
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIncreasing the activity of the Cu/CuAl2O4/Al2O3 catalyst for the RWGS through preserving the Cu2+ ions†
Ali M.
Bahmanpour
 a,
Benjamin P.
Le Monnier
a,
Yuan-Peng
Du
a,
Florent
Héroguel
a,
Jeremy S.
Luterbacher
a and
Oliver
Kröcher
*ab
a,
Benjamin P.
Le Monnier
a,
Yuan-Peng
Du
a,
Florent
Héroguel
a,
Jeremy S.
Luterbacher
a and
Oliver
Kröcher
*ab
aInstitute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne (EPFL), Lausanne 1015, Switzerland
bPaul Scherrer Institut (PSI), Villigen 5232, Switzerland. E-mail: oliver.kroecher@psi.ch
First published on 7th January 2021
Abstract
Cu–Al spinel oxide is a highly active catalyst for CO2 conversion to CO. However, it suffers from low surface area. By depositing a silica layer, we protected the catalyst surface and preserved the Cu2+ ions during the calcination process. These ions form well-dispersed Cu sites which participate in the reaction.
Reducing fossil-derived carbon dioxide emissions and their associated environmental effects is one of today's major challenges. Carbon recycling is more vital than ever in order to limit global warming and the resulting climate change.1,2 In addition, CO2 is a cheap source of carbon, the efficient usage of which can ensure a secure sustainable economy. Therefore, mitigation of CO2 and its conversion to value-added chemicals has been the objective of many research projects.
Methanol is an important bulk chemical that can be produced through direct conversion of CO2. However, due to the low activity and high thermal stability of CO2, only low yields are obtained. In contrast, synthesis of methanol and other chemicals from CO is well-established and well-studied.3,4 In many of the direct CO2 hydrogenation routes to chemicals, CO is actually formed as a reaction intermediate.5,6 The hydrogenation of CO2 to CO is known as the reverse water gas shift reaction (RWGSR) and has gained more attention in recent years. Noble metal-based catalysts have shown high activity towards this reaction. However, due to their high cost, efforts have been focused on finding an inexpensive substitute. Cu/ZnO/Al2O3 can actively catalyse the reaction. However, it suffers from severe deactivation.7,8 Recently, we have demonstrated that copper aluminate (CuAl2O4, spinel structure) is as an active catalyst for the RWGSR.7,9 This catalyst is commonly prepared through co-precipitation of the constituting metal precursors followed by a high-temperature calcination step (≥900 °C). The high temperature causes the reaction of Cu and Al2O3 to form CuAl2O4 but it also leads to particle sintering and a low surface area.10 Variation of the chemical composition of the support has been used as another method for preserving metal ions and particles on the catalyst surface.11,12 Another way to inhibit particle sintering is to deposit a protective layer on the catalyst surface.13,14 In most cases, the protective layer acts as a physical barrier, which immobilizes the nanoparticles.15,16 In some cases, it also alters the chemical properties of the support or even enhances the overall catalyst activity due to advantageous changes of the catalyst structure13,14 despite the layer covering some of the active sites. Layer deposition techniques could be improved by several strategies to inhibit or suppress site blockage, which can minimize this negative effect.17,18
In this study, we added a silica layer through a controlled surface sol–gel method to protect Cu nanoparticles impregnated on Al2O3 during the high temperature calcination step that forms the desired copper aluminate phase (catalyst labelled as CuAlSi-900). The schematic is shown in Fig. 1a. In order to study the effect of the silica layer's addition, we have also prepared a control catalyst using the same procedure without depositing the silica layer (catalyst labelled as CuAl-900). The experimental methods for catalyst preparation are described fully in the (ESI†). The EDSX-STEM image (Fig. 1b) shows the successful deposition of a uniform silica layer as well as the high dispersion of the Cu particles. The image for CuAl-900 is presented in the ESI,† (Fig. S1).
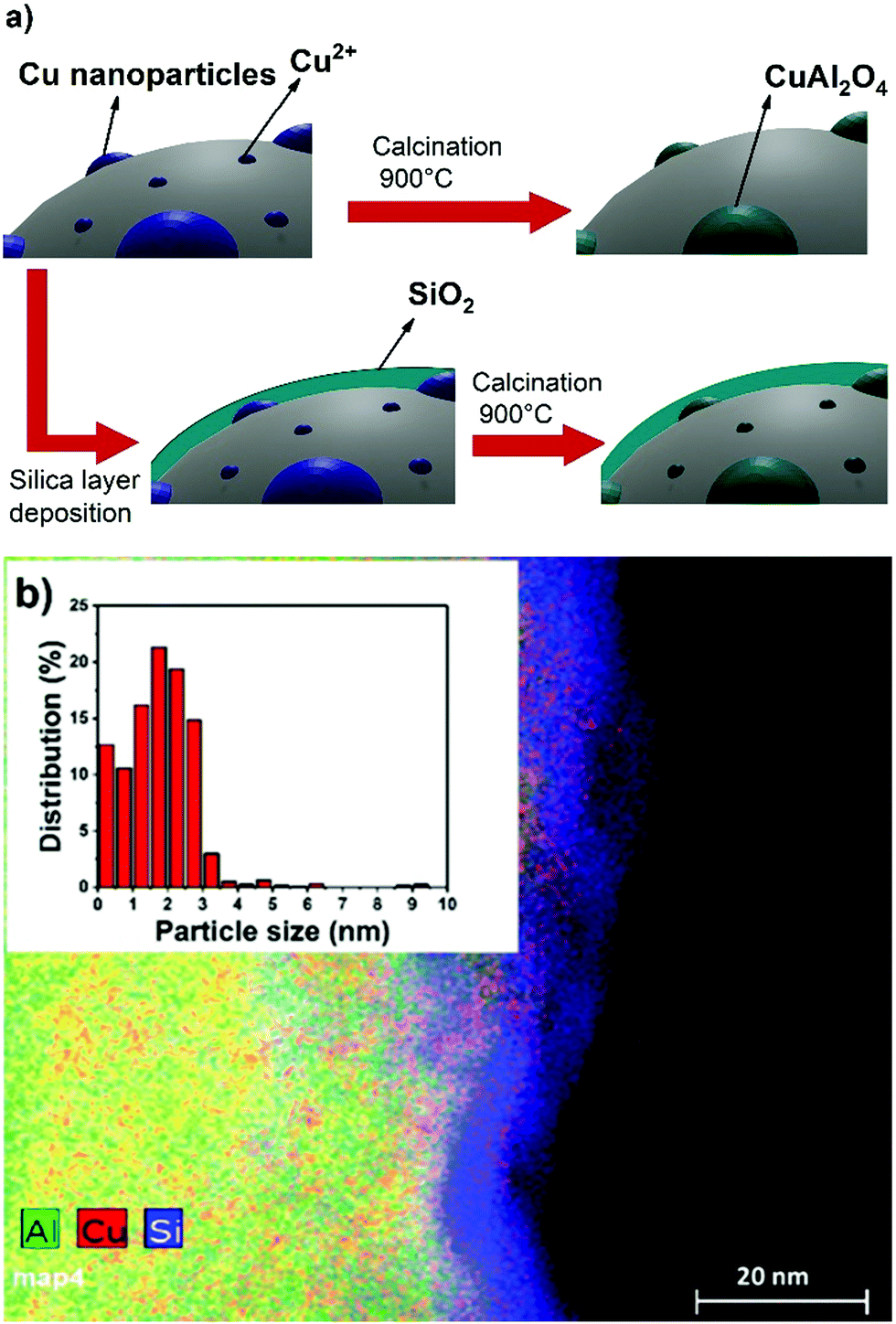 | ||
| Fig. 1 (a) Schematic presentation of the catalyst preparation process. (b) STEM and particle size distribution of the CuAlSi-900 catalyst. | ||
The catalyst activities for CO2 hydrogenation to CO are shown in Fig. 2. By depositing a silica layer, not only did the initial activity of the catalyst not deteriorate due to the potential blockage of some of the active sites, but appears to have been enhanced by about 10% compared to the control catalyst. The stability of the CuAlSi-900 catalyst is presented in Fig. S2 (ESI†) and its activity is compared with other catalysts in the literature (Table S1, ESI†). The reproducibility of the catalyst syntheses and activity tests was checked and the second set of results closely matched the first (Fig. S3, ESI†). This confirmed the protective effect of the silica layer and proved the reliability and low sensitivity of the preparation method, which is important for its potential application in industrial processes. The catalyst samples were further characterized to identify the source of the higher activity.
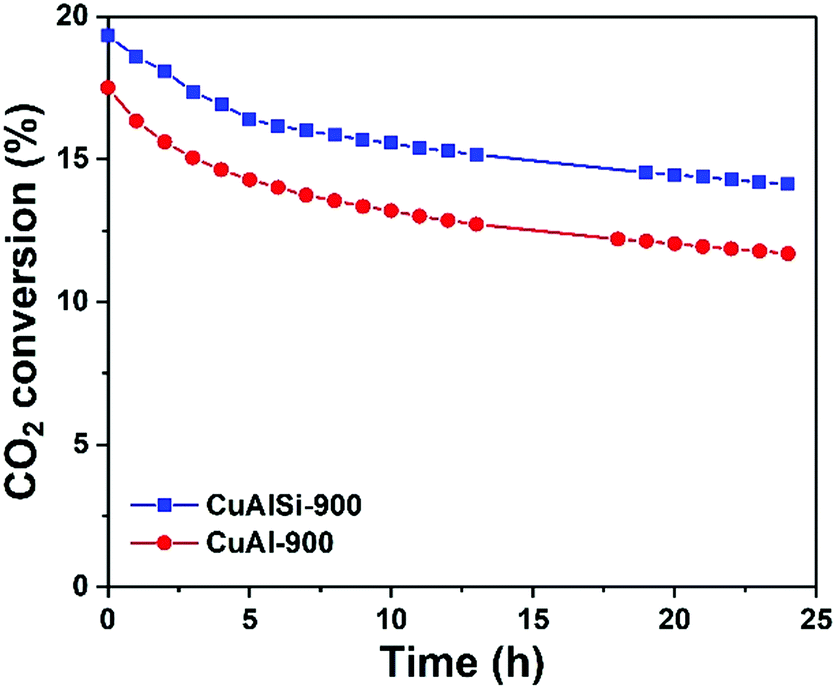 | ||
Fig. 2 Catalyst activity of CuAl-900 and CuAlSi-900. T = 450 °C, P = 1 bar, WHSV = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1. 000 mL g−1 h−1. | ||
X-ray diffraction (XRD) measurements (Fig. S4, ESI†) did not show any Cu-based crystals, which implied that the most of the nanoparticles were smaller than the detection threshold (ca. 2 and 3 nm), which was confirmed by the Cu particles size distribution determined by multiple images analysis using ImageJ software (Fig. 1b, inset). However, the XRD patterns did show the formation of α-alumina peaks on the control catalyst CuAl-900. Therefore, high temperature treatment partly converted γ-alumina into α-alumina. This phenomenon did not occur on CuAlSi-900, presumably due to the presence of the protective silica layer which acted as a stabilizer for γ-alumina.19,20
α-alumina is known to have a lower surface area compared to γ-alumina.21 Therefore, deposition of the protective silica layer helped to maintain the surface area of the catalyst as was further verified using N2 physisorption (Fig. S5 and Table S2, ESI†). The BET surface area of both catalysts before the high-temperature calcination was similar (∼116 m2 g−1). The pore volume of the CuAlSi catalyst was lower compared to CuAl (0.13 mL g−1 as opposed to 0.22 mL g−1) since the non-calcined deposited silica layer potentially plugged some of the pores (5 and 6 nm pore range, Fig. S6, ESI†). However, after calcination at 900 °C the BET surface area of CuAl-900 catalyst dropped to 63 m2 g−1, presumably due to the sintering of the γ-alumina crystals and formation of the low-surface area α-alumina. In contrast, for the CuAlSi-900 catalyst, the surface area dropped only to 85 m2 g−1. We attributed the higher surface area of the CuAlSi-900 to the inhibition of the formation of low-surface area α-alumina crystals as well as the protection of the γ-alumina crystals from sintering, which can be inferred from the lower intensity of the γ-alumina crystal peaks in its XRD pattern compared to the CuAl-900 catalyst (Fig. S4, ESI†). Moreover, the pore volume of the CuAlSi-900 increased to 0.17 mL g−1 after calcination of the silica layer, which might have cracked the overcoat, creating larger pores (Fig. S6, ESI†), or removed leftover organic precursors, while the CuAl-900 lost some of its pore volume (0.19 mL g−1 after calcination, Table S2, ESI†).
X-ray photoelectron spectroscopy (XPS) was applied in order to study the potential formation of CuAl2O4 on the catalyst surfaces (Fig. S7, ESI†). For both catalysts, the same surface Cu/Al molar ratio (0.012) was found (Table S2, ESI†). We also noted the presence of tetrahedral coordinated Cu on the surface of both catalysts (the binding energy of ∼935 eV on Cu 2p), which confirms the presence of the CuAl2O4 phase.22
In an attempt to identify the Cu species on both catalysts, temperature-programmed reduction (H2-TPR) tests (Fig. 3a) and electron paramagnetic resonance (EPR) tests (Fig. 3b) were conducted. Three peaks were identified through H2-TPR tests for both catalysts. The first peak (∼250–310 °C) is assigned to the reduction of very well-dispersed Cu–O entities and the second peak (∼550 °C) is assigned to reduction of CuO in the bulk. The third peak (∼670–740 °C) is assigned to the reduction of the CuAl2O4 phase in both catalysts.23,24 The formation of the well-dispersed Cu–O entities is known to be either caused by isolated Cu2+ ions, which are present on the catalyst surface and strongly interact with their support due to their ionic character, or by weak magnetic associates, or two- or three-dimensional Cu clusters.25 For the sample with the surface silica layer, the peak in the TPR profile representing the well-dispersed Cu–O entities appeared at higher temperatures (251 °C in CuAl-900 and 307 °C in CuAlSi-900) while the third peak, which represents the reduction of the CuAl2O4 phase, shifted to lower temperatures (from 737 °C in CuAl-900 to 677 °C in CuAlSi-900). The apparent upward shift of the peak for well-dispersed Cu–O shows the reluctance of these entities to react with hydrogen due to their stronger interaction with CuAl2O4. This implies that, unlike CuAl-900 catalyst, this peak is mostly caused by the isolated Cu2+ ions on this catalyst rather than the weak magnetic associates, or two- and three- dimensional clusters. Therefore, the appearance of this peak at higher temperatures is due to the higher contribution of the isolated Cu2+ ions on the CuAlSi-900 catalyst surface since they are more prominent. This is found to be a dispersion effect and not a new site creation. The downward shift of the peak for CuAl2O4 reduction to lower temperatures shows the higher reducibility of CuAl2O4 due to the spillover effect caused by the higher interaction of CuAl2O4 with the isolated Cu2+ ions forming well-dispersed and small Cu islands after reduction.26,27 This phenomenon shows that there is a strong interaction between copper ions and the copper aluminate that was formed.
 | ||
| Fig. 3 (a) H2-TPR profiles and (b) EPR spectra for CuAl-900 and CuAlSi-900 catalysts. | ||
To confirm this hypothesis, we used EPR to characterise the catalyst. The hyperfine structure, which appears due to the lack of spin–spin interaction, is almost equally well-resolved for both samples indicating that the Cu2+ ions, which are present on both catalysts, are isolated. This is in line with the relatively low Cu loading for both samples (1 wt%), which helps ion dispersion. Whereas the degree of isolation of the Cu2+ ions is comparable for both catalysts, the higher intensity of the spectra of CuAlSi-900 reveals a higher concentration of Cu2+ ions in this sample. This means that the isolated Cu2+ ions were better preserved in the CuAlSi-900 catalyst during the high-temperature calcination process, probably due to the effect of the interfacial sites.
In our previous study, we had shown that the CuAl2O4 phase leads to particularly favourable CO2 adsorption and that most of the adsorbed CO2 molecules remain on the CuAl2O4 surface as spectators due to their strong adsorption on the oxygen vacancies.9 Only the adsorbed CO2 intermediates (either in form of carbonates or formates) which are adsorbed next to surface hydrogen will be further hydrogenated and desorb from the catalyst surface as CO.28,29 Therefore, we suggest that the role of the isolated surface Cu2+ ions is to form highly dispersed metallic Cu entities, which act as the H2 binding sites during the reaction. Since the reaction temperature was kept at 450 °C, and based on our H2-TPR results, the isolated Cu2+ ions were likely reduced under operation conditions. In order to test the effect of these dispersed ions, a temperature-programmed surface reaction (TPSR) was conducted using the CuAlSi-900 catalyst (Fig. 4). In one test, the catalyst was only activated under Ar at 300 °C to avoid Cu2+ reduction while in the other test, the catalyst was pre-reduced using a H2/Ar flow at 400 °C before the test. When the catalyst was not pre-reduced, the formation of CO started at ∼268 °C (Fig. 4a), which almost matches the start of the first reduction peak of H2-TPR experiment. For the pre-reduced catalyst (Fig. 4b), however, the CO evolution starts already at ∼228 °C. We surmise that, since the Cu2+ ions were already reduced, they could facilitate the reaction at these lower temperatures. Based on the H2-TPR profiles, the bulk CuO and the CuAl2O4 phase were likely not reduced at this temperature. Therefore, the active sites of the CuAlSi-900 catalyst are consisted of CuAl2O4, which were found to be active in CO2 adsorption, as well as dispersed Cu0 entities, active for H2 adsorption and dissociation, which formed due to the reduction of the Cu2+ ions on the catalyst surface during the reaction.
 | ||
| Fig. 4 TPSR test for CuAlSi-900 (a) without pre-reduction and (b) with pre-reduction of the catalyst at 400 °C. | ||
The formation of high-surface area CuAl2O4 phase, also known as the Cu–Al spinel oxide phase, which act as strong CO2 adsorption sites, is complicated by the high-temperature calcination step which causes severe sintering. Here, we have used a silica layer as a protective overcoat for this step in order to maintain the surface area of the catalyst. We observed that not only was the surface area partly preserved after calcination by using the protective layer, but the well-dispersed, isolated Cu2+ ions also saw their stability increase during the calcination step. These ions were then reduced during the reaction to form Cu entities, which acted as adsorbed H2 sources for the activation of the adsorbed CO2 on the CuAl2O4 surface.
This research project is part of the Swiss Competence Center for Energy Research SCCER BIOSWEET of the Swiss Innovation Agency Innosuisse. JSL, BPLM and YPD were supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Starting grant: CATACOAT, No 758653). The authors would like to acknowledge Dr Mounir Mensi for XPS analysis, Dr Pascal Schouwink for XRD analysis, Dr Andrzej Sienkiewicz for EPR analysis, Mr Kasper Sohald for his assistance with the experiments, and Mr Sylvain Coudret for ICP-OES analysis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- J. C. Zachos, G. R. Dickens and R. E. Zeebe, Nature, 2008, 451, 279–283 CrossRef CAS.
- G. C. Chinchen, P. J. Denny, J. R. Jennings, M. S. Spencer and K. C. Waugh, Appl. Catal., 1988, 36, 1–65 CrossRef CAS.
- R. G. Herman, K. Klier, G. W. Simmons, B. P. Finn, J. B. Bulko and T. P. Kobylinski, J. Catal., 1979, 56, 407–429 CrossRef CAS.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3, 2094–2100 CrossRef CAS.
- I. A. Fisher and A. T. Bell, J. Catal., 1996, 162, 54–65 CrossRef CAS.
- A. M. Bahmanpour, F. Héroguel, M. Kılıç, C. J. Baranowski, L. Artiglia, U. Röthlisberger, J. S. Luterbacher and O. Kröcher, ACS Catal., 2019, 9, 6243–6251 CrossRef CAS.
- X. Zhang, X. Zhu, L. Lin, S. Yao, M. Zhang, X. Liu, X. Wang, Y.-W. Li, C. Shi and D. Ma, ACS Catal., 2017, 7, 912–918 CrossRef CAS.
- A. M. Bahmanpour, F. Héroguel, M. Kılıç, C. J. Baranowski, P. Schouwink, U. Röthlisberger, J. S. Luterbacher and O. Kröcher, Appl. Catal., B, 2020, 266, 118669 CrossRef.
- G. Li, C. Gu, W. Zhu, X. Wang, X. Yuan, Z. Cui, H. Wang and Z. Gao, J. Cleaner Prod., 2018, 183, 415–423 CrossRef CAS.
- T. Sun, P. Zhang, W. Chen, K. Wang, X. Fu, T. Zheng and J. Jiang, Chem. Commun., 2020, 56, 798–801 RSC.
- T. Sun, Y. Li, T. Cui, L. Xu, Y.-G. Wang, W. Chen, P. Zhang, T. Zheng, X. Fu, S. Zhang, Z. Zhang, D. Wang and Y. Li, Nano Lett., 2020, 20, 6206–6214 CrossRef CAS.
- S. H. Krishna, L. Zhang, I. Hermans, G. W. Huber, T. F. Kuech and J. A. Dumesic, J. Catal., 2020, 389, 111–120 CrossRef CAS.
- L. Cao and J. Lu, Catal.: Sci. Technol., 2020, 10, 2695–2710 RSC.
- H. O. Otor, J. B. Steiner, C. García-Sancho and A. C. Alba-Rubio, ACS Catal., 2020, 10, 7630–7656 CrossRef CAS.
- B. P. Le Monnier, F. Wells, F. Talebkeikhah and J. S. Luterbacher, Adv. Mater., 2019, 31(52), 1904276 CrossRef CAS.
- T. Van Cleve, D. Underhill, M. Veiga Rodrigues, C. Sievers and J. W. Medlin, Langmuir, 2018, 34, 3619–3625 CrossRef CAS.
- F. Liu, C. Okolie, R. M. Ravenelle, J. C. Crittenden, C. Sievers, P. C. A. Bruijnincx and B. M. Weckhuysen, Appl. Catal., A, 2018, 551, 13–22 CrossRef CAS.
- Y. Saito, T. Takei, S. Hayashi, A. Yasumori and K. Okada, J. Am. Ceram. Soc., 1998, 81, 2197–2200 CrossRef CAS.
- T. Chopin, G. Dovergne and J.-L. Le Loarer, US Pat., US5273949A, 1993 Search PubMed.
- D. S. Maciver, H. H. Tobin and R. T. Barth, J. Catal., 1963, 2, 485–497 CrossRef CAS.
- R. B. Mane and C. V. Rode, Green Chem., 2012, 14, 2780–2789 RSC.
- M. Jabłońska, Chem. Pap., 2015, 69, 1141–1155 Search PubMed.
- M. Jabłońska, A. M. Beale, M. Nocuń and R. Palkovits, Appl. Catal., B, 2018, 232, 275–287 CrossRef.
- W.-P. Dow, Y.-P. Wang and T.-J. Huang, J. Catal., 1996, 160, 155–170 CrossRef CAS.
- M.-S. Kim, S.-H. Chung, C.-J. Yoo, M. S. Lee, I.-H. Cho, D.-W. Lee and K.-Y. Lee, Appl. Catal., B, 2013, 142–143, 354–361 CrossRef CAS.
- F. Liao, Y. Huang, J. Ge, W. Zheng, K. Tedsree, P. Collier, X. Hong and S. C. Tsang, Angew. Chem., Int. Ed., 2011, 50, 2162–2165 CrossRef CAS.
- L. F. Bobadilla, J. L. Santos, S. Ivanova, J. A. Odriozola and A. Urakawa, ACS Catal., 2018, 8, 7455–7467 CrossRef CAS.
- X. Wang, H. Shi, J. H. Kwak and J. Szanyi, ACS Catal., 2015, 5, 6337–6349 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, catalyst characterization data, XRD patterns, XPS spectra, and N2 physisorption data. See DOI: 10.1039/d0cc07142k |
| This journal is © The Royal Society of Chemistry 2021 |