
High temperature quantum tunnelling of magnetization and thousand kelvin anisotropy barrier in a Dy2 single-molecule magnet†
Haipeng
Wu‡
 ab,
Min
Li‡
ab,
Zhengqiang
Xia
*a,
Vincent
Montigaud
c,
Olivier
Cador
*c,
Boris
Le Guennic
*c,
Hongshan
Ke
a,
Wenyuan
Wang
a,
Gang
Xie
a,
Sanping
Chen
*a and
Shengli
Gao
a
ab,
Min
Li‡
ab,
Zhengqiang
Xia
*a,
Vincent
Montigaud
c,
Olivier
Cador
*c,
Boris
Le Guennic
*c,
Hongshan
Ke
a,
Wenyuan
Wang
a,
Gang
Xie
a,
Sanping
Chen
*a and
Shengli
Gao
a
aKey Laboratory of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi’an, 710127, China. E-mail: northwindy@126.com; sanpingchen@126.com
bDepartment of Chemistry and Chemical Engineering, Jinzhong University, Jinzhong, 030619, China
cUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, Rennes F-35000, France. E-mail: olivier.cador@univ-rennes1.fr; boris.leguennic@univ-rennes1.fr
First published on 26th November 2020
Abstract
We report here a dinuclear DyIII iodine-bridged single-molecule magnet self-assembled by cis/trans coordination chemistry that displays a large anisotropy barrier of ca. 1300 K and a hysteresis opening temperature of 16 K. High temperature quantum tunnelling of magnetization is observed up to 56 K in zero-field and explained by the combination of the large anisotropy barrier and the local transverse field at the trans site. The results provide a model for thorough understanding of the effect of electronic structure on the magnetic behavior of lanthanide complexes.
The strong magnetic anisotropy of lanthanide complexes often induces single-molecule magnet (SMM) behavior,1 which spurs their various applications in prototype magnetic devices.2,3 Among the lanthanide based SMMs, DyIII is acclaimed as an unrivalled candidate owing to the strongly oblate spheroidal shape of its electron density and the presence of Kramers doublets.4 The anisotropy of DyIII is therefore enhanced in axial coordination environments.5 The design of DyIII complexes with low coordination numbers presents a particularly efficient avenue to realize a high degree of axiality but remains quite synthetically challenging. The pitfall in lanthanide chemistry is the significant quantum tunnelling of the magnetization (QTM) between the MJ levels that plays crucial roles in the magnetic properties.6 The existence of fast QTM often causes the reduction or even loss of memory effect of the magnetization at zero field, limiting their potential applications.7 But the QTM can be reduced by consideration on the local symmetry around the lanthanide ions.8 It has been shown that high CF symmetries, such as C∞v, D4d, S8, D5h, D6d, and D∞h, etc. eliminate transverse CFs that enlarge CF splitting and reduce QTM.8 However, in a real system, the symmetry is most likely to be lower than ideal, and the high CF symmetry is mostly obtained from more coordinatively saturated molecules instead of low-coordinate systems. How to control structural factors and take advantage of the symmetry strategies to suppress QTM while maintaining the magnetic axiality in low-coordinate systems is still a current challenge that needs to be further explored.
Octahedral six-coordinate complexes, as the threshold of the regular coordination number (7–12) of lanthanide complexes towards the low-coordinate ones and with a well-defined axial and equatorial distribution, are excellent candidates to realize performant SMMs. Increasing efforts have been made to understand magnetic relaxation and optimize the electrostatic distribution of DyIII in pseudo-octahedral geometry.9–14 We have recently reported the cis/trans stereoisomers in a distorted octahedral bromine-bridged DyIII dimer [Dy(Cy3PO)2(μ-Br)(Br)2]2·2C7H8 (1·2C7H8) with a large effective barrier of 684 K.12 Interestingly, it is characterized by the very weak magnetic interaction between DyIII ions as a consequence of the mutual orthogonal anisotropy arrangement and the bridging bromide ions. In this system the magnetic relaxation is dominated by the strong single ion anisotropy of the trans-Br4O2 site.12 We have decided to take advantage of this situation as we go down the periodic table: to substitute bromine with iodine. Weaker coordinating anions such as iodine (1) minimize the in-plane CF at the trans site and, in principle, should maximize the axial anisotropy and (2) minimize the superexchange interaction between DyIII centers. It is striking that the energy barrier (ca. 1300 K) of the new dinuclear DyIII SMM [Dy(Cy3PO)2(μ-I)(I)2]2·4C7H8 (2·4C7H8) is the highest among the reported polymetallic complexes obtained via conventional coordination chemistry approaches while QTM is visible up to ca. 56 K. Solid-state magnetic site dilution and ab initio calculations allow rationalizing these prominent features.
The addition of DyI3 and Cy3PO at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio in ethyl ether produced a white suspension solution. The volatiles were removed under reduced pressure and faint yellow crystals of 2·4C7H8 were obtained by recrystallization from toluene (Scheme S1, ESI†). It crystallizes in the monoclinic space group of P21/n (Table S1, ESI†). The asymmetric unit contains two unique stereoisomeric [Dy(Cy3PO)2I4] molecules with metal ions bridged by two μ2-I ions and each DyIII center is hexacoordinated adopting a distorted octahedral geometry (Fig. 1). Dy1 is ligated by two strong Cy3PO ligands in the trans position which are in the cis position for Dy2. The Dy–O distances (2.172–2.195 Å) are considerably shorter than the Dy–I bonds (2.985–3.192 Å) (Table S2, ESI†). Consequently, the coordination polyhedra are strongly distorted with CShMs values for an Oh symmetry at 2.79 and 2.56 (Table S3, ESI†).15 In 1, the distances are equivalent for Dy–O (2.188–2.199 Å) but much shorter for Dy–Br (2.735–2.950 Å) and less distorted polyhedra were observed (CShMs values at 1.921 and 1.342).12 The intramolecular Dy–Dy distance (4.80 Å) is longer than in 1 (4.46 Å) while maintaining the same Dy–X–Dy angles. Within the lattice, the shortest intermolecular Dy⋯Dy distance is 10.661 Å (Fig. S1, ESI†).
:2 molar ratio in ethyl ether produced a white suspension solution. The volatiles were removed under reduced pressure and faint yellow crystals of 2·4C7H8 were obtained by recrystallization from toluene (Scheme S1, ESI†). It crystallizes in the monoclinic space group of P21/n (Table S1, ESI†). The asymmetric unit contains two unique stereoisomeric [Dy(Cy3PO)2I4] molecules with metal ions bridged by two μ2-I ions and each DyIII center is hexacoordinated adopting a distorted octahedral geometry (Fig. 1). Dy1 is ligated by two strong Cy3PO ligands in the trans position which are in the cis position for Dy2. The Dy–O distances (2.172–2.195 Å) are considerably shorter than the Dy–I bonds (2.985–3.192 Å) (Table S2, ESI†). Consequently, the coordination polyhedra are strongly distorted with CShMs values for an Oh symmetry at 2.79 and 2.56 (Table S3, ESI†).15 In 1, the distances are equivalent for Dy–O (2.188–2.199 Å) but much shorter for Dy–Br (2.735–2.950 Å) and less distorted polyhedra were observed (CShMs values at 1.921 and 1.342).12 The intramolecular Dy–Dy distance (4.80 Å) is longer than in 1 (4.46 Å) while maintaining the same Dy–X–Dy angles. Within the lattice, the shortest intermolecular Dy⋯Dy distance is 10.661 Å (Fig. S1, ESI†).
 | ||
| Fig. 1 Molecular structure of 2. Hydrogen atoms are omitted for clarity. For unlabeled atoms: P green, and C grey. | ||
Static magnetic susceptibility measurements on 2·4C7H8 are carried out at 1000 Oe between 2 and 300 K. The room-temperature χMT value is equal to 27.71 cm3 K mol−1 in agreement with the expected values for two free DyIII ions (28.34 cm3 K mol−1) (Fig. S4, ESI†). χMT decreases monotonically on cooling down with a low temperature limit that is close to 24.01 cm3 K mol−1. A small drop below 5 K is visible with a value of 22.95 cm3 K mol−1 at 2 K. At this stage, there is no evidence of interactions (dipolar and/or superexchange) between DyIII magnetic moments. The M vs. H curve at 2 K saturates at a value close to 10 Nβ in agreement with the stabilization of the MJ = ±15/2 using component of the 6H15/2 ground state multiplet (Fig. S4, ESI†). The slow relaxation of the magnetization is evidenced through ac susceptibility measurements. A maximum shows up on the out-of-phase component of the ac susceptibility below 70 K in zero external dc field (Fig. 2a and Fig. S5, ESI†). The maximum shifts to low frequency on lowering the temperature but becomes temperature independent below 56 K, displaying a tunnelling frequency of 412 Hz. At each temperature, the frequency dependence of the ac signals is analyzed quantitatively with an extended Debye model (Fig. S6 and Table S4, ESI†).16 Important information is extracted at this stage: (1) a narrow distribution of the relaxation times (α) of 0.05–0.18 is obtained, in agreement with a single relaxation process and (2) the relaxation time follows an Arrhenius law combined with a thermally independent process (τ−1 = τQTM−1 + τ0−1exp(−Ueff/T)) with Ueff = 1290 K, τ0 = 1.26 × 10−12 s and τQTM = 3.88 × 10−4 s (Fig. 2b and 3). The non-relaxing fraction (χS/χT) is close to 40% in all the temperature range (Table S4, ESI†). This last characteristic suggests that only one DyIII site is slowly relaxing and that the second one is not involved. Clearly, the energy barrier jumped from ∼700 K in 1·2C7H8 up to ∼1300 K in 2·4C7H8.12 This proves that our approach which consists in minimizing CF in the plane rather than maximizing it along one axis is valid for DyIII. However, QTM in 2·4C7H8 is faster than in 1·2C7H8 (Fig. S7, ESI†).12 The application of an external dc field annihilates the thermally independent process and shifts the relaxation process to lower frequencies (Fig. S8, ESI†). The relaxation becomes so slow on cooling that zero-field cooled (ZFC) and field-cooled (FC) curves measured at a sweep rate of 0.5 K min−1 diverge at 12 K, which reveals the onset of the memory effect (Fig. 2c). On Fig. S8 (ESI†) the relaxation is the slowest at 1 kOe and the frequency dependence at various temperatures of the ac signals has been recorded at this optimum field (Fig. S9, ESI†) and analyzed in the framework of the generalized Debye model (Table S5, ESI†). The non-relaxing fraction remains close to 40% and now the Raman process (τ−1 = CTn) becomes the fastest at low temperature. The best fit is obtained with C = 1.04 × 10−4 s−1 K−n, n = 3.31, Ueff = 1323 K, τ0 = 7.3 × 10−12 s (Fig. 2b). As expected, τ0 and Ueff are almost unchanged between zero external dc field and 1 kOe while QTM is too slow to be numerically extracted.
 | ||
| Fig. 2 (a) Frequency-dependent out-of-phase χM′′ ac susceptibility signals for 2 at zero external dc field. (b) Temperature dependences of the relaxation time for 2 at zero external dc field (●), 2 at 1 kOe (Δ) and 2@Y at zero external dc field (○) with the best fitted curves: in red QTM + Orbach and in blue Raman + Orbach. (c) ZFC–FC magnetization curves at 2 kOe for 2·4C7H8 at a sweep rate of 0.5 K min−1. (d) Normalized M vs. H hysteresis profiles of 2@Y and 2 at 2 K. In inset: (top left) zoomed-in view at the origin of the 2 K hysteresis; (bottom right) zoomed-in view at the origin of the 16 K hysteresis. | ||
The disappearance of QTM at low temperature with the magnetic field is common in DyIII SMMs.17 Hyperfine coupling, dipolar and/or superexchange interactions are known to be the sources of this thermally independent effect.18–20 Dilution in a diamagnetic matrix and isotopic enrichment are standard tools employed to eliminate QTM.21,22 However, in polynuclear complexes, a fortiori in dimers, the doping with non-magnetic metal ions (YIII) may result in either Dy2 complexes within a matrix of diamagnetic dimers or heteronuclear Dy–Y units. In the present case, 5% of DyIII ion has been injected in the diamagnetic YIII matrix to yield 2@Y with a final ratio Dy/Y: 1/15.2. The magnetic investigation in zero external field on 2@Y reveals a low frequency shift of χM′′ peak at low temperature compared with 2·4C7H8 (Fig. S10, ESI†). The QTM is then efficiently released by the dilution. Remarkably, the characteristic relaxation times superimpose with those of the condensed 2·4C7H8 at 1 kOe (Table S6 and Fig. 2b, ESI†). Here again, 40% of the magnetic moment is non relaxing. The best fit is obtained with Raman + Orbach processes (Ueff = 1311 K, τ0 = 1.08 × 10−12 s, C = 6.69 × 10−4 s−1 K−n, n = 2.82). The dilution experiment indicates that intramolecular and/or intermolecular magnetic interactions are highly responsible for the prevalence of fast tunnelling in 2·4C7H8. However, there is no possible discrimination between the two types of dilution since we cannot dilute magnetic dimers into a diamagnetic matrix in frozen solution while ensuring the integrity of the complex in solution.
In-field magnetic hysteresis is clearly observed for 2·4C7H8 up to 14 K, for a sweep rate of 0.12 kOe s−1 (Fig. S11, ESI†) which matches with the divergence of ZFC–FC curves at 12 K. The closing at the origin whatever the temperature is clearly due to the QTM fast relaxation. In contrast, the magnetic hysteresis curves for 2@Y is open at the origin up to 16 K (Fig. S12, ESI†). The normalized hysteresis loops are represented for both 2@Y and 2·4C7H8 at 2 K on Fig. 2d highlighting remanence (24% of saturation magnetization) and coercivity (360 Oe) achieved through the dilution.
Ab initio complete active-space self-consistent field (CASSCF) calculations were performed on the X-ray structure of 2, using the MOLCAS 8.2 and SINGLE_ANISO programs (see the ESI,† for computational details). Calculations were carried out on each asymmetric DyIII center of the dimer unit (namely Dy1/Dy2) while the second metal ion was replaced by a diamagnetic YIII ion. The resulting energy spectrum and g-tensors (gXX, gYY, gZZ) of the eight lowest Kramers Doublets (KD) of 2 are reported in Table S7 (ESI†). Dy1 and Dy2 show a strong ground state anisotropy with a main gZZ component at 19.9 for Dy1 and only 19.1 for Dy2 (gXX = gYY = 0.00 for Dy1; gXX = 0.03 and gYY = 0.07 for Dy2). The orientations of the magnetic axes for the KD ground states are reported in Fig. 3. Due to the large intermolecular Dy–Dy distances (10.66 Å), only intramolecular dipolar magnetic interactions are calculated. The intramolecular dipolar term was computed in the effective spin ![[S with combining tilde]](https://www.rsc.org/images/entities/i_char_0053_0303.gif) = 1/2 formalism using the POLY_ANISO routine (see Experimental section for details).23,24 The resulting intramolecular dipolar constant is J = −0.012 cm−1. This very weak antiferromagnetic coupling is explained by the orthogonal configuration of the anisotropy axes in the KD ground states (92°). In this frame, the computed molar magnetic susceptibility and magnetization curve at 2 K are in good agreement with the experimental data (Fig. S4, ESI†).
= 1/2 formalism using the POLY_ANISO routine (see Experimental section for details).23,24 The resulting intramolecular dipolar constant is J = −0.012 cm−1. This very weak antiferromagnetic coupling is explained by the orthogonal configuration of the anisotropy axes in the KD ground states (92°). In this frame, the computed molar magnetic susceptibility and magnetization curve at 2 K are in good agreement with the experimental data (Fig. S4, ESI†).
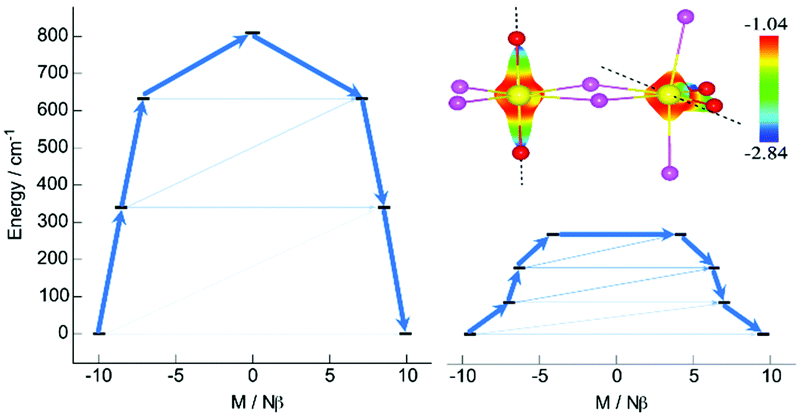 | ||
| Fig. 3 Computed magnetization blocking barriers of Dy1 (left) and Dy2 (right) of 2. The four lowest KDs (black lines) are represented according to their magnetic moment along the main magnetic axis. The arrows are drawn according to the mean values of the transversal matrix element of the transition magnetic moment. The total electrostatic potential at 1.6 Å of each Dy center is also represented (top right). | ||
The electrostatic potential (Fig. 3), computed from the ab initio LoProp charges using the home-made software CAMMEL,25,26 governs the DyIII anisotropy axis orientation that follows the most negative potential induced by the O atoms of the Cy3PO ligands (Fig. 3 and Fig. S13, ESI†). The energy spectra of the ground state multiplet 6H15/2 for each DyIII ion are reported in Fig. 3. First, the total splitting is twice larger for Dy1 (1365 K) than for Dy2 (685 K). Second, the gap between the ground state KD and the first excited KD state is four times larger for Dy1 (488 K) than for Dy2 (122 K). The computed transition moments between each state (Fig. 3) for both Dy1 and Dy2 shows two drastically different relaxation pathways. For Dy2, the non-zero transversal components of the ground state g-tensor and a mixed low-lying 1st excited state (Table S8, ESI†) lead to sizable QTM and thermally assisted QTM suggesting a fast relaxation mechanism. In contrast for Dy1, the strong Ising-type anisotropy of the ground and 1st excited state (Table S8, ESI†) results in a relaxation path through the third excited state at 1165 K (Fig. 3), which corresponds to the effective energy barrier (Orbach) in almost perfect agreement with the experiment (∼1300 K). The computed results support the rationale that at high temperature 2 relaxes via an over-barrier (Orbach) mechanism associated with the large single ion anisotropy of the trans-DyI4O2 site.
It is noticed that the change of the ancillary halogen donors connecting DyIII is shown not only to induce differences in the octahedrally distorted DyX4O2 coordination sphere that adjusts the magnetic anisotropy and the superexchange interaction but also to modify the packing and intermolecular dipolar interactions. The principal axes of monoclinic 2 are canted by 45° between neighboring molecules. Whereas 1 crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group12 without a canted angle between two independent molecules (Fig. S14, ESI†). Although there is no possibility to discriminate the origin of QTM, the large tilting of the principle axes between neighboring molecules should enhance the transverse components of fluctuating magnetic fields, thus improving tunnelling of the magnetization in 2.
space group12 without a canted angle between two independent molecules (Fig. S14, ESI†). Although there is no possibility to discriminate the origin of QTM, the large tilting of the principle axes between neighboring molecules should enhance the transverse components of fluctuating magnetic fields, thus improving tunnelling of the magnetization in 2.
In fact, 2·4C7H8 displays the best performance of six-coordinate SMMs with an effective barrier of up to 1300 K and hysteresis loops open up to 16 K (Table S9, ESI†). This strong anisotropy combined with weak coercivity appears to be a common feature in the limited reported examples of D3h and Oh geometries.9–14,27–29 Compared with two widely studied families of high-performance pentagonal bipyramidal and metallocene dysprosium single-ion magnets,10,30–32 it has been found that higher charge symmetry and weaker thermal vibration of atoms favor stronger coercivity. The uneven and the monodentate coordinated transverse sites diminish the CF symmetry and increase the molecular flexibility, which contributes to the off-diagonal elements of the matrix representations, thereby enhancing QTM.
In summary, we observed a large anisotropy barrier of 1300 K and magnetic hysteresis opened up to 16 K in a dimetallic octahedral DyIII system along with significant QTM up to 56 K. Strong QTM in the presence of a large barrier is associated with the magnetic environment around Dy1. The exceptional properties originate from the combination of large single-ion anisotropy at the trans site and cancelation of the magnetic interactions through near-orthogonality of the magnetic axes. The magnetic dilution proved that the dipolar coupling accounts for the significant zero-field QTM. Comparative analysis of DyIII complexes with different configurations suggests that the synthesis of high axial low coordination systems with uniform molecule anisotropy orientations, high charge symmetry and restricted molecular vibrations are expected to achieve a new breakthrough for the development of SMMs.
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Grant no. 21673180, 21727805, 21803042 and 21973074), the Natural Science Basic Research Program of Shaanxi (Grant No. 2017JZ002, 2018JM5180 and 2019JQ-067) and the 64th China Postdoctoral Science Foundation Funded Project (2018M643706). V. M. is thankful to ERC (project no. 725184) for funding. B. L. G. and V. M. thank the French GENCI/IDRIS-CINES center for high-performance computing resources.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. N. Woodruff, R. E. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- R. Vincent, S. Klyatskaya, M. Ruben, W. Wernsdorfer and F. Balestro, Nature, 2012, 488, 357–360 CrossRef CAS PubMed.
- S. Thiele, F. Balestro, R. Ballou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Science, 2014, 344, 1135–1138 CrossRef CAS PubMed.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- L. Ungur and L. F. Chibotaru, Inorg. Chem., 2016, 55, 10043–10056 CrossRef CAS PubMed.
- J. R. Friedman, M. P. Sarachik, J. Tejada and R. Ziolo, Phys. Rev. Lett., 1996, 76, 3830–3833 CrossRef CAS PubMed.
- F. Ortu, D. Reta, Y.-S. Ding, C. A. P. Goodwin, M. P. Gregson, E. J. L. McInnes, R. E. P. Winpenny, Y.-Z. Zheng, S. T. Liddle, D. P. Mills and N. F. Chilton, Dalton Trans., 2019, 48, 8541–8545 RSC.
- J. L. Liu, Y. C. Chen and M. L. Tong, Chem. Soc. Rev., 2018, 47, 2431–2453 RSC.
- M. Gregson, N. F. Chilton, A.-M. Ariciu, F. Tuna, I. F. Crowe, W. Lewis, A. J. Blake, D. Collison, E. J. L. McInnes, R. E. P. Winpenny and S. T. Liddle, Chem. Sci., 2016, 7, 155–165 RSC.
- B.-C. Liu, N. Ge, Y.-Q. Zhai, T. Zhang, Y.-S. Ding and Y.-Z. Zheng, Chem. Commun., 2019, 55, 9355–9358 RSC.
- M. Li, H. Wu, Z. Xia, L. Ungur, D. Liu, L. F. Chibotaru, H. Ke, S. Chen and S. Gao, Inorg. Chem., 2020, 59, 7158–7166 CrossRef CAS PubMed.
- M. Li, H. Wu, Z. Xia, V. Montigaud, O. Cador, B. Le Guennic, H. Ke, W. Wang, G. Xie and S. Chen, Chem. Commun., 2019, 55, 14661–14664 RSC.
- M. J. Giansiracusa, S. Al-Badran, A. K. Kostopoulos, G. F. S. Whitehead, D. Collison, F. Tuna, R. E. P. Winpenny and N. F. Chilton, Dalton Trans., 2019, 48, 10795–10798 RSC.
- X.-L. Ding, Y.-Q. Zhai, T. Han, W.-P. Chen, Y.-S. Ding and Y.-Z. Zheng, Chem. – Eur. J. DOI:10.1002/chem.202003931.
- M. Llunell, D. Casanova, J. Cirera, J. M. Bofill, P. Alemany and S. Alvarez, SHAPE (Version 2.1), 2013 Search PubMed.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, 2006 Search PubMed.
- M. Li, H. Wu, Q. Yang, H. Ke, B. Yin, Q. Shi, W. Wang, Q. Wei, G. Xie and S. Chen, Chem. – Eur. J., 2017, 23, 17775–17787 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita and W. Wernsdorfer, Angew. Chem., Int. Ed., 2005, 44, 2931–2935 CrossRef CAS PubMed.
- P. H. Lin, T. J. Burchell, R. Clerac and M. Murugesu, Angew. Chem., Int. Ed., 2008, 47, 8848–8851 CrossRef CAS.
- T. Han, M. J. Giansiracusa, Z.-H. Li, Y.-S. Ding, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, Chem. – Eur. J., 2020, 26, 6773–6777 CrossRef CAS PubMed.
- F. Pointillart, K. Bernot, S. Golhen, B. Le Guennic, T. Guizouarn, L. Ouahab and O. Cador, Angew. Chem., Int. Ed., 2015, 54, 1504–1507 CrossRef CAS PubMed.
- E. Moreno-Pineda, M. Damjanović, O. Fuhr, W. Wernsdorfer and M. Ruben, Angew. Chem., Int. Ed., 2017, 56, 9915–9919 CrossRef CAS PubMed.
- L. F. Chibotaru, L. Ungur and A. Soncini, Angew. Chem., Int. Ed., 2008, 47, 4126–4129 CrossRef CAS.
- L. Ungur, W. Van den Heuvel and L. F. Chibotaru, New J. Chem., 2009, 33, 1224–1230 RSC.
- G. Huang, G. Fernandez-Garcia, I. Badiane, M. Camarra, S. Freslon, O. Guillou, C. Daiguebonne, F. Totti, O. Cador, T. Guizouarn, B. L. Guennic and K. Bernot, Chem. – Eur. J., 2018, 24, 6983–6991 CrossRef CAS PubMed.
- The CAMMEL code is available under GNU General Public License v3.0 and can be downloaded at https://github.com/rmarchal1/CAMME.
- K. L. Harriman, J. L. Brosmer, L. Ungur, P. L. Diaconescu and M. Murugesu, J. Am. Chem. Soc., 2017, 139, 1420–1423 CrossRef CAS PubMed.
- T. Han, Y.-S. Ding, Z.-H. Li, K.-X. Yu, Y.-Q. Zhai, N. F. Chilton and Y.-Z. Zheng, Chem. Commun., 2019, 55, 7930–7933 RSC.
- R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. McInnes, L. F. Chibotaru and R. E. Winpenny, Nat. Chem., 2013, 5, 673–678 CrossRef CAS PubMed.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- K.-X. Yu, J. G. C. Kragskow, Y.-S. Ding, Y.-Q. Zhai, D. Reta, N. F. Chilton and Y.-Z. Zheng, Chemistry, 2020, 6, 1777–1793 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details, structural data, additional magnetic figures and ab initio studies. CCDC 1901845. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc06993k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |