
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid build up of nanooptomechanical transduction in single crystals of a ruthenium-based SO2 linkage photoisomer†
Jacqueline M.
Cole
 *abcd,
David J.
Gosztola
d,
Jose de J.
Velazquez-Garcia
a,
SuYin
Grass Wang
e and
Yu-Sheng
Chen
e
*abcd,
David J.
Gosztola
d,
Jose de J.
Velazquez-Garcia
a,
SuYin
Grass Wang
e and
Yu-Sheng
Chen
e
aCavendish Laboratory, Department of Physics, University of Cambridge, J. J. Thomson Avenue, Cambridge, CB3 0HE, UK. E-mail: jmc61@cam.ac.uk
bISIS Neutron and Muon Facility, STFC Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Didcot, OX11 0QX, UK
cDepartment of Chemical Engineering and Biotechnology, University of Cambridge, West Cambridge Site, Philippa Fawcett Drive, Cambridge, CB3 0AS, UK
dArgonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, USA
eNSF's ChemMatCARS Beamline, The University of Chicago, Advanced Photon Source, Lemont, Illinois 60439, USA
First published on 8th December 2020
Abstract
Single-crystal nanooptomechanical transduction occurs in [Ru(SO2)(NH3)4(H2O)]chlorobenzenesulfonate2, reaching maximal levels within 40 s at 100 K when photostimulated by 505 nm light. Its in situ light-induced crystal structure reveals the molecular origins of this optical actuation: 26.0(3)% of the η1-SO2 ligand photoconverts into an η1-OSO photoisomer which, in turn, induces a 49.6(9)° arene ring rotation in its neighbouring counter ion.
Materials that display optical switching in crystalline media are of interest for a range of solid-state applications, from optical sensing1 to optical data storage.2 A sub-class of optical switches exhibit single-crystal nanooptomechanical transduction, whereby a light-driven switching process in one molecule or ion stimulates mechanical motion in a neighbouring molecule or ion. Nanooptomechanical transducers may be employed in light-driven molecular machinery3–5 or nanotechnology;6 when observed in single-crystal form, they have prospective use in futuristic circuitry for quantum computers.7 Such functionality in materials is rare, especially in single crystals, which offer a high-quality solid-state medium for single-photon control. Only a few materials have demonstrated such function;8–13 this includes a family of ruthenium-based complexes that undergo SO2-linkage photoisomerisation in single crystals.8–11 The generic formula for these complexes is trans-[Ru(SO2)(NH3)4X]m+ Yn where X is the ligand that lies trans to the SO2-linkage isomer and Y is the counter ion, with the integer values of m and n being dictated by the charge-balancing requirements of X and Y. Certain combinations of X and Y afford nanooptomechanical transduction.8–11 A concerted two-step molecular mechanism is responsible for this functionality, whereby an S-bound η1-SO2 ligand photoisomerises to an O-bound η1-OSO photoisomer. The non-bound oxygen in this photoisomer protrudes from the cation within the crystal lattice, in a direction such that it lies uncomfortably close to one of the counter ions. Pending the anion contains an arene ring, it rotates in order to alleviate the crystal-lattice strain that results from the close proximity of the η1-OSO ligand.
This paper reports the discovery of a new nanooptomechanical transducer in this family of ruthenium-SO2 based complexes: trans-[Ru(SO2)(NH3)4(H2O)]chlorobenzenesulfonate2 (1). Scheme 1 presupposes its two-step operational mechanism based on our previous work.8–11 This is confirmed by its light-induced crystal structure which is revealed by in situ light-induced single-crystal X-ray diffraction, a recently developed technique that has become known as photocrystallography.14–17 Concerted single-crystal optical absorption spectroscopy and microscopy is then employed to determine the visible absorption characteristics of a single crystal of 1 as well as reveal its anticipated photochromic effects. Kinetic aspects of this optical metrology will reveal that the nanooptomechanical transduction in 1 builds up macroscopically very rapidly.
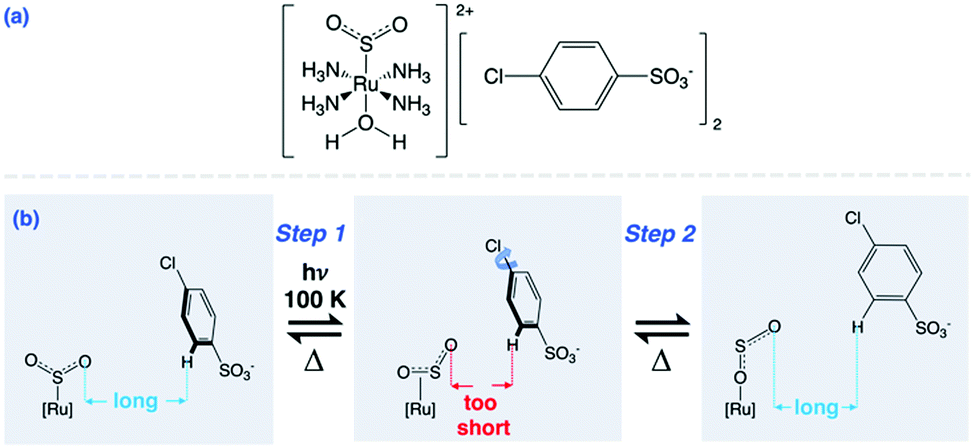 | ||
| Scheme 1 (a) Dark-state chemical diagram of 1; (b) η1-SO2 to η1-OSO photoisomerisation in 1 which induces nanooptomechanical transduction via arene ring rotation to reduce interionic strain. | ||
1 was synthesised from trans-[Ru(SO2)(NH3)4Cl]Cl, which was prepared according to a literature procedure.18 5 mg (16 mmol) of this precursor was dissolved in a mixture of Na2CO3 (1 M, 350 mL) and methanol (350 mL), into which a distilled water/methanol (1/1, v/v) solution of p-chlorobenzenesulfonic acid (100 mL, 2 M; >90% purity, Sigma Aldrich) was added dropwise. Red plate-like crystals precipitated from solution which were collected by filtration and washed with methanol.
The light-induced crystal structure of 1 was determined using synchrotron-based X-ray photocrystallography at the ChemMatCARS beamline of the Advanced Photon Source, Argonne National Laboratory, IL, USA. See ESI† for full crystallography details; just the relevant photostimulation procedure is given here. A reference dataset for the dark-state crystal structure was first obtained from a 50 × 25 × 25 μm3 single crystal. A 505 nm light (Thorlabs M505L3 LED) was then shone onto the crystal for 2.5 h while it was held at 100 K on the diffractometer. This light was switched off before acquiring data for the light-induced crystal structure. For control purposes, ambient lighting in the experimental hutch of the beamline was extinguished while all data were being collected.
The 505 nm light-induced crystal structure of 1 is shown in Fig. 1. 26.0(3)% of its dark-state η1-SO2 ligand has photoconverted into an O-bound η1-OSO photoisomer. 22.4(5)% of the arene ring in one of the chlorobenzenesulfonate counter ions has rotated by 49.6(9)° in response to the formation of the photoisomer. This is the nanooptomechanical transduction. The dark-state structure remains the dominant residue, as is witnessed in the majority of cases for this family of complexes. The clean conversion from the η1-SO2 isomer to the η1-OSO photoisomer is notable since η1-OSO formation in this series of complexes is generally accompanied by a discernible fraction of the side-bound η2-(OS)O photoisomer.8–10 To this end, there is only one other reported instance of a clean conversion which occurs in trans-[Ru(SO2)(NH3)4(3-phenylpyridine)]Cl2·H2O where 52(3)% of the η1-OSO photoisomer is shown to form.19 Its small counter ions render it to behave as an optical switch. Thus, 1 is the only reported nanooptomechanical transducer whose η1-OSO photoisomer is not polluted by another type of photoisomer. The practical ramifications of such a clean conversion could ultimately lead 1 to be more favourable than other complexes in prospective applications.
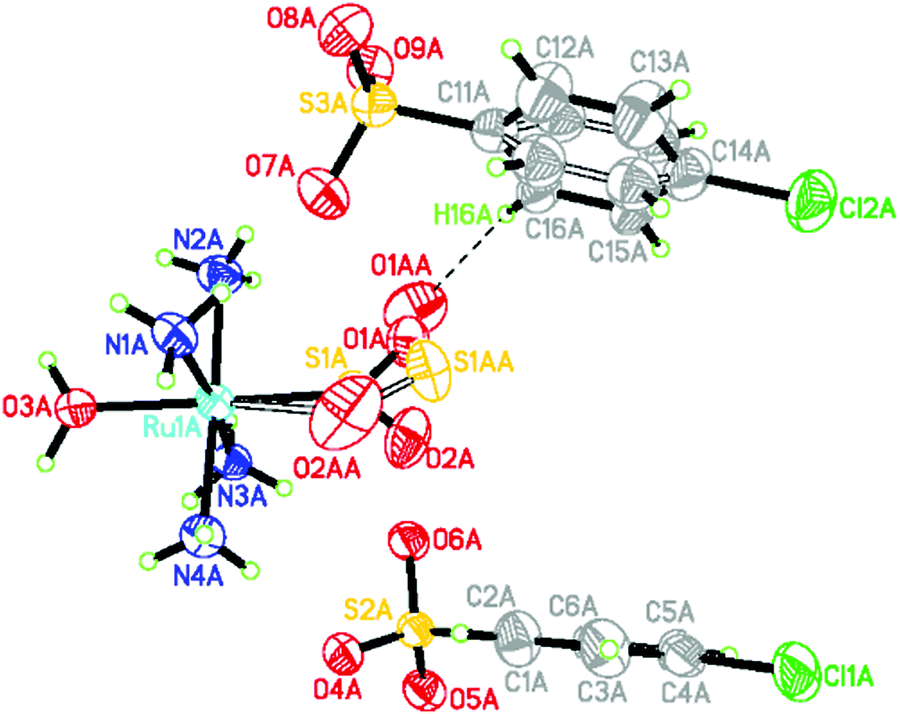 | ||
| Fig. 1 Light-induced crystal structure of 1. Disordered species have hollow bonds. The close O1AA⋯H16A contact (2.265(7) Å) between the η1-OSO ligand and arene ring has a dashed line. | ||
The bond geometry of the light-induced structure of 1 at 100 K is very similar to that of the 90 K structure of trans-[Ru(SO2)(NH3)4(H2O)]tosylate2,20 aside from the SO2 and an arene ring in one of the counter ions which are affected by nanooptomechanical transduction in 1. The SO2 ligand in trans-[Ru(SO2)(NH3)4(H2O)]tosylate2 photoconverts into an η2-(OS)O photoisomer at 100 K, and thus behaves as an optical switch rather than a transducer. The two complexes chemically differ by only one substituent: a Cl rather than methyl substituent in each counter ion. This structural comparison illustrates nicely how a subtle detail of the counter ions can completely alter the optical properties in this series of complexes.
Despite taking all of the usual experimental precautions that are usually successful in our photocrystallography work, the reference dark-state crystal structure of 1 was polluted by some photoinduced species. Thereby, the ‘dark-state’ structure displayed 12.0(3)% of the η1-OSO photoisomer and 17.6(4)% of a corresponding arene ring disorder which displays 48.7(9)° rotation. One expects a pure η1-SO2 configuration and ordered arene ring in its true dark state, judging from all other materials in this family of complexes. This expectation is at least corroborated by the much lower photoconversion fractions found in this ‘dark’ structure compared to the deliberately intended light-induced structure of 1.
On the face of it, the light pollution is curious since only ambient light from the experimental hutch of the synchrotron beamline would have impinged upon the sample, once it had been cooled to 100 K where it might be susceptible to photoisomerisation; and even then, the crystal was only exposed to this ambient light for the few minutes that was required to centre the crystal on the diffractometer. The ambient lighting had been switched off for the entire data collection, as per a standard control measure that is employed in our photocrystallography experiments. A pure dark-state structure could not be obtained despite best efforts. This was unusual. So, a series of concerted single-crystal optical absorption spectroscopy and microscopy experiments on this same crystal of 1 were employed in order to understand why.
Single-crystal optical absorption spectroscopy and microscopy were performed on 1, using the custom-built micro-spectroscopy system described by Cole and co-workers.19,21 For micro-spectroscopy details, see ESI.† The crystal was photosimulated by the 505 nm LED source at 100 K. The 505 nm light-excitation power measured at the 20×, 0.3 NA objective (Olympus UMPlanFL) was 360 μW, giving an estimated 261 μW mm−2 illuminating the field of view.
Single-crystal optical absorption spectra of 1, acquired after t minutes of light exposure at 100 K, are shown in Fig. 2. These results reveal that the photoisomeric fraction in 1 build up very fast: the crystal essentially attains its maximum optical absorbance after only 40 s of 505 nm light exposure. This compares with at least several hours of light exposure that is normally required to maximise photoconversion levels in these types of complexes.19,21 The build up of nanooptomechanical transduction in 1 will likewise be very fast, judging from the highly correlated percentage growth of the η1-OSO species and that of the rotated arene ring; and given that the arene ring rotates as a direct consequence of η1-OSO formation.8 The rapid build up of nanooptomechanical transduction explains our inability to obtain a pure dark-state crystal structure of 1.
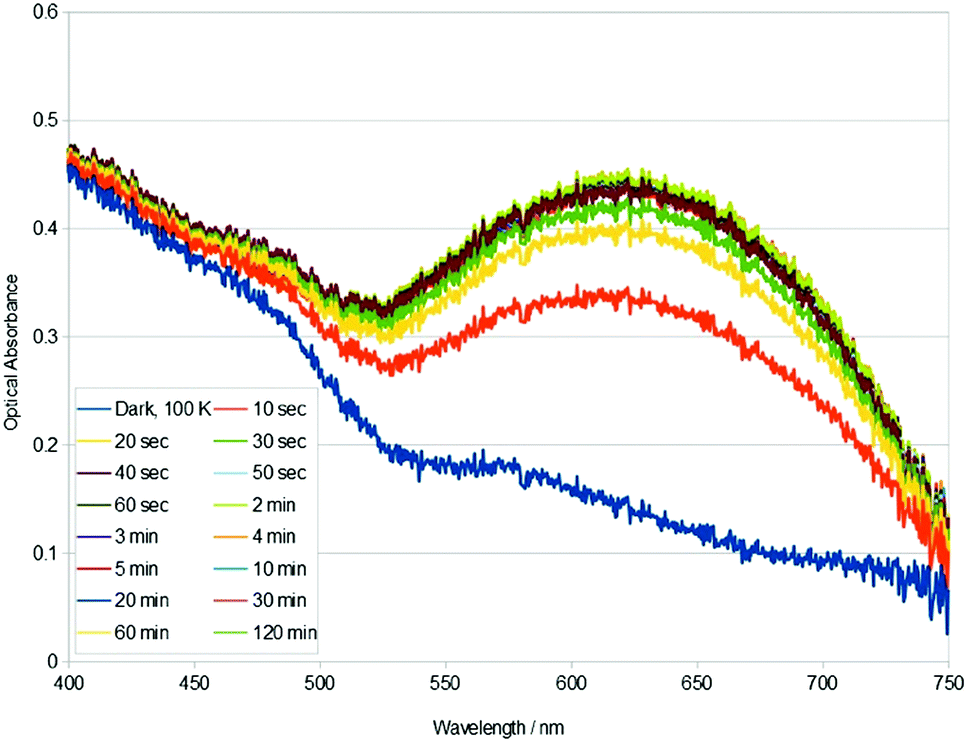 | ||
| Fig. 2 Single-crystal optical absorption spectra of 1, acquired after t minutes of 505 nm light exposure at 100 K. A reference dark-state spectrum of 1 at 100 K is provided. | ||
The rate constant associated with the photoisomerisation process in 1 was calculated using the light-induced area under each curve in Fig. 2 as a proxy metric for the concentration of photoisomer species. The calculation proceeded in four steps, as described in the ESI.† This yielded a rate constant, k = 7.90 × 10−3 s−1, for the η1-SO2 (dark) to η1-OSO (light) photoisomerisation process in 1, from a linear fit whose coefficient of determination, R2 = 0.991. This is the first time that k has been evaluated for the photoisomerisation process in this family of complexes; yet, we have observed qualitatively that it is a faster process than other complexes in the series.8–10
Metal–ligand charge-transfer (MLCT) effects tend to dominate the optical absorption of ruthenium-based coordination complexes.22–24 Changes in the optical absorption profile of 1 in response to light exposure (Fig. 2) are therefore expected to be indicative of MLCT bands that are influenced by η1-OSO and dark η1-SO2 isomers, and the arene ring rotation, given the light-induced crystal structure of 1. The light-induced rise of the broad band in the 525–750 nm spectral region of 1 (Fig. 2) is echoed in the optical absorption spectral changes observed for other complexes in this family of materials, in those that form η2-(OS)O or η1-OSO photoisomers.19,21 However, this rise in optical absorption is followed by a rise of additional bands in these other complexes of this family of materials. Crystals of such complexes that display η2-(OS)O photoisomerisation adopt almost ‘black-body’ absorption within the visible spectrum, upon external light exposure.21 In contrast, the rise in optical absorption of 1 ceases after the formation of the broad band shown in Fig. 2. trans-[Ru(SO2)(NH3)4(3-phenylpyridine)]Cl2·H2O represents the only previous report of a η1-SO2 to η1-OSO photoisomerisation that is not accompanied by η2-(OS)O formation.19 Its changes in optical absorption in response to 505 nm light exposure are broadly similar to those of 1, at least within the part of the crystal that lies under the surface that is shown to peel macroscopically; yet, this peeling effect in that crystal, its twinned nature, and its lack of nanooptomechanical transduction behaviour, render difficult a like-for-like comparison of its optical absorption spectra with those of 1. Fig. 2 would therefore seem to provide a representative optical absorption profile for a complex in this series of materials that exclusively exhibits the η1-OSO photoisomer together with nanooptomechanical transduction.
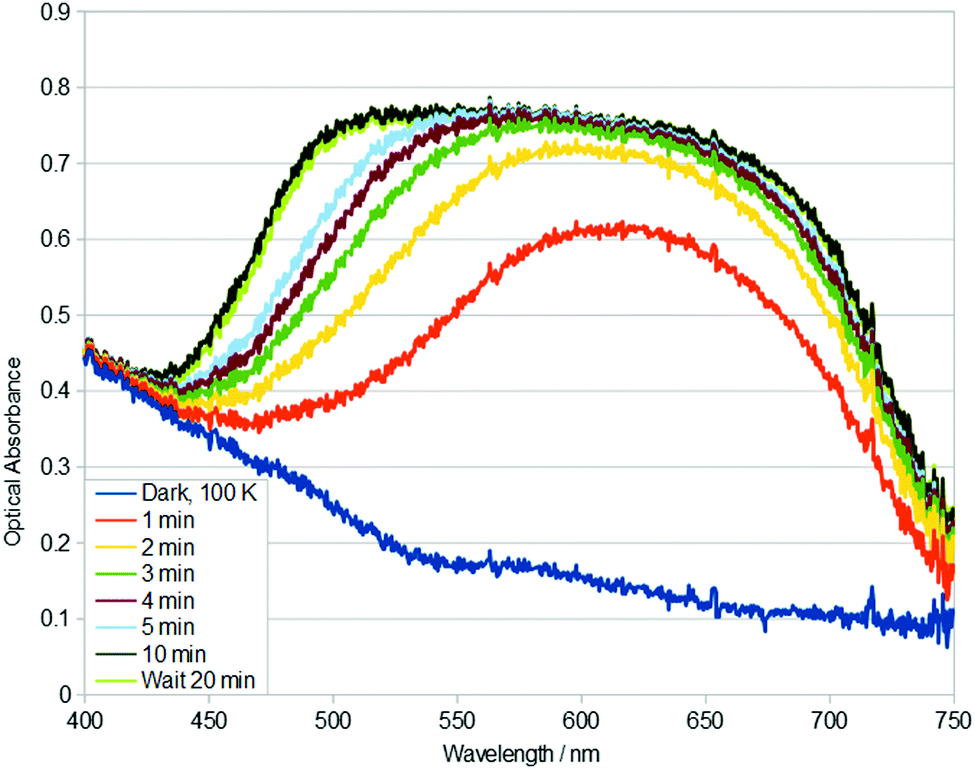 | ||
| Fig. 3 Single-crystal optical absorption spectra of 1, acquired after t minutes of 405 nm light exposure at 100 K. A reference dark-state spectrum of 1 at 100 K is given. | ||
Optical microscopy images that accompany these optical spectroscopy findings show that the crystal of 1 is photochromic, transforming from an orange to bluey-grey colour over the duration of 505 nm light exposure (see Video, ESI†). These photochromic effects corroborate the optical absorption spectral profiles observed.
The light-induced structure of 1 thermally decays back to its dark state; its decay rate was too fast to measure reliably with our instrument set up, although it was observed that full decay occurred within 2 minutes at T = 160 K.
Given the rapid build up of nanooptomechanical transduction in 1 at 100 K, even in the ambient light conditions of the experimental hutch of the synchrotron for just a few minutes, the optical absorption spectral characteristics of 1 were explored using two additional wavelengths of external light stimulation: 405 nm and 455 nm. Single-crystal optical absorption spectra of 1 acquired after t minutes of 405 nm light exposure at 100 K are displayed in Fig. 3. The same rise in optical absorption within the 525–750 nm spectral region is observed for 1 upon 405 nm and 505 nm light stimulation. Yet, this rise is slower with 405 nm light activation, and then a new charge-transfer (CT) band opens up in its 450–550 nm region, yielding almost ‘black-body’ absorption in the visible spectrum. 455 nm light stimulus also exhibits this CT band (see ESI†).
This ‘black-body’ like absorption is seen in members of this family of complexes that exhibit η2-(OS)O photoisomers.21 This suggests that the growth of this low-wavelength CT band could be MLCT that arises from η2-(OS)O photoisomer formation. Indeed, η1-OSO photoisomers usually form together with a discernible fraction of η2-(OS)O photoisomers. The 405 nm and 455 nm stimulated photoisomers thermally decay to their dark state at 160 K (see ESI†).
Rate constants could not be determined for the 405 nm stimulated photoisomerisation or thermal decay of 1, because the process was not first order over the full data range. However, plots of temperature versus ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k were approximately linear for two disparate sets of data, before and after the onset or deterioration of the new CT band, respectively. cf. k = 3.174 × 10−3 s−1 (R2 = 0.936) or k = 6.586 × 10−4 s−1 (R2 = 0.984) for t = 0–3 min or t = 3–5 min of 405 nm light exposure; while k = 3.398 × 10−3 s−1 (R2 = 0.952) or k = 7.838 × 10−3 s−1 (R2 = 1.000) for t = 0–6 min or t = 8–12 min time lapse after the crystal of 1 had reached 160 K, respectively. These bifurcated results further corroborate the notion that a new CT band opens up in the 405 nm data, while it is absent in the 505 nm data. The 455 nm data behave similarly to the 405 nm data (see ESI†).
k were approximately linear for two disparate sets of data, before and after the onset or deterioration of the new CT band, respectively. cf. k = 3.174 × 10−3 s−1 (R2 = 0.936) or k = 6.586 × 10−4 s−1 (R2 = 0.984) for t = 0–3 min or t = 3–5 min of 405 nm light exposure; while k = 3.398 × 10−3 s−1 (R2 = 0.952) or k = 7.838 × 10−3 s−1 (R2 = 1.000) for t = 0–6 min or t = 8–12 min time lapse after the crystal of 1 had reached 160 K, respectively. These bifurcated results further corroborate the notion that a new CT band opens up in the 405 nm data, while it is absent in the 505 nm data. The 455 nm data behave similarly to the 405 nm data (see ESI†).
The rate constant for 505 nm light exposure of 1 at 100 K is faster that that at 405 nm. Its photoisomerisation process will thus dictate the nature of the ‘dark’ structure that resulted from modest exposure to ambient light conditions at 100 K.
In summary, nanooptomechanical transduction from 505 nm light activation of a single crystal of 1 has been showcased in this work. Its rapid build up in single crystals of 1 may have practical implications for its ultimate prospects in solid-state optical device technology. Further work is required to confirm the nature of the extra CT band that is seen with 405 nm or 455 nm light stimulation. Pending confirmation that it is the anticipated η2-(OS)O photoisomer, it is exciting to consider the prospect that η1-OSO and η2-(OS)O photoisomerisation could be alternated in the same crystal using different light sources.
J. M. C. is grateful for the BASF/Royal Academy of Engineering Research Chair in Data-Driven Molecular Engineering of Functional Materials, which is partly supported by the Science and Technology Facilities Council via the ISIS Neutron and Muon Facility. J. M. C. also thanks the 1851 Royal Commission of the Great Exhibition for the 2014 Fellowship in Design, hosted by Argonne National Laboratory where work done was supported by the U.S. Department of Energy (DOE) Office of Science, Office of Basic Energy Sciences, and used research resources of the Center of Nanoscale Materials and the Advanced Photon Source (APS), Office of Science User Facilities operated for the DOE Office of Science by Argonne National Laboratory, supported by the U.S. DOE, all under contract no. DE-AC02-06CH11357. Within the APS, the NSF's ChemMatCARS Sector 15 is supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation (NSF), under grant no. NSF/CHE-1346572 and NSF/CHE-1834750. Use of the PILATUS 3X CdTe 1M detector is supported by the NSF under grant no. NSF/DMR-1531283. J. d. J. V.-G. acknowledges the National Council of Science and Technology of Mexico (CONACyT) and the Cambridge Trust for a PhD Scholarship (217553).
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Liu, F. Pagliano, R. van Veldhoven, V. Pogoretskiy, Y. Jiao and A. Fiore, Nat. Commun., 2020, 11, 2407 CrossRef CAS PubMed.
- M. Imlau, Th. Woike, R. Schieder and R. A. Rupp, Europhys. Lett., 2001, 53, 471–477 CrossRef CAS.
- N. Koumura, R. Zijlstra, R. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- N. Ruangsupapichat, M. M. Pollard, S. R. Harutyunyan and B. L. Feringa, Nat. Chem., 2011, 3, 53–60 CrossRef CAS PubMed.
- J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, ACS Nano, 2015, 9, 7746–7768 CrossRef CAS PubMed.
- L. Midolo, A. Schliesser and A. Fiore, Nat. Nanotechnol., 2018, 13, 11–18 CrossRef CAS PubMed.
- J. Bochmann, A. Vainsencher, D. D. Awschalom and A. N. Cleland, Nat. Phys., 2013, 9, 712–716 Search PubMed.
- S. O. Sylvester and J. M. Cole, Adv. Mater., 2013, 25, 3324–3328 CrossRef CAS PubMed.
- S. O. Sylvester and J. M. Cole, J. Phys. Chem. Lett., 2013, 4, 3221–3226 CrossRef CAS.
- S. O. Sylvester, J. M. Cole, P. G. Waddell, H. Nowell and C. Wilson, J. Phys. Chem. C, 2014, 118, 16003–16010 CrossRef CAS.
- K. T. Mukaddem, J. M. Cole, K. A. Beyer and S. O. Sylvester, J. Phys. Chem. C, 2020, 124, 10094–10104 CrossRef CAS.
- F. Tong, W. Xu, T. Guo, B. F. Lui, R. C. Hayward, P. Palffy- Muhoray, R. O. Al-Kaysi and C. J. Bardeen, J. Mater. Chem. C, 2020, 8, 5036–5044 RSC.
- R. O. Al-Kaysi, F. Tong, M. Al-Haidar, L. Zhu and C. J. Bardeen, Chem. Commun., 2017, 53, 2622–2625 RSC.
- P. Coppens, D. V. Fomitchev, M. D. Carducci and K. Culp, J. Chem. Soc., Dalton Trans., 1998, 6, 865–872 RSC.
- J. M. Cole, Chem. Soc. Rev., 2004, 33, 501–513 RSC.
- J. M. Cole, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 259–271 CrossRef CAS PubMed.
- J. M. Cole, Analyst, 2011, 136, 448–455 RSC.
- L. H. Vogt, J. L. Katz and S. E. Wiberley, Inorg. Chem., 1965, 4, 1157–1163 CrossRef CAS.
- J. M. Cole, J. d. J. Velazquez-Garcia, D. J. Gosztola, S.-Y. G. Wang and Y.-S. Chen, Chem. Mater., 2019, 31, 4927–4935 CrossRef CAS.
- A. Y. Kovalevsky, K. A. Bagley and P. Coppens, J. Am. Chem. Soc., 2002, 124, 9241–9248 CrossRef CAS PubMed.
- J. M. Cole, J. d. J. Velazquez-Garcia, D. J. Gosztola, S.-Y. G. Wang and Y.-S. Chen, Inorg. Chem., 2018, 57, 2673–2677 CrossRef CAS PubMed.
- E. Tfouni, Coord. Chem. Rev., 2000, 196, 281–305 CrossRef CAS.
- R. S. Lumpkin, E. M. Kober, L. A. Worl, Z. Murtaza and T. J. Meyer, J. Phys. Chem., 1990, 94, 239–243 CrossRef CAS.
- J. J. Rack, Coord. Chem. Rev., 2000, 253, 78–85 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2035266 and 2035267. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc06755e |
| This journal is © The Royal Society of Chemistry 2021 |