
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpontaneous macrocyclization through multiple dynamic cyclic aminal formation†
Daniel
Carbajo‡
a,
Antonio Jesús
Ruiz-Sánchez‡
 bc,
Francisco
Nájera
bc,
Ezequiel
Pérez-Inestrosa
*bc and
Ignacio
Alfonso
*a
bc,
Francisco
Nájera
bc,
Ezequiel
Pérez-Inestrosa
*bc and
Ignacio
Alfonso
*a
aDepartment of Biological Chemistry, Institute of Advanced Chemistry of Catalonia, IQAC-CSIC c/Jordi Girona 18-26, Barcelona, 08034, Spain. E-mail: ignacio.alfonso@iqac.csic.es
bUniversidad de Málaga-IBIMA, Departamento de Química Orgánica, Campus de Teatinos s/n, Málaga-29071, Spain. E-mail: inestrosa@uma.es
cAndalusian Centre for Nanomedicine and Biotechnology-BIONAND, Parque Tecnológico de Andalucía, c/Severo Ochoa, 35, Málaga-29590, Spain
First published on 2nd January 2021
Abstract
The use of aminals in dynamic covalent chemistry is slightly underexplored, probably due to their inherent instability. Here we report the spontaneous [2+2] macrocyclization of tetrakis(aminals). Their unexpected stability and structural modularity, the dynamic nature of the connections and their water tolerance make them appealing systems for future applications as stimulus-responsive materials.
Dynamic covalent chemistry (DCvC) has experienced an important development in the last two decades, based on the use of reversible covalent bonds to generate exchanging mixtures under equilibrium.1,2 Examples of reversible reactions used in DCvC are boronates,3 acetals,4 hydrazones,5 disulfides,6 imines7 and aminals.8 In this context, aminals are characterized by the connectivity of two amino groups to the same carbon atom,9 being the nitrogenated equivalents of acetals, hence they are also called N,N-acetals. However, stable aminals from primary amines are rare, since they exist in solution in equilibrium with other species, such as the corresponding imines and cyclic oligomers, which complicates their isolation.10–18 More stable cyclic aminals have been obtained from the condensation of N,N-disubstituted 1,n-diamines. They are usually generated under dehydrating conditions to shift the equilibrium to the product.9,19 Moreover in some cases, the product precipitation in water allows the preparation of cyclic aminals in high yield and high purity without a catalyst.20
Tuning the dynamic equilibrium between aminals and their open forms has allowed their application as protecting groups of aldehydes.21 Interestingly, since aminal formation is reversible, they generate in solution dynamic mixtures from diamines and carbonyl compounds. This reversible reaction has been applied to the synthesis of open-chain unsymmetrical aminals,14 the controlled evaporation of volatile carbonyl compounds,8,22 induce movement through a molecular track23 and the preparation of a dynamic chemical network that self-directs its amplification.24
Inspired by a previous research25 we envisioned to study the reaction between amino and aldehyde building blocks allowing a large structural and functional diversity, including the possible formation of dynamic aminals. To that, we chose bis(1,3)-diamines derived from 3,3′-diaminopivalic acid26 (Scheme 1a, 1a–c) as the amino partner. On the other hand, we used terephthaldehyde (2a) and isophthaldehyde (2b) as simple scaffolds to react with any of the four amino groups of the other building blocks. Considering our previous experience, we also selected different aromatic diacids (Scheme 1b, 4a–4d) as potential templates to favour the reaction, as the distance between carboxylic groups was suitable to interact with amide NHs while the aromatic rings could establish a π-stacking with 2a,b. Reactions of equimolecular mixtures of 1 + 2 in methanol initially lead to complex mixtures, as indicated by NMR. Nevertheless, they evolve to the formation of a main unique compound (i.e.3a from 1a and 2a, Scheme 1a). As can be inferred from the NMR spectra (Fig. 1), the obtained compound shows a highly symmetric structure with sharp signals and the presence of diastereotopic methylenes. The process seemed very general since similar results were obtained (though at different rates) with the combination of any of the amines 1a–c and any dialdehyde 2a,b (Scheme 1a). The NMR and MALDI-TOF MS data allowed us to identify the products as the corresponding [2+2] macrocyclic tetraaminals. Even when we added more equivalents of aromatic dialdehydes, we still observed the (faster) formation of the same [2+2] macrocycle, leaving unreacted dialdeyhde in the mixture.
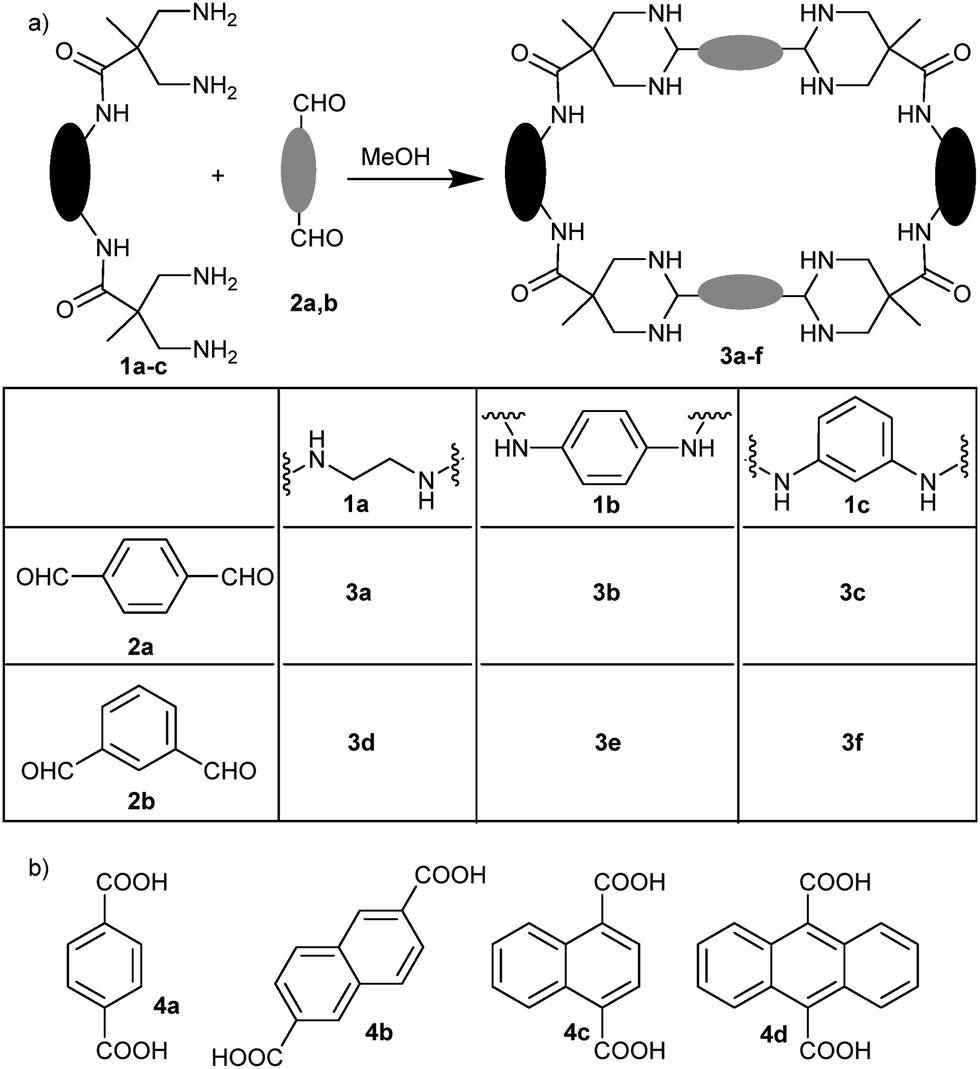 | ||
| Scheme 1 (a) General macrocyclization reaction through cyclic aminal formation; (b) dicarboxylic acids assayed as potential templates. | ||
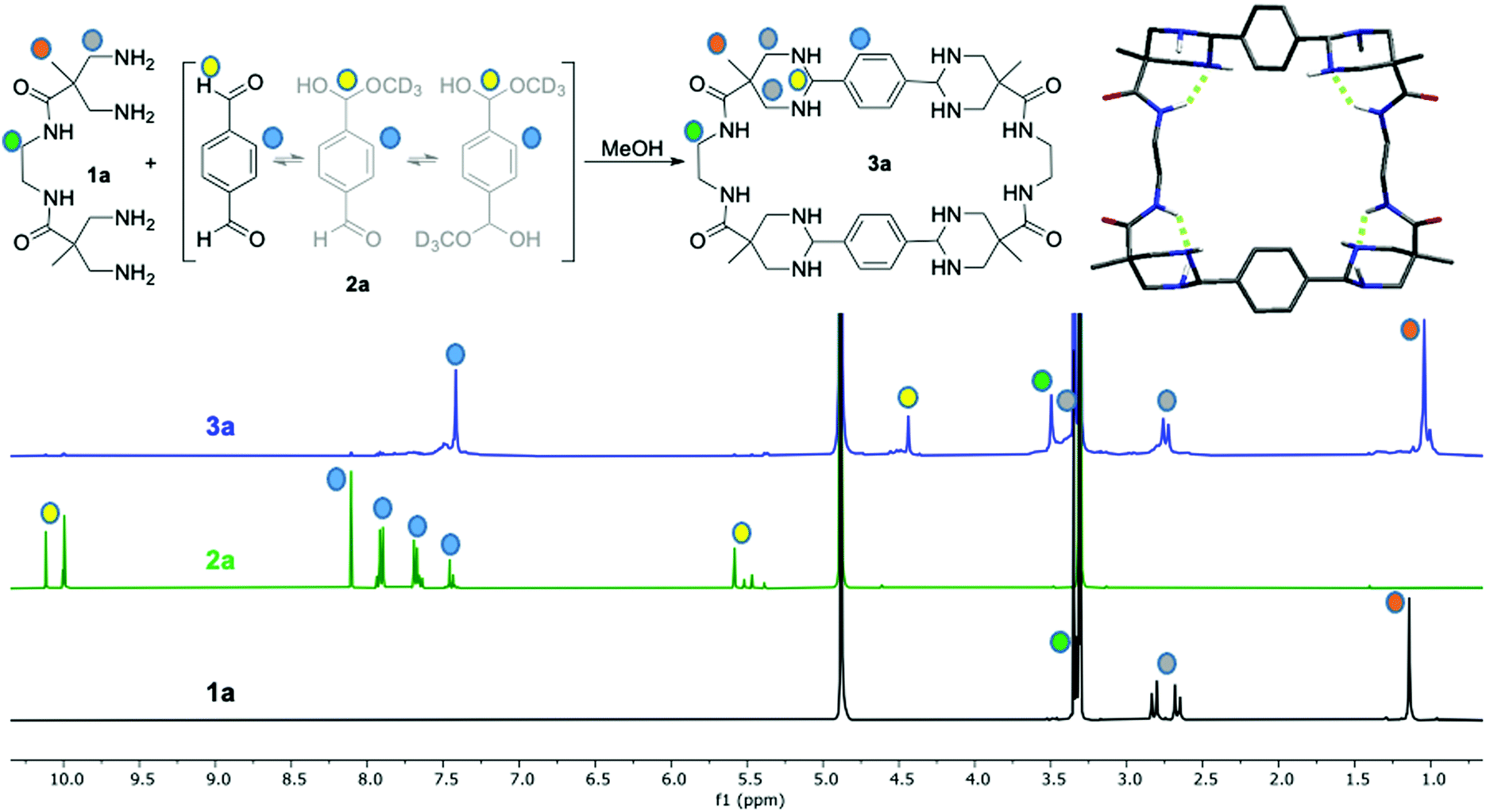 | ||
| Fig. 1 NMR spectra (400 MHz, CD3OD) of 1a, 2a and 3a showing the proton signal assignments. Note 2a in CD3OD solution is in equilibrium with its hemiacetal forms. The inset shows the D2 molecular model for 3a (hydrogens atoms bonded to carbons have been omitted and the hydrogen bonds detected are in green). | ||
This observation indicates that the [2+2] macrocycle presents high structural stability, as it overcomes both the lability of aminal bond and the trend to polymerize that may come from additional aldehyde equivalents. Indeed, even having up to eight stereogenic carbons, a single isomer was mainly formed. Accordingly, the macrocyclization requires a precise combination of the generated stereocentres, so the most suitable stereoisomer is selected in all the cases.
The 1H-NMR time monitoring of the reaction between 1a and 2a (Fig. S13, ESI†) revealed a dynamic correction process. The aldehyde proton signal decreases while aromatic proton signals became complex due to the initial desymmetrization of this moiety. As far as the reaction proceeded, the aromatic proton signals simplified until a main symmetrical compound is clearly detected. Likewise, a signal around δ 5.5 ppm appears at the beginning of the reaction due to the hemiacetal formation upon reaction with methanol solvent. This hemiacetal signal at δ 5.5 ppm disappears as long as a signal at δ = 4.41 ppm grows due to the formation of the cyclic aminal. On the other hand, the proton signals of the corresponding tetraamine building block, 3,3′-diaminopivalic moieties, initially split in complex patterns, indicating a mixture of species. Over time they collapse and converge to a unique set of signals with different chemical shift from the original. As an indication of the increased rigidity generated upon cyclization, the diastereotopic methylene protons signals split to δ 2.72 and 3.31 ppm, respectively. Bidimensional NMR experiments as 1H–13C HSQC, HMBC and NOESY, as well as mass spectrometry confirmed the tetraaminal macrocycle formation (Fig. 1 and Fig. S14–S17, ESI†).
Although all three compounds 1a–c led to the corresponding macrocycles 3a–f with the studied aromatic dialdehydes 2a,b, the reaction rate varied depending on the scaffold core. For example, the time required for reaction completion with dialdehyde 2a depends on the structure of the tetramine, being: 1a (1.5 days) < 1b (3 days) < 1c (one week). We concluded that the more flexible ethylene core of 1a can better fold to form the final macrocycle. Meanwhile, the rigid aromatic core of 1b allows the cyclization more slowly than 1a.
However, the slowest reaction with the rigid derivative 1c suggested that the meta disposition is less preorganized for the macrocyclization in this case. Actually, the [3+3] macrocycle was detected as a by-product with 3c (Fig. S27, ESI†). This trend is also reflected regarding the dialdehyde structure, 2b leading to slower reaction than 2a.
In order to obtain a three dimensional structure of the corresponding macrocycles, molecular mechanics (AMBER) conformational searches guided by the experimentally observed NOEs were carried out, followed by DFT minimizations. Thus, we identified several energetically accessible local minima. Considering the high flexibility of large macrocycles, the symmetry observed in the NMR spectra can be reasonably explained by the fast exchange between these conformations, as additionally supported by the theoretical prediction of the 1H and 13C NMR chemical shifts (see ESI†). For simplicity only the D2 symmetric minimum of 3a is shown in Fig. 1, the other conformations can be found in the ESI.† This structure shows a specific stereoisomer in the six-membered ring aminal, which is stabilized by a hydrogen bond between the amide NH and one aminal nitrogen (see Atoms in molecules analysis in the ESI,† Tables S1–S8).27 For 3b we obtained a symmetric minimum that also agrees with the experimental data (Fig. 2, up), while all the macrocycles containing meta-substituted aromatic rings rendered folded figure-of-eight geometries (Fig. 2, down), likely explaining their more difficult cyclization.
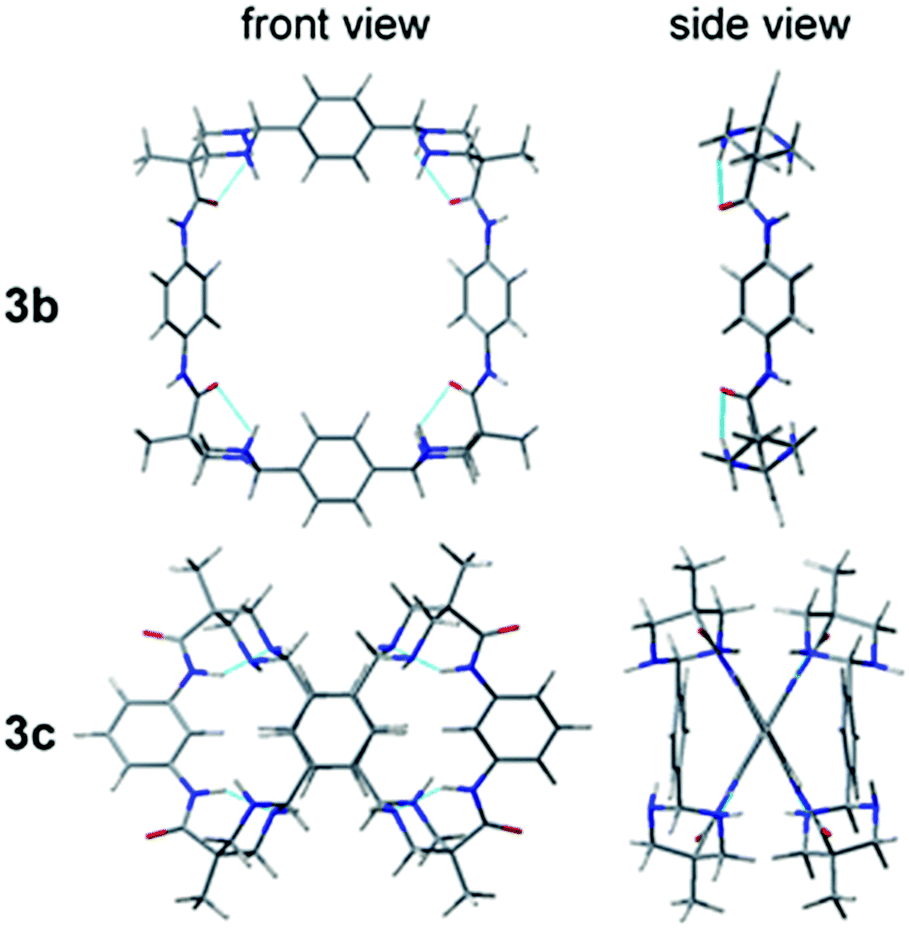 | ||
| Fig. 2 Molecular models for 3b with para-substituted aromatic rings (C2v symmetry) and 3cmeta-substituted aromatic rings (folded as a figure-of-eight conformation). | ||
In contrast with similar cyclizations,25 there is no need of any template to obtain the [2+2] macrocycles. Nevertheless, we hypothesized the addition of aromatic dicarboxylic acids to catalyse the reaction. Interestingly, the presence of naphthalene-derived diacids reduced the time to reach equilibrium in 3a (Fig. S57, ESI†): 4 hours with 4c and 24 hours with 4b. The enhancement effect of 4a was much weaker and no changes were observed with 4d.
All the reactions were carried out in MeOH containing residual water, so the aminal formation that leads to the macrocycle tolerates its presence, an unusual feature for this type of compounds. We then decided to systematically increase the amount of water for 3a and, quite remarkably, the [2+2] macrocycle efficiently assembles even in 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 D2O:CD3OD. Interestingly, when the pH was decreased below 4, the macrocycles split up instantly and only starting material was recovered in a matter of seconds.
:5 D2O:CD3OD. Interestingly, when the pH was decreased below 4, the macrocycles split up instantly and only starting material was recovered in a matter of seconds.
When we mixed preformed cycles 3a and 3b, no changes were detected by NMR within 3 days (Fig. S60 and S63, ESI†). As soon as we added some free 1a/1b (0.3 equiv. each), apparition of signals corresponding to mixed macrocycles was detected. This indicates that there is no exchange between species once the macrocycle is closed, but this is possible as soon as there is free amine in the medium. These cyclic tetraaminals retained their dynamic behaviour8 since the addition of an equimolar amount of another building block to any pre-formed macrocycle led to the formation of the corresponding mixed macrocycles in a proportion close to the statistical one (Fig. S62 and S64, ESI†). The same proportion was obtained by mixing the starting building blocks, suggesting that the macrocyclization is thermodynamically controlled.
It is worth to highlight that, to the best of our knowledge, only secondary amines tend to form stable aminals rather than mixtures of different species. For example, reaction between 1,3-diaminopropane and 2a generates a mixture from different imine macrocycles to hexahydropyrimidin derivates,28 providing complex 1H-NMR spectra. In this sense, to unravel the role of the macrocyclization in the aminal formation, we performed the same reaction with a monomeric version of the tetraamines (diamines 5a or 5b in Scheme 2). We mixed any of the building blocks with dialdehyde 2a in the same conditions as previously used, monitoring the course of the reaction by NMR. The spectroscopic data clearly showed the efficient formation of the corresponding symmetric bisaminal derivatives (6a or 6b, respectively) as a single stereoisomer. The NOE cross-peaks revealed that the obtained isomer corresponds to the one observed in the macrocyclic compounds. Thus, the aminals are also stable in the open-chain derivatives, suggesting that the six-membered ring structure of the aminal could be playing an important role in the reaction, facilitating the formation of the [2+2] macrocycle.
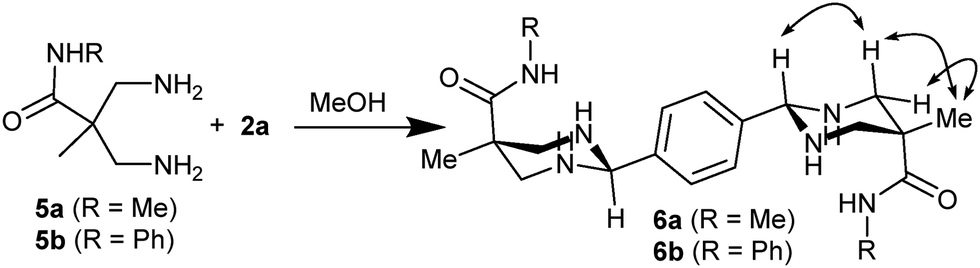 | ||
| Scheme 2 Reaction with diamines 5a,b. Observed NOEs in 6a,b are shown as double-headed arrows. | ||
In conclusion, we have obtained tetraaminal macrocycles through the reaction of primary bis(1,3-diamine) entities with aromatic dialdehydes. Despite the large amount of possibilities within the corresponding dynamic equilibria, a main [2+2] macrocycle is obtained in all the cases as the thermodynamic product. The aminal bond retains its dynamic nature that can be tuned by addition of competing amines (through exchange) and the presence of acid (leading to hydrolysis). The observation of a kinetic template effect, the dynamic nature of the covalent bonds and the aqueous compatibility of these systems open the door to their use in stimuli-responsive materials with appealing properties.29
The authors thank financial support from the Spanish Ministry of Science and Innovation (MCI/AEI/FEDER, RTI2018-096182-B-I00, CTQ2016-75870-P, PID2019-104293GB-I00), EuroNanoMed 2019 (PCI2019-111825-2), AGAUR (2017 SGR 208), Institute of Health Carlos III (ISCIII) RETIC ARADYAL (RD16/0006/0012), Universidad de Málaga-Junta de Andalucía (UMA18-FEDERJA-007), and UMA Postdoctoral Grant (CAPTACIÓN TALENTO 2015) for A. J. R.-S. Computer resources were provided by the SCBI (Supercomputing and Bioinformatics Centre) of the UMA. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Chem. Soc. Rev., 2017, 46, 2543–2554 RSC.
- S. Lascano, K. Da Zhang, R. Wehlauch, K. Gademann, N. Sakai and S. Matile, Chem. Sci., 2016, 7, 4720–4724 RSC.
- R.-C. Brachvogel and M. von Delius, Eur. J. Org. Chem., 2016, 3662–3670 CrossRef CAS.
- S. J. Sonawane, R. S. Kalhapure and T. Govender, Eur. J. Pharm. Sci., 2017, 99, 45–65 CrossRef CAS.
- N. Phan, E. G. Percástegui and D. W. Johnson, ChemPlusChem, 2020, 85, 1270–1282 CrossRef CAS.
- I. Janica, V. Patroniak, P. Samorì and A. Ciesielski, Chem. – Asian J., 2018, 13, 465–481 CrossRef CAS.
- B. Buchsnée Levrand, G. Godin, A. Trachsel, J. Y. De Saint Laumer, J. M. Lehn and A. Herrmann, Eur. J. Org. Chem., 2011, 681–695 CrossRef.
- L. Duhamel, Supplement F: The Chemisrty of Amino, Nitroso and Nitro Compounds and their Derivatives: Part 2, John Wiley & Sons, Ltd, Chichester, UK, 2010, vol. 2, pp. 849–907 Search PubMed.
- R. W. Layer, Chem. Rev., 1963, 63, 489–510 CrossRef CAS.
- J. Hine and K. W. Narducy, J. Am. Chem. Soc., 1973, 95, 3362–3368 CrossRef CAS.
- G. Moad and S. J. Benkovic, J. Am. Chem. Soc., 1978, 100, 5495–5499 CrossRef CAS.
- A. J. Hoefnagel, H. V. Bekkum and J. A. Peters, J. Org. Chem., 1992, 57, 3916–3921 CrossRef CAS.
- A. R. Katritzky, K. Yannakopoulou and H. Lang, J. Chem. Soc., Perkin Trans. 2, 1994, 1867–1870 RSC.
- A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5470 CrossRef.
- J. M. Locke, R. Griffith, T. D. Bailey and R. L. Crumbie, Tetrahedron, 2009, 65, 10685–10692 CrossRef CAS.
- M. Chan-Huot, S. Sharif, P. M. Tolstoy, M. D. Toney and H. H. Limbach, Biochemistry, 2010, 49, 10818–10830 CrossRef CAS.
- J. K. Clegg, J. Harrowfield, Y. Kim, Y. H. Lee, J.-M. Lehn, W. T. Lim and P. Thuéry, Dalton Trans., 2012, 41, 4335–4357 RSC.
- M. A. Ramírez, G. Ortiz, G. Levín, W. McCormack, M. M. Blanco, I. A. Perillo and A. Salerno, Tetrahedron Lett., 2014, 55, 4774–4776 CrossRef.
- V. Jurčík and R. Wilhelm, Tetrahedron, 2004, 60, 3205–3210 CrossRef.
- T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., New York, USA, 1999, vol. 9 Search PubMed.
- G. Godin, B. Levrand, A. Trachsel, J.-M. Lehn and A. Herrmann, Chem. Commun., 2010, 46, 3125–3127 RSC.
- P. Kovaříček and J. M. Lehn, Chem. – Eur. J., 2015, 21, 9380–9384 CrossRef.
- C. Chen, J. Tan, M. C. Hsieh, T. Pan, J. T. Goodwin, A. K. Mehta, M. A. Grover and D. G. Lynn, Nat. Chem., 2017, 9, 799–804 CrossRef CAS.
- I. Alfonso, M. Bolte, M. Bru, M. I. Burguete, S. V. Luis and J. Rubio, J. Am. Chem. Soc., 2008, 130, 6137–6144 CrossRef CAS.
- A. J. Ruiz-Sanchez, P. Mesa-Antunez, N. Barbero, D. Collado, Y. Vida, F. Najera and E. Perez-Inestrosa, Polym. Chem., 2015, 6, 3031–3038 RSC.
- I. Mata, I. Alkorta, E. Molins and E. Espinosa, Chem. – Eur. J., 2010, 16, 2442–2452 CrossRef CAS.
- C. X. Ren, B. H. Ye, H. L. Zhu, J. X. Shi and X. M. Chen, Inorg. Chim. Acta, 2004, 357, 443–450 CrossRef CAS.
- A. Chao and D. Zhang, Macromolecules, 2019, 52, 495–503 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc07184f |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2021 |