
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into phosphatase-activated chemical defense in a marine sponge holobiont†
Takahiro
Jomori
a,
Kenichi
Matsuda
 ab,
Yoko
Egami
a,
Ikuro
Abe
cd,
Akira
Takai
e and
Toshiyuki
Wakimoto
*ab
ab,
Yoko
Egami
a,
Ikuro
Abe
cd,
Akira
Takai
e and
Toshiyuki
Wakimoto
*ab
aFaculty of Pharmaceutical Sciences, Hokkaido University, Kita 12, Nishi 6, Sapporo 060-0812, Japan
bGlobal Station for Biosurfaces and Drug Discovery, Global Institution for Collaborative Research and Education (GI-CoRE), Hokkaido University, Kita 12, Nishi 6, Sapporo 060-0812, Japan
cGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
dCollaborative Research Institute for Innovative Microbiology, The University of Tokyo, Yayoi 1-1-1, Bunkyo-ku, Tokyo 113-8657, Japan
eDepartment of Physiology, Asahikawa Medical University, 1-1-1 Midorigaoka Higashi 2 jo, Asahikawa 078-8510, Japan
First published on 6th October 2021
Abstract
Marine sponges often contain potent cytotoxic compounds, which in turn evokes the principle question of how marine sponges avoid self-toxicity. In a marine sponge Discodermia calyx, the highly toxic calyculin A is detoxified by the phosphorylation, which is catalyzed by the phosphotransferase CalQ of a producer symbiont, “Candidatus Entotheonella” sp. Here we show the activating mechanism to dephosphorylate the stored phosphocalyculin A protoxin. The phosphatase specific to phosphocalyculin A is CalL, which is also encoded in the calyculin biosynthetic gene cluster. CalL represents a new clade and unprecedently coordinates the heteronuclear metals Cu and Zn. CalL is localized in the periplasmic space of the sponge symbiont, where it is ready for the on-demand production of calyculin A in response to sponge tissue disruption.
Introduction
Marine sponges often contain highly potent cytotoxic compounds, and some play a role in chemical defense.1–3 However, these cytotoxic compounds usually target molecules or signaling pathways that are common and essential to all eukaryotic organisms, including the sponges themselves.4,5 This fact evokes the principle question of how the marine sponges avoid self-toxicity caused by the accumulated cytotoxins.6We previously reported the biosynthetic gene cluster (BGC) of calyculin A, which is the major cytotoxin originally isolated from a Japanese marine sponge, Discodermia calyx (Fig. 1).7,8 Its potent cytotoxicity is attributable to the specific inhibitory activity against protein phosphatases 1 and 2A.9–11 The structure features a variety of functionalities, including tetraene, oxazole, phosphate and dimethylamino groups. The main framework of this molecule is biosynthesized by a hybrid pathway of polyketide synthase (PKS) and non-ribosomal peptide synthetase (NRPS), followed by post-assembly line modifications such as nitrile formation.8 The detailed mechanisms of the tailoring reactions at the later stage of calyculin biosynthesis have remained largely unknown, but we previously reported that a phosphotransferase, CalQ (Fig. 1), is involved in the phosphorylation process unique to the calyculin pathway.8 Notably, CalQ, encoded in the calyculin BGC, appends an additional phosphate group on the intrinsic phosphate group of calyculin A to generate phosphocalyculin A, a previously unknown derivative. Since the phosphate group in calyculin A is essential to exert the specific inhibition of protein phosphatases 1 and 2A, the cytotoxicity of phosphocalyculin A is significantly diminished by this post-assembly line modification.8–11 This bioconversion process offers a clue for the elucidation of the resistance mechanism of the host sponge, conferred by the symbiotic bacterium “Candidatus Entotheonella” sp. producing calyculin A.8
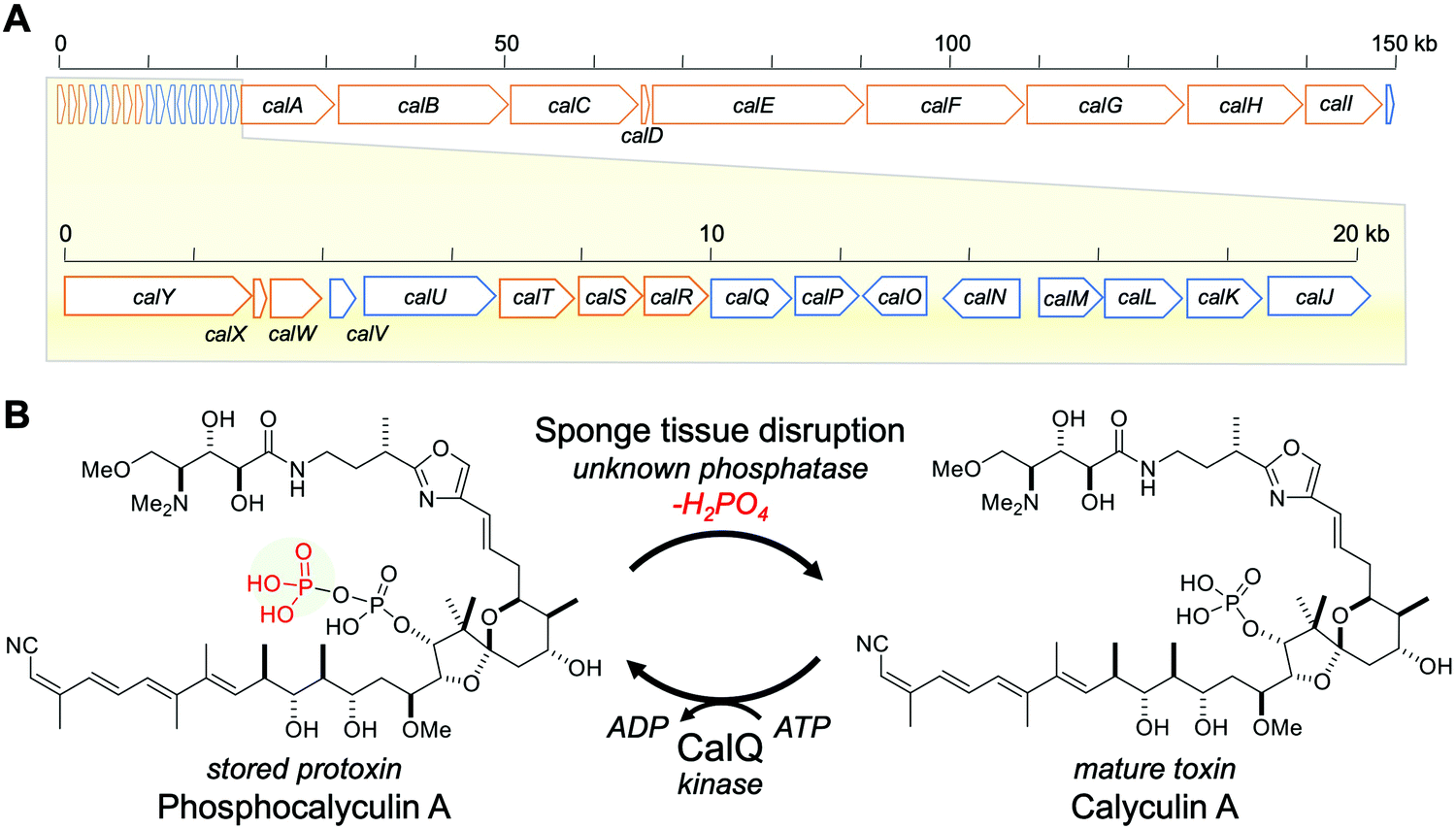 | ||
| Fig. 1 Calyculin A biosynthetic gene cluster and bioconversion process between calyculin A and phosphocalyculin A. (A) Calyculin biosynthetic gene cluster. The ORFs encoding the NRPS and PKS responsible for the assembly line of calyculin biosynthesis are highlighted in orange. The other ORFs in blue are putative tailoring enzymes except for CalQ, which has been identified as the calyculin A-specific phosphotransferase. The putative functions of calJ–Y genes are listed in Table S1, ESI.† (B) The activation/inactivation mechanism of phosphocalyculin A/calyculin A through dephosphorylation/phosphorylation in the marine sponge D. calyx. | ||
Thus, phosphocalyculin A was first discovered as the product of the CalQ-catalyzed reaction. This is because the homogenization of the wet sponge, even with organic solvents, stimulates the rapid conversion of phosphocalyculin A to calyculin A, which resulted in the disappearance of the phosphocalyculin A in the extract.8 To circumvent this conversion process, the fresh sponge was flash-frozen in liquid nitrogen, and then the frozen sponge was freeze-dried to remove water. The methanol extract of the freeze-dried sponge contained phosphocalyculin A as the dominant metabolite, in place of calyculin A.8 Furthermore, the crude enzyme fraction of the sponge D. calyx efficiently dephosphorylated phosphocalyculin A to generate calyculin A in a few minutes, while the heat-denatured fraction had diminished activity. Thus, this highly efficient bioconversion process is obviously catalyzed by an unidentified enzyme in the sponge-microbe association (Fig. 1B).8
This bioconversion proceeds through the wound-activated mechanism of the toxic secondary metabolite, as often found in higher plants and exemplified by the cyanogen glycosides, which are defense chemicals that generate hydrogen cyanide.12 The wound-activated chemical defense system prevails not only in terrestrial higher plants, but also in mushrooms, marine algae, and a few marine sponges.12–21 To achieve both self-resistance and chemical defense, this process requires a suite of activating and deactivating enzymes. In the case of the phosphatase inhibitor, calyculin A, the deactivation and activation are accomplished by adding and removing a phosphate group on calyculin A itself. However, the activating enzyme catalyzing the dephosphorylation of phosphocalyculin A has not been identified yet. In particular, the origin of the phosphatase is interesting to illuminate how calyculin biogenesis is triggered for the purpose of chemically defending the sponge holobiont. In this study, we purified phosphocalyculin A phosphatase from the sponge, and confirmed that the phosphatase is encoded in the calyculin BGC and therefore produced by the bacterial symbiont Entotheonella.
Results and discussion
Isolation of phosphocalyculin A phosphatase from the sponge D. calyx
We first attempted to isolate a native phosphatase specific to phosphocalyculin A from the sponge holobiont, since the origins of the phosphatase in the sponge-microbe association remain enigmatic. The crude enzyme solution of the D. calyx sponge showed an intense phosphatase activity against phosphocalyculin A, which could be reproducibly detected by the malachite green assay even after one month of storage at 4 °C. To distinguish between the phosphocalyculin-specific phosphatase and protein phosphatases, we utilized the malachite green assay in conjunction with the conventional phosphatase assay with the chromogenic substrate p-nitrophenyl phosphate (pNPP), which excludes the fraction containing protein phosphatases derived from the marine sponge.The sponge (1.8 kg wet weight) was extracted with buffer, and the extract was desalted by ultrafiltration. We performed several preliminary fractionation methods, such as ammonium sulfate fractionation, and precipitation by using organic solvents, but all attempts were inefficient, and the activity was distributed broadly in the fractions. Therefore, the desalted extract was directly fractionated by anion exchange column chromatography. The phosphatase activity specific to phosphocalyculin A was detected in the flow-through fraction, which was then further fractionated on a cation exchange column. Fortunately, the phosphatase was retained in the column and eluted with 250 mM NaCl, and exhibited phosphatase activity against phosphocalyculin A, but not pNPP. The results indicated that the phosphocalyculin-specific phosphatase is a basic protein and the concomitant acidic proteins could be efficiently removed at the early fractionation stage by an anion exchange column. Encouraged by these results, the active fraction was fractionated on a Phenyl-Sepharose column, followed by gel filtration with Sephacryl S200, which resulted in a 660-fold purification of the phosphatase as compared to the origin of cell free extract. Finally, after separation on a MonoS column, the active fraction was further purified by HPLC with a gel filtration column (Shodex PROTEIN LW-803) to afford the native phosphocalyculin phosphatase, which was the major spot detected by 2D gel electrophoresis (Fig. S1, ESI†). The 2D gel electrophoresis profile indicated that the isoelectric point (pI) of the native phosphatase is ca. 8.3–9.5 and the molecular weight is ca. 45 kDa, which is consistent with the elution profile of the active fraction in the gel filtration chromatography (Fig. 2A).
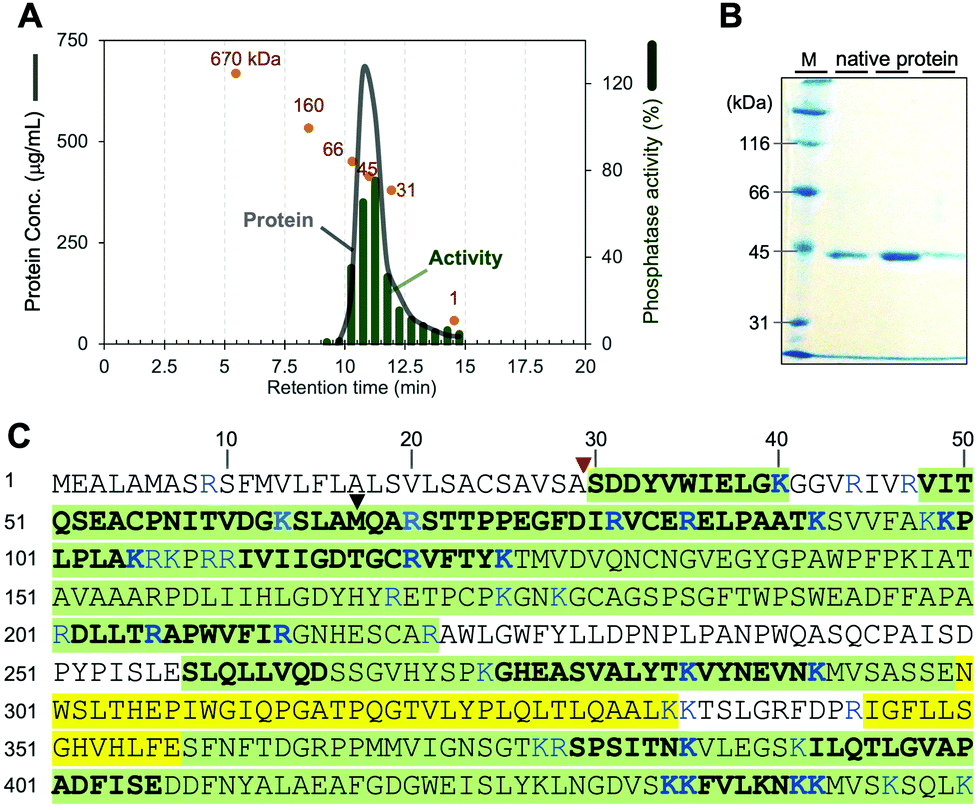 | ||
| Fig. 2 Identification of the native phosphocalyculin A phosphatase. (A) Chromatogram of LW-803 gel filtration. The elution time of the native phosphatase was 10.7 min, suggesting that the molecular weight is 45 ± 3 kDa (orange spots indicate the profile of each calibration marker: 670–1 kDa). (B) 10% SDS-PAGE of the purified native protein (CBB stained). Lane M is molecular weight markers. The three lanes represent fractions eluted between 10.5–12.0 min. The protein bands were subjected to the PMF analysis (see Fig. S2, ESI†). (C) Amino acid sequences of native phosphatase and recombinant CalL detected by PMF. The peptide fragments detected from both the recombinant and native CalL proteins, highlighted in green. The fragments only detected with recombinant CalL are highlighted in yellow. Plain regions were not detected from both proteins. The fragments detected by LC–MS/MS are shown as bold letters. The MS/MS fragmentation patterns of the digested amino acids of both native and recombinant CalL are compared in Fig. S2 (ESI†). The signal peptidase I cleavage site and the start methionine in the previously predicted ORF are pointed out with red and black triangles, respectively. | ||
CalL, the native phosphatase specific to phosphocalyculin A
Based on the physico-chemical properties of the native phosphatase, we reinvestigated the modification enzymes encoded in the calyculin BGC originating from Ca. Entotheonella sp. There is one open reading frame (ORF) (CalL, 43 kDa, Protein ID: BAP05586.1) annotated as a calcineurin-like metallophosphoesterase encoded in the upstream region, along with three phosphotransferases including CalQ (Fig. 1A). To our surprise, the isoelectric points of all of the other proteins were acidic or neutral, except for CalL (pI = 9.01, Table S1, ESI†). To determine whether the phosphocalyculin-specific phosphatase is identical to CalL, the band detected in the SDS-PAGE (Fig. 2B) of the native phosphatase fraction was excised, digested with trypsin and subjected to peptide mass fingerprinting (PMF) by liquid chromatography–electrospray ionization–tandem mass spectrometry (LC–ESI–MS/MS). As expected, the peptide fragments with amino acid sequences corresponding to CalL were successfully detected from the band of the native phosphatase (Fig. 2C), and this identification was further corroborated by the peptide fragments detected from the recombinant CalL. However, the PMF unexpectedly suggested that the native CalL contains an additional N-terminal region (Fig. S2C, ESI†), which was inconsistent with the ORF predicted from the originally reported DNA sequence.8 This observation prompted us to resequence the upstream region of calL, and we discovered that the adenine encoded at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 214–base pair (bp) was incorrectly duplicated (Fig. S3, ESI†). The revised sequence allowed us to find a new start codon within a more upstream region than the previous sequence (Fig. 2C and Fig. S4, ESI†). To further confirm the N-terminus of the native form of CalL, we carefully inspected the PMF data of the native phosphatase, by using reference sequences cleaved at every single amino acid from the theoretical N-terminus of the new ORF. As a result, no peptide fragment from the theoretical N-terminus to A29 was detected, but among the detected fragments, we found an N-terminally truncated peptide fragment (′30-SDDYVWIELGK-40′, Fig. 2C and Fig. S2A, ESI†) that corresponded to the distal end. The detected fragment at the C-terminal end of the native phosphatase was identical to the ORF of CalL (45 kDa) (Fig. 2C and Fig. S2, ESI†). Therefore, we reasoned that the N-terminally truncated CalL, encoded upstream of the calyculin assembly-line (Fig. 1A), is identical to the native phosphatase specific to phosphocalyculin A.
214–base pair (bp) was incorrectly duplicated (Fig. S3, ESI†). The revised sequence allowed us to find a new start codon within a more upstream region than the previous sequence (Fig. 2C and Fig. S4, ESI†). To further confirm the N-terminus of the native form of CalL, we carefully inspected the PMF data of the native phosphatase, by using reference sequences cleaved at every single amino acid from the theoretical N-terminus of the new ORF. As a result, no peptide fragment from the theoretical N-terminus to A29 was detected, but among the detected fragments, we found an N-terminally truncated peptide fragment (′30-SDDYVWIELGK-40′, Fig. 2C and Fig. S2A, ESI†) that corresponded to the distal end. The detected fragment at the C-terminal end of the native phosphatase was identical to the ORF of CalL (45 kDa) (Fig. 2C and Fig. S2, ESI†). Therefore, we reasoned that the N-terminally truncated CalL, encoded upstream of the calyculin assembly-line (Fig. 1A), is identical to the native phosphatase specific to phosphocalyculin A.
Functional analysis of recombinant CalL
CalL is composed of an N-terminal signal peptide sequence fused with a catalytic domain belonging to the metallophosphoesterase (MPE) superfamily.22 According to signal sequence prediction,23 the N-terminal signal peptide is likely to be involved in the protein localization, in good agreement with the N-terminal truncation to generate the peptide fragment ′30-SDDYVWIELGK-40′, detected in the PMF data. To confirm this finding, we implemented a functional analysis of recombinant CalL. Due to the possible cleavage of the N-terminal signal peptide by the signal peptidases of the heterologous host, the C-terminal Strep tag II-fused CalL (CalL-strep) was heterologously expressed in Escherichia coli BL21(DE3). The fraction containing CalL-strep was purified with Strep-Tactin® agarose, followed by PROTEIN LW-803 column chromatography (Fig. S5, ESI†). The purified CalL-strep not only showed phosphatase activity against phosphocalyculin A (Fig. S6A, ESI†), but also migrated as a major spot on the 2D gel electrophoresis, which was superimposable onto that of the native CalL (Fig. S1, ESI†). Furthermore, to identify the signal peptidase cleavage site, the purified CalL-strep was subjected to PMF in the same manner as the native CalL. As expected, the 30–40 peptide fragment was detected from the recombinant CalL as well as the native CalL (Fig. 2C, 30′-SDDYVWIELGK-40′, in comparison with the MS/MS spectra of native CalL in Fig. S2A, ESI†). A VXA motif at residues 27–29, which is well-known as the signal peptidase I (SPase I) recognition site, is present at the adjacent upstream position of the cleavage site between residues 29–30.24The steady-state kinetic parameters of native and recombinant CalL were also comparable (Table S3 and Fig. S7, ESI†), strongly validating that CalL is the phosphocalyculin-specific phosphatase. As compared with the kinetic parameters of the phosphotransferase CalQ, the kcat/Km value of CalL is three orders of magnitude higher, which corroborates the highly efficient bioconversion process in response to sponge tissue disruption, even with the co-existence of CalQ (Table S3, ESI†). Thus, we concluded that CalL is the phosphatase triggering the wound-activated generation of calyculin A in the sponge holobiont.
Biochemical properties of CalL
The amino acid sequence of CalL shares only minimal homology with other functionally characterized phosphatase groups (<20%), according to the basic logical alignment search tool (BLAST). However, the metal-binding residues in CalL are highly conserved and identical to those of purple acid phosphatases (PAPs), as shown in Fig. 3. The phylogenetic analysis was implemented with the seed alignment of the MPE superfamily, using the neighbor-joining algorithm in molecular evolutionary genetics analysis-X (MEGA-X) (Pfam code: PF00149, Fig. S8, ESI†).25,26 Indeed, the PAP clade is the most similar phosphatase clade to CalL among MPEs, but the phylogenetic tree suggested that CalL and its homologs form an independent clade. To elucidate the characteristics of the newly generated CalL clade, we evaluated the biochemical properties of the recombinant CalL-strep.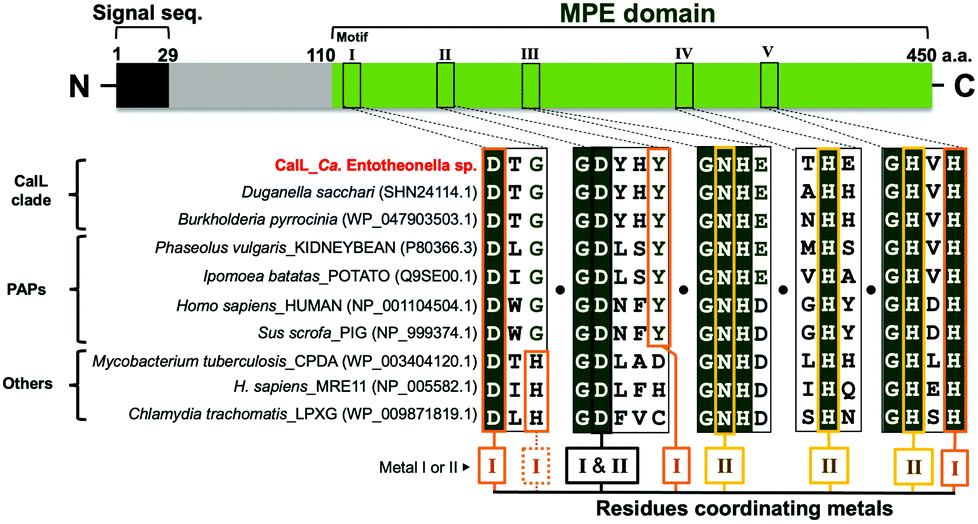 | ||
| Fig. 3 Alignment of the five metal-coordinating motifs. Amino acid sequences of CalL, its homologs, and representative enzymes (purple acid phosphatases, pyrophosphatases; and nucleases) are aligned and five metal-coordinating motifs (I–V) were showed. The MPE domain has two catalytic metals at the active site, and each is coordinated by four residues (residues for metal I, orange box; metal II, yellow box; I and II, black box). The NCBI codes were described in parentheses after species names. | ||
According to the elution time of the gel filtration chromatography (Fig. 2A and Fig. S5, ESI†), the recombinant and native CalL exist as the monomer form, which is the same as the mammalian PAP, but differs from a plant PAP composed of homodimeric proteins.27,28 Although PAPs prefer acidic pH condition in general,27,28 the highest activity of recombinant CalL was observed at neutral pH 7.25 (Fig. S6B, ESI†). The inhibition profile of CalL was investigated, using various phosphatase inhibitors (Fig. S6C, ESI†). Imidazole, an alkaline phosphatase inhibitor, sodium fluoride, a non-specific inhibitor of Ser/Thr protein phosphatases,29,30 and sodium tartrate, an acid phosphatase inhibitor, did not affect the CalL activity at all.31 While the calcium-specific chelator EGTA had a subtle effect on the activity, the di- and trivalent metal-chelating agent EDTA decreased its activity to 20% at 10 mM. These data supported the proposal that CalL is a metalloenzyme and a calcium ion is not utilized as a catalytic metal.22 The most effective inhibitors of CalL were phosphate and sodium molybdate, which are representative inhibitors of PAPs.31 Therefore, the profile of CalL showed the characteristics of the PAP group, but not those of any other phosphatases.
Catalytic metal center in CalL
The presence of a bimetallic center at the active site of MPEs is crucial for their dephosphorylation activity.22 Although the metal-binding residues are highly conserved, the repertories of metal ions among MPEs are diverse.22 The PAPs with metal-binding residues identical to those of CalL are distinguished from other phosphatases by their characteristic purple color and acidic pH optima. This purple color (absorbance within the 550–560 nm visible region) is caused by the charge transfer from the phenolic hydroxy group of tyrosine to Fe(III) at the bimetallic “Fe(III) and Metal(II)” catalytic center of the enzyme (where the divalent metal is Fe, Mn or Zn).22,27,28,31 However, even though this tyrosine residue at the site II metal-binding motif is conserved in CalL (Fig. 3), no absorbance in the visible region was observed for both the native and recombinant CalL solutions. To clarify whether the putative metal-binding residues of CalL are important for the metal-binding as well as its activity, the amino acid residues at the metal I - and II - binding sites (I: D116A, Y168F, Y168A, H352A, II: N215A, H305A, H352A, I and II: D165A) were mutated. As a result, the phosphatase activities of all mutants were significantly diminished (Fig. S9A, ESI†). Therefore, all of the putative metal-coordinating residues of CalL are essential for its activity, as in the cases of other phosphatases.22To determine the catalytic metals of CalL, the metal contents of native and recombinant CalL, together with its mutants, were analyzed qualitatively by inductively coupled plasma–mass spectrometry (ICP–MS). Neither Fe nor Mn, which are commonly coordinated in PAP active sites, was detected. Therefore, Fe is not the catalytic metal in CalL, in agreement with the fact that CalL is not purple colored. Instead, in both the native and recombinant CalL, Cu was the most abundant, followed by Zn (Fig. S9B, ESI†). To confirm the ICP–MS results, we also quantified the copper and zinc concentrations in recombinant CalL-strep using Metallo assay kits. The molar ratios of wild-type CalL to Cu and Zn were 0.74 and 0.88, respectively, while they were less than 0.15 in the D116A, Y168F, and D168A mutants (Table S4, ESI†). The ratio of Cu and Zn below 1.0 is probably due to the contaminating apo proteins that were not eliminated during the purification step. Based on the ICP–MS data and metallic assay results, CalL most likely harbors the heteronuclear metals Cu and Zn at the active site, which is unprecedented among the MPE superfamily members.
Localization of recombinant CalL and its reaction place in the producer
Considering the wound-activated mechanism of this bioconversion, we reasoned that the specific localization of CalL would be crucial for the compartmentalization of the bioconversion process. To obtain clues toward the localization of CalL, the amino acid sequences of CalL were analyzed with the well-established localization predictor of bacterial proteins, InterPro.23 The prediction tool showed that the signal peptide sequence at the N-terminus of CalL would be recognized by the SecYEG translocase, which would drag it into the periplasm.32–34 To evaluate this hypothesis, the subcellular localization of recombinant CalL-strep in E. coli BL21(DE3) was investigated. In order to purify CalL-strep from whole cell protein extracts, a two-step purification was generally required to remove the chaperones (GroES, 60 kDa; and DnaK, 70 kDa; Fig. S10, ESI†) derived from the heterologous expression host. However, CalL-strep was purified efficiently from the periplasmic fraction, without any endogenous chaperone contamination, by using only the strep-affinity column (Fig. S10, ESI†), indicating that CalL accumulates in the periplasmic space. After translocation by the SecYEG complex, the N-terminus of the CalL preprotein would be recognized by SPase I and cleaved between residues 29 and 30, and then successively released into the periplasmic space of the heterologous expression host E. coli.On the other hand, CalQ is expected to be cytosolic according to the localization predictors,23 which led us to hypothesize that both phosphocalyculin A and CalQ are localized in the cytoplasm of the producer cells. To confirm the place where the dephosphorylation reaction occurs, the sponge homogenate was subjected to density gradient centrifugation to fractionate the sponge and bacterial cells, and then the localization of calyculin A in conjunction with the phosphatase activity of each fraction was examined. As shown in Fig. S11 (ESI†), prominent phosphatase activity was detected in the Entotheonella fraction, in which calyculin A also accumulated.
We performed an imaging analysis by using two different Entotheonella cells associated with the same genus of sponges, Discodermia kiiensis and D. calyx, respectively (Fig. S12, ESI†). The Entotheonella cells associated with the marine sponge D. kiiensis are useful as a negative control, since D. kiiensis does not show wound-activated changes of any major metabolites in the extracts. The Entotheonella cells were prepared from the freeze-dried D. calyx sponge specimens and soaked in artificial seawater. Subsequently, the cells were treated with malachite green reagents and then observed by microscopy (Fig. 4). The green color was exclusively observed in the Entotheonella cells associated with D. calyx, while the Entotheonella cells associated with D. kinesis were not stained. This result suggests that the substrate phosphocalyculin A is preserved in the cytoplasm. Upon sponge tissue wounding, the inner membrane of the Entotheonella cells is disrupted, as demonstrated by the scanning electron microscopy (SEM) shown in Fig. S13 (ESI†), thus exposing phosphocalyculin A to the periplasmic space and triggering CalL-catalytic dephosphorylation.
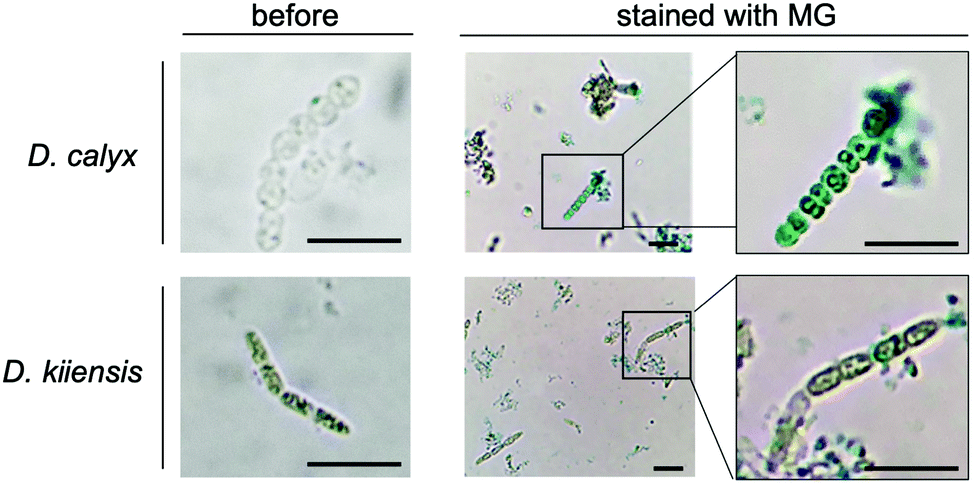 | ||
| Fig. 4 Localization of CalL and its reaction place in the producer cells. To visualize the accumulation of free phosphoric acid in filamentous bacterial cells, cells were stained with malachite green reagent (MG). The left panels are the images of filamentous bacterial cells before staining. The right panels show high-magnification images of stained cells. Scale bars, 10 μm. | ||
Activated chemical defense strategies are prevalent in plants, such as terrestrial higher plants, mushrooms, and aqueous algae.12–15 The advantage of these systems is the on-demand production of a toxic metabolite, which is not suitable for constitutive storage due to its toxicity to the producer organisms.8 Calyculin A is the dominant metabolite produced by the PKS and NRPS hybrid pathway in the symbiont Entotheonella, and it is also potentially harmful to the host sponge because of its potent cytotoxicity. To circumvent self-toxicity, a mechanism to regulate the calyculin A cytotoxicity would be required for the sponge, D. calyx. The previously identified biosynthetic gene cluster of calyculin A and the encoded resistance gene calQ provided insight into the well-regulated, wound-induced process of calyculin A generation.8 The phosphotransferase CalQ detoxifies calyculin A and generates phosphocalyculin A, as a protoxin stored in the sponge holobiont. To trigger the activated chemical defense, an activating mechanism, in particular the enzyme responsible for the activation process, is necessary.
In this study, we isolated the activating enzyme that specifically dephosphorylates the protoxin phosphocalyculin A and identified it as CalL, encoded in the calyculin biosynthetic gene cluster in Ca. Entotheonella. CalL is a metalloprotein belonging to the binuclear MPE superfamily, with a signal peptide sequence at its N-terminus. The MPEs share the catalytic center that requires two adjacently positioned transition metal ions to catalyze the hydrolytic reaction.22 The characteristics of CalL, such as the metal-binding motifs, are diagnostic for PAPs as shown in Fig. 3, except for the pH optima and the sensitivity against EDTA, which has no effect on PAP at 10 mM.31 Although the tyrosine residue in motif II (Fig. 3) is conserved in both CalL and PAPs, the metal contents of CalL were different from the Fe(III) and Mn(II) of PAPs. Even in cultures of E. coli expressing the recombinant CalL in the presence of a high concentration of Fe(III), the purple color of CalL was not observed. The results demonstrated that the Cu and Zn ions are the catalytic metals in both the wild-type recombinant and native CalL proteins, according to the ICP–MS data and the quantitative colorimetric assay. This is the first report to describe the heteronuclear Cu–Zn catalytic center in the MPE superfamily.22 Previously, Mitić et al. reported that the Fe(III) in PAP can be exchanged with a divalent Mn ion. Although the Mn-mutant was no longer purple, it retained the dephosphorylation activity.35 This report demonstrated that CalL is also capable of chelating Cu and Zn, and showing its activity without a purple color. One of the prominent examples of a Cu/Zn metalloprotein is Cu/Zn superoxide dismutase (SOD), a scavenger of peroxide generated by oxidative stress,36 in which the metal-coordinating residues of SOD coincide with those of MPE.37 Our phylogenetic analysis of MPE domains indicated that CalL and its homologs represent a new clade that is independent from PAPs and any other phosphatases. The physiological roles of this enzyme family have remained unknown, except for CalL.
The phosphorylation-involved activation/deactivation process resembles the biosynthetic pathway of antibacterial aminoglycosides and NRPs, such as viomycin in Actinomycetes.38–41 Streptomycin and viomycin are produced as phosphorylated protoxins, and are excreted into the extracytosolic space to confer self-resistance to antibiotics. The well-known phosphotransferases StrA and Vph are responsible for the phosphorylation of streptomycin and viomycin, respectively.39,40 On the other hand, the streptomycin-producing Streptomyces species express a phosphatase (StrK) that is highly related to alkaline phosphatases, such as PhoA in E. coli.41 It specifically cleaves both streptomycin-6-phosphate and, more slowly, streptomycin-3′′-phosphate. Likewise, VioS, encoded in the viomycin BGC as a homolog of StrK, is responsible for the dephosphorylation of viomycin phosphate.39 All of these phosphatases are considered to be exported to extracytosolic spaces, as corroborated by the N-terminal signal peptide sequence featured in these phosphatases. Streptomycin and viomycin are exported in the phosphorylated form and then activated outside the cell by StrK and VioS, respectively.39,40 This mechanism of resistance, translocation and reactivation is required for the biosynthesis of these ribosome targeting antibiotics, which are highly toxic to the free-standing Gram-positive bacteria. This prevalent regulatory system of antibiotic biosynthesis is also found in Gram-negative bacterial producers of the genotoxin colibactin, which is biosynthesized from pre-colibactin and activated by the ClbP peptidase in the periplasm.42
In the case of the sponge-derived cytotoxin, given that calyculin A does not exhibit antibacterial activity against any Gram-negative and positive bacteria, the calyculin biosynthetic pathway involving deactivation and reactivation processes is rather beneficial for the host sponge, D. calyx. A suite of protoxins and activation enzymes coexists, and they are compartmentalized in the dynamic machinery, which would confer chemical defense as well as self-resistance for the sponge itself. To compartmentalize CalL, the N-terminal signal sequence plays an important role in the periplasmic localization of CalL, as is the case for the N-terminal signal sequences of StrK and VioS.39,40 The N-terminal signal sequence (a.a. residues: 1–29) in CalL is composed of a hydrophobic helix structure (residues: 13–24) and an A/V/SXA motif (residues: 27–29). The former is essential for anchoring to the inner-membrane and the latter is recognized by SPase I. A ribosomally synthesized polypeptide bearing a signal sequence is transported through a general secretory (Sec) pathway in the cytosol and moved to the inner-membrane as an unfolded pre-protein, which is then translocated toward the surface of the periplasmic space, where SPase I cleaves the signal peptide to release the mature protein into the periplasmic space.32–34 This Sec-SPase I pathway is also present in “Ca. Entotheonella sp.”, since SPase I homologs (Protein sequence IDs: PON19655.1, ETX08252.1, ETW99059.1, and WP_179131489.1) are encoded in the genomes of Ca. Entotheonella serta, gemina, factor, and palauensis, respectively.43–46 Therefore, we envisioned that CalL is also localized within the periplasmic space, with the aid of the signal peptide and through the Sec pathway in the sponge symbiont Ca. Entotheonella sp. Indeed, we detected the cleaved mature form of CalL with not only the native enzyme, but also the recombinant protein, validating that the N-terminal sequence of CalL is also recognized by the Sec-SPase I pathway in the heterologous host E. coli BL21(DE3).
Conclusions
We have demonstrated that the phosphatase CalL, which originates in the Entotheonella symbiont of D. calyx, initiates the on-demand activation of calyculin A in response to environmental stimuli. The highly facile and site-specific activation process may provide significant opportunities not only for understanding the chemical defense strategies in marine sponge holobionts, but also for the on-site exposure of calyculin A targeted to cancer cells in combination with an enzyme-activated prodrug therapy.47 Further research will be required to elucidate the intercellular cross-talk shared by symbiont bacterial cells and sponge cells involved in the wound-activated system.Author contributions
Conceptualization: TJ, KM, IA, AT, TW, Methodology: TJ, YE, KM, IA. AT, TW, Investigation: TJ, YE. KM, Visualization: TJ, YE, KM, Supervision: IA, AT, TW, Writing—original draft: TJ, TW, Writing—review & editing: TJ, KM, IA, AT, TW.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. Yoshimitsu Hamano and Dr Chitose Maruyama (Fukui Prefectural University) for providing mass spectra. We also thank Dr Masanori Yasui (Electron Microscope Laboratory, Research Faculty of Agriculture, Hokkaido University) for technical assistance. This work was partly supported by Global Station for Biosurfaces and Drug Discovery, a project of Global Institution for Collaborative Research and Education at Hokkaido University (KM and TW), the Japan Agency for Medical Research and Development JP19ae0101045 (TW), Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan JP16703511 (TW), JP18056499 (TW), and JP19178402 (KM).References
- J. T. Wright, K. Benkendorff and A. R. Davis, J. Exp. Mar. Biol. Ecol., 1997, 213, 199–213 CrossRef.
- M. D. Tianero, J. N. Balaich and M. S. Donia, Nat. Microbiol., 2019, 4, 1149–1159 CrossRef CAS PubMed.
- M. Rust, E. J. N. Helfrich, M. F. Freeman, P. Nanudorn, C. M. Field, C. Rückert, T. Kündig, M. J. Page, V. L. Webb, J. Kalinowski, S. Sunagawa and J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9508–9518 CrossRef CAS PubMed.
- L. A. Risinger and L. Du, Nat. Prod. Rep., 2020, 37, 634–652 RSC.
- T. Wakimoto, Y. Egami and I. Abe, Nat. Prod. Rep., 2016, 33, 751–760 RSC.
- S. Rohde, S. Nietzer and P. J. Schupp, PLoS One, 2015, 10, e0132236 CrossRef PubMed.
- Y. Kato, N. Fusetani, S. Matsuaga, K. Hashimoto, S. Fujita and T. Furuya, J. Am. Chem. Soc., 1986, 108, 2780–2781 CrossRef CAS.
- T. Wakimoto, Y. Egami, Y. Nakashima, Y. Wakimoto, T. Mori, T. Awakawa, T. Ito, H. Kenmoku, Y. Asakawa, J. Piel and I. Abe, Nat. Chem. Biol., 2014, 10, 648–655 CrossRef CAS PubMed.
- H. Ishihara, B. L. Martin, D. L. Brautigan, H. Karaki, H. Ozaki, Y. Kato, N. Fusetani, S. Watabe, K. Hashimoto, D. Uemura and D. J. Hartshorne, Biochem. Biophys. Res. Commun., 1989, 159, 871–877 CrossRef CAS PubMed.
- T. Wakimoto, S. Matsunaga, A. Takai and N. Fusetani, Chem. Biol., 2002, 9, 309–319 CrossRef CAS PubMed.
- A. Kita, S. Matsunaga, A. Takai, H. Kataiwa, T. Wakimoto, N. Fusetani, M. Isobe and K. Miki, Structure, 2002, 10, 715–724 CrossRef CAS PubMed.
- A. Mithöfer and W. Boland, Annu. Rev. Plant Biol., 2012, 12, 431–450 CrossRef PubMed.
- C. Lenz, J. Wick, D. Braga, M. García-Altares, G. Lackner, C. Hertweck, M. Gressler and D. Hoffmeister, Angew. Chem., Int. Ed., 2020, 59, 1450–1454 CrossRef CAS PubMed.
- V. J. Paul and K. L. Van Alstyne, J. Exp. Mar. Biol. Ecol., 1992, 160, 191–203 CrossRef CAS.
- V. J. Paul, M. P. Puglisi and R. Ritson-Williams, Nat. Prod. Rep., 2006, 23, 153–180 RSC.
- R. Teeyapant and P. Proksch, Naturwissenschaften, 1993, 80, 369–370 CrossRef CAS.
- C. Thoms, R. Ebel and P. Proksch, J. Chem. Ecol., 2006, 32, 97–123 CrossRef CAS PubMed.
- B. Lipowicz, N. Hanekop, L. Schmitt and P. Proksch, Mar. Drugs, 2013, 11, 3046–3067 CrossRef PubMed.
- C. Thoms, M. Wolff, K. Padmakumar, R. Ebel and P. Proksch, Z. Naturforsch., C: J. Biosci., 2004, 59, 113–122 CrossRef CAS PubMed.
- P. Ettinger-Epstein, C. A. Motti, R. Nys, A. D. Wright, C. N. Battershill and D. M. Tapiolas, J. Nat. Prod., 2007, 70, 648–651 CrossRef CAS PubMed.
- C. Thoms and P. J. Schupp, J. Chem. Ecol., 2008, 34, 1242–1252 CrossRef CAS PubMed.
- N. Matange, M. Podobnik and S. S. Visweswwariah, Biochem. J., 2015, 467, 201–216 CrossRef CAS PubMed.
- M. Blum, H. Y. Chang, S. Chuguransky, T. Grego, S. Kandasaamy, A. Mitchell, G. Nuka, T. Paysan-Lafosse, M. Qureshi, S. Raj, L. Richardson, G. A. Salazar, L. Williams, P. Bork, A. Bridge, J. Gough, D. H. Haft, I. Letunic, A. Marchler-Bauer, H. Mi, D. A. Natale, M. Necci, C. A. Orengo, A. P. Pandurangan, C. Rivoire, C. J. A. Sigrist, I. Sillitoe, N. Thanki, P. D. Thomas, S. C. E. Tosatto, C. H. Wu, A. Bateman and R. D. Finn, Nucleic Acids Res., 2021, 49, D344–D354 CrossRef CAS PubMed.
- H. S. Payne, S. Bonissone, S. Wu, N. R. Brown, N. D. Ivankov, D. Frishman, L. Paša-Tolić, D. R. Smith and A. P. Pevzner, mBio, 2012, 3, e00339–12 CrossRef PubMed.
- R. D. Finn, A. Bateman, J. Clements, P. Coggill, R. Y. Eberhardt, S. R. Eddy, A. Heger, K. Hetherington, L. Holm, J. Mistry, E. L. L. Sonnhammer, J. Tate and M. Punta, Nucleic Acids Res., 2014, 42, D222–D230 CrossRef CAS PubMed.
- S. Kumar, G. Stecher, M. Li, C. Knyaz and K. Tamura, Mol. Biol. Evol., 2018, 35, 1547–1549 CrossRef CAS PubMed.
- L. W. Guddat, A. S. McAlpine, D. Hume, S. Hamilton, J. de Jersey and J. L. Martin, Structure, 1999, 7, 757–767 CrossRef CAS PubMed.
- M. Olczak, B. Morawiecka and W. Watorek, Acta Biochim. Pol., 2003, 50, 1245–1256 CrossRef CAS PubMed.
- C. Brunel and G. Cathala, Biochim. Biophys. Acta, 1972, 268, 415–421 CrossRef CAS.
- J. A. Gordon, Methods Enzymol., 1991, 201, 477–482 CAS.
- S. L. Yeung, C. Cheng, T. K. Lui, J. S. Tsang, W. T. Chan and B. L. Lim, Gene, 2009, 440, 1–8 CrossRef CAS PubMed.
- I. Gelis, A. M. Bonvin, D. Keramisanou, M. Koukaki, G. Gouridis, S. Karamanou, A. Economou and C. G. Kalodimos, Cell, 2007, 131, 756–769 CrossRef CAS PubMed.
- S. M. Auclair, M. K. Bhanu and D. A. Kendall, Protein Sci., 2012, 21, 13–25 CrossRef CAS PubMed.
- K. E. Chatzi, M. F. Sardis, S. Karamanou and A. Economou, Biochem. J., 2013, 449, 25–37 CrossRef CAS PubMed.
- N. Mitić, C. J. Noble, L. R. Gahan, G. R. Hanson and G. Schenk, J. Am. Chem. Soc., 2009, 131, 8173–8179 CrossRef PubMed.
- R. J. Carrico and H. F. Deutsch, J. Biol. Chem., 1970, 245, 723–727 CrossRef CAS.
- R. Rakhit and A. Chakrabartty, Biochim. Biophys. Acta, 2006, 1762, 1025–1037 CrossRef CAS PubMed.
- W. Piepersberg, Biotechnology, 1995, 28, 531–570 CAS.
- G. M. Thomas, A. Y. Chan and G. S. Ozanick, Antimicrob. Agents Chemother., 2003, 47, 2823–2830 CrossRef PubMed.
- C. K. Lim, M. C. Smith, J. Petty, S. Baumberg and J. C. Wootton, J. Gen. Microbiol., 1989, 135, 3289–3302 CAS.
- K. Mansouri and W. Pipersberg, Mol. Gen. Genet., 1991, 228, 459–469 CrossRef CAS PubMed.
- D. Dubois, O. Baron, A. Cougnoux, J. Delmas, N. Pradel, M. Boury, B. Bouchon, M. A. Bringer, M. J. P. Nougayrède, E. Ostwald and R. Bonnet, J. Biol. Chem., 2011, 286, 35562–35570 CrossRef CAS PubMed.
- M. C. Wilson, T. Mori, C. Rückert, A. R. Uria, M. J. Helf, K. Takada, C. Gernert, U. A. Steffens, N. Heycke, S. Schmitt, C. Rinke, E. J. Helfrich, A. O. Brachmann, C. Gurgui, T. Wakimoto, M. Kracht, M. Crüsemann, U. Hentschel, I. Abe, S. Matsunaga, J. Kalinowski, H. Takeyama and J. Piel, Nature, 2014, 506, 58–62 CrossRef CAS PubMed.
- R. Ueoka, A. R. Uria, S. Reiter, T. Mori, P. Karbaum, E. E. Peters, E. J. N. Helfrich, B. I. Morinaka, M. Gugger, H. Takeyama, S. Matsunaga and J. Piel, Nat. Chem. Biol., 2015, 11, 705–712 CrossRef CAS PubMed.
- G. Lackner, E. E. Peters, E. J. N. Helfrich and J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E347–E356 CrossRef CAS PubMed.
- T. Mori, J. K. B. Cahn, M. C. Wilson, R. A. Meoded, V. Wiebach, A. F. C. Martinez, E. J. N. Helfrich, A. Albersmeier, D. Wibberg, S. Dätwyler, R. Keren, A. Lavy, C. Rückert, M. Ilan, J. Kalinowski, S. Matsunaga, H. Takeyama and J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1718–1723 CrossRef CAS PubMed.
- S. K. Sharma and K. D. Bagshawe, Adv. Drug Delivery Rev., 2017, 118, 2–7 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00163a |
| This journal is © The Royal Society of Chemistry 2021 |