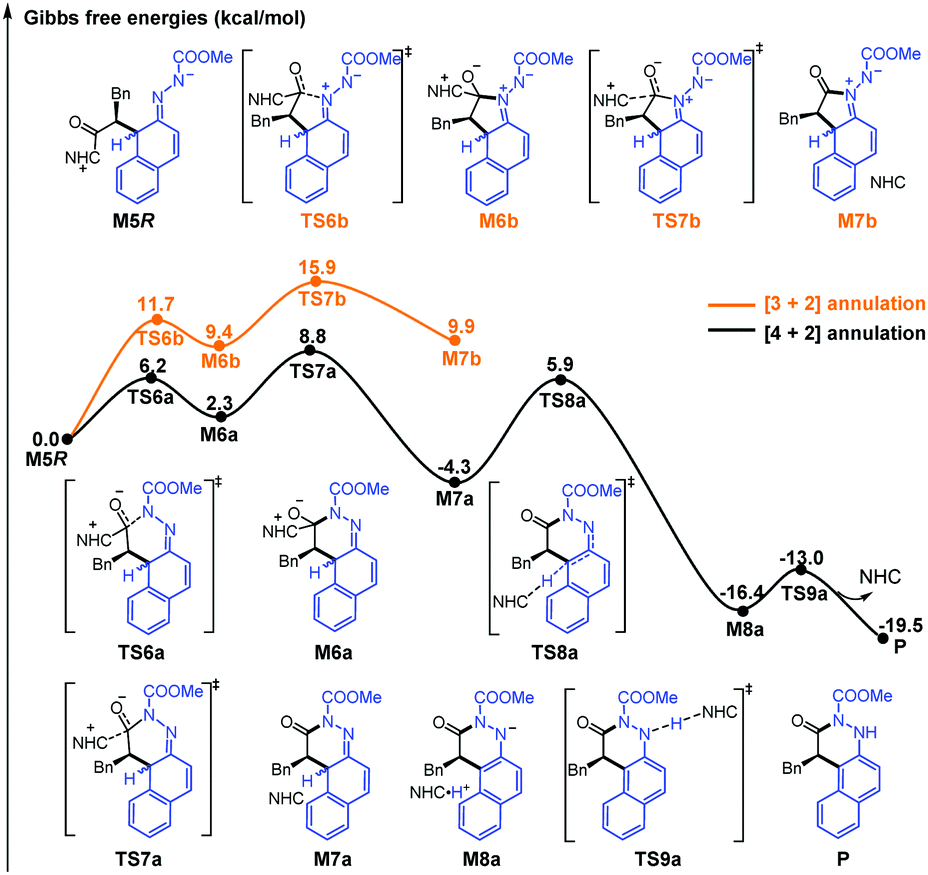 | ||
| Fig. 3 Relative Gibbs free energy profiles of the most energetically favorable pathways for the possible [3+2] and [4+2] cyclization reactions. | ||
Two RC-first and two PT-first mechanisms were then considered for the chemoselective annulation step, and totally more than 16 possible pathways had been considered in this study (see Fig. S5–S8, ESI†). The highest energy barriers of the best pathways of the RC-first and PT-first mechanisms corresponding to the [4+2] cyclization are 10.2 and 26.7 kcal mol−1, whereas those of the [3+2] cyclization are 15.9 and 21.7 kcal mol−1, respectively. Hence, it should be more favorable for the RC-first mechanism regardless of the formation of the [3+2] or [4+2] cyclization product under NHC catalysis. The most favorable pathways associated with the RC-first mechanism for the [3+2] and [4+2] cyclization reactions are shown and compared in Fig. 3. It can be concluded that the local nucleophilicity of the N atom close to the ester group is stronger than that of the other N atom of the azo group (see Table S3 of ESI†), and thus the [4+2] cyclization occurs more easily. This is also consistent with the experimental observation,12 and we only considered the [4+2] cyclization pathway in the following process.
Subsequently, we investigated two types of mechanisms for the rearomatization of intermediate M6a, in which the NHC acts as an LB and an MFB (Scheme 2). As shown in Fig. S5 (ESI†), we found that the energy barrier of the pathway in which NHC acts as an LB is extremely high (37.6 kcal mol−1) in the rearomatization process. Therefore, we can safely exclude this pathway. As shown in Fig. 3, ring-closure occurs viaTS6a with an energy barrier of 6.2 kcal mol−1 to form M6a, which dissociates the NHC to form intermediate M7aviaTS7a with an energy barrier of 6.5 kcal mol−1. The in situ generated NHC can act as an MFB to realize C(sp3)–H deprotonation viaTS8a with an energy barrier of 10.2 kcal mol−1, which is followed by final proton transfer for the formation of the final product and regeneration of the NHC catalyst viaTS9a with an energy barrier of 3.4 kcal mol−1. Other possible DIPEA-mediated deprotonation pathways were also considered and excluded (see the ESI†). Considering the computational error reported by Singleton and Plata,36 we have additionally calculated the proton transfer processes by using different DFT methods and explicit solvents to ensure the reliability of the conclusion, and more details and discussions can be found in part 9 of the ESI.†
Although the basicity of NHC should not be weaker than that of the base additive such as DIPEA,37,38 NHC is not proposed as the possible BB catalyst in most of the previous experimental and theoretical studies. Thus, it should be very important to further explore why the transition state has relatively lower energy when the NHC rather than DIPEA abstracts the proton. To uncover this puzzle, the additional NCI analyses on the deprotonation transition state associated with NHC or DIPEA as BB were further performed. As depicted in Fig. S10 of the ESI,† the results indicate that the NHC mainly provides LP⋯π and C–H⋯π interactions to stabilize the deprotonation transition state, and thus we think that NHC should be denoted as a multiple functional base rather than a simple BB in this process. Based on the above results, the NHC should act as an MFB catalyst in the rearomatization process, which is remarkably different from the well-known LB role of NHCs in previous reports.
In summary, the possible reaction mechanisms and selectivities of NHC-catalyzed dearomatization/rearomatization of azonaphthalene with α-chloroaldehyde have been systematically investigated by DFT methods. The C–C bond formation step is determined to be the stereoselectivity-determining step, and the pathway associated with the R-configurational product is predominant in terms of both the kinetics and thermodynamics. According to the energy profiles of different pathways, the RC-first [4+2] annulation pathway is more favorable than the [3+2] annulation pathway. All of the calculated results are in agreement with the experimental results.12 Furthermore, AIM analysis shows that C–H⋯O and C–H⋯π interactions are the key factors for controlling the stereoselectivity. Note that the NHC was found to be an MFB in this special case for the first time. The obtained mechanistic insight will guide the understanding of the new role of the NHC catalyst, and thus prompt the development of new NHC catalytic strategies in the future.
Financial support from the National Natural Science Foundation of China (No. 21903072, 21773214, 21822303, and 21772020), the Startup Fund of Zhengzhou University of Light Industry (No. 2017BSJJ036), the Backbone Teacher Project (2020GGJS016), the 111 Project (D20003), and the National Supercomputing Center in Zhengzhou.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS.
- J. B. McManus and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 2880–2883 CrossRef CAS.
- Y. Liu, Y. L. S. Tse, F. Y. Kwong and Y. Y. Yeung, ACS Catal., 2017, 7, 4435–4440 CrossRef CAS.
- L. Qiao, Z. W. Duan, X. N. Wu, D. H. Li, Q. Q. Gu and Y. K. Liu, Org. Lett., 2018, 20, 1630–1633 CrossRef CAS.
- C. Ma, F. Jiang, F. T. Sheng, Y. Jiao, G. J. Mei and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 3014–3020 CrossRef CAS.
- L. W. Qi, S. Y. Li, S. H. Xiang, J. Wang and B. Tan, Nat. Catal., 2019, 2, 314–323 CrossRef CAS.
- L. W. Qi, J. H. Mao, J. Zhang and B. Tan, Nat. Chem., 2018, 10, 58–64 CrossRef CAS.
- C. Ma, J. Y. Zhou, Y. Z. Zhang, G. J. Mei and F. Shi, Angew. Chem., Int. Ed., 2018, 57, 5398–5402 CrossRef CAS.
- H. Zhao, H. Yuan, Y. Zhang, R. Li and W. Li, Org. Lett., 2019, 21, 6557–6561 CrossRef CAS.
- Z. Wu and J. Wang, Chem. Sci., 2019, 10, 2501–2506 RSC.
- Y. Wang, C. Du, Y. Y. Wang, X. K. Guo, L. Fang, M. P. Song, J. L. Niu and D. H. Wei, Adv. Synth. Catal., 2018, 360, 2668–2677 CrossRef CAS.
- G. J. Wang, Q. Q. Shi, W. Y. Hu, T. Chen, Y. Y. Guo, Z. L. Hu, M. H. Gong, J. C. Guo, D. H. Wei, Z. Q. Fu and W. Huang, Nat. Commun., 2020, 11, 946 CrossRef CAS.
- X. H. Wang, Y. Wang, J. S. Song and D. H. Wei, Org. Chem. Front., 2020, 7, 1113–1121 RSC.
- Q. Q. Deng, S. J. Li, D. H. Wei and Y. Lan, Org. Chem. Front., 2020, 7, 1828–1836 RSC.
- S. Li, Z. Tang, Y. Wang, D. Wang, Z. Wang, C. Yu, T. Li, D. Wei and C. Yao, Org. Lett., 2019, 21, 1306–1310 CrossRef CAS.
- N. Liu, Y. F. Xie, C. Wang, S. J. Li, D. H. Wei, M. Li and B. Dai, ACS Catal., 2018, 8, 9945–9957 CrossRef CAS.
- Y. Y. Wang, D. H. Wei, Y. Wang, W. J. Zhang and M. S. Tang, ACS Catal., 2016, 6, 279–289 CrossRef CAS.
- X. Li, J. Xu, S. J. Li, L. B. Qu, Z. J. Li, Y. G. R. Chi, D. H. Wei and Y. Lan, Chem. Sci., 2020, 11, 7214–7225 RSC.
- R. B. Sunoj, Acc. Chem. Res., 2016, 49, 1019–1028 CrossRef CAS.
- D. Jansen, J. Gramüller, F. Niemeyer, T. Schaller, M. C. Letzel, S. Grimme, H. Zhu, R. M. Gschwind and J. Niemeyer, Chem. Sci., 2020, 11, 4381–4390 RSC.
- T. Niemi, I. Fernández, B. Steadman, J. K. Mannisto and T. Repo, Chem. Commun., 2018, 54, 3166–3169 RSC.
- C. Trujillo, I. Rozas, A. Botte and S. J. Connon, Chem. Commun., 2017, 53, 8874–8877 RSC.
- S.-Y. Zhu, Y. Zhang, X.-F. Chen, J. Huang, S.-H. Shi and X.-P. Hui, Chem. Commun., 2019, 55, 4363–4366 RSC.
- R. C. Samanta, S. De Sarkar, R. Fröhlich, S. Grimme and A. Studer, Chem. Sci., 2013, 4, 2177–2184 RSC.
- S. J. Li, X. Li, H. Mo, L. B. Qu, D. Wei and Y. Lan, Chem. Commun., 2020, 56, 4732–4735 RSC.
- Y. Li, H. Chen, L.-B. Qu, K. N. Houk and Y. Lan, ACS Catal., 2019, 9, 7154–7165 CrossRef CAS.
- S. Liu, T. Zhang, L. Zhu, F. Liu, R. Bai and Y. Lan, Org. Lett., 2020, 22, 2124–2128 CrossRef CAS.
- L.-C. Yang, Y.-N. Wang, R. Liu, Y. Luo, X. Q. Ng, B. Yang, Z.-Q. Rong, Y. Lan, Z. Shao and Y. Zhao, Nat. Chem., 2020, 12, 860–868 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- R. E. Plata and D. A. Singleton, J. Am. Chem. Soc., 2015, 137, 3811–3826 CrossRef CAS.
- N. Wang, J. Xu and J. K. Lee, Org. Biomol. Chem., 2018, 16, 8230–8244 RSC.
- Z. Li, X. Li and J.-P. Cheng, J. Org. Chem., 2017, 82, 9675–9681 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, energy profile of other possible pathways; Cartesian coordinates, energies of all the stationary points; and imaginary frequencies of transition states. See DOI: 10.1039/d0cc07260e |
| This journal is © The Royal Society of Chemistry 2021 |