
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDi-tert-butyldiphosphatetrahedrane as a building block for phosphaalkenes and phosphirenes†
Gabriele
Hierlmeier
 ,
Maria K.
Uttendorfer
and
Robert
Wolf
*
,
Maria K.
Uttendorfer
and
Robert
Wolf
*
Universität Regensburg, Institut für Anorganische Chemie, Regensburg 93040, Germany. E-mail: robert.wolf@ur.de
First published on 12th February 2021
Abstract
The remarkable ‘mixed’ diphosphatetrahedrane (tBuCP)2 (1) – which is both the elusive dimeric form of the phosphaalkyne tBuCP and an isolobal analogue of the important industrial feedstock P4 – was recently isolated for the first time; however, its chemistry remains unexplored. Herein we report that treatment of 1 with various N-heterocyclic carbenes readily yields unusual, unsaturated organophosphorus motifs. These results demonstrate the significant potential of 1 as a building block for the synthesis of previously unknown organophosphorus compounds.
Organophosphorus compounds (OPCs) are of high industrial and academic importance due to their widespread applications, e.g. as specialty chemicals, pharmaceuticals or ligands in homogeneous catalysis.1,2 The development of suitable synthetic building blocks is a high priority for broadening the range of available OPCs. Traditional reagents available are chlorinated P compounds (e.g. PCl3, PCl5, and OPCl3), monophosphane (PH3),3 and, in very selected cases, phosphoric acid and phosphates.4 In addition, low-coordinate phosphorus compounds such as phosphaalkynes R–C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P (R = alkyl, aryl; see Fig. 1a)5–8 and, more recently, phosphacyanate salts have also received attention as versatile building blocks.9–11
P (R = alkyl, aryl; see Fig. 1a)5–8 and, more recently, phosphacyanate salts have also received attention as versatile building blocks.9–11
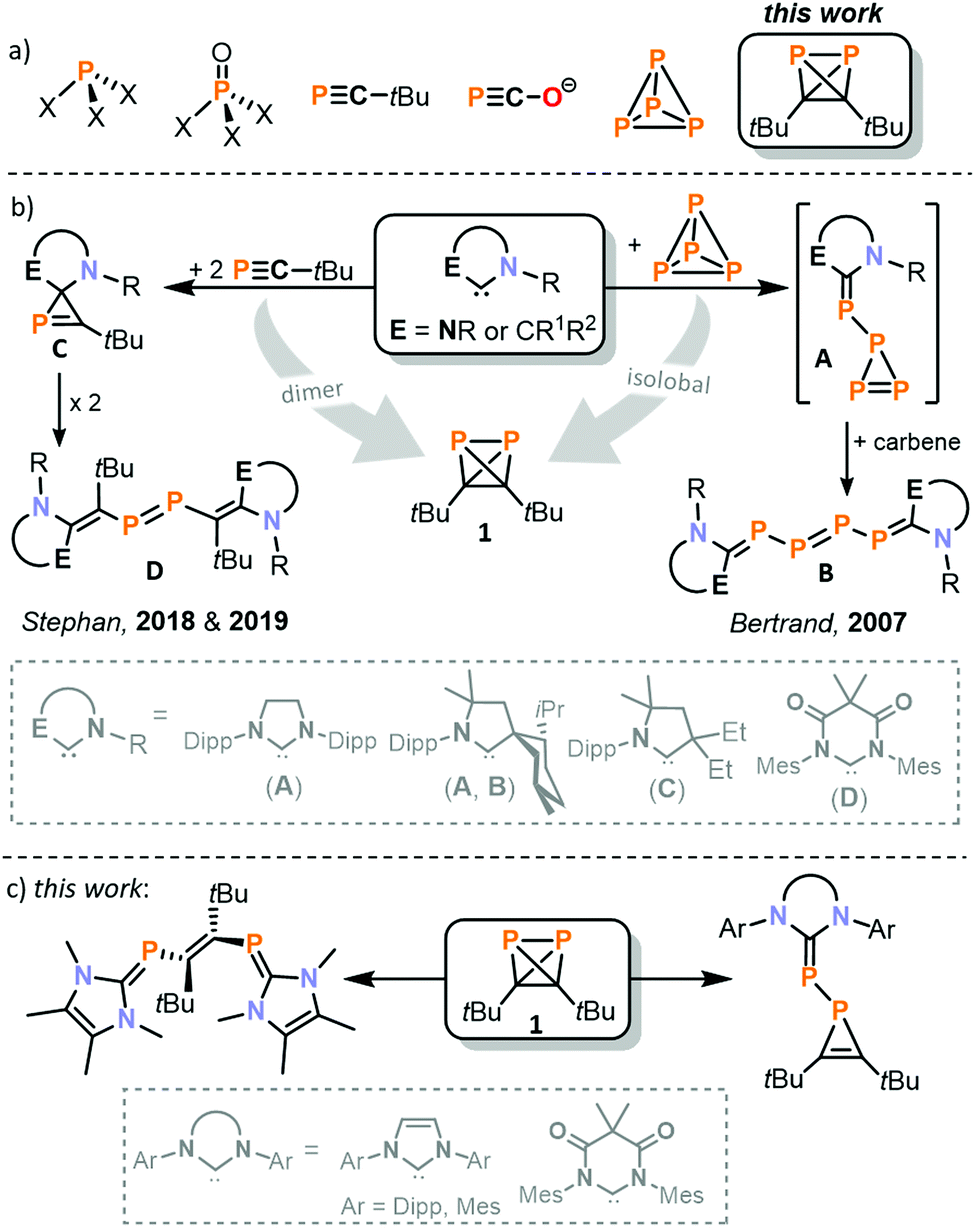 | ||
| Fig. 1 (a) Examples of common building blocks used for the synthesis of organophosphorus compounds. X = H, Cl; (b) reactivity of carbenes toward tBuCP, P4 and (tBuCP)2;17,18,21,22 (c) reactivity of 1 toward carbenes. | ||
Nearly all organophosphorus building blocks are ultimately derived from white phosphorus (P4) as the single common precursor. Therefore, fundamental reactivity studies on P4 are of high importance.12 As early as 1963, Rauhut and Semsel reported the synthesis of (cyclo)polyphosphides and primary phosphines (e.g. phenylphosphine) after hydrolytic work-up by reacting P4 with organolithium and –magnesium compounds.13,14 This approach has gained renewed interest recently through the work of Lammertsma and Xu.15,16 Bertrand and co-workers studied the reactivity of stable carbenes towards P4 (see Fig. 1b).17–20 Both acyclic and N-heterocyclic carbenes (NHCs) were used, and the nature of the obtained products strongly depends on the steric and electronic properties of the carbene. Using a bulky menthyl- and diisopropylphenyl-substituted cyclic alkyl aminocarbene (MentCAAC) or 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene (SIPr), the triphosphirene A was generated.17,18 This species was trapped and characterised as a cycloaddition product with 2,3-dimethylbutadiene. Upon addition of another equivalent of carbene, A converts to the tetraphosphene B. For other NHCs, an aggregation of P4 to larger polyphosphorus clusters and the degradation even down to monophosphorus fragments was observed.19,20
Stephan and co-workers synthesised compounds similar to A and B by reacting various NHCs with tert-butylphosphaalkyne (see Fig. 1b).21,22 The phosphirene structure C is stable and can be isolated when a diamidocarbene is used.21 Using a specific cyclic alkylamino carbene (CAAC), the dimerisation of the phosphirene is observed, resulting in the formation of a diphosphene D.22
We recently discovered di-tert-butyldiphosphatetrahedrane (tBuCP)2 (1), which is a rare example of a neutral tetrahedrane comprising two distinct p-block elements and the long-sought-after dimer of tBuCP.23–25 The ready accessibility of 1via a simple nickel-catalysed process, and its isolobal relationship with P4, prompted us to study its reaction chemistry. For our initial investigations, we chose carbenes to examine whether 1 behaves more like the isolobal P4 molecule or the parent monomer tBuCP.
In order to assess the impact of the steric and electronic properties of the NHC, a range of known NHCs were reacted with 1. With 1,3-di-iso-propyl-4,5-dimethylimidazolin-2-ylidene (iPr2ImMe) and menthyl-substituted MentCAAC, 31P{1H} NMR spectroscopic monitoring only showed the clean formation of the ladderane (tBuCP)4 (identified by a singlet at −23.0 ppm).26 (tBuCP)4 is the formal dimer of 1 and we identified this phosphaalkyne tetramer as the decomposition product of 1.23 However, the reaction of 1 with TMC (2,3,4,5-tetramethylimidazolin-2-ylidene) proceeds differently, affording a deep orange precipitate after 5 minutes when 1 is added to a solution of TMC in benzene. When dissolved in THF-d8, this solid gives rise to a singlet at −28.4 ppm in the 31P{1H} NMR spectrum. Dissolution of the precipitate in THF, filtration and subsequent recrystallisation from THF yielded crystals suitable for single crystal X-ray crystallography, which revealed the molecular structure of [(TMC)PCtBu]2 (2, Fig. 2b).
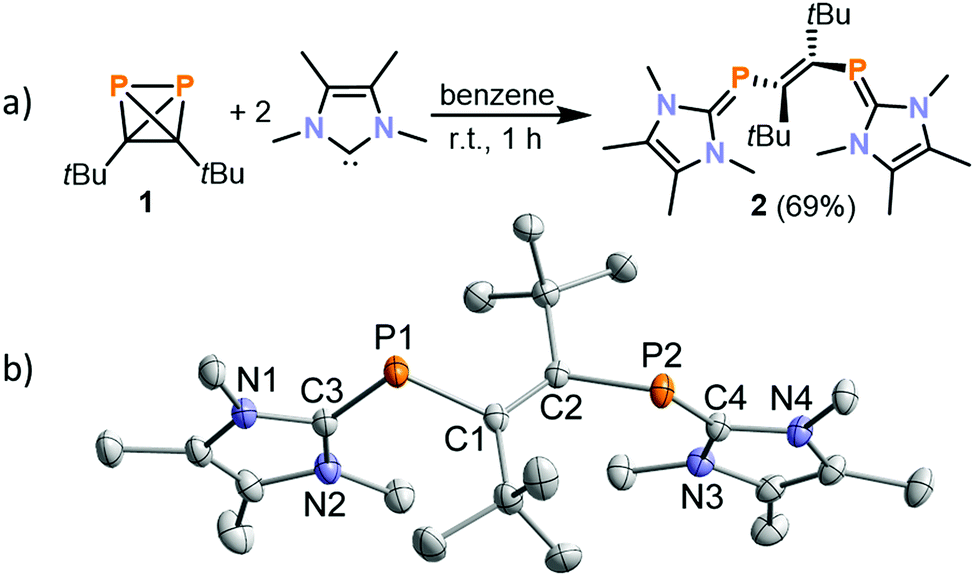 | ||
| Fig. 2 (a) Reaction of (tBuCP)2 with TMC; (b) molecular structure of 2 in the solid state. Thermal ellipsoids are set at 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: C1–C2 1.366(2), P1–C1 1.8637(16), P2–C2 1.8630(15), P1–C3 1.7673(17), P2–C4 1.7660(16), N1–C3 1.376(2), N2–C3 1.368(2), N3–C4 1.371(2), N4–C4 1.371(2), C3–P1–C1 108.69(8), C4–P2–C2 108.18(7), C2–C1–P1 120.54(13), C1–C2–P2 120.37(12). | ||
Compound 2 can be described as a vinyl-bridged bis(phosphaalkene) formed by P–P bond cleavage of the (tBuCP)2 tetrahedron.27 Related reactions of tBuCP and NHCs reported by Stephan and co-workers afford diphosphenes (e.g.D, Fig. 1b). The formation of 2 from 1 and TMC thus highlights the distinct reactivity of 1 compared to tBuCP.22
The crystallographic analysis of 2 suggests the presence of a C1–C2 double bond (1.366(2) Å) in an E configuration. The P–C bonds of the TMC units (P1–C3 1.7673(17) Å and P2–C4 1.7660(16) Å) are elongated compared to common C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P double bonds,28 while the P–C bond lengths of the vinyl group (P1–C1 1.8637(16) Å and P2–C2 1.8630(15) Å) are in the range commonly observed for P–C single bonds.29,30 The P1–C3 and P2–C4 distances are similar to the values observed for (IMes)PPh (1.763(6) Å) and (TMC)PPh (1.794(3) Å).31,32 Due to the presence of two amino substituents at carbon, such “inversely polarised phosphaalkenes” show only a partial double bond character and an inverse polarity of the PC bond compared to more common hydrocarbyl-substituted phosphaalkenes.33 Notably, the PC and CC bonds in 2 are not conjugated with plane to plane twist angles of 51° (C1–P1–C3 vs. P1–C1–C2) and 53° (C2–P2–C4 vs. C1–C2–P2).
P double bonds,28 while the P–C bond lengths of the vinyl group (P1–C1 1.8637(16) Å and P2–C2 1.8630(15) Å) are in the range commonly observed for P–C single bonds.29,30 The P1–C3 and P2–C4 distances are similar to the values observed for (IMes)PPh (1.763(6) Å) and (TMC)PPh (1.794(3) Å).31,32 Due to the presence of two amino substituents at carbon, such “inversely polarised phosphaalkenes” show only a partial double bond character and an inverse polarity of the PC bond compared to more common hydrocarbyl-substituted phosphaalkenes.33 Notably, the PC and CC bonds in 2 are not conjugated with plane to plane twist angles of 51° (C1–P1–C3 vs. P1–C1–C2) and 53° (C2–P2–C4 vs. C1–C2–P2).
The molecular structure of 2 was well-reproduced by DFT calculations on the BP86-D3BJ/def2-TZVP level. The presence of an inverse electron density distribution (Pδ−–Cδ+) on the phosphalkene PC π-bonds of 2 is supported by an analysis of the relevant intrinsic bond orbitals (IBO analysis, Fig. 3 top).33 Notably, the P–C σ-bond also features a slightly distorted electron density distribution, which is polarised to carbon in this case. In addition, low Mayer bond orders for these PC bonds (1.40 for each bond) indicate a significant contribution of the resonance structure 2-II as expected for inversely polarised phosphaalkenes (Fig. 3 bottom).
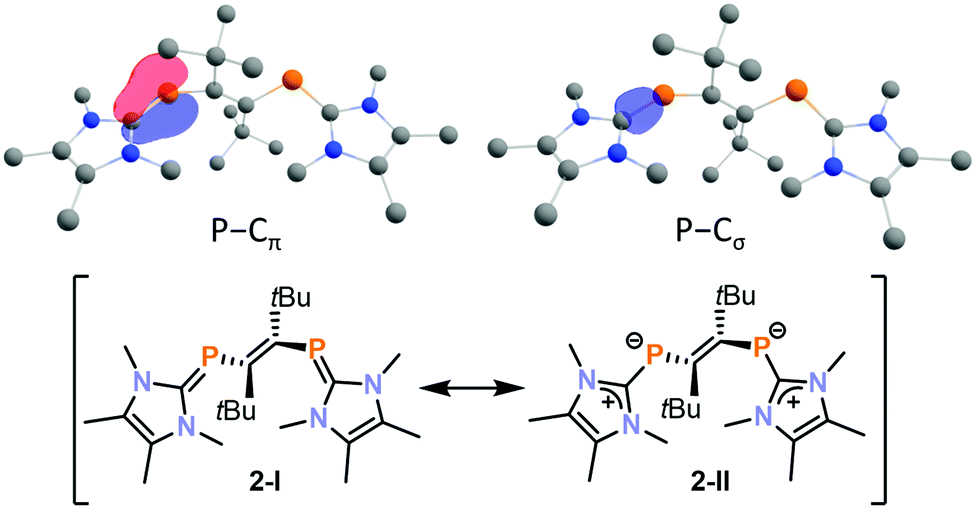 | ||
| Fig. 3 Intrinsic bond orbital of 2 showing the inversely polarised PC π- and σ-bonds (top) and resonance structures of 2 (bottom). | ||
Bis(phosphaalkene) 2 was isolated in good yield of 69% as a pure, bright orange solid. The 31P{1H} NMR spectrum of 2 in THF-d8 exhibits a singlet resonance at −28.4 ppm, which is consistent with chemical shifts reported for (IMes)PPh (−23.0 ppm) and (TMC)PPh (−53.5 ppm).31,32 The 1H NMR spectrum shows one broad resonance assigned to the tBu group and two broad signals for the Me substituents on the TMC backbone. These signals are further split into two sets of signals upon cooling (see ESI,† for details). This fluxional behaviour is presumably caused by slow rotation around the PC bonds at low temperature. In agreement with the 1H NMR data, 13C{1H} NMR data recorded at ambient temperature show only one set of resonances for the tBu groups and one set of resonances for the TMC unit. The UV/Vis absorption spectrum of 1 in THF reveals three bands at 270, 340 and 450 nm, accounting for its orange colour. TD-DFT calculations at the wB97X-D3 def2-SVP level of theory show that the absorption band in the visible part of the spectrum (450 nm) is caused by a HOMO–LUMO transition (nP → π*C–C; see the ESI,† for a density difference plot).
An initial reactivity study demonstrates the high lability of the TMC moieties of 2. Reactions with different metal complexes ([AuCl(tht)] (tht = tetrahydrothiophene), [(p-cymene)RuCl2]2, [(cod)RhCl]2 (cod = 1,5-cyclooctadiene), Ag[Al{OC(CF3)3}4]) and heterocumulenes (e.g. diphenyldiazomethane (Ph2CN2) or phenylisothiocyanate (Ph–NCS)) result in formation of (tBuCP)4. The latter reactions additionally afford TMC adducts of the heterocumulene, e.g. (TMC)N–NCPh2 and (TMC)C(S)(NPh) (see the ESI,† for characterisation data).
It was anticipated that by increasing the size of the NHC it might be possible to favour formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct with 1, in contrast to the 2:1 adduct observed using TMC. Thus, 1 was reacted with bulky aryl-substituted NHCs IMes (1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene), IPr (1,3-bis(2,6-di-iso-propylphenyl)imidazolin-2-ylidene), and MesDAC (1,3-bis-(2,4,6-trimethylphenyl)-4,6-diketo-5,5-dimethylpyrimidin-2-ylidene, see Fig. 1). 31P{1H} NMR spectra of the reaction mixtures reveal the presence of a distinct AX spin system in each case, which was subsequently assigned to 1H-phosphirenes 3a–c (vide infra). (tBuCP)4 was detected by 31P NMR as the only side product. Prolonged stirring at room temperature for two weeks (for IMes and MesDAC) or three weeks (for IPr) led to complete consumption of the NHC, and the products were subsequently crystallised from saturated n-hexane solutions.
:1 adduct with 1, in contrast to the 2:1 adduct observed using TMC. Thus, 1 was reacted with bulky aryl-substituted NHCs IMes (1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene), IPr (1,3-bis(2,6-di-iso-propylphenyl)imidazolin-2-ylidene), and MesDAC (1,3-bis-(2,4,6-trimethylphenyl)-4,6-diketo-5,5-dimethylpyrimidin-2-ylidene, see Fig. 1). 31P{1H} NMR spectra of the reaction mixtures reveal the presence of a distinct AX spin system in each case, which was subsequently assigned to 1H-phosphirenes 3a–c (vide infra). (tBuCP)4 was detected by 31P NMR as the only side product. Prolonged stirring at room temperature for two weeks (for IMes and MesDAC) or three weeks (for IPr) led to complete consumption of the NHC, and the products were subsequently crystallised from saturated n-hexane solutions.
Single-crystal X-ray diffraction studies revealed the molecular structures of 3a–c (Fig. 4) which result from P–C bond cleavage of 1. The molecular structure of 3a is very similar to those of 3b and 3c and so for simplicity only 3a will be discussed below. The structural data of 3a indicate the presence of a P1–P2 single bond (2.2200(3) Å) and a short C1C2 double bond (1.2960(19) Å) with a low C2–P2–C1 angle (41.03(6) °). The C11–P1 bond is rather short (1.7647(12) Å) compared to the P2–C1 and P2–C2 bonds (1.8502(13) and 1.8479(13) Å) and compares well to the P1–C3 bond length in 2 (1.7673(17) Å). Overall, these bond metric data are comparable to previously reported phosphirene transition metal complexes.34–37
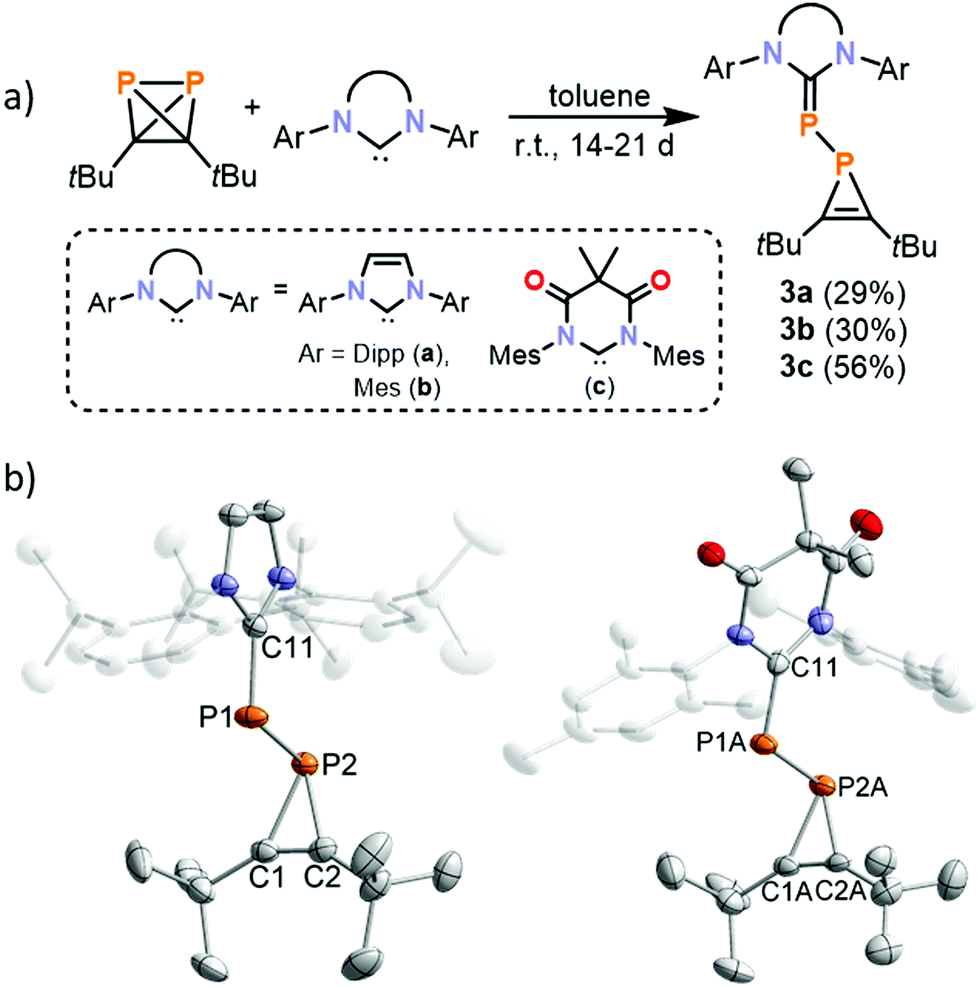 | ||
| Fig. 4 (a) Reaction of (tBuCP)2 with aryl-substituted carbenes IMes, IPr and MesDAC; (b) molecular structures of 3a and 3c in the solid state. Thermal ellipsoids are set at 50% probability level. Hydrogen atoms and positional disorder (in 3c) are omitted for clarity. Selected bond lengths [Å] and angles [°] for 3a: P1–P2 2.2200(4), P2–C1 1.8502(13), P2–C2 1.8479(13), P1–C11 1.7647(12), C1–C2 1.2960(19), C2–P2–C1 41.03(6) C1–P2–P1 98.09(4), C2–P2–P1 97.92(4), C2–C1–P2 69.39(8), C1–C2–P2 69.58(8), C11–P1–P2 109.55(4); 3c: P1A–P2A 2.234(2), P2A–C1A 1.862(6), P2A–C2A 1.854(7), P1A–C11 1.759(5), C1A–C2A 1.290(10), C2A–P2A–C1A 40.6(3), C1A–P2A–P1A 94.7(2), C2A–P2A–P1A 96.8(2), C2A–C1A–P2A 69.3(4), C1A–C2A–P2A 70.0(4), C11–P1A–P2A 111.37(19). | ||
Phosphinophosphirenes related to 3a–c are scarce, and their synthesis usually requires metal coordination of the phosphino group.35–37 An uncomplexed phosphinophosphirene was described by Bertrand and co-workers.38 This species is formed as a minor by-product of the photolysis of bis(di-iso-propylamino)phosphino(trimethylsilyl)diazomethane with tBuCP and can also be isolated in high yield by reaction of bis(di-iso-propylamino)trimethylstannylphosphine with a chlorophosphirene.
The formation of 3a–c from 1 is in stark contrast to analogous reactions of NHCs [1,3-bis(2,4,6-trimethylphenyl)imidazolidin-2-ylidene (SIMes), IMes, and IPr] with tBuCP. In these reactions, the formation of a 2H-phosphirene was observed for MesDAC,21 a triphosphole for IMes and a carbene-stabilised (tBuCP)6 framework for IPr.39 It should also be noted that three-membered heterocycles related to 3a–c are formed in reactions of carbenes with other tetrahedranes. The triphosphirene A (Fig. 1) is obtained from one equivalent of a CAAC or SIPr and P4;17,18 a related cyclopropene is formed by reaction of tetra-tert-butyltetrahedrane (tBuC)4 and tetracycanoethylene.40
Compounds 3a–c were isolated as yellow solids in 29%–56% yield after removal of the side product (tBuCP)4 by sublimation and re-crystallisation. 1H and 13C{1H} NMR spectra of 3a and 3b dissolved in C6D6 showed the presence of one set of signals for the carbene unit, while the spectra of 3c show two sets of signals, indicating a hindered rotation around the P–C bond in solution at ambient temperature. For each compound, the 31P{1H} NMR spectra show two signals of an AX spin system with chemical shifts of −172.3/−39.4 (3a), −170.0/−47.2 (3b) and −162.9/138.9 (3c, see the ESI†). The doublet observed at high field is assigned to the phosphirene moiety. The observed 1JPP coupling constants (1JPP = 296 Hz for 3a, 1JPP = 299 Hz for 3b, 1JPP = 312 Hz for 3c) and the 1JC–P coupling constants of the phosphirene P-atom to the carbon ring atoms (50–52 Hz) are consistent with a covalent P–P single bond. UV/Vis absorption spectra of 3a–c show an intense absorption band at 360 nm tailing into the visible region. This accounts for the bright yellow colour of these solids. DFT calculations on a truncated model compound (IPh)PP(CtBu)2 (IPh = 1,3-diphenylimidazolin-2-ylidene) show that the electronic transition corresponding to this wavelength is attributed to the HOMO–LUMO transition from the p-orbital of the phosphorus atom connected to the imidazoliumyl substituent to an empty p-orbital of the former carbene C atom (see ESI,† for density difference plot). It seems plausible, that 1H-phosphirenes are intermediates in the formation of bisphosphaalkenes such as 2. However, 3b does not react with TMC to afford a bis(phosphaalkene); instead, a complex reaction mixture was obtained according to a 31P{1H} NMR spectrum.
In summary, the outcome of reactions of di-tert-butyldiphosphatetrahedrane 1 with N-heterocyclic carbenes is strongly influenced by the steric and electronic properties of the NHC. TMC, the smallest NHC used for this study, cleaves the P–P bond of 1, selectively forming a bis(phosphaalkene) 2. Bulkier NHCs IMes, IPr and MesDAC afford phosphirenes 3a–cvia P–C bond cleavage. While the full mechanistic details of this dichotomous reactivity still need to be elaborated, the divergent reaction behaviour is likely attributed to the different sterics of the NHCs used. Importantly, the reactivity of 1 is clearly distinguished from its monomer tBuCP and resembles P4, and the results suggest that 1 has significant potential for the preparation of hitherto unknown organophosphorus compounds. Further reactivity studies of 1 are in hand.
Financial support by the Fonds der Chemischen Industrie (Kekulé Fellowship for G. H.) and the European Research Council (CoG 772299) is gratefully acknowledged. We thank Peter Coburger for valuable advice on DFT calculations and Sebastian Bestgen and Daniel Scott for helpful comments on the manuscript.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. E. C. Corbridge, Phosphorus, Chemistry, Biochemistry and Technology, Elvesier, 2000 Search PubMed.
- W. Schipper, Eur. J. Inorg. Chem., 2014, 1567 CrossRef CAS.
- M. Bispinghoff and H. Grützmacher, Chimia, 2016, 70, 279 CrossRef CAS.
- M. B. Geeson and C. C. Cummins, Science, 2018, 359, 1383 CrossRef CAS.
- M. Regitz, Chem. Rev., 1990, 90, 191 CrossRef CAS.
- A. Chirila, R. Wolf, J. C. Slootweg and K. Lammertsma, Coord. Chem. Rev., 2014, 270–271, 57 CrossRef CAS.
- W. Rösch and M. Regitz, Angew. Chem., Int. Ed. Engl., 1984, 23, 900 CrossRef.
- W. Rösch and M. Regitz, Synthesis, 1987, 689 CrossRef.
- A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064 CrossRef CAS.
- D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 831 RSC.
- X. Chen, S. Alidori, F. F. Puschmann, G. Santiso-Quinones, Z. Benkő, Z. Li, G. Becker, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 1641 CrossRef CAS.
- J. E. Borger, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, Chem. – Eur. J., 2017, 23, 11738 CrossRef CAS.
- M. M. Rauhut and A. M. Semsel, J. Org. Chem., 1963, 28, 471 CrossRef CAS.
- M. M. Rauhut and A. M. Semsel, J. Org. Chem., 1963, 28, 473 CrossRef CAS.
- J. E. Borger, M. S. Bakker, A. W. Ehlers, M. Lutz, J. C. Slootweg and K. Lammertsma, Chem. Commun., 2016, 52, 3284 RSC.
- L. Xu, Y. Chi, S. Du, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2016, 55, 9187 CrossRef CAS.
- J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 7052 CrossRef CAS.
- J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2007, 129, 14180 CrossRef CAS.
- O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530 CrossRef CAS.
- C. D. Martin, C. M. Weinstein, C. E. Moore, A. L. Rheingold and G. Bertrand, Chem. Commun., 2013, 49, 4486 RSC.
- L. L. Liu, J. Zhou, L. L. Cao, R. Andrews, R. L. Falconer, C. A. Russell and D. W. Stephan, J. Am. Chem. Soc., 2018, 140, 147 CrossRef CAS.
- L. L. Liu, L. L. Cao, J. Zhou and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 273 CrossRef CAS.
- G. Hierlmeier, P. Coburger, M. Bodensteiner and R. Wolf, Angew. Chem., Int. Ed., 2019, 58, 16918 CrossRef CAS.
- P(CtBu)3: M.-L. Y. Riu, R. L. Jones, W. J. Transue, P. Müller and C. C. Cummins, Sci. Adv., 2020, 6, eaaz3168 CrossRef CAS.
- (AsP3): B. M. Cossairt, M.-C. Diawara and C. C. Cummins, Science, 2009, 323, 602 CrossRef CAS.
- B. Geissler, S. Barth, U. Bergsträsser, M. Slany, J. Durkin, P. B. Hitchcock, M. Hofmann, P. Binger, J. F. Nixon, P. von Ragué Schleyer and M. Regitz, Angew. Chem., Int. Ed. Engl., 1995, 34, 484 CrossRef CAS.
- P. L. Floch, Coord. Chem. Rev., 2006, 250, 627 CrossRef.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326 CrossRef.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186 CrossRef.
- A. J. Arduengo III, J. C. Calabrese, A. H. Cowley, H. V. Rasika Dias, J. R. Goerlich, W. J. Marshall and B. Riegel, Inorg. Chem., 1997, 36, 2151 CrossRef.
- A. J. Arduengo III, H. V. Rasika Dias and J. C. Calabrese, Chem. Lett., 1997, 143 CrossRef.
- L. Weber, Eur. J. Inorg. Chem., 2000, 2425 CrossRef CAS.
- F. Mathey, Chem. Rev., 1990, 90, 997 CrossRef CAS.
- J. Foerstner, A. Kakoschke, D. Stellfeldt, H. Butenschön and R. Wartchow, Organometallics, 1998, 17, 893 CrossRef CAS.
- J. Simon, G. J. Reiß, U. Bergsträßer, H. Heydt and M. Regitz, Eur. J. Inorg. Chem., 2001, 2067 CrossRef CAS.
- A. Jayaraman and B. T. Sterenberg, Organometallics, 2013, 32, 745 CrossRef CAS.
- M. Sanchez, R. Réau, C. J. Marsden, M. Regitz and G. Bertrand, Chem. – Eur. J., 1999, 1, 274 CrossRef.
- L. L. Liu, J. Zhou, Y. Kim, L. L. Cao and D. W. Stephan, Dalton Trans., 2019, 48, 14242 RSC.
- G. Maier, K.-A. Schneider, K.-D. Malsch, H. Irngartinger and A. Lenz, Angew. Chem., Int. Ed. Engl., 1982, 21, 437 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2040301–2040306. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc07103j |
| This journal is © The Royal Society of Chemistry 2021 |