
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioinspired scaffolds that sequester lead ions in physically damaged high efficiency perovskite solar cells†
Muhamad Z.
Mokhtar
 *a,
Jiangyu
He
a,
Menghan
Li
a,
Qian
Chen
a,
Jack Chun Ren
Ke
a,
David J.
Lewis
a,
Andrew G.
Thomas
ab,
Ben F.
Spencer
ab,
Saif A.
Haque
c and
Brian R.
Saunders
*a
*a,
Jiangyu
He
a,
Menghan
Li
a,
Qian
Chen
a,
Jack Chun Ren
Ke
a,
David J.
Lewis
a,
Andrew G.
Thomas
ab,
Ben F.
Spencer
ab,
Saif A.
Haque
c and
Brian R.
Saunders
*a
aDepartment of Materials, University of Manchester, Manchester, M13 9PL, UK. E-mail: muhamadzulhasif.mokhtar@manchester.ac.uk; brian.saunders@manchester.ac.uk
bPhoton Science Institute and The Henry Royce Institute, University of Manchester, Manchester, M13 9PL, UK
cDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, Wood Lane, W12 0BZ, UK
First published on 22nd December 2020
Abstract
Hydroxyapatite nanoparticles (HAP NPs) are blended with TiO2 NPs to prepare mixed mesoporous scaffolds which are used to prepare high efficiency perovskite solar cells (PSCs) with a best power conversion efficiency (PCE) of 20.98%. HAP not only increases the PCE but also limits the concentration of Pb released in water from intentionally broken PSCs by ion sequestration thereby potentially offering a promising in-device fail-safe system.
Perovskite solar cells (PSCs) have attracted enormous attention since the seminal publication of Miyasaka et al. in 2009.1 Unprecedented increases in the power conversion efficiencies (PCEs) in just over a decade have resulted in values reaching 25.5%.2 Hybrid organic–inorganic perovskites have a high absorption coefficient, micron-scale exciton diffusion lengths3 and a favourable bandgap for light harvesting. They also offer low processing costs, making PSCs potentially attractive for application.4,5 Consequently, PSCs have received much interest for commercial exploitation.6 Two concerns that need to be considered for mass exploitation are stability and the possibility of Pb escaping to the environment.7 Major improvements in stability have been achieved8 and attention is turning toward reducing the possibility that Pb might be released if modules undergo catastrophic failure followed by immersion in water (for example, as a result of hail damage).
The CH3NH3PbI3 PSC photoactive layer can contain ∼30% of Pb.9 Hailegnaw et al. investigated the effect of rain on Pb leaching from PSCs after catastrophic failure.10 They suggested that the amount of Pb that could be released from a perovskite (PVK) solar facility would be less than that emitted from a coal-fired power station generating the same amount of power. Because Pb is toxic and can accumulate in bones11 strategies for fail-safe, efficient PSCs that prevent release of Pb are of increasing interest. Jiang et al. established self-healing polymer-based encapsulation that reduced leakage of Pb from damaged modules exposed to water.12 Li et al. applied a coating of phosphonic-acid based polymer and achieved Pb sequestration from high efficiency devices.13 The polymer layers swelled when Pb was absorbed. However, the mechanical integrity and longevity of the Pb capture might be compromised in the case of a discarded or broken module. Here, we demonstrate a bioinspired potential solution to lead sequestration using hydroxyapatite (Ca10(PO4)6(OH)2, HAP).
HAP is the main component of bone, is easily synthesised via solution processing and absorbs Pb at high levels.14 HAP is also strong. Here, we use HAP nanoparticles (NPs) blended with TiO2 NPs as scaffolds for efficient PSCs. Our results show that HAP increases the PCE whilst also enabling capture of soluble Pb in the event of catastrophic device failure and immersion in water. We show that HAP NPs are potentially attractive for fail-safe in-device Pb capture for damaged PSCs.
The HAP NPs used in this study as a Pb-sequestering agent are prepared using a scalable hydrothermal synthesis (see Experimental details in the ESI†). They had lengths and thicknesses in the range of 25–50 nm and 5–10 nm, respectively, as shown by TEM (Fig. S1, ESI†). The XRD profile (Fig. S2, ESI†) agrees with those reported elsewhere15,16 and the FTIR spectrum also matches that reported for HAP17 (Fig. S3, ESI†). An adsorption isotherm was measured for Pb2+ and fitted with the Langmuir model. The HAP NPs adsorbed Pb2+ strongly (Fig. S4, ESI†). The measured HAP absorption capacity was 1350 mg g−1 which compares favourably with the literature. Additional discussion regarding HAP characterisation and Pb uptake is provided in the ESI.†
To prepare scaffolds we mixed dispersions of HAP NPs and TiO2 NPs (particle size = 30 nm18). The mixed dispersions were spin-coated and then annealed to give HAP/TiO2 scaffolds (See Experimental details, ESI†). We identify the scaffolds with respect to the nominal wt% HAP in the scaffold based on the composition of HAP/TiO2 mixed dispersion used for spin-coating. SEM images of the scaffolds are shown in Fig. 1A–D. (Larger area SEM images are shown in Fig. S5 (ESI†).) The TiO2 and HAP NPs appear white and grey, respectively. This is due to differences in electron contrast. The densities of TiO2 and HAP are 3.84 and 3.16 g mL−1,19,20 respectively. Moreover, as the HAP concentration increases the average size of the pores within the scaffold increases (see Fig. S6, ESI†). The average pore sizes for the 0%, 30% and 70% scaffolds are 36, 52 and 62 nm, respectively. For the 100% HAP scaffold large interconnected HAP-free regions (Fig. 1D) are present. The average scaffold layer thicknesses estimated for the 0, 30, 70 and 100% HAP scaffolds are 334, 305, 183 and 93 nm, respectively, based on the SEM cross-sectional images of the PVK films (see Fig. 2). We attribute this trend to the tendency of the HAP NPs to form aggregates rather than well-separated individual NPs as the HAP concentration increased (See the grey features in Fig. 1 and Fig. S5, ESI†). Transmittance spectra were measured for the scaffolds (Fig. S7A, ESI†). The average transmittance over the range of 350–800 nm was 76.0% for the 0% HAP scaffold. The average transmittance values for the 30%, 70% and 100% HAP scaffolds were slightly lower and were 74.1%, 70.8% and 72.1%, respectively (Fig. S7B, ESI†). The decrease in the average transmittance values is due to increased light scattering from the HAP NPs.
 | ||
| Fig. 1 SEM images of scaffolds prepared using (A) 0% HAP, (B) 30% HAP, (C) 70% HAP and (D) 100% HAP. The scale bars shown in (D) apply to all images. | ||
 | ||
| Fig. 2 SEM top surface images, cross-sections and grain size distributions for PVK films prepared using (A) 0% HAP, (B) 30% HAP, (C) 70% HAP and (D) 100% HAP scaffolds. The yellow arrows highlight PbI2-rich grains. All scale bars are 1000 nm. | ||
We employed our scaffolds as substrates to prepare (FAPbI3)1−x(MAPbBr3)x PVK films (See Experimental details, ESI†). The method used followed an earlier procedure that gave high PCE planar devices,21 which we adapted to our mesoporous devices. The earlier study reported residual PbI2 in their films.21 In contrast to that work, here PbI2-rich grains are present both on the surface and the lower film regions as revealed by surface and cross-section SEM images. (see arrows in Fig. 2.) This implies that PbI2 was not only produced by annealing21 but also from unreacted PbI2. XRD profiles confirm that both FAPbI3 and PbI2 were present (Fig. S8A, ESI†). It is noted that residual PbI2 is beneficial for PSC performance.22 UPS data were obtained (Fig. S9, ESI†) and are discussed in the ESI.† All the films are n-type with only small differences in energy levels. The morphologies and grain size distributions for the PVK films are shown in Fig. 2. The average grain sizes for the PVK films prepared using the 0, 30, 70 and 100% HAP scaffolds are 470, 675, 745 and 690 nm, respectively. This trend agrees with the full-width at half-maximum values obtained from the XRD profiles (Fig. S8B, ESI†). Hence, the grain size increased with the inclusion of HAP. Furthermore, the cross-sectional SEM images (Fig. 2) reveal that the grains extend vertically to the scaffold layer. Because HAP NPs created pores within the meso-TiO2 scaffolds (from Fig. 1) there was more room for unrestricted PVK grain growth to occur. Hence, increased space within the scaffolds is the likely cause for the increased grain size observed in the present study.23,24 The average PVK capping layer thicknesses measured from the SEM cross-sections of the 0%, 30%, 70% and 100% HAP systems are 311, 358, 702 and 615 nm, respectively.
Solar cells were constructed using the HAP/TiO2 scaffolds and the architecture is shown in Fig. 3A. Photocurrent density–voltage (J–V) curves for the best performing devices in each class are shown in Fig. 3B. The overall best performing device was obtained using 70% HAP and had a PCE of 20.98%. Fig. 3C shows the PCE values for the various PSCs with different concentrations of HAP. The Jsc, Voc and FF values are shown in Fig. S10 (ESI†). (All the device data are shown in Table S1, ESI†). Remarkably, the best PCE improved from 17.76% to 20.98% as the concentration of HAP in the scaffold increased from 0 to 70 wt% The PCE decreased to 14.15% for 100% HAP. Snaith et al. showed that insulating scaffolds of Al2O3 could also be used to prepare efficient PSCs.25 HAP has a band gap >6 eV and is insulating.26 The PCE trend in Fig. 3C is dominated by changes FF and to a lesser extent Voc (see Fig. S10D–F, ESI†). The larger grain sizes for the systems prepared with HAP (Fig. 2) imply fewer grain boundaries, which should result in less carrier recombination.27 Grain boundaries are a well-known source of defects.28 An increase in grain size should therefore increase FF (and Voc) due to less recombination. Space-charge limiting current (SCLC) measurements were therefore conducted (Fig. S11, ESI†). The data show that the 70% system has the lowest defect concentration (Nd = 4.6 × 1015 cm−3). (See SCLC discussion and Fig. S11C in the ESI†). The hysteresis index (HI) for the cells is highest (2.24%) for the 100% HAP cell and is 2.18% for the 70% HAP system and 1.77% for 0% HAP (Table S1, ESI†). A low HI is an additional potential benefit offered by these new devices prepared using HAP-based scaffolds.
 | ||
| Fig. 3 (A) Solar cell architecture. (B) J–V curves and performance data for the best cell. (C) PCE statistical data measured for the cells. (D) Ambient PCE stability (40–75% RH). (E and F) show UV-visible and PL spectra for the films. | ||
Device stability was measured for the non-encapsulated cells stored under ambient conditions (Fig. 3D). (Stability data for Jsc, Voc and FF are shown in Fig. S12, ESI.†) The 0% to 70% HAP devices had similar stability retaining >85% of their initial PCEs over 38 days. In contrast, the 100% HAP device retained only 60% of its initial PCE. The latter scaffold is expected to be the least efficient for removing electrons due to the insulating nature of HAP. Accordingly, a higher concentration of reactive O2− can be expected which would decrease stability.29
To further probe the properties of the PVK films we measured the UV-visible spectra (Fig. 3E). There is a strong increase of the absorbance over the 760 nm and 900 nm range as the HAP content of the scaffolds increase. This is due to a capping layer thickness increase (above) and more scattering from the scaffold (Fig. S7, ESI†). Furthermore, an increase of the HAP concentration causes the PVK absorption edge to redshift to >800 nm. The band gaps (Eg) for the films were estimated from Tauc plots (see Fig. S13, ESI†). The Egs for the PVK films prepared using 0%, 30%, 70% and 100% HAP are, respectively, 1.545, 1.531, 1.518 and 1.472 eV. The latter is a 73 meV red-shift due to replacing TiO2 NPs with HAP NPs. This is likely due to the increased grain size30 noted above. The red shift was also apparent from the PL spectra (see Fig. 3F). There is also a decrease of PL intensity with increasing HAP concentration in the scaffolds. This may be due to trap-assisted non-radiative recombination caused by HAP as the PL probed the scaffold region of the photoactive layer. To explore the possibility of an interaction between HAP and PVK model reactions were conducted and FTIR evidence for Pb absorption in HAP was found (see discussion and Fig. S14 in the ESI†). Based on the data we suggest that the superior performance for the devices prepared using 70% HAP scaffold is due to the combination of relatively large PVK grain size, optimum PbI2/FAPI ratio and sufficient TiO2/PVK interface for efficient charge transfer (see PCE mechanism discussion in the ESI†).
Complete devices (0% to 70% HAP) were tested by water-challenge failure simulations. We studied devices prepared using 30% and 70% HAP because they had the highest PCE values (Fig. 3C). The 0% device was used as a control. The HAP-containing devices were encapsulated with an extra layer of HAP on top of the Au electrode as depicted in Fig. 4A. The encapsulating HAP layer had a thickness of 20 μm (identified as E20) or 40 μm (E40). We measured J–V data for the former devices (Fig. S15, ESI†). The best cell had a PCE of 18.70%. The PCE decrease compared to Fig. 3B is due to the additional contacts needed for the back electrode. The cells were subsequently either broken at one or two points (Fig. 4A). For example, 70%E20-1 and 70%E20-2 are cells prepared with the 70% HAP scaffold and broken at the front (one-point break) or front and back surfaces (two-point break), respectively.
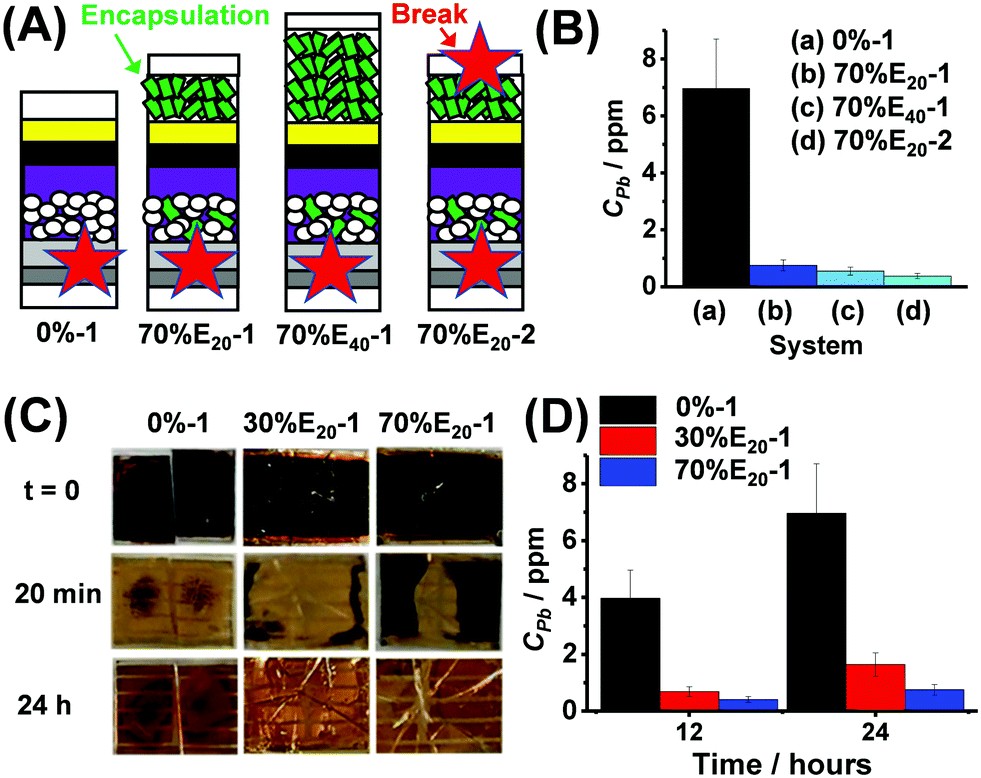 | ||
| Fig. 4 (A) Device architectures used for failure tests. The labels shown in Fig. 3A apply to this figure. The breakage points are shown (star). (B) Initial study of Pb release after 24 h using different architectures (See text). (C) Photographs of failure tests. The times after immersion in water are shown. (D) Concentration of Pb released 12 or 24 h after cell breakage. | ||
We examined the effects of different architectures and breakage points (Fig. 4A) on the released Pb concentration in water (CPb) after 24 h (see Fig. 4B). The control device (0%-1, i.e., HAP-free) resulted in a CPb of 7.0 ppm after 24 h (Fig. 4B). A major decrease in Pb release was achieved (i.e., much lower CPb values) when HAP was used in the scaffold and encapsulating layer. The one-point broken 70% devices with 20 and 40 μm HAP encapsulation layers (70%E20-1 and 70%E40-1) released CPb of 0.75 and 0.55 ppm, respectively. The two-point broken device (70%E20-2) had the lowest CPb, with only 0.38 ppm. The latter is a decrease in released CPb by a factor of ∼20. This is likely due to better involvement of the encapsulating HAP in Pb capture. We next focussed on one-point breakage experiments as they more closely resemble real failure (e.g., from hail). Photographs of broken 0%-1, 30%E20-1 and 70%E20-1 cells after water immersion for 0–24 h are shown in Fig. 4C. The black colour of PVK is rapidly lost and replaced by the yellow colour due to PbI2. Interestingly, this process was slowest for the devices containing HAP and, in particular, 70%E20-1. The release data (Fig. 4D) confirm that using HAP in the scaffold/encapsulation layers decreased CPb.
X-ray photoelectron spectroscopy (XPS) was used to probe the surface composition of HAP after 24 h for the experiments shown Fig. 4D. The surface compositions of the HAP encapsulation layer and the scaffold were probed for the broken 70%E20-1 device. Fig. S16 (ESI†) shows the Pb 4f and P 2p spectra of the samples, giving rise to Pb 4f7/2,5/2 and P 2p3/2,1/2 doublets. These data show that Pb was present in both the encapsulation layer and the scaffold. The binding energy (BE) of the Pb 4f7/2 peaks of both scaffold and encapsulating HAP surfaces is located at approximately 139.2 eV, which is ascribed to the bonding formation between Pb2+ and PO3−4.31 This BE value also matches that reported for Pb absorbed by HAP32 and is much higher than typical values for Pb–I bonds in halide perovskites (ca. 138.6 eV)33 as well as PbI2 (ca. 138.9 eV).34 This suggests HAP does not simply adsorb Pb-containing degraded species from the perovskites; instead, Pb forms new bonding with phosphate (PO3−4) in lead hydroxyapatite.
In summary, we have shown that HAP NPs, a natural biomaterial, blended with TiO2 NPs in scaffolds for PSCs gives a larger grain size, higher absorbance, improved device PCE and good stability. Moreover, HAP is able to substantially decrease the concentration of Pb released into water in the event of catastrophic failure due to immobilisation within a Pb hydroxyapatite matrix. Our simple and scalable bioinspired in-device sequestration approach has potential for application and may assist with the large scale deployment of PSCs.
The authors would like to thank the EPSRC for funding (EP/R020590/1 and EP/R020574/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- NREL Best Research-cell Efficiencies, http://www.nrel.gov/pv/cell-efficiency.html, Accessed December 12, 2020.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341 CrossRef CAS.
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036–3103 CrossRef CAS.
- Y. Rong, Y. Hu, A. Mei, H. Tan, M. I. Saidaminov, S. I. Seok, M. D. McGehee, E. H. Sargent and H. Han, Science, 2018, 361, eaat8235 CrossRef.
- B. J. Kim, S. Lee and H. S. Jung, J. Mater. Chem. A, 2018, 6, 12215–12236 RSC.
- A. Krishna, M. A. Akhavan Kazemi, M. Sliwa, G. N. M. Reddy, L. Delevoye, O. Lafon, A. Felten, M. T. Do, S. Gottis and F. Sauvage, Adv. Funct. Mater., 2020, 30, 1909737 CrossRef CAS.
- S.-H. Turren-Cruz, A. Hagfeldt and M. Saliba, Science, 2018, 362, 449 CrossRef CAS.
- J. Yan and B. R. Saunders, RSC Adv., 2014, 4, 43286–43314 RSC.
- B. Hailegnaw, S. Kirmayer, E. Edri, G. Hodes and D. Cahen, J. Phys. Chem. Lett., 2015, 6, 1543–1547 CrossRef CAS.
- H. Hu, M. Rabinowitz and D. Smith, Environ. Health Perspect., 1998, 106, 1–8 CrossRef CAS.
- Y. Jiang, L. Qiu, E. J. Juarez-Perez, L. K. Ono, Z. Hu, Z. Liu, Z. Wu, L. Meng, Q. Wang and Y. Qi, Nat. Energy, 2019, 4, 585–593 CrossRef CAS.
- X. Li, F. Zhang, H. He, J. J. Berry, K. Zhu and T. Xu, Nature, 2020, 578, 555–558 CrossRef CAS.
- Q. Y. Ma, S. J. Traina, T. J. Logan and J. A. Ryan, Environ. Sci. Technol., 1993, 27, 1803–1810 CrossRef CAS.
- S. T. Ramesh, N. Rameshbabu, R. Gandhimathi, P. V. Nidheesh and M. Srikanth Kumar, Appl. Water Sci., 2012, 2, 187–197 CrossRef CAS.
- Y. Han, X. Wang and S. Li, J. Nanopart. Res., 2009, 11, 1235–1240 CrossRef CAS.
- B. O. Fowler, Inorg. Chem., 1974, 13, 194–207 CrossRef CAS.
- https://www.greatcellsolar.com/shop/30-nr-d.html, Accessed 02/04/20.
- K. A. Hing, S. M. Best and W. Bonfield, J. Mater. Sci.: Mater. Med., 1999, 10, 135–145 CrossRef CAS.
- C. R. Ottermann and K. Bange, Thin Solid Films, 1996, 286, 32–34 CrossRef CAS.
- Q. Jiang, Z. Chu, P. Wang, X. Yang, H. Liu, Y. Wang, Z. Yin, J. Wu, X. Zhang and J. You, Adv. Mater., 2017, 29, 1703852 CrossRef.
- X.-X. Gao, W. Luo, Y. Zhang, R. Hu, B. Zhang, A. Züttel, Y. Feng and M. K. Nazeeruddin, Adv. Mater., 2020, 32, 1905502 CrossRef CAS.
- M. T. Hörantner, W. Zhang, M. Saliba, K. Wojciechowski and H. J. Snaith, Energy Environ. Sci., 2015, 8, 2041–2047 RSC.
- Q. Lian, M. Z. Mokhtar, D. Lu, M. Zhu, J. Jacobs, A. B. Foster, A. G. Thomas, B. F. Spencer, S. Wu, C. Liu, N. Hodson, B. Smith, A. Alkaltham, O. Alkhudhari, T. M. Watson and B. R. Saunders, ACS Appl. Mater. Interfaces, 2020, 12, 18578–18589 CrossRef CAS.
- S. Guarnera, A. Abate, W. Zhang, J. M. Foster, G. Richardson, A. Petrozza and H. J. Snaith, J. Phys. Chem. Lett., 2015, 6, 432–437 CrossRef CAS.
- M. Tsukada, M. Wakamura, N. Yoshida and T. Watanabe, J. Mol. Catal. A: Chem., 2011, 338, 18–23 CAS.
- C. Bi, Q. Wang, Y. Shao, Y. Yuan, Z. Xiao and J. Huang, Nat. Commun., 2015, 6, 7747 CrossRef CAS.
- S. Yang, J. Dai, Z. Yu, Y. Shao, Y. Zhou, X. Xiao, X. C. Zeng and J. Huang, J. Am. Chem. Soc., 2019, 141, 5781–5787 CrossRef CAS.
- D. Bryant, N. Aristidou, S. Pont, I. Sanchez-Molina, T. Chotchunangatchaval, S. Wheeler, J. R. Durrant and S. A. Haque, Energy Environ. Sci., 2016, 9, 1655–1660 RSC.
- T. Hwang, S. Lee, J. Kim, J. Kim, C. Kim, B. Shin and B. Park, Nanoscale Res. Lett., 2017, 12, 57 CrossRef.
- T. T. Eighmy, A. E. Kinner, E. L. Shaw, J. D. Eusden Jr and C. A. Francis, Surf. Sci. Spectra, 1999, 6, 202–209 CrossRef CAS.
- Z. Zhang, M. Li, W. Chen, S. Zhu, N. Liu and L. Zhu, Environ. Pollut., 2010, 158, 514–519 CrossRef CAS.
- C.-R. Ke, D. J. Lewis, A. S. Walton, Q. Chen, B. F. Spencer, M. Mokhtar, C. L. Compean-Gonzalez, P. O'Brien, A. G. Thomas and W. R. Flavell, ACS Appl. Energy Mater., 2019, 2, 6012–6022 CrossRef CAS.
- B. Philippe, B.-W. Park, R. Lindblad, J. Oscarsson, S. Ahmadi, E. M. J. Johansson and H. Rensmo, Chem. Mater., 2015, 27, 1720–1731 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional discussion, additional figures and a table. See DOI: 10.1039/d0cc02957b |
| This journal is © The Royal Society of Chemistry 2021 |