
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntibody drug conjugates with hydroxamic acid cargos for histone deacetylase (HDAC) inhibition†
Claudio
Cianferotti
 a,
Valentina
Faltoni
a,
Elena
Cini
a,
Elena
Ermini
a,
Francesca
Migliorini
a,
Elena
Petricci
a,
Maurizio
Taddei
*a,
Laura
Salvini
b,
Gianfranco
Battistuzzi
c,
Ferdinando Maria
Milazzo
c,
Anna Maria
Anastasi
c,
Caterina
Chiapparino
c,
Rita
De Santis
c and
Giuseppe
Giannini
*c
a,
Valentina
Faltoni
a,
Elena
Cini
a,
Elena
Ermini
a,
Francesca
Migliorini
a,
Elena
Petricci
a,
Maurizio
Taddei
*a,
Laura
Salvini
b,
Gianfranco
Battistuzzi
c,
Ferdinando Maria
Milazzo
c,
Anna Maria
Anastasi
c,
Caterina
Chiapparino
c,
Rita
De Santis
c and
Giuseppe
Giannini
*c
aDipartimento di Biotecnologie, Chimica e Farmacia, Università di Siena, Via A. Moro 2, 53100 Siena, Italy. E-mail: maurizio.taddei@unisi.it
bFondazione Toscana Life Science, Via Fiorentina 1, 53100, Siena, Italy
cR&D Alfasigma S.p.A., Via Pontina, Km. 30.400, 00071 Pomezia, Roma, Italy. E-mail: Giuseppe.Giannini@alfasigma.com
First published on 12th January 2021
Abstract
Antitumor hydroxamates SAHA and Dacinostat have been linked to cetuximab and trastuzumab through a non-cleavable linker based on the p-mercaptobenzyl alcohol structure. These antibody drug conjugates (ADCs) were able to inhibit HDAC in several tumour cell lines. The cetuximab based ADCs block human lung adenocarcinoma cell proliferation, demonstrating that bioconjugation with antibodies is a suitable approach for targeted therapy based on hydroxamic acid-containing drugs. This work also shows that ADC-based delivery might be used to overcome the classical pharmacokinetic problems of hydroxamic acids.
Hydroxamic acids are a peculiar type of organic molecules featuring a large variety of biological activities. They are potent enzyme inhibitors binding lipoxygenase, hydroxylase, matrix-metalloproteinase, histone deacetylase, carbonic anhydrase, ribonucleotide reductase and several other enzyme families.1 Besides this wide range of inhibition activities, hydroxamic acids have found therapeutic applications in cancer,2 cardiovascular diseases, including hypertension,3 tuberculosis,4 HIV infection,5 and Alzheimer's disease.6 Most of their biological activity is related to the complexation of metals present in enzymes through interaction with N-hydroxy and carbonyl groups.7 They are excellent bidentate ligands for metals such as iron, nickel and zinc. However, translation of this exceptional potential to real drugs has been highly limited by several drawbacks such as mutagenicity, poor pharmacokinetics and several off-target interactions, resulting in ventricular repolarization impairment, thrombocytopenia, gastrointestinal toxicity and several other adverse side effects.8
Hydroxamic acids are privileged structures to block histone deacetylase (HDAC) enzymes, and HDAC inhibitors (HDACis) have emerged as one of the most likely classes of multifunctional anticancer drugs.9 Cell cycle arrest, apoptosis, angiogenesis regulation, activation of tumour suppressor genes and oncogene suppression are some of the down-stream effects observed upon pharmacological treatment with HDACis.3
Although acting on the pharmacophore shape through the HDACis structure modification might improve the target selectivity,10 an attractive alternative would be redirecting the inhibitor to a suitable target using selective delivery. Few examples of hydroxamic acid pro-drug delivery systems have been described to date.11
One of the most efficient methods for drug delivery is the antibody–drug conjugate (ADC) strategy, where the antibody drives the drug inside the cell expressing the antibody receptor.12 Once internalized, the antibody–drug linker is cleaved, and the drug is released inside the malignant cells.13 The FDA has approved half a dozen ADCs, all of them carrying highly cytotoxic payloads like vedotin, emtansine and ozogamicin, and many others are under clinical trials.14 It is worth noting that trastuzumab emtansine (T-DM1, Kadcyla) is the only product approved for the treatment of solid tumours.
The current linker technology allows charging antibodies mostly with drugs carrying amines, alcohols or phenols through carbamates or carbonates, while the bioconjugation of drugs with other functional groups is still an undeveloped issue. It is not surprising that the only example of hydroxamic acid linked to antibodies deals with bifunctional chelates bearing hydroxamate arms for the radiometal labeling of monoclonal antibodies.15
Following our long-standing interest in HDAC inhibition,16 we report here the first example of ADC cargos carrying the FDA approved Vorinostat/SAHA (1) and Dacinostat/NVP-LAQ824 (4),17 a highly potent HDAC pan-inhibitor with IC50 = 32 nM (Scheme 1). These products were conjugated with cetuximab (Ctx) or trastuzumab (Trast), and a preliminary study on their biological activity confirmed the efficient targeting of HDACs within the tumour cells.
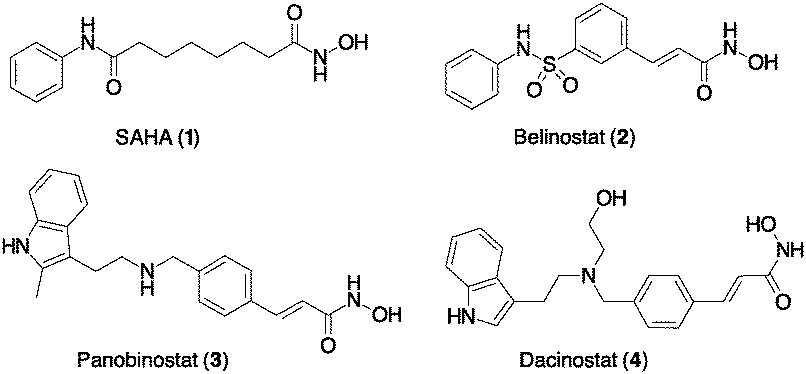 | ||
| Scheme 1 Hydroxamic acids as power HDAC inhibitors. Compounds 1–3 have been approved by the FDA. | ||
Based on our experience with bioconjugation,18 we started to explore the possibility to link SAHA with mAbs through non-cleavable linkers. Many non-cleavable linkers are based on 3-thiol–maleimide conjugates. During the lysosomal ADC metabolism, a retro Michael-type reaction or a thiol exchange of thioether succinimide occurs in response to thiol-containing environments, enabling drug release.19 Compound 10 (Scheme 2a) was designed as a potential cargo for antibody conjugation through the amidation of antibody lysines, via N-hydroxysuccinimide (NHS) activation. Thus, Michael acceptor succinimide 5 was reacted with freshly prepared p-mercaptobenzyl alcohol (PMBA, 6) with the formation of the thioether–maleimide derivative 7. Alkylation of 7 through the corresponding bromide was accomplished with SAHA in the presence of NaOH, resulting in compound 8 in acceptable yields.
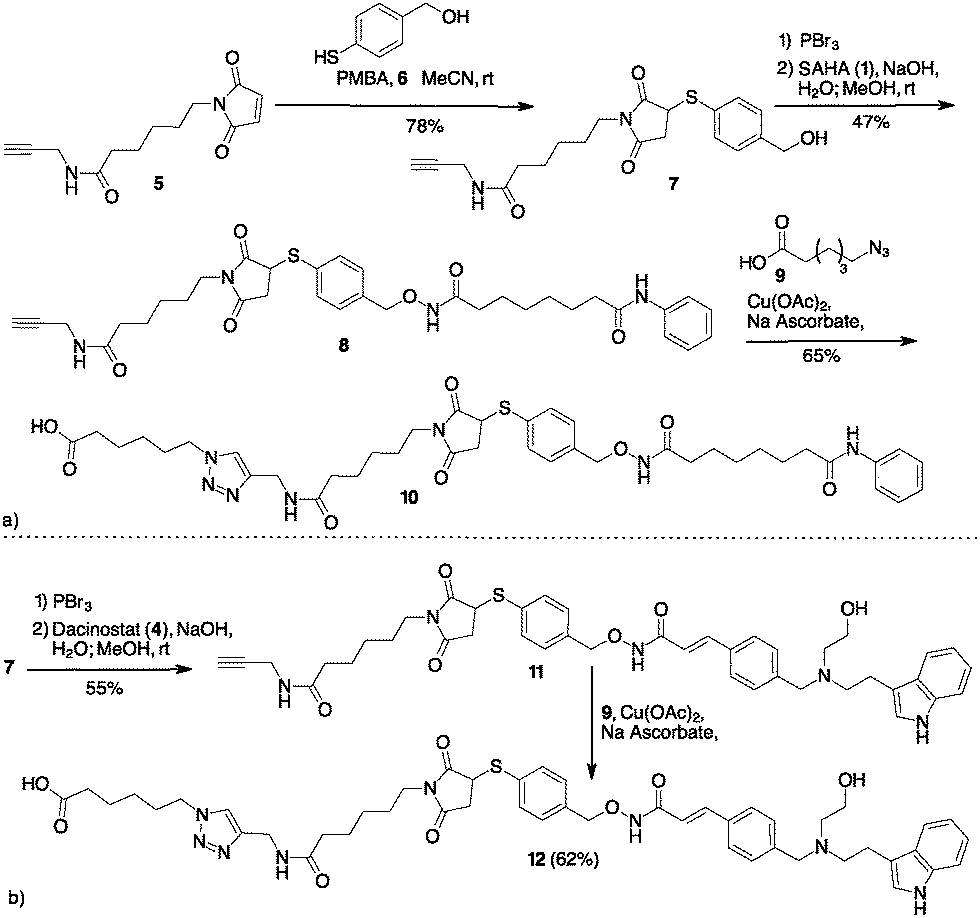 | ||
| Scheme 2 (a) Preparation of SAHA containing cargo for lysine bioconjugation. (b) Preparation of Dacinostat containing cargo for lysine bioconjugation. | ||
Compound 8 was subject to a Cu catalysed Huisgen reaction with 6-azidohexanoic acid 9 in the presence of Cu(II) acetate and sodium ascorbate in DMF/water, providing the desired compound 10 in 65% yield (Scheme 2a). A similar approach permitted to link Dacinostat (4) that was transformed into product 11 with 55% overall yield starting from 7 (Scheme 2b). Cu catalyzed Huisgen cycloaddition furnished 12 in 62% yield.
The synthesis of a Cathepsin B cleavable linker 16 started with the amide 13 composed by dipeptide Fmoc-Val-Cit-OH and p-amino benzyl alcohol (PABA) (Scheme 3). Compound 13 was converted into the corresponding bromide 14 with PBr3.18 The final introduction of SAHA proceeded in aqueous NaOH to afford product 15. Compounds 10, 12 and 15 were stable in PBS and in the human plasma at 37 °C for 24 h (see the ESI,† page 8).
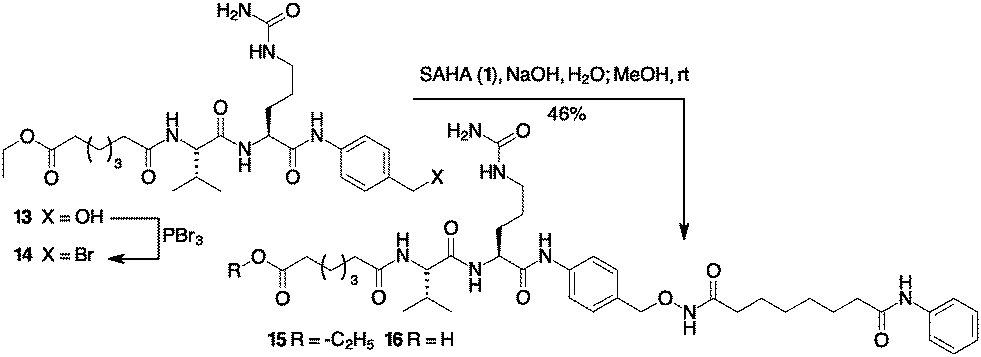 | ||
| Scheme 3 Preparation of Cathepsin B sensitive cargo containing SAHA. | ||
Cargos 10, 12 and 16 (obtained through hydrolysis of 15 with LiOH) were transformed into the corresponding NHS-esters (ESI,† page 7) and reacted with Ctx, a monoclonal antibody (mAb) specific for the epidermal growth factor receptor (EGFR), and Trast, an mAb targeting the ErbB2 receptor. Conjugation through the stable amide bond afforded ADCs 17–20 (Scheme 4).
Purification of 17–20 was carried out by dialysis, and the drug antibody ratio (DAR) was determined by MALDI analysis. The data show an acceptable degree of homogeneity for ADCs linked through lysine to intact antibodies (Fig. S1–S4, ESI†). The binding of the new ADCs (17–20) to EGFR or ErbB2 receptors on tumour cells was assessed by flow cytometry (FACS analysis) on A549 (human lung carcinoma), SKBR3 (human breast carcinoma) and Capan-1 (human pancreas carcinoma) cell lines, expressing different levels of receptors. All ADCs interact with their specific receptor target with a potency comparable to the unconjugated mAbs (Fig. S5, ESI†).
Investigation of ADC internalization was then carried out on the same cell lines by high content screening (HCS) imaging analysis.
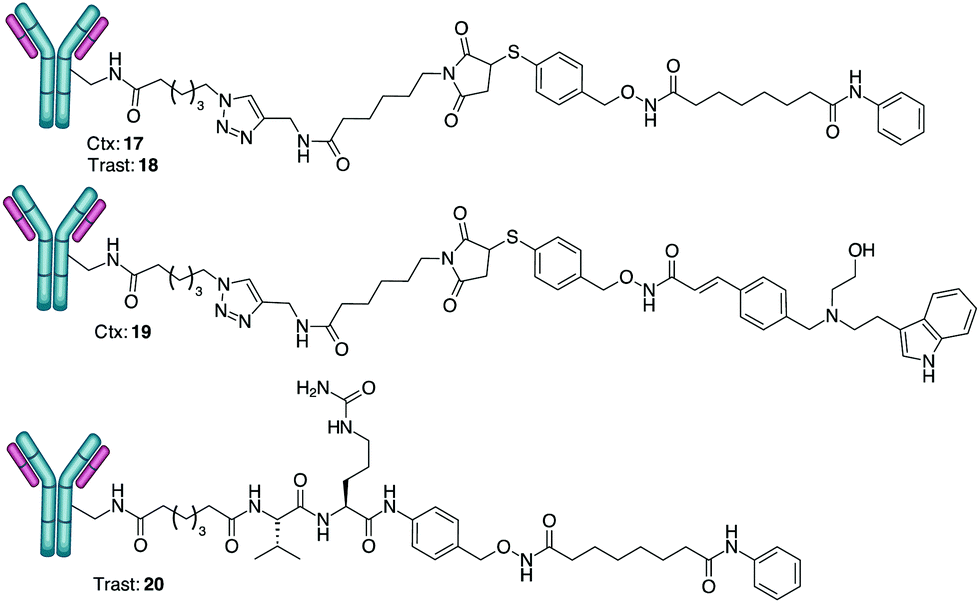 | ||
| Scheme 4 Conjugation of hydroxamates with cetuximab (Ctx) and trastuzumab (Trast). DAR determined by MALDI analysis: 17 DAR = 4 (±0.5); 18 DAR = 5 (±0.5); 19 DAR 3 (±0.5); 20 DAR 6 (±0.5) (average of three experiments). | ||
Results show that all the ADCs are internalized after binding to EGFR or ErbB2 receptors depending on the target receptor expression level. Internalization was equal with respect to the unconjugated Ctx or Trast, resulting in accumulation within the internal vesicles of the multivesicular bodies and subsequent translocation to the lysosomal compartment (Fig. S6, ESI†).
Release of hydroxamic acids 1 and 4 occurred upon treatment of ADCs 17–20 with human hepatic microsomes. After 72 h of incubation at 37 °C, quantitative HPLC/HRMS analysis showed the presence of peaks at m/z 264.1472 and 349.1792, respectively, attributed exclusively to compounds 1 and 4 in concentrations corresponding to more than 70% of release of the payload present in the starting ADC.20
Inhibition of the HDAC activity was determined through HCS-imaging analysis, monitoring the levels of acetylated histone H3 and acetylated α-tubulin in A549 (human lung adenocarcinoma) cells, as the indicators of specific inhibition of nuclear class I HDACs and of cytoplasmic HDAC6, respectively (Fig. 1). Results showed that ADCs 17, 19 and 20 induced a relevant increase in the acetylation level of α-tubulin and histone H3, as a consequence of direct enzymatic inhibition, while no significant effect was noted in cells treated with Ctx or Trast alone (Fig. 1). Neither cetuximab nor trastuzumab is endowed with HDAC inhibitor activity. So, an increase in the acetylation degree of α-tubulin- or histone H3 is not expected in the cells treated with these drugs. In fact, the extent of acetylation observed in A549 cells following treatment with the above drugs was observed to be similar or apparently a little lower (depending on the fields observed) with respect to the basal level of untreated cells (as shown in Fig. 1). ADC 18 induced an increased acetylation of both α-tubulin and histone H3, although with lower potency with respect to the others. In these cells, the cetuximab-based ADCs 17 and 19 would seem to act with higher potency, probably due to the highest expression of EGFR as compared to ERbB2 receptors on the cell surface. Increased acetylation of histone H3 and α-tubulin was detected through western blot analysis of the total protein lysate. The results of the densitometric analysis of the specific band intensity, after normalization to β-actin signals, showed that the acetylation enhanced upon treatment of tumour cells with ADCs as compared to the vehicle-treated cells and cells treated with Ctx and Trast alone (Fig. S7, ESI†). These results demonstrate that the ADCs are efficiently internalized by the tumour cells releasing the hydroxamic acids required for epigenetic modulation. Similar results were observed on other human tumor (pancreas, melanoma, and breast) cell lines (data not shown).
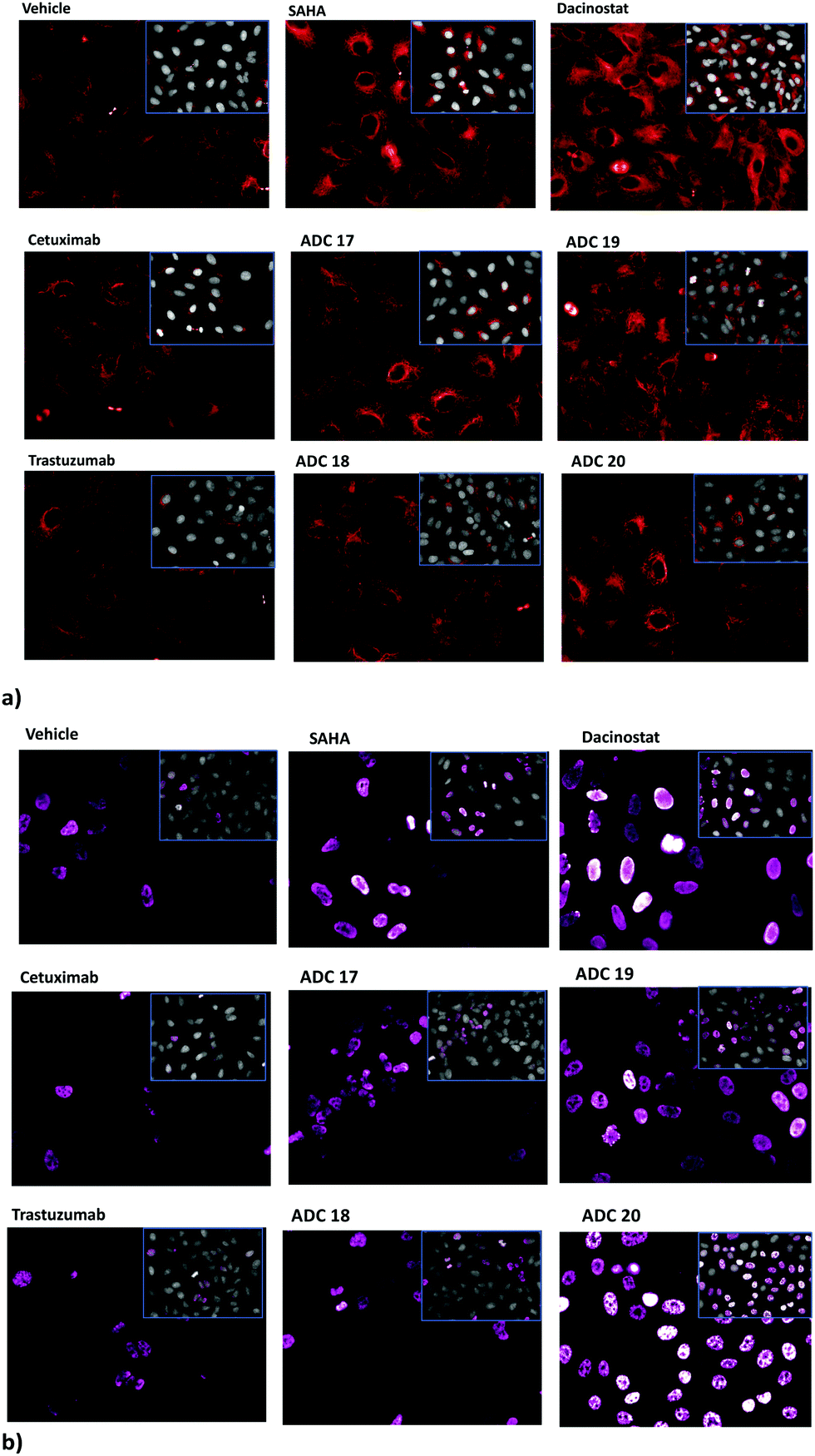 | ||
| Fig. 1 Effect of SAHA, Dacinostat, Cet and Trast, compared to ADCs 17–20, on the acetylation of α-tubulin (a) and histone H3 (b) in A549 cells. Cells cultivated for 3 h with reference drugs (100 nM) or antibodies (5 μg mL−1) and stained with anti-acetylated-α-tubulin (red) or anti-acetylated-H3 histone (pink). Insets show the Draq5 dye-stained nucleus and cytoplasm. Fluorescence imaging by HCS Operetta. Data not normalized. Each image is representative of at least 5 fields of duplicate wells. Magnification 60×. | ||
The anti-proliferative activity of the conjugates 17 and 19 was evaluated in A549 cells upon the treatment, for 6 days, with several doses of ADCs and with equivalent doses of Ctx compared to the unconjugated SAHA and Dacinostat (Fig. 2). ADC 19 inhibited cell proliferation very efficiently in a dose-dependent manner and with quite a similar potency (IC50 = 90 ± 3.4 nM) to that of the reference Dacinostat (IC50 = 33 ± 1.7 nM).21 ADC 17 (IC50 = 1440 ± 17 nM) was less efficacious than ADC 19.22 Interestingly, ADC 17 retains a certain percentage of activity in a range of concentrations (100–500 nm) in which SAHA resulted utterly inactive.
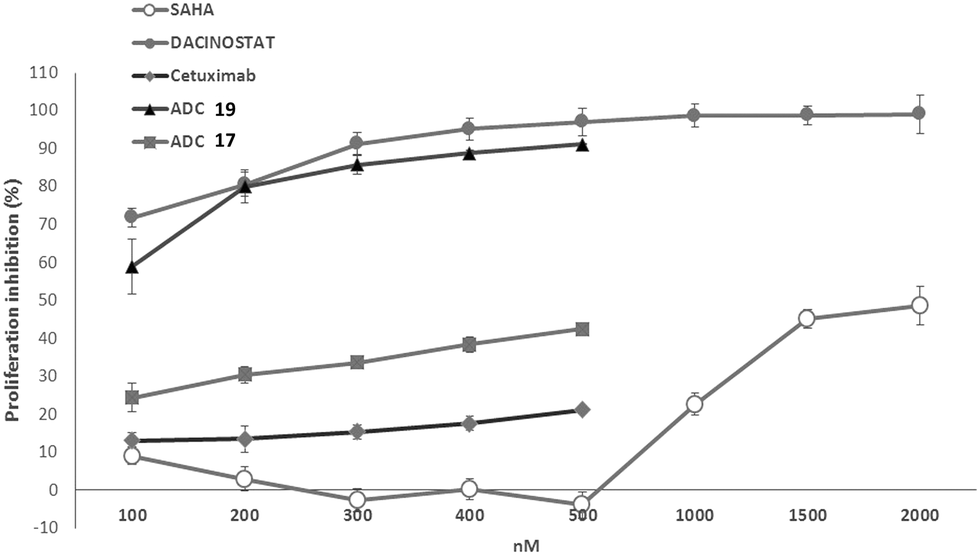 | ||
| Fig. 2 Antiproliferative activity of ADCs 17 and 19 on A549 cells upon 6 days of treatment. Data are the mean (±SD) of percentage inhibition (n = 5). | ||
In conclusion, we have developed a new class of ADCs for the targeted delivery of hydroxamic acids. The new linkers based on PMBA can release the hydroxamic acid after metabolic degradation demonstrating that bioconjugation with mAbs can be employed to develop more effective targeted therapy based on hydroxamate HDACis.23
This work was supported in part by the AIRC (under IG 2017 – ID. 20758 project – P.I. Taddei Maurizio) and by the project “Development and application of QM/MM technologies for the design of light responsive proteins or protein-mimics based on rhodopsin architecture” within the program “Dipartimenti di Eccellenza – 2018–2022 MIUR (Rome, Italy).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) S. P. Gupta, Chem. Rev., 2015, 115, 6427–6490 CrossRef CAS; (b) E. M. F. Muri, M. J. Nieto, R. D. Sindelar and J. S. Williamson, Curr. Med. Chem., 2002, 9, 1631–1653 CrossRef CAS.
- M. Manal, M. J. N. Chandrasekar, J. Gomathi Priya and M. J. Nanjan, Bioorg. Chem., 2016, 67, 18–42 CrossRef CAS.
- R. Sangwan, R. Rajan and P. K. Mandal, Eur. J. Med. Chem., 2018, 158, 620–706 CrossRef CAS.
- T. van den Bosch, M. Kwiatkowski, R. Bischoff and F. J. Dekker, Epigenomics, 2017, 9, 1013–1028 CrossRef CAS.
- R. Badia, J. Grau, E. Riveira-Muñoz, E. Ballana, G. Giannini and J. A. Esté, Antiviral Res., 2015, 123, 62–69 CrossRef CAS.
- P. Narayan and M. Dragunow, Adv. Exp. Med. Biol., 2017, 978, 321–336 CrossRef CAS.
- J. M. Roosenberg, Y. M. Lin, Y. Lu and M. J. Miller, Curr. Med. Chem., 2000, 7, 159–197 CrossRef CAS.
- (a) A. Rambaldi, C. M. Dellacasa, G. Finazzi, A. Carobbio, M. L. Ferrari, P. Guglielmelli, E. Gattoni, S. Salmoiraghi, M. C. Finazzi, S. Di Tollo, C. D’Urzo, A. M. Vannucchi, G. Barosi and T. Barbui, Br. J. Haematol., 2010, 150, 446–455 CAS; (b) S. Subramanian, S. E. Bates, J. J. Wright, I. Espinoza-Delgado and R. L. Piekarz, Pharmaceuticals, 2010, 3, 2751–2767 CrossRef CAS.
- (a) Y. Sun, Y. Sun, S. Yue, Y. Wang and F. Lu, Curr. Top. Med. Chem., 2019, 18, 2420–2428 CrossRef; (b) C. M. Grunewald, W. A. Schulz, M. A. Skowron, M. J. Hoffmann and G. Niegisch, Transl. Cancer Res., 2018, 7, 1151–1160 CrossRef CAS.
- A. V. Bieliauskas and M. K. H. Pflum, Chem. Soc. Rev., 2008, 37, 1402 RSC.
- (a) W. Fan, L. Zhang, Q. Jiang, W. Song, F. Yan and L. Zhang, Eur. J. Med. Chem., 2020, 203, 112628 CrossRef CAS; (b) K. Zuwala, A. A. A. Smith, M. Tolstrup and A. N. Zelikin, Chem. Sci., 2016, 7, 2353–2358 RSC; (c) W. Szymanski, M. E. Ourailidou, W. A. Velema, F. J. Dekker and B. L. Feringa, Chem. – Eur. J., 2015, 21, 16517–16524 CrossRef CAS; (d) S. A. Senevirathne, K. E. Washington, J. B. Miller, M. C. Biewer, D. Oupicky, D. J. Siegwart and M. C. Stefan, J. Mater. Chem. B, 2017, 5, 2106–2114 RSC; (e) P. Šilhár, L. M. Eubanks, H. Seki, S. Pellett, S. Javor, W. H. Tepp, E. A. Johnson and K. D. Janda, J. Med. Chem., 2013, 56, 7870–7879 CrossRef.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS.
- (a) M. R. Gordon, M. Canakci, L. Li, J. Zhuang, B. Osborne and S. Thayumanavan, Bioconjugate Chem., 2015, 26, 2198–2215 CrossRef CAS; (b) R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS.
- K. Tsuchikama and Z. An, Protein Cell, 2018, 9, 33–46 CrossRef CAS.
- (a) S. Ait-Mohand, C. Denis, G. Tremblay, M. Paquette and B. Guérin, Org. Lett., 2014, 16, 4512–4515 CrossRef CAS; (b) A. Safavy, M. B. Khazaeli, M. Kirk, L. Coward and D. J. Buchsbaum, Bioconjugate Chem., 1999, 10, 18–23 CrossRef CAS.
- (a) G. Giannini, L. Vesci, G. Battistuzzi, D. Vignola, F. M. Milazzo, M. B. Guglielmi, M. Barbarino, M. Santaniello, N. Fanto, M. Mor, S. Rivara, D. Pala, M. Taddei, C. Pisano and W. Cabri, J. Med. Chem., 2014, 57, 8358–8377 CrossRef CAS; (b) M. Taddei, S. Ferrini, L. Giannotti, M. Corsi, F. Manetti, G. Giannini, L. Vesci, F. M. Milazzo, D. Alloatti, M. B. Guglielmi, M. Castorina, M. L. Cervoni, M. Barbarino, R. Foderà, V. Carollo, C. Pisano, S. Armaroli and W. Cabri, J. Med. Chem., 2014, 57, 2258–2274 CrossRef CAS; (c) C. B. Botta, W. Cabri, E. Cini, L. De Cesare, C. Fattorusso, G. Giannini, M. Persico, A. Petrella, F. Rondinelli, M. Rodriquez, A. Russo and M. Taddei, J. Med. Chem., 2011, 54, 2165–2182 CrossRef CAS; (d) M. Rodriquez, S. Terracciano, E. Cini, G. Settembrini, I. Bruno, G. Bifulco, M. Taddei and L. Gomez-Paloma, Angew. Chem., Int. Ed., 2006, 45, 423–427 CrossRef CAS.
- S. Remiszewski, Curr. Med. Chem., 2003, 10, 2393–2402 CrossRef CAS.
- E. Cini, V. Faltoni, E. Petricci, M. Taddei, L. Salvini, G. Giannini, L. Vesci, F. M. Milazzo, A. M. Anastasi, G. Battistuzzi and R. De Santis, Chem. Sci., 2018, 9, 6490–6496 RSC.
- (a) H. Wu, P. J. Levalley, T. Luo, A. M. Kloxin and K. L. Kiick, Bioconjugate Chem., 2018, 29, 3595–3605 CrossRef CAS; (b) A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS.
- A. J. Bessire, T. E. Ballard, M. Charati, J. Cohen, M. Green, M. H. Lam, F. Loganzo, B. Nolting, B. Pierce, S. Puthenveetil, L. Roberts, K. Schildknegt and C. Subramanyam, Bioconjugate Chem., 2016, 27, 1645–1654 CrossRef CAS.
- T. Beckers, C. Burkhardt, H. Wieland, P. Gimmnich, T. Ciossek, T. Maier and K. Sanders, Int. J. Cancer, 2007, 121, 1138–1148 CrossRef CAS.
- P. Agarwal and C. R. Bertozzi, Bioconjugate Chem., 2015, 26, 176–192 CrossRef CAS.
- S. Shen and A. P. Kozikowski, ChemMedChem, 2016, 11, 15–21 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc06131j |
| This journal is © The Royal Society of Chemistry 2021 |