
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cell permeable bimane-constrained PCNA-interacting peptide†
Aimee J.
Horsfall
 abc,
Beth A.
Vandborg
ad,
Zoya
Kikhtyak
e,
Denis B.
Scanlon
ab,
Wayne D.
Tilley
e,
Theresa E.
Hickey
e,
John B.
Bruning
*ad and
Andrew D.
Abell
*abc
abc,
Beth A.
Vandborg
ad,
Zoya
Kikhtyak
e,
Denis B.
Scanlon
ab,
Wayne D.
Tilley
e,
Theresa E.
Hickey
e,
John B.
Bruning
*ad and
Andrew D.
Abell
*abc
aInstitute of Photonics and Advanced Sensing (IPAS), The University of Adelaide, Adelaide, South Australia 5005, Australia. E-mail: john.bruning@adelaide.edu.au; andrew.abell@adelaide.edu.au
bSchool of Physical Sciences, The University of Adelaide, Adelaide, South Australia 5005, Australia
cAustralian Research Council Centre of Excellence for Nanoscale BioPhotonics (CNBP), Australia
dSchool of Biological Sciences, The University of Adelaide, Adelaide, South Australia 5005, Australia
eDame Roma Mitchell Cancer Research Laboratories, Adelaide Medical School, Faculty of Health and Medical Sciences, University of Adelaide, Adelaide, South Australia 5005, Australia
First published on 21st July 2021
Abstract
The human sliding clamp protein known as proliferating cell nuclear antigen (PCNA) orchestrates DNA-replication and -repair and as such is an ideal therapeutic target for proliferative diseases, including cancer. Peptides derived from the human p21 protein bind PCNA with high affinity via a 310-helical binding conformation and are known to shut down DNA-replication. Here, we present studies on short analogues of p21 peptides (143–151) conformationally constrained with a covalent linker between i, i + 4 separated cysteine residues at positions 145 and 149 to access peptidomimetics that target PCNA. The resulting macrocycles bind PCNA with KD values ranging from 570 nM to 3.86 μM, with the bimane-constrained peptide 7 proving the most potent. Subsequent X-ray crystallography and computational modelling studies of the macrocyclic peptides bound to PCNA indicated only the high-affinity peptide 7 adopted the classical 310-helical binding conformation. This suggests the 310-helical conformation is critical to high affinity PCNA binding, however NMR secondary shift analysis of peptide 7 revealed this secondary structure was not well-defined in solution. Peptide 7 is cell permeable and localised to the cell cytosol of breast cancer cells (MDA-MB-468), revealed by confocal microscopy showing blue fluorescence of the bimane linker. The inherent fluorescence of the bimane moiety present in peptide 7 allowed it to be directly imaged in the cell uptake assay, without attachment of an auxiliary fluorescent tag. This highlights a significant benefit of using a bimane constraint to access conformationally constrained macrocyclic peptides. This study identifies a small peptidomimetic that binds PCNA with higher affinity than previous reported p21 macrocycles, and is cell permeable, providing a significant advance toward development of a PCNA inhibitor for therapeutic applications.
Introduction
Proliferating cell nuclear antigen (PCNA) interacts with more than 200 proteins to act as an essential mediator of DNA-replication and -repair processes.1–3 As such, it has been identified as a key therapeutic target for the treatment of diseases defined by aberrant DNA-replication, including many cancers.4–7 The cell cycle regulator protein, p21, binds PCNA with the highest known affinity of all PCNA interacting proteins, through a motif referred to as the PCNA-interacting protein (PIP) box.7–10 p21 is an intrinsically disordered protein, however on binding PCNA, it adopts a well-defined single 310-helical turn to insert three hydrophobic residues (Met147, Phe150 and Tyr151) into a cleft on the PCNA surface (Fig. 1).11 This 310-helix is observed in the binding conformation of almost all PCNA-interacting proteins or peptides, and is often flanked by two short β-strands.7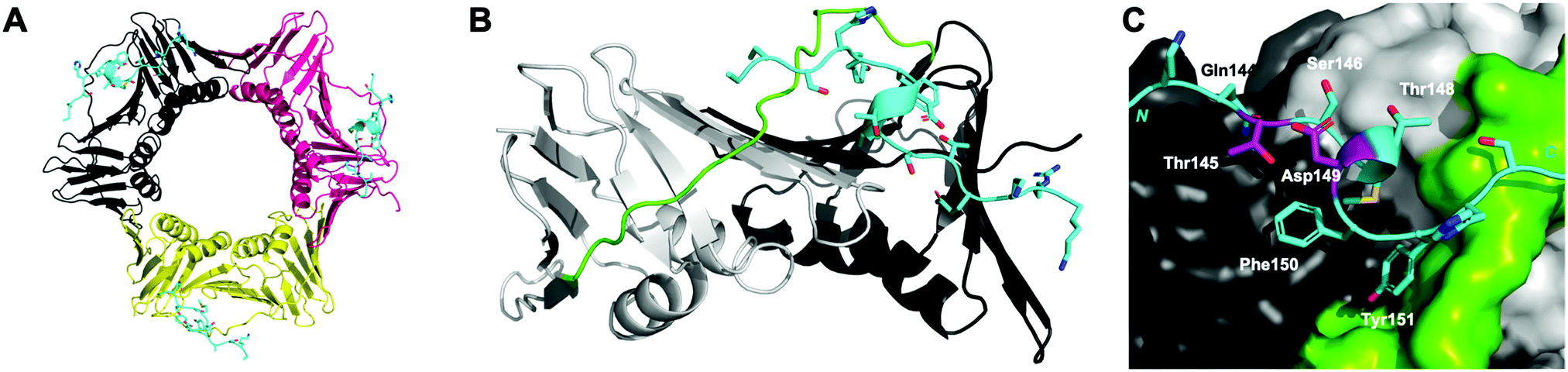 | ||
| Fig. 1 p21141–155 (cyan) bound to PCNA (PDB: 7KQ1). (A) Ring-shaped PCNA with three peptides (cyan) bound to the PIP-box binding site. PCNA monomers (three) shown in shades of grey, pink and yellow. (B) Single PCNA subunit with two domains shown in shades of grey and inter-domain connecting loop (IDCL) in green. p21141–155 shown in cyan. (C) PCNA shown as surface representation (grey) with IDCL highlighted (green). p21141–155 shown in cartoon and side-chains as sticks. PIP-box residues are labelled and the residues (145 and 149) to be modified and reacted to form a constraint are highlighted in purple. | ||
A p21 peptide (residues 139–160) derived from the C-terminus of p21 contains the PIP-box QTSMTDFY, and has been reported to bind PCNA with an affinity of ∼5 nM.7,8,12,13 This peptide competitively inhibits interaction between the major processive polymerase δ and PCNA to shut down SV40 DNA replication in vitro.14 Thus, p21-derived peptides provide an optimal template to design anti-cancer therapeutics that target PCNA. A conformation favourable to binding can be stabilised in such a peptide by covalently linking two appropriately spaced amino-acid side-chains, either by reaction together, or reaction with a bifunctional reagent.15–18 To this end, we previously reported the first macrocyclic peptidomimetic of p21139–152, where a lactam bridge was introduced between a lysine or 2,4-diaminobutyric acid residue at position 145, and a glutamic acid residue at 149 to provide two peptidomimetics referred to as ACR1 and ACR2, respectively.19 The lactam bridge was shown by NMR to stabilise a 310-helix in solution, where the 310-helix was better defined in the smaller macrocycle ACR2, than in ACR1. Interestingly, ACR1 bound PCNA with higher affinity than ACR2, indicating the well-defined 310-helix in solution did not correlate with higher PCNA affinity. A crystal structure of ACR2 bound to PCNA suggested an unfavourable interaction of the lactam linker amide bond of ACR2 and Phe150 of the peptide, which may account for the lower affinity for PCNA. Consequently, we set out to investigate a variety of different linkers in a short p21 peptide (p21143–154, peptide 1) in order to study how different peptide linkers influence the secondary structure and affinity of macrocyclic peptides bound to PCNA. A range of different macrocycles can be rapidly accessed from the same parent peptide containing two cysteines (peptide 2) using dithiol bis-alkylation chemistry.18,20 Here, five macrocyclic p21 peptide derivatives were synthesised (peptides 3–7) and their affinity for PCNA determined by surface plasmon resonance (SPR). The conformation of the macrocycles bound to PCNA was studied by X-ray crystallography and computational modelling, to investigate how the linkers interact with the PCNA surface, and how the resulting peptide structures relate to binding affinity for PCNA. These studies reveal a bimane-constrained peptide with high affinity for PCNA that is cell permeable and is a promising lead for development of a potential therapeutic that targets PCNA.
Results/discussion
Design & synthesis
The peptide p21139–160 is reported to bind PCNA with high specificity and provides an ideal template to develop PCNA inhibitors. It is known that the p21139–160 sequence can be shortened without drastically impacting affinity for PCNA13,19 and consequently a short variant, p21143–154 (1), was chosen as the starting scaffold for this study for ease of synthesis and to improve synthetic yields. The sequence was modified to include two cysteine residues at positions 145 and 149, in place of threonine and aspartic acid of p21143–154 (Fig. 1C, purple), to give peptide 2. Positions 145 and 149 were chosen for modification as these side-chains are close in space when bound to PCNA (Fig. 1C), and were successfully linked in a p21 lactam bridge peptide in our earlier study to stabilise the 310-helical binding conformation.19 This structure-informed design gave rise to an i − i + 4 constraint, although i − i + 3 constraints are more commonly reported for 310-helices. The cysteine-containing peptide can be derivatised into a range of macrocycles, where the cysteine residues are linked by dithiol bis-alkylation (Scheme 1). Three alkyl linkers (propyl, butyl and trans-butenyl),21,22 the aromatic m-xylene linker,20,22 and our fluorescent bimane linker23 were chosen as a chemically distinct range of linkers which vary in length and rigidity, in order to investigate how the different linkers impact peptide structure and PCNA binding affinity. | ||
Scheme 1 Peptide synthesis scheme (1–7) and generation of multiple cyclic peptides from a single parent utilising cysteine bis-alkylation to give macrocycles 3–7. (a) Successive Fmoc-amino-acid coupling and Fmoc-deprotection steps to assemble the peptide sequence. Coupling: HATU (5 equiv.), DIPEA (10 equiv.), Fmoc-AA-OH (5 equiv.), DMF, 1 h. Deprotection: 20% piperidine with 0.1 M HOBt, DMF, 10 min. (b) Acetylation: Ac2O (50 equiv.), DIPEA (50 equiv.), DMF, 15 min. (c) Cleavage: 92.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5:2.5 TFA/TIPS/DODT/H2O, 2 h. (d) Mmt deprotection: 2% TFA in DCM, 1 min × 40. (e) NaI (17.5 equiv.), TCEP (0.5 equiv.), DMF, N2, 15 min. (f) DIPEA (35 equiv.), DMF, 20 min. (g) 1,3-Dibromopropane (3.5 equiv.), MW 2 min, 125 °C. (h) 1,4-Dibromobutane (3.5 equiv.), MW 2 min, 125 °C. (i) trans-1,4-Dibromo-2-butene (2 equiv.), DIPEA (4 equiv.), DMF, 3 h. (j) Dibromo-m-xylene (2 equiv.), DIPEA (4 equiv.), DMF, 3 h. (k) Dibromobimane (2 equiv.), DIPEA (4 equiv.), DMF, 3 h. :2.5:2.5:2.5 TFA/TIPS/DODT/H2O, 2 h. (d) Mmt deprotection: 2% TFA in DCM, 1 min × 40. (e) NaI (17.5 equiv.), TCEP (0.5 equiv.), DMF, N2, 15 min. (f) DIPEA (35 equiv.), DMF, 20 min. (g) 1,3-Dibromopropane (3.5 equiv.), MW 2 min, 125 °C. (h) 1,4-Dibromobutane (3.5 equiv.), MW 2 min, 125 °C. (i) trans-1,4-Dibromo-2-butene (2 equiv.), DIPEA (4 equiv.), DMF, 3 h. (j) Dibromo-m-xylene (2 equiv.), DIPEA (4 equiv.), DMF, 3 h. (k) Dibromobimane (2 equiv.), DIPEA (4 equiv.), DMF, 3 h. | ||
All peptides were synthesised by a standard Fmoc/tBu solid-phase peptide synthesis (SPPS) protocol detailed in the Experimental section (see ESI†) and shown in Scheme 1. For the peptide macrocycles, the linear amino-acid sequence RQCSMTCFYHSK was assembled on a solid-support, and the N-terminal amine capped by reaction with acetic anhydride to give resin-bound 2a (Scheme 1). The cysteine side-chain protecting groups, 4-methoxytrityl (Mmt), were then selectively deprotected on-resin by repetitive 1 min treatment with 2% TFA in DCM. This provided a resin-bound peptide with two free thiols, which were subsequently reacted with either 1,3-dibromopropane, 1,4-dibromobutane, trans-1,4-dibromobut-2-ene, m-dibromoxylene or dibromobimane (Scheme 1) under literature conditions (see Experimental section in ESI†).21,23,24 Reaction of the resin-bound peptides with 1,3-dibromopropane or 1,4-dibromobutane required addition of NaI to generate the more reactive alkyliodides in situ,21 whereas reaction with trans-1,4-dibromobut-2-ene, m-dibromoxylene or dibromobimane proceeds under mild basic conditions (in DMF with DIPEA).25 The resulting macrocyclic peptides were then cleaved from the resin by treatment with 92.5% TFA containing scavengers (2.5% of each TIPS, DODT & H2O) for 2 h to give the crude macrocyclic peptides 3–7 (Scheme 1). The peptides were purified by semi-preparative RP-HPLC, and the identity and purity confirmed by HRMS and analytical RP-HPLC (>90%), respectively. Full details and characterisation data are described in the Experimental section (see ESI†).
Binding affinity
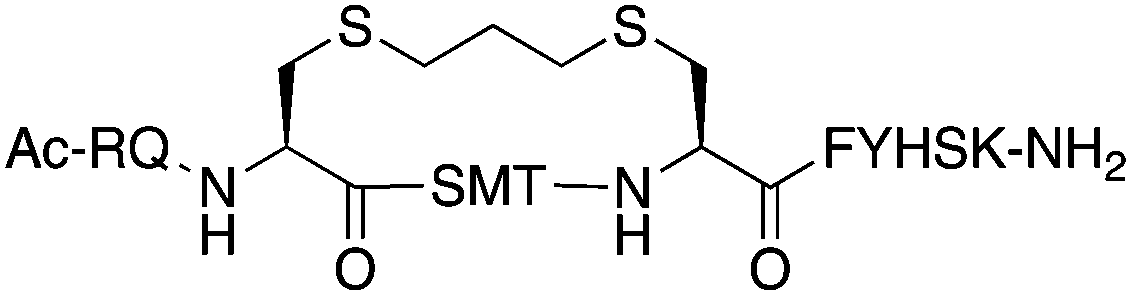
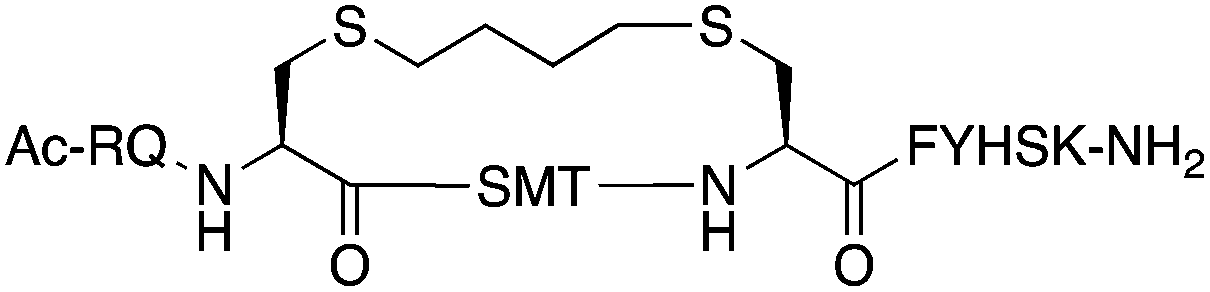
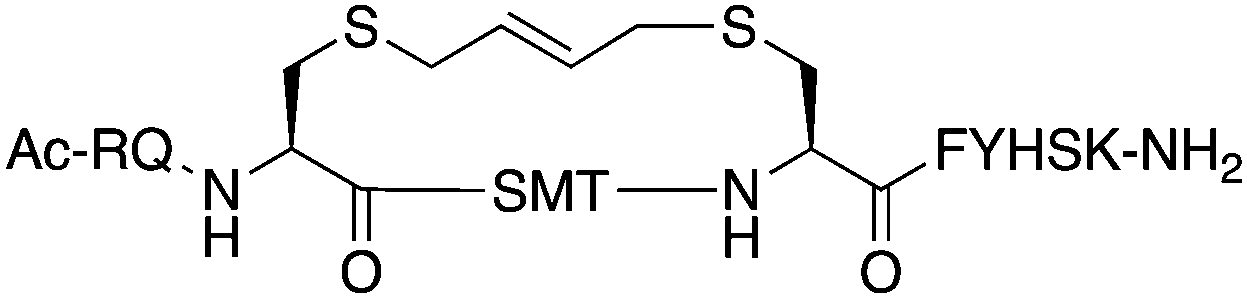
The binding affinity of the parent peptide 1, linear cysteine-containing precursor 2 and five macrocyclic peptides (3–7), for PCNA was determined by SPR (Table 1). Peptide 1 bound PCNA with a KD value of 102 nM, in line with reported affinities of short p21 peptides for PCNA.8,9,13,19,26 The cysteine-modified peptide 2 displayed significantly reduced PCNA affinity, a result that is consistent with earlier reports that polar residues at positions 145 and 149 in p21 (Thr and Asp) stabilise the binding conformation via hydrogen bonding, which in turn enhances binding affinity for PCNA.13,27,28 The affinity of the macrocyclic peptides 3–7 for PCNA ranged from 570 nM to 3.86 μM, which indicates all macrocycles were able to interact with PCNA in contrast to the precursor peptide 2. The affinity in all cases was lower than the native peptide 1, in line with observations made for our previously reported lactam macrocycles.19 Macrocyclic peptide 3, with its propyl linker, gave the second highest affinity for PCNA at 769 nM, whereas the larger butyl linker of peptide 4 (8 atom linker cf. 7 atoms of 3) resulted in reduced affinity for PCNA with a KD value of 1.99 μM. The more rigid trans-butenyl-based linker in peptide 5 resulted in a KD value for PCNA of 2.82 μM, and the binding affinity for PCNA of aromatic m-xylene linked peptide 6 (9 atom linker) was lower again at 3.86 μM. Considered together, these observations suggest that the longer and more rigid linkers result in a lower binding affinity for PCNA. The bimane-linked peptide 7 proved to be a particularly potent binder with a KD value of 570 nM, which is only 5-fold lower affinity than the native peptide 1 and is the highest affinity macrocyclic peptide to bind PCNA reported to date. The bimane linker contains 9 atoms but the bimane moiety, in contrast to the xylene ring, is not rigid and is able to flex along the plane of symmetry that bisects the two nitrogen atoms, which may explain in part the observed higher PCNA affinity.| Peptide | Sequence | Linker (atom length) | Affinity KD ± SE (μM) | χ 2 |
|---|---|---|---|---|
| 1 | H-RQTSMTDFYHSK-NH2 | — | 0.102 ± 0.005 | 0.070 |
| 2 | Ac-RQCSMTCFYHSK-NH2 | — | NS | — |
| 3 |
|
Propyl (7) | 0.769 ± 0.078 | 0.250 |
| 4 |
|
Butyl (8) | 1.99 ± 0.140 | 0.160 |
| 5 |
|
trans-Butenyl (8) | 2.82 ± 0.080 | 0.106 |
| 6 |

|
m-Xylene (9) | 3.86 ± 0.035 | 0.527 |
| 7 |
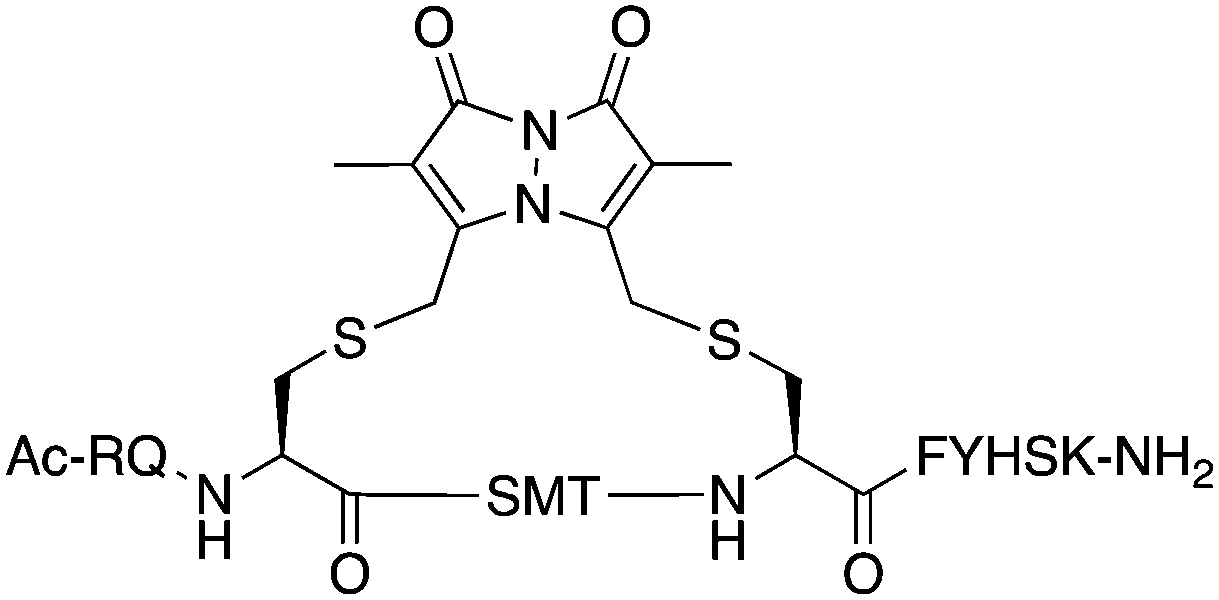
|
Bimane (9) | 0.570 ± 0.030 | 0.118 |
| 8 | FITC-βA-RQTSMTDFYHSK-NH2 | — | — | — |
| 9 |
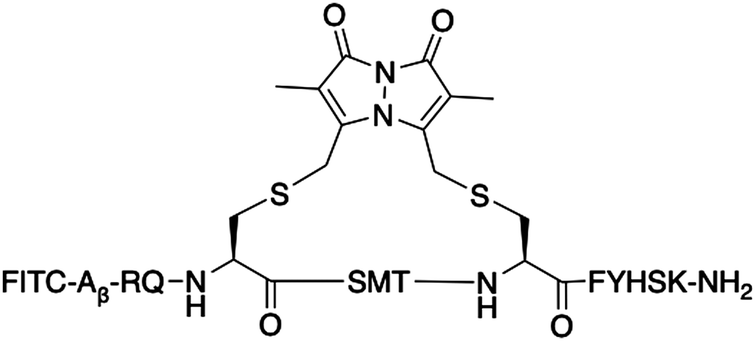
|
Bimane (9) | 25.2 ± 1.90 | 0.055 |
Structural analysis
Next, the binding conformation of macrocycles 3–7 bound to PCNA was investigated by X-ray crystallography and computational modelling studies to better understand the structural mechanisms responsible for the difference in PCNA binding affinities. Co-crystal structures of peptide 3 (PDB: 7M5L), 5 (PDB: 7M5M) and 6 (PDB: 7M5N) bound to PCNA were solved and are shown in Fig. 2 and 3, as well as Fig. S2, S4 and S5 (ESI†). Data collection and refinement statistics are summarised in Table S2 (ESI†). Attempts to obtain a co-crystal structure of peptides 1, 4 and 7 bound to PCNA were unsuccessful at this time. Peptide 1 was however modelled onto the PCNA surface, to confirm the short peptide interacted with the protein surface in the same manner (see Fig. S1, ESI†), and was constructed from the previously published structure of p21141–155 bound to PCNA (PDB: 7KQ1).13 The remaining macrocycles (4 and 7) were computationally modelled onto the PCNA surface, where the crystal structure of 3 (PDB: 7M5L) was used as the starting structure. Peptide 3 was energy minimised on the PCNA surface in order to verify the computational method, and indicates a high degree of similarity to the crystal structure (Fig. S2F and G, ESI†). Macrocycles 4 and 7 were then modelled onto the PCNA surface and energy minimised in the same way and are shown in Fig. 2, 3 and Fig. S3, S6 (ESI†).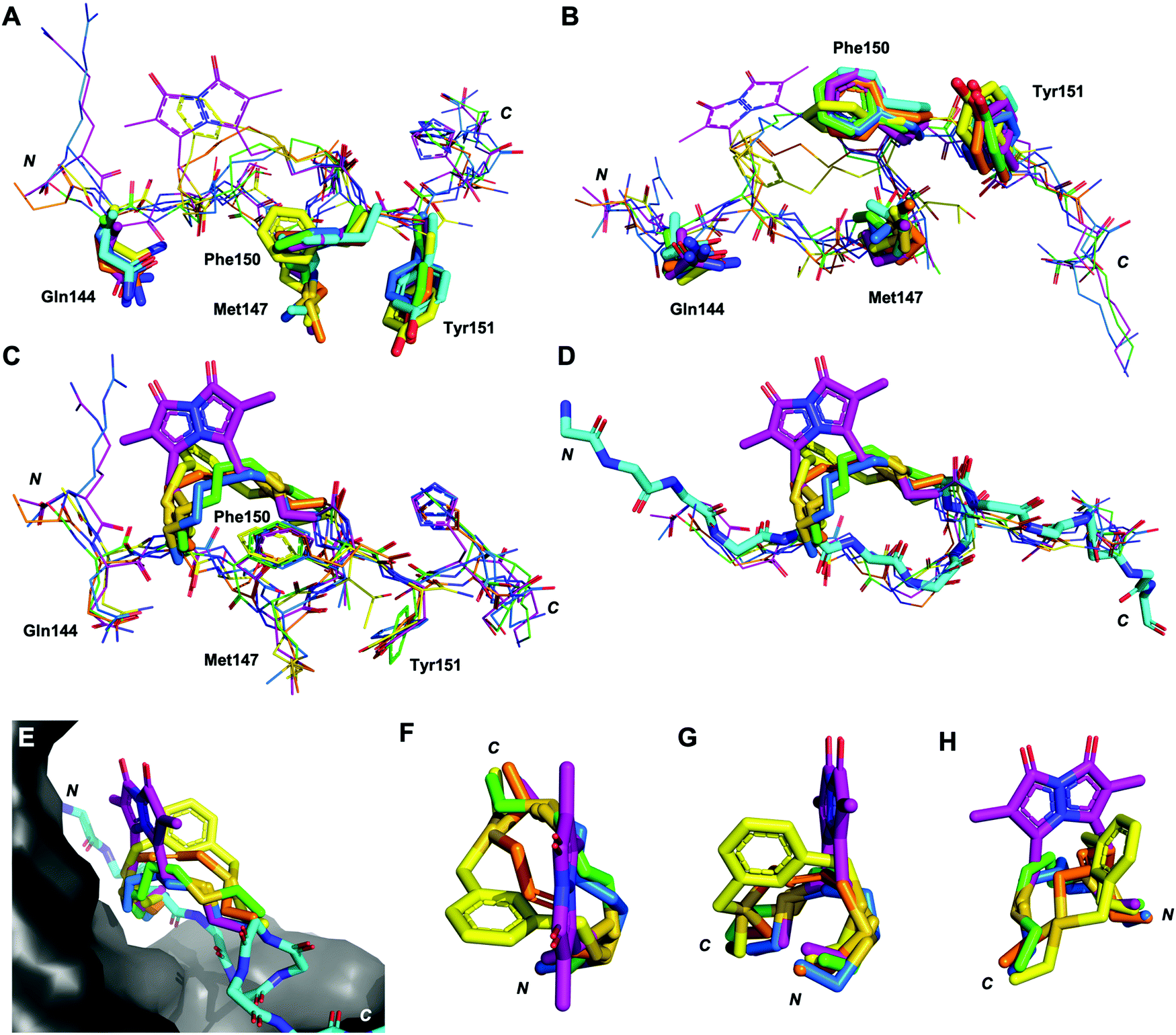 | ||
| Fig. 2 Differences in the linker position, and impact of the linker on backbone conformation. Peptides p21141–155 (light blue, 7KQ1), 3 (green, co-crystal 7M5L), 4 (blue, computationally modelled), 5 (orange, co-crystal 7M5M), 6 (yellow, co-crystal 7M5N) and 7 (purple, computationally modelled). (A and B) The conserved residues (labelled, shown as sticks) adopt similar conformations in all macrocycles. (C) The linkers (shown as sticks) induce subtle changes in the backbone of the peptide, in particular the direction of the amides. (D) The overall backbone conformation of the macrocycles is still able to mimic the p21141–155 conformation. (E) The linker length and rigidity alters where the linker sits relative to the PCNA surface. (F–H) Conformation of the peptide linkers relative to one another. | ||
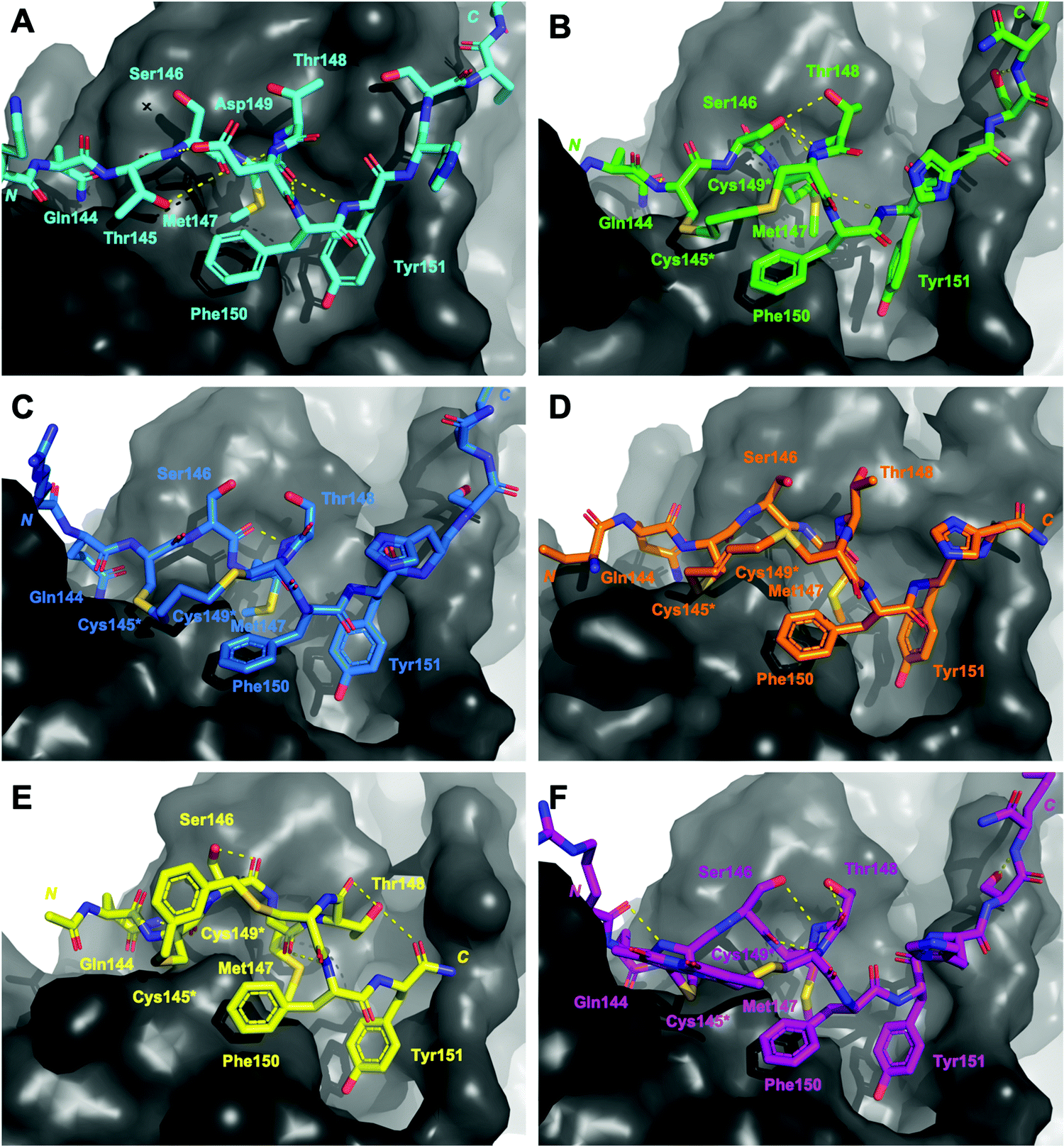 | ||
| Fig. 3 Structures of peptides (stick representation) bound to PIP-box binding site on PCNA (grey, surface). Intramolecular polar interactions are shown as yellow dashed lines. (A) p21141–155 (light blue), PDB: 7KQ1.13 (B) Peptide 3 – with propyl linker (green), co-crystal structure (PDB: 7M5L). (C) Peptide 4 – with butyl linker (blue), computationally modelled. (D) Peptide 5 – with trans-butenyl linker (orange) co-crystal structure (PDB: 7M5M). (E) Peptide 6 – with m-xylene linker (yellow), co-crystal structure (PDB: 7M5N). (F) Peptide 7 – with bimane linker (purple), computationally modelled. | ||
The structures of all five macrocyclic peptides bound to PCNA show the conserved PIP-box residues Gln144, Met147, Phe150 and Tyr151 inserted onto the PCNA surface, in a similar mode to the p21 native peptide structures (7KQ1,13Fig. 2A–D). The structures of macrocyclic peptides 3–7 bound to PCNA indicate a percentage buried surface area (%BSA) of 60% for the PIP-box residues collectively (Table S3, ESI†), suggesting similar structures overall. These observations together indicate the linker, in all cases, has not compromised the ability of the macrocycles to interact with PCNA. The difference in the linker conformations is shown in Fig. 2C–H. Interestingly, the main-chain hydrogen bond between residue 146 (i) and 149 (i + 3), that defines the classic 310-helical binding conformation of PCNA-binding peptides, is only observed for peptide 7 which possesses the highest binding affinity of a macrocycle for PCNA, to date.
The co-crystal structure of the propyl-linked peptide 3 bound to PCNA shows the conserved Gln144 side-chain makes an intermolecular 3.2 Å hydrogen bond with Ala252 (Fig. S2A–D, ESI†) and a 2.9 Å intramolecular hydrogen bond to the Cys145* amide (Fig. 3B, green), where * indicates the modified cysteine residue. Additionally, the Cys145* amide makes a 2.9 Å mainchain hydrogen bond to the carbonyl of Pro253. The aromatic residues Phe150 and Tyr151 are positioned similarly to the analogous residues in p21141–155 (Fig. 2), however Tyr151 does not make a hydrogen bond with Gln131 as in p21141–155. The Met147 carbonyl makes an intramolecular hydrogen bond to the amide NH of Tyr151 (3.4 Å) and also to Phe150 (2.4 Å), which defines an unconventional α-helical PCNA binding conformation (cf. 310-helix from 146–149 for p21141–155). The Ser146 side-chain makes a 2.4 Å and 3.1 Å intramolecular bond to the amides of Thr148 and Cys149* respectively, to further stabilise this conformation. The propyl linker sits above Pro253 of PCNA (4.4 Å) and Phe150 of the peptide (4.4 Å) to provide favourable hydrophobic interactions (Fig. 3B, green) which may be in part responsible for the relatively high affinity of 3 (769 nM).
The co-crystal structure of trans-butenyl-linked peptide 5 bound to PCNA shows the conserved hydrophobic residues (Met147, Phe150, Tyr151) are in a similar conformation to the analogous residues in p21141–155 (Fig. 2); however, the %BSA for Met147 is notably lower at 70% than in p21141–155, or the other macrocycles (100%, Table S3, ESI†). Three intermolecular hydrogen bonds are observed between peptide 5 and PCNA to anchor the macrocycle onto the surface (Fig. S4, ESI†). These are between: the Met147 amide NH and the mainchain carbonyl of His44 (2.3 Å); the Gln144 side-chain to the Ala252 carbonyl (2.8 Å) and the Pro253 carbonyl and Cys145* amide NH (3.4 Å). A weak 3.1 Å interaction between the Cys145* sulfur and Pro253 carbonyl is also observed. A 2.2 Å intramolecular hydrogen bond between Thr148 carbonyl and amide of Phe150 defines a γ-turn (Fig. 3D, orange). The butenyl linker of peptide 5 is raised off the PCNA surface and the only interaction with the PCNA surface is from the aforementioned intermolecular bond of the Cys145* sulfur and Pro253. The carbon-chain of the linker in macrocycle 5 does not appear to interact with the protein surface, and does not stabilise a 310-helix, which may together account for the lower affinity of peptide 5 (2.82 μM) compared to peptide 3 (769 nM).
The co-crystal structure of peptide 6 bound to PCNA shows the conserved Gln144 and Met147 positioned similar to those in p21141–155, however the aromatic residues are notably shifted (Fig. 2). Tyr151 protrudes into the hydrophobic cleft where it is 3.5 Å from Gln131 located in the bottom of the cleft and interacts with the Tyr133 phenol at 3.1 Å (Fig. S5, ESI†). The m-xylene linker sits up off the PCNA surface and does not make any clear interactions, except the Cys145* amide makes a weak 3.3 Å with Pro253 (Fig. S5, ESI†). A weak intramolecular 3.0 Å hydrogen bond between the Met147 carbonyl and Phe150 amide defines a 310-helical turn, however this is not at the same location as in native p21141–155 (i.e. 146 to 149). Additionally, this conformation does not appear well stabilised as there are only three other intramolecular interactions present. The first between the Ser146 side-chain and carbonyl; second between the Thr148 side-chain and carbonyl; and third between the Thr148 side-chain and the Tyr151 carbonyl (Fig. 3E, yellow). The sequence that flanks the PIP-box is not resolved in this co-crystal structure which may also suggest a weakly defined structure, and may account for the lower affinity of 6 (KD 3.86 μM), than for peptides 3–5 and 7.
The computational model of peptide 4 bound to PCNA shows the conserved residues are positioned similarly to p21141–155, except the Tyr151 %BSA is reduced to 70% compared to 90–100% in p21141–155 and peptides 3, 5–7 (Table S3, ESI†). The Gln144 side-chain makes a 3.0 Å hydrogen bond with Ala252, however this is the only intermolecular hydrogen bond made by a PIP-box residue in peptide 4 (Fig. S3, ESI†). One intramolecular 2.0 Å interaction is observed between the Ser146 carbonyl and Thr148 amide (Fig. 3C, blue). The overall lack of secondary interactions likely contributes to the lower affinity observed for 4 (1.99 μM) compared to 3 (769 nM). The butyl linker is >4 Å from the PCNA surface and does not make any clear interactions with PCNA, though a weak 3.0 Å interaction between the Cys145* sulfur and Pro253 carbonyl may be present.
The computational model of peptide 7 bound to PCNA (Fig. 2, purple) shows the backbone is shifted compared to p21141–155, indicated by the decreased distance between the mainchain amide of Met147 and the mainchain carbonyl atom of Val45 in PCNA (4.5 Å to 4.1 Å). This shift does not impact the overall %BSA of the sidechain in the hydrophobic pocket which remains at 100% (Table S3, ESI†). Peptide 7 is the only macrocycle that adopts the classical 310-helix binding conformation, where the carbonyl of Ser146 makes a 2.6 Å hydrogen bond to Cys149* amide (Fig. 3F, purple). This conformation is further stabilised by four additional intramolecular bonds which exist between the Ser146 side-chain and Thr148 amide (3.1 Å), the Arg143 carbonyl to Cys145* amide (2.7 Å); the Thr148 side-chain to carbonyl (2.7 Å); and the Ser153 side-chain to the Lys154 carbonyl (2.4 Å) (Fig. 3F). This stabilised 310-helical binding conformation likely contributes to the high binding affinity of peptide 7 for PCNA observed (570 nM). Interestingly, there are no significant intermolecular hydrogen bonds made between peptide 7 and PCNA, except for a 2.7 Å hydrogen bond of Arg143 to Asp257 and a weak 3.2 Å interaction of the bimane carbonyl and the main-chain carbonyl of Glu256 (Fig. S6, ESI†).
The classical 310-helical conformation seen for PCNA-binding peptides was only observed here for PCNA-bound macrocycle 7. Consequently, the solution-phase structure of the high PCNA affinity peptide 7 was investigated by NMR to investigate whether the bimane linker in 7 pre-organises the peptide backbone into this conformation. Secondary shifts of the bimane-linked peptide 7 were determined for the α-proton (Hα), amide proton (NH), α-carbon (Cα) and carbonyl carbon (CO) for each residue (Fig. 4 and Table S6, ESI†). The secondary shift for each signal (Hα, NH, Cα, CO; Fig. 4) was calculated as the difference between the observed resonance in macrocycle 7 (Table S5, ESI†) and the observed resonance in the linear precursor peptide 2 for each residue (Table S4, ESI†). The secondary shifts in the region of interest (i.e. between residues 146 and 149), were almost all shifted in the direction expected for helical structure, that is, negative proton (<−0.1 ppm) and positive carbon secondary shifts (>1 ppm). Random coil structure is indicated by secondary shifts close to 0 ppm. Large negative NH secondary shifts for residues 148–150, in addition to negative Hα and positive Cα secondary shifts for residues 147 and 148 together suggest helical structure in this region (Fig. 4). Furthermore, the CO secondary shifts of residues 145–147 are positive, which is in the direction anticipated for helical structure, although these are small in magnitude. Overall, the secondary shifts for residues 148–149 are in the direction expected for helical structure, however these shifts are quite small, which suggests the peptide is still flexible and samples a variety of conformers. Circular dichroism of peptide 7 displays a deep minimum near 200 nm (Fig. S8, ESI†) which is also consistent with a flexible structure. The computationally modelled structure of 7 bound to PCNA indicates the peptide backbone adopts the classic 310-helix defining hydrogen bonds expected for a PIP-box peptide when bound to PCNA. This importantly demonstrates that the bimane linker does not restrict the peptide from adopting this key conformation, consistent with previous reports.25
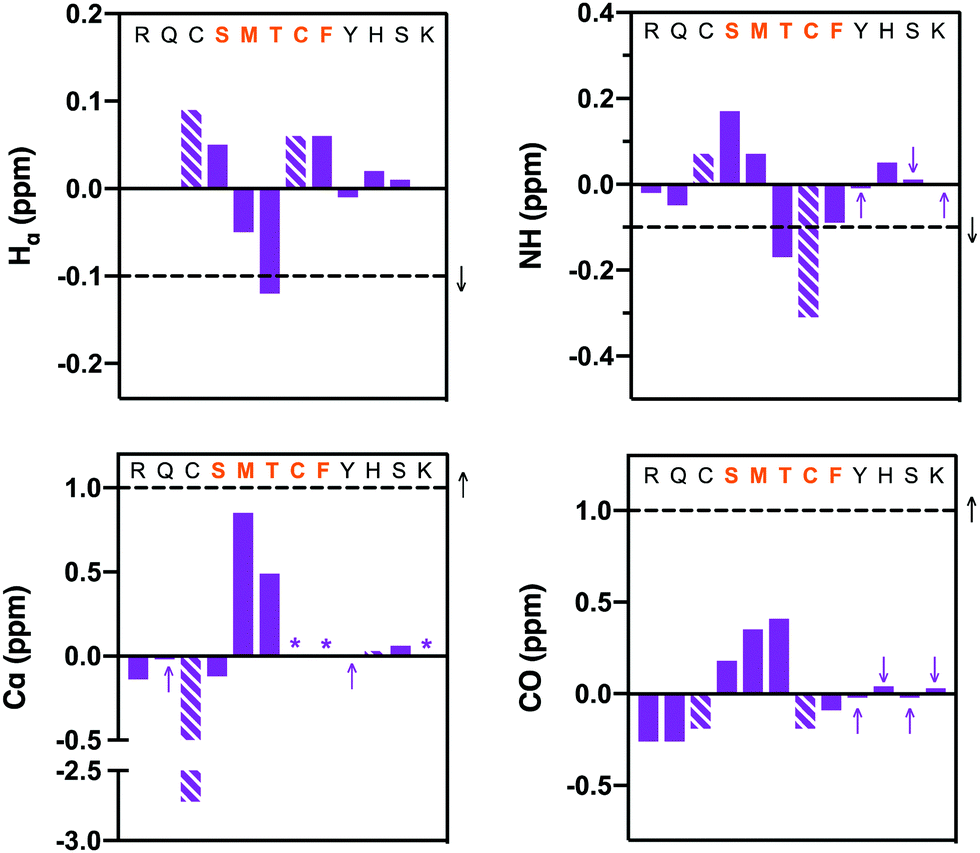 | ||
| Fig. 4 NMR structural data (10% aq. D2O, pH ∼ 5) for peptide 7: Hα, NH, Cα and carbonyl (CO) carbon secondary shifts are calculated relative to the corresponding resonances of peptide 2. Each column represents a consecutive residue. Dashed columns represent bimane-modified cysteine residues. The sequence at the top of each graph shows the segment where helical structure is anticipated in orange. Purple arrows highlight values that are close to zero on the side of zero the value lies. Stars (*) represent data that could not be reliably extracted from the spectrum. The horizontal dashed lines and black arrow indicate the generally accepted threshold (and direction) to indicate helical structure. Three consecutive residues surpassing the horizontal dashed lines strongly indicates the presence of helical structure. | ||
Cell imaging
The bimane-linker is inherently fluorescent, which equips peptide 7 with the additional advantage that it can be directly subjected to a cell uptake assay and imaged by confocal microscopy without further derivatisation. Breast cancer cells (MDA-MB-468) were treated with 10 μM of peptide 7 in order to determine whether the high affinity bimane-constrained peptide was cell permeable. Two controls peptides were prepared: a linear p21143–154 peptide with an N-terminal fluorescein appended (peptide 8, Table 1); along with a derivative of peptide 7 that is macrocyclised with the bimane linker and also includes an N-terminal fluorescein (peptide 9, Table 1). Peptides 8 and 9 were subjected to the same cell uptake experiment to compare to the cell uptake of the linear and cyclised analogues, and the impact of a fluorescein tag. Cells were seeded into culture dishes, incubated for 48 h, then treated with the peptide for 24 h. After treatment, media containing the peptide was removed and cells were washed with ice-cold PBS to remove all traces of media and peptide, fixed with 4% formaldehyde and imaged by confocal fluorescence microscopy. The nucleus, fluorescently tagged with genetically-expressed mKate, was imaged at ex. 594 nm, em. 600–700 nm and these images overlaid with the peptide fluorescence images (Fig. 5). Peptide 8 treated cells were imaged at ex. 488 nm, and em. 490–534 nm (GREEN channel in Fig. 5), which revealed no green fluorescence corresponding to the fluorescein-tagged peptide, indicating the linear peptide was not able to enter the cells. In contrast, peptide 7 treated cells were imaged with ex. 405 nm, and em. 410–485 nm (BLUE channel in Fig. 5) to reveal blue fluorescence within the cytoplasm, corresponding to the bimane fluorophore, indicating that the bimane-cyclised peptide is cell permeable. The fluorescence signal is mostly punctate, suggesting a significant portion of intracellular 7 is endosomally trapped.29,30 Macrocyclic peptides generally have increased cell permeability compared to the linear analogue.31–34 This is the first example of a biologically relevant bimane-constrained peptide able to enter cells and be directly imaged using the bimane fluorescence. Imaging peptide 9 on both the GREEN and BLUE channels, revealed blue punctate fluorescence throughout the cytoplasm, as for peptide 7. Green fluorescence was also observed throughout the cytoplasm for the peptide 9 treated cells, which is colocalised with the blue fluorescence. Together this indicates that peptide 9 is also cell permeable, with a similar distribution to peptide 8. The green fluorescence was somewhat diffuse, suggesting that some free fluorescein may be present. Auxiliary fluorophores can alter the secondary structure, target binding and cell permeability of the peptide and consequently the activity in imaging assays may not accurately reflect true behaviour of the parent peptide. For this reason, there is a need to develop and utilise technologies that incorporate imaging modality directly into the therapeutic scaffold (such as the bimane) to allow direct imaging of the compound of interest. Here we demonstrate that an N-terminal fluorescein tag appended to peptide 7, as in peptide 9, dramatically decreases the binding affinity for the protein target (PCNA, Table 1), though in this case the cellular uptake and distribution is not significantly impacted. | ||
| Fig. 5 Breast cancer cells MDA-MB-468 treated with 10 μM of peptide 7, peptide 8 or peptide 9, then fixed and imaged by confocal microscopy. Peptide 7 is cell permeable as shown by punctate blue fluorescence throughout the cytoplasm. Peptide 8 is not cell permeable, where no green fluorescence is evident in the cell image. Peptide 9 is cell permeable, with both blue and green fluorescence present throughout the cell cytoplasm. The BLUE channel (ex. 405 nm, em. 410–485 nm) indicates bimane fluorescence (peptides 7 and 9); the GREEN channel (ex. 488 nm, em. 490–534 nm) indicates the FITC fluorophore (peptides 8 and 9); the bottom panels show the BLUE and GREEN channel fluorescence overlaid with mKate fluorescence (mKate, ex. 594 nm, em. 600–700 nm) that marks the nucleus. | ||
Conclusions
Two cysteine residues were incorporated at positions 145 and 149 of a p21 peptide (143–151, peptide 1) known to bind PCNA to provide peptide 2, which was then derivatised into five different macrocycles by dithiol bisalkylation. This gave rise to peptides with a propyl (3), butyl (4), trans-butenyl (5), m-xylene (6) and bimane (7) linker installed between the cysteine side-chains. The affinity of the resulting peptides 1–7 for PCNA was determined by SPR, with KD values for the macrocycles (3–7) ranging from 570 nM to 3.86 μM. The bimane-constrained macrocyclic peptide 7 is the highest affinity peptidomimetic reported to bind PCNA, with a KD value of 570 nM. Peptide 7 was the only macrocycle that adopted a classical 310-helical binding conformation upon binding PCNA, which suggests this conformation is likely responsible for the high affinity binding. However, it is interesting that the remaining four peptide macrocycles (3–6) still bind PCNA, but do not adopt this conformation upon binding. The 310-helical structure of high affinity peptide 7 was not well-defined in solution, suggesting that the binding conformation is not substantially preorganised prior to the peptide binding PCNA. However, it is significant that the bimane linker does not preclude PCNA binding, and still allows this key conformation to be adopted upon binding. This observation is in line with our previous study19 which suggests that pre-defining a rigid peptide backbone may not improve PCNA binding affinity, and a linker that affords some flexibility is preferable to enable the peptide to adopt its ideal conformation on binding. Additionally, the inherently fluorescent peptide 7 is cell permeable and in contrast, a fluorescein-tagged linear p21 peptide of the same length (peptide 8) was not cell permeable. This highlights how macrocyclization can improve cell permeability of short peptides. Furthermore, these results emphasise the utility of the bimane moiety as a peptide linker as it can influence peptide structure, and the resulting peptides can be directly imaged without further derivatisation to investigate cell uptake of a bimane peptidomimetic. This allows the behaviour of the molecule of therapeutic interest to be assessed, instead of a related analogue that includes a fluorescent tag; and eliminates the need for an additional synthetic step. Attachment of an auxiliary fluorophore such as fluorescein to a peptide, can impair the target binding affinity (as seen for peptide 9) and may alter cellular permeability or intracellular distribution relative to the untagged analogue. In summary, this study identifies a short, cell permeable, high affinity PCNA-binding peptidomimetic, as a significant advance towards a pre-clinical anti-cancer therapeutic. Future work will focus on imparting nuclear permeability to macrocyclic peptides, such as peptide 7, in order to determine the ability of such p21-peptidomimetics to inhibit DNA-replication in cancer cells.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research was supported by the Australian Research Council Centre of Excellence for Nanoscale BioPhotonics (CNBP) (CE140100003). A. J. H. and B. A. V. are supported by Australian Government Research Training Program Stipends (RTPS). This research was undertaken in part using the MX135 and MX236 beamlines at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector. The facilities of the OptoFab node of the Australian National Fabrication Facility (ANFF) and associated Commonwealth and SA State Government funding are also gratefully acknowledged. This work was supported by grants from the National Health and Medical Research Council of Australia (W. D. T., T. E. H., ID 1084416, ID 1130077), the National Breast Cancer Foundation (NBCF; W. D. T.; ID PS-15-041) and a Movember & National Breast Cancer Foundation Collaboration Initiative grant (MNBCF-17-012 to W. D. T., T. E. H.). T. E. H. is currently supported by an NBCF Fellowship (IIRS-19-009).References
- T. Tsurimoto, Front. Biosci., 1999, 4, d849–858 CrossRef CAS PubMed.
- G. Maga and U. Hubscher, J. Cell Sci., 2003, 116, 3051–3060 CrossRef CAS PubMed.
- G. L. Moldovan, B. Pfander and S. Jentsch, Cell, 2007, 129, 665–679 CrossRef CAS PubMed.
- S. N. Naryzhny and H. Lee, FEBS Lett., 2007, 581, 4917–4920 CrossRef CAS PubMed.
- I. Stoimenov and T. Helleday, Biochem. Soc. Trans., 2009, 37, 605–613 CrossRef CAS PubMed.
- D. Zhongyun, M. Wortman, Z. Tan and K. Dillehay, WO 2012/033938A2, 2012.
- A. J. Horsfall, A. D. Abell and J. B. Bruning, ChemBioChem, 2019, 21, 442–450 CrossRef PubMed.
- D. I. Zheleva, N. Z. Zhelev, P. M. Fischer, S. V. Duff, E. Warbrick, D. G. Blake and D. P. Lane, Biochemistry, 2000, 39, 7388–7397 CrossRef CAS PubMed.
- J. B. Bruning and Y. Shamoo, Structure, 2004, 12, 2209–2219 CrossRef CAS PubMed.
- E. M. Boehm and M. T. Washington, BioEssays, 2016, 38, 1117–1122 CrossRef CAS PubMed.
- J. M. Gulbis, Z. Kelman, J. Hurwitz, M. O’Donnell and J. Kuriyan, Cell, 1996, 87, 297–306 CrossRef CAS PubMed.
- E. Gibbs, Z. Kelman, J. M. Gulbis, M. O’Donnell, J. Kuriyan, P. M. J. Burgers and J. Hurwitz, J. Biol. Chem., 1997, 272, 2373–2381 CrossRef CAS PubMed.
- A. J. Horsfall, B. A. Vandborg, W. Kowalczyk, T. Chav, D. B. Scanlon, A. D. Abell and J. B. Bruning, J. Biol. Chem., 2021, 296, 100773 CrossRef CAS.
- E. Warbrick, D. P. Lane, D. M. Glover and L. S. Cox, Curr. Biol., 1995, 5, 275–282 CrossRef CAS PubMed.
- T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020–13041 CrossRef CAS PubMed.
- A. P. Higueruelo, H. Jubb and T. L. Blundell, Curr. Opin. Pharmacol., 2013, 13, 791–796 CrossRef CAS PubMed.
- Q. Chu, R. E. Moellering, G. J. Hilinski, Y.-W. Kim, T. N. Grossmann, J. T. H. Yeh and G. L. Verdine, MedChemComm, 2015, 6, 111–119 RSC.
- D. P. Fairlie and A. D. de Araujo, Biopolymers, 2016, 106, 843–852 CrossRef CAS PubMed.
- K. L. Wegener, A. E. McGrath, N. E. Dixon, A. J. Oakley, D. B. Scanlon, A. D. Abell and J. Bruning, Chem. – Eur. J., 2018, 24, 11325–11331 CrossRef CAS.
- L. Peraro, T. R. Siegert and J. A. Kritzer, Methods Enzymol., 2016, 580, 303–332 CAS.
- G. Zhang, F. Barragan, K. Wilson, N. Levy, A. Herskovits, M. Sapozhnikov, Y. Rodriguez, L. Kelmendi, H. Alkasimi, H. Korsmo, M. Chowdhury and G. Gerona-Navarro, Angew. Chem., Int. Ed., 2018, 57, 17073–17078 CrossRef CAS PubMed.
- H. Jo, N. Meinhardt, Y. Wu, S. Kulkarni, X. Hu, K. E. Low, P. L. Davies, W. F. DeGrado and D. C. Greenbaum, J. Am. Chem. Soc., 2012, 134, 17704–17713 CrossRef CAS PubMed.
- A. J. Horsfall, K. R. Dunning, K. L. Keeling, D. B. Scanlon, K. L. Wegener and A. D. Abell, ChemBioChem, 2020, 21, 3423–3432 CrossRef CAS PubMed.
- A. D. de Araujo, H. N. Hoang, W. M. Kok, F. Diness, P. Gupta, T. A. Hill, R. W. Driver, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 6965–6969 CrossRef CAS PubMed.
- A. J. Horsfall, D. P. McDougal, D. B. Scanlon, J. B. Bruning and A. D. Abell, ChemBioChem, 2021, 22 DOI:10.1002/cbic.202100241.
- A. J. Kroker and J. B. Bruning, Biochemistry, 2015, 54, 3483–3493 CrossRef CAS.
- K. N. Choe and G.-L. Moldovan, Mol. Cell, 2016, 65, 380–392 CrossRef CAS PubMed.
- G. Kontopidis, S.-Y. Wu, D. I. Zheleva, P. Taylor, C. McInnes, D. P. Lane, P. M. Fischer and M. D. Walkinshaw, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1871–1876 CrossRef CAS PubMed.
- T. B. Potocky, A. K. Menon and S. H. Gellman, J. Biol. Chem., 2003, 278, 50188–50194 CrossRef CAS PubMed.
- P. Ramoino, A. Diaspro, M. Fato and C. Usai, Molecular Regulation of Endocytosis, 2012, ch. 6 DOI:10.5772/46061.
- P. M. Cromm, J. Spiegel, P. Kuchler, L. Dietrich, J. Kriegesmann, M. Wendt, R. S. Goody, H. Waldmann and T. N. Grossmann, ACS Chem. Biol., 2016, 11, 2375–2382 CrossRef CAS PubMed.
- Y. Tian, Y. Jiang, J. Li, D. Wang, H. Zhao and Z. Li, ChemBioChem, 2017, 18, 2087–2093 CrossRef CAS.
- S. R. Perry, T. A. Hill, A. D. de Araujo, H. N. Hoang and D. P. Fairlie, Org. Biomol. Chem., 2018, 16, 367–371 RSC.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y. S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS.
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Williamson and T. Caradoc-Davies, J. Synchrotron Radiat., 2015, 22, 187–190 CrossRef CAS PubMed.
- D. Aragao, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00113b |
| This journal is © The Royal Society of Chemistry 2021 |