
Chirality from scratch: enantioselective adsorption in geometrically controlled lateral nanoconfinement†
Johannes
Seibel
 *a,
Zeno
Tessari
a,
David B.
Amabilino
b and
Steven
De Feyter
*a
*a,
Zeno
Tessari
a,
David B.
Amabilino
b and
Steven
De Feyter
*a
aDepartment of Chemistry, Division of Molecular Imaging and Photonics, KU Leuven – University of Leuven, Celestijnenlaan 200F, B-3001 Leuven, Belgium. E-mail: johannes.seibel@kuleuven.be; steven.defeyter@kuleuven.be
bSchool of Chemistry & The GSK Carbon Neutral Laboratories for Sustainable Chemistry, The University of Nottingham, Triumph Road, Nottingham NG7 2TU, UK
First published on 25th November 2020
Abstract
Chiral symmetry breaking in molecular adsorption at the solid/liquid interface by lateral geometric nanoconfinement is demonstrated. The chiral nanoconfinement is created at the interface of achiral covalently modified highly-oriented pyrolytic graphite and a racemate by in situ scanning probe lithography. Enantioselective adsorption of chiral molecules is achieved by adjusting the relative orientation between the nanoconfining walls and substrate symmetry direction.
The influence of geometrical shape on stereoselective binding is well established at the molecular level in host–guest binding systems in solution1,2 but largely unexplored at larger scales. The emergence of homochirality in biological systems may have arisen from enantioselective processes at interfaces in these size ranges.3,4 From an application perspective, understanding enantiospecific interactions is crucial for the development of many research areas, such as chiral separation techniques, enantioselective heterogeneous catalysis5 and chiral functional materials in general.6
Visualizing chirality at interfaces provides huge insight into these process, and on atomically flat conductive surfaces, scanning tunnelling microscopy (STM)7 can provide sub-molecular resolution detail of enantiospecific interactions by imaging the adsorption and organization of chiral molecules.8 The creation of homochiral surfaces is of particular interest, which is typically achieved by the adsorption of enantiopure chiral molecules,9 although alternative approaches exist.10–12
However, the selective adsorption of only one enantiomer from a racemic mixture remains a major challenge. The formation of one enantiomorph in molecular systems forming a conglomerate could be suppressed based on enantiospecific hydrogen-bonds13 or van der Waals (vdW) interactions14 by the addition of enantiopure molecules. Another way to achieve enantioselective adsorption of chiral molecules is the use of intrinsically chiral surfaces.15 For example, chiral metal surfaces have been used successfully for the enantioselective adsorption16 and desorption17 of amino acids. The described approaches, however, rely on either molecular chirality or intrinsically chiral surfaces. Another way to induce chirality is symmetry breaking by external forces like electric or magnetic field or by introducing additional confinement. In three dimensions, nanoconfinement has been shown to significantly affect the formation and induction of chiral liquid-crystal phases.18 Chiral induction at surfaces and interfaces based on symmetry breaking using external forces has been achieved in the two-dimensional (2D) crystallization of 4-octyl-4′-cyanobiphenyl using an oriented magnetic field.19 The symmetry breaking elements to create chirality were the surface confinement, the substrate symmetry and a magnetic field orientation parallel to the interface.
We recently demonstrated that lateral confinement on a surface can be used as symmetry breaking element to create chirality in 2D crystallization, though for a prochiral molecule.20 Now, we show that this approach can be used to achieve enantioselective adsorption from a solution containing a racemic adsorbate. The lateral confinement is achieved by the formation of 2D nanocorrals, that are clean areas on a substrate of controlled shape, size and orientation surrounded by a covalently bound layer. The formation of these nanocorrals is realized by covalent functionalization of the basal plane of highly oriented pyrolytic graphite (HOPG) by reaction with the diazonium salt derived from 3,5-bis-tert-butylaniline that grafts the 3,5-bis-tert-butylphenyl (3,5-TBP) unit, followed by the local removal of the grafted TBP groups using an STM based nanolithography protocol20 called nanoshaving. Molecules in the solution phase exclusively adsorb and crystallize in these nanocorrals, forming self-assembled molecular networks (SAMNs).21 The nanoshaving is carried out at the liquid–solid interface. Therefore, while a well-defined area of the pristine HOPG surface is gradually exposed (Fig. 1a), SAMN formation can occur simultaneously. Further experimental details of this in situ nanoshaving protocol are described in the supporting information. The chiral preference is created by controlling the relative orientation of the two achiral symmetry breaking elements determining surface confinement, i.e. the HOPG surface and the square-shaped nanocorrals (Fig. 1a). Chiral nanoconfinement conditions are created when relative orientation of the square nanocorrals with respect to high-symmetry substrate directions do not coincide with certain angles (vide infra).
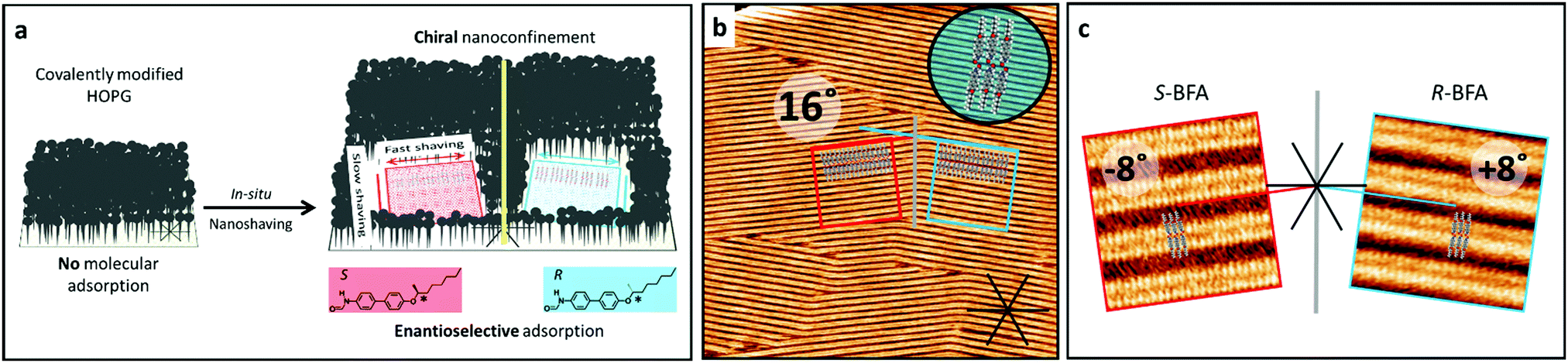 | ||
| Fig. 1 A cartoon showing the molecular structure of BFA and the creation of chiral nanoconfinement (see text) (a). The STM image in (b) shows the self-assembled structure of racemic BFA on bare HOPG, where it appears as a row-like structure with two different orientations that include an angle of 16°. The orientations of the molecules within the assembly and the chiral confinement are indicated (not to scale). High-resolution STM images show that the row structure is composed of head-to-head dimers and includes an angle of +8° or +8°, respectively, with respect to the HOPG substrate (c). The bright protrusions in the STM image correspond to the aromatic core of the molecules, while the alkyl-chains are in the darker area between the bright rows. The exact alignment of the alkyl-chains, however, could not be determined. Imaging parameters: 200 nm × 200 nm (b), 10 nm × 10 nm (c), Vb = 0.700 V, It = 70 pA. | ||
Here, we use this approach for the enantioselective adsorption of one enantiomer from a solution containing a racemate. The proposed mechanism at work involves a two-stage domain selection; (i) the nucleation stage in which the enantioselection takes place, followed by (ii) domain growth from the chiral seed (Fig. 1). Based on known selective domain adsorption in nanocorrals,20–22 a molecular system showing enantioselective adsorption in nanocorrals was postulated to require the following properties: (I) Anisotropy in the interactions along the two unit cell vectors of the SAMN and (II) conglomerate formation with different angles of the two enantiomorphs with respect to the underlying graphite lattice. As a model compound to demonstrate enantioselective adsorption, 4-[(S/R-1-methylheptyl)oxy]-4′-biphenylform-amide (BFA)23 was chosen. At high BFA concentrations (15 mM in 1-phenyloctane (PO)) row-like structures are formed, with two different orientations with respect to a high-symmetry substrate direction (Fig. 1b). These rows consist of head-to-head dimers (revealed by high-resolution STM images) that have an angle of +8° or −8°, respectively, between the row elongation direction and the high-symmetry substrate direction. A tentative model of the assembly shows that hydrogen bond formation is possible between the dimers as well as along the rows and vdW interactions along the rows (Fig. 1c). Enantiopure BFA (15 mM in PO) forms the same row-like structure on pristine HOPG, with one of the orientations and unit cell parameters formed by the racemate (Fig. S1, ESI†). That is, the rows of the enantiopure molecules have an angle of either −8° (S-BFA) or +8° (R-BFA), respectively, with respect to a high-symmetry HOPG lattice direction, while in the racemate domains with one or the other angle are both observed. Furthermore, high-resolution STM images show the same contrast and unit cell parameters for the structures formed by the enantiopure molecules as well as the racemate. Thus, the racemate assembles as a racemic conglomerate on pristine HOPG, i.e. the enantiomers separate into enantiomorphous domains. Note that this assignment is based on the domain orientations with respect to the underlying HOPG lattice, as we were not able to resolve other enantiomeric features of the molecules in the STM images.
Based on the intermolecular interactions in the 2D crystal structure, i.e. attractive interactions along the row structure, and previous results on domain selection,22 nanoshaving experiments were carried out with the fast shaving direction of square-shaped nanocorrals parallel to the row structure, i.e. with an angle of ±8° with respect to the substrate lattice (Fig. 1). This approach should preferentially lead to the formation of SAMNs in which the molecular rows run parallel to the fast shaving direction, thereby leading to enantioselective adsorption, and limiting the formation of rotational domains, i.e. domains at 60° or 120° where enantioselective adsorption is not expected.
The results of the enantiomer selection, summarized in Fig. 2, show that we were able to efficiently select the formation of one domain, i.e. the selective adsorption of only one enantiomer from a racemate. Thereby, the targeted enantiomer can be chosen simply by choosing the orientation of the square shaped nanocorrals with respect to the HOPG lattice as indicated in Fig. 1. In addition to the targeted enantiomer, which was found in about 62% of the corrals, its mirror domain containing the opposite enantiomer appeared in 10% of the corrals. Other rotational domains were found in 21% of the corrals and the remaining 7% contained more than one domain. Comparing these relative domain appearances with the theoretical random distribution of 16.67% for each of the six possible orientations clearly shows selectivity for the targeted enantiomer.
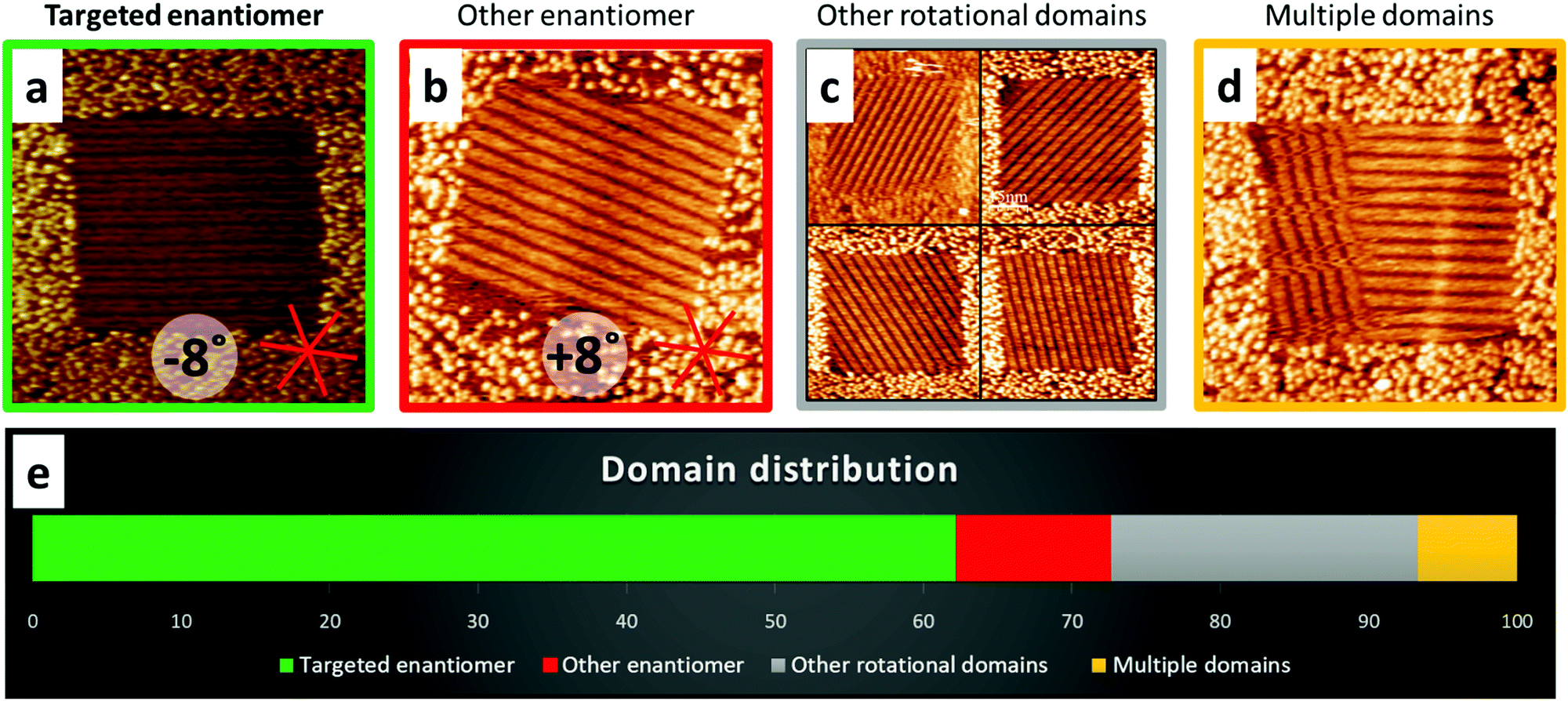 | ||
| Fig. 2 Representative STM images (a)–(d) of the different domains observed in chiral in situ nanocorrals and domain distribution when using a 15 mM racemic BFA solution in PO. The targeted domain (a) has the row structure running parallel to the top nanocorral border, i.e. the fast shaving direction and can be observed in about 62% of the nanocorrals. In addition to the targeted domain, the other enantiomer (b, 10%), rotational (c, 21%) or multiple domains (d, 7%) are observed. The statistics are from overall 621 nanocorrals. The HOPG orientation shown in a applies to all STM images shown. Imaging parameters: 70 nm × 70 nm, Vb = 0.700 V, It = 70 pA. | ||
The domain statistics are the sum of several independent measurement sessions with different samples and STM tips. Thereby, we observed that the selectivity varied between different sessions, in particular the relative amount of rotational domains (Fig. S2, ESI†). Changing the nanocorral shape from quadratic to rectangular or the nanoshaving speed did not have a significant impact on the session-to-session variance (Fig. S3, ESI†), leaving the STM tip condition as a factor affecting the selection efficiency. The possibility of the STM being intrinsically chiral may additionally affect the selection efficiency.24 The concentration window in which the enantioselective adsorption can be observed is very limited, because the nanocorrals often remain empty at lower concentrations22 and gel formation occurs at higher concentrations at room temperature for this particular compound (Fig. S4, ESI†). As a qualitative observation, selectivity often seemed to decrease over time within a session, i.e. after several hours of nanoshaving and imaging with the same tip. Therefore, the reason could be either a degrading tip, or slow adsorption of contaminations (Fig. S5, ESI†) between the grafted molecules, which then slows down the BFA assembly and reduces selectivity.
To further support that the lateral chiral confinement is the main factor responsible for the observed selectivity, control experiments with squares aligned with a high-symmetry HOPG substrate direction (in which the relative orientation of the assembled molecules with respect to the confinement is the same for both enantiomorphs) were carried out (Fig. 3a and c). In these achiral nanocorrals, domains with the smallest angle with respect to the fast-shaving direction formed preferentially, but, as expected for parity, without a significant chiral bias towards one of the enantiomers. Nanocorrals with multiple domains appeared more frequently in achiral nanocorrals, indicating that the mismatch between domain orientation and fast-shaving direction of the nanocorrals leads to more nucleation events. Additionally, rotational domain selection from enantiopure solutions (15 mM in PO) was performed. As to be expected, selecting rotational domains is possible with a very high efficiency and the mirror domain is not observed (Fig. 3c and d). Compared to the domain formation from a racemate, the number of rotational domains observed is also significantly lower. However, when using enantiopure BFA (15 mM) the polymorph which is formed at lower concentrations22 is found occasionally, which was not the case with the racemate (Fig. S6, ESI†).
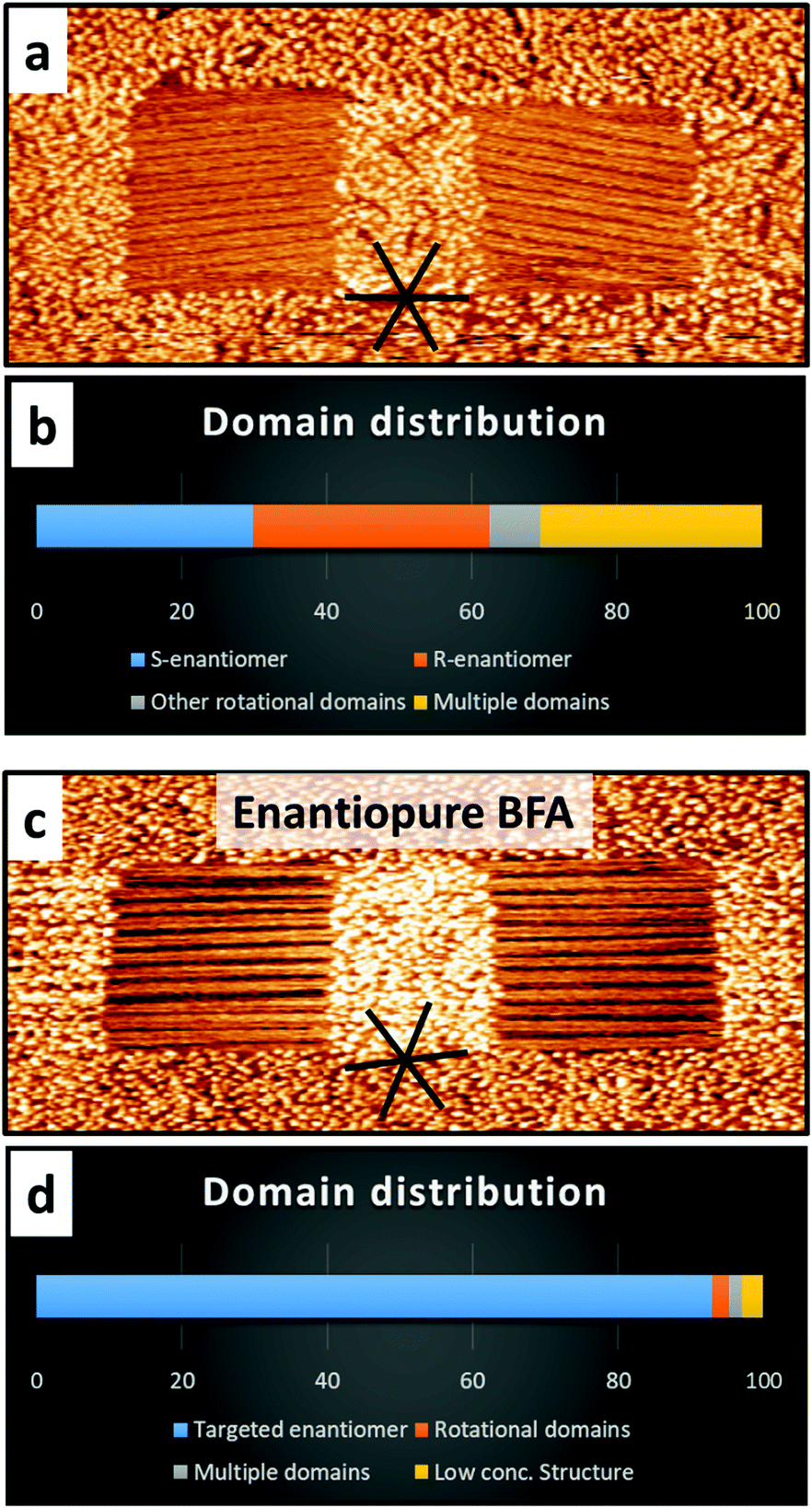 | ||
| Fig. 3 Representative STM image and corresponding statistics of the domain formation in squares aligned with a high-symmetry HOPG substrate direction, from a racemate (a and b, 301 nanocorrals). While domains with the smallest angle between the row structure and fast-shaving direction are formed preferentially, no chiral bias towards one enantiomer is found. Adsorption from enantiopure solution shows a very high selectivity towards the rotational domain parallel to the fast-shaving direction (c and d, 169 nanocorrals). Imaging parameters: 160 nm × 80 nm. Vb = 0.700 V, It = 70 pA. | ||
Anisotropic interactions in the 2D crystal structure are believed to play a major role in the domain selection process.22 A similar reasoning can be applied here, considering the directionality of the hydrogen bonds that form between the head-to-head dimers and potentially along the row in the 2D packing in the nanoconfined regions. Between adjacent rows, however, vdW interactions with limited intermolecular contact are present as there is no interdigitation of the alkyl chains. As a result, the formation of a single long row, which can assemble if the fast-shaving direction is parallel to the row direction, is expected to be favoured in the narrow rectangular nanocorral in the initial stages of its formation by nanoshaving. On the other hand, the formation of several adjacent short rows, which would be the case in the formation of other rotational domains, is less favoured (Fig. S7, ESI†). Similarly, the assembly of the mirror domain with an off-angle of ±16° with respect to the targeted enantiomer is less favoured. In other words, the domain selection is predominantly a kinetic effect, i.e. the first stable domain assembling during the nanoshaving process is the one assembling in the entire nanocorral.
In summary, we have shown the enantioselective adsorption from a solution of a racemate of chiral molecules in chiral lateral nanoconfinement at the solid/liquid interface. Chiral symmetry breaking was achieved by the combination of surface symmetry and additional lateral geometric confinement during nucleation. One enantiomer can be selected simply by choosing the relative orientation between the (achiral) substrate and the square-shaped region where monolayer growth is nanoconfined. Therefore, spatial confinement alone can provide chiral symmetry breaking to achieve enantioseparation in the assembly of chiral molecules.
The authors gratefully acknowledge financial support from the Fund of Scientific Research Flanders (FWO), KU Leuven – Internal Funds, and FWO under EOS 30489208. J. S. acknowledges financial support through a Marie Skłodowska-Curie Individual Fellowship (EU project 789865 – EnSurf). D. B. A. thanks the EPSRC (project EP/M005178/1) and the School of Chemistry at the University of Nottingham for funding. Z. T. acknowledges financial support through a FWO PhD fellowship fundamental research (fellowship 1182620N).
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1–73 CrossRef CAS.
- X. X. Zhang, J. S. Bradshaw and R. M. Izatt, Chem. Rev., 1997, 97, 3313–3361 CrossRef CAS.
- M. Zhang, G. Qing and T. Sun, Chem. Soc. Rev., 2012, 41, 1972–1984 RSC.
- A. González-Campo and D. B. Amabilino, Top. Curr. Chem., 2013, 333, 109–156 CrossRef.
- T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS.
- K. Ariga, T. Mori, T. Kitao and T. Uemura, Adv. Mater., 2020, 1905657 CrossRef CAS.
- M. Stöhr, Supramolecular Chemistry, John Wiley & Sons, Ltd, Chichester, UK, 2012 Search PubMed.
- F. Zaera, Chem. Soc. Rev., 2017, 46, 7374–7398 RSC.
- M. Ortega Lorenzo, C. J. Baddeley, C. Muryn and R. Raval, Nature, 2000, 404, 376–379 CrossRef.
- M. Parschau, S. Romer and K. H. Ernst, J. Am. Chem. Soc., 2004, 126, 15398–15399 CrossRef CAS.
- S. Haq, N. Liu, V. Humblot, A. P. J. Jansen and R. Raval, Nat. Chem., 2009, 1, 409–414 CrossRef CAS.
- I. Destoop, E. Ghijsens, K. Katayama, K. Tahara, K. S. Mali, Y. Tobe and S. De Feyter, J. Am. Chem. Soc., 2012, 134, 19568–19571 CrossRef CAS.
- C. Roth, D. Passerone and K. H. Ernst, Chem. Commun., 2010, 46, 8645–8647 RSC.
- J. Seibel, O. Allemann, J. S. Siegel and K. H. Ernst, J. Am. Chem. Soc., 2013, 135, 7434–7437 CrossRef CAS.
- J. D. Horvath and A. J. Gellman, Top. Catal., 2003, 25, 9–15 CrossRef CAS.
- Y. Yun and A. J. Gellman, Langmuir, 2015, 31, 6055–6063 CrossRef CAS.
- J. D. Horvath and A. J. Gellman, J. Am. Chem. Soc., 2002, 124, 2384–2392 CrossRef CAS.
- S. Shadpour, A. Nemati, J. Liu and T. Hegmann, ACS Appl. Mater. Interfaces, 2020, 12, 13456–13463 CrossRef.
- A. M. Berg and D. L. Patrick, Angew. Chem., Int. Ed., 2005, 44, 1821–1823 CrossRef CAS.
- J. Seibel, L. Verstraete, B. E. Hirsch, A. M. Braganca and S. De Feyter, J. Am. Chem. Soc., 2018, 140, 11565–11568 CrossRef CAS.
- L. Verstraete, J. Greenwood, B. E. Hirsch and S. De Feyter, ACS Nano, 2016, 10, 10706–10715 CrossRef CAS.
- J. Seibel, D. B. Amabilino and S. De Feyter, Angew. Chem., Int. Ed., 2019, 58, 12964–12968 CrossRef CAS.
- W. Mamdouh, H. Uji-i, A. Gesquière, S. De Feyter, D. B. Amabilino, M. M. S. Abdel-Mottaleb, J. Veciana and F. C. De Schryver, Langmuir, 2004, 20, 9628–9635 CrossRef CAS.
- H. L. Tierney, C. J. Murphy and E. C. H. Sykes, Phys. Rev. Lett., 2011, 106, 1–4 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional STM data. See DOI: 10.1039/d0cc06845d |
| This journal is © The Royal Society of Chemistry 2021 |