
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFaster, better, and cheaper: harnessing microfluidics and mass spectrometry for biotechnology
Noel S.
Ha
 ab,
Markus
de Raad
c,
La Zhen
Han
cd,
Amber
Golini
cd,
Christopher J.
Petzold
ab and
Trent R.
Northen
*abcd
ab,
Markus
de Raad
c,
La Zhen
Han
cd,
Amber
Golini
cd,
Christopher J.
Petzold
ab and
Trent R.
Northen
*abcd
aBiological Systems and Engineering, Lawrence Berkeley National Laboratory, Berkeley, CA, USA. E-mail: trnorthen@lbl.gov
bUS Department of Energy Joint BioEnergy Institute, Emeryville, CA, USA
cEnvironmental Genomics and Systems Biology, Biosciences, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
dUS Department of Energy Joint Genome Institute, Berkeley, CA, USA
First published on 20th July 2021
Abstract
High-throughput screening technologies are widely used for elucidating biological activities. These typically require trade-offs in assay specificity and sensitivity to achieve higher throughput. Microfluidic approaches enable rapid manipulation of small volumes and have found a wide range of applications in biotechnology providing improved control of reaction conditions, faster assays, and reduced reagent consumption. The integration of mass spectrometry with microfluidics has the potential to create high-throughput, sensitivity, and specificity assays. This review introduces the widely-used mass spectrometry ionization techniques that have been successfully integrated with microfluidics approaches such as continuous-flow system, microchip electrophoresis, droplet microfluidics, digital microfluidics, centrifugal microfluidics, and paper microfluidics. In addition, we discuss recent applications of microfluidics integrated with mass spectrometry in single-cell analysis, compound screening, and the study of microorganisms. Lastly, we provide future outlooks towards online coupling, improving the sensitivity and integration of multi-omics into a single platform.
 Virtual group photo: Amber Golini (Top left), Noel Ha (Top center), Markus de Raad (Top right), Christopher Petzold (Bottom left), Trent Northen (Bottom center), La Zhen Han (Bottom right) | Dr. Ha obtained his BS in Materials Science at Sung Kyun Kwan University followed by a MS in Biomedical Engineering from the University of California, Irvine and a PhD in Bioengineering from the University of California, Los Angeles with Prof. Michael van Dam. He is currently a post-doctoral fellow at Lawrence Berkeley National Laboratory within the Biological Systems and Engineering Division (BSE) and working with Dr. Trent Northen. Dr. Ha's research is focused on the development of microfluidic bioanalytical technologies for high-throughput quantitative bioassays in a wide range of applications including drug discovery and synthetic biology. Dr. de Raad earned his masters degree in Drug Innovation from Utrecht University followed by a PhD in Pharmaceutics from Utrecht University with Prof. Enrico Mastrobattista. He then did his post-doctoral research at Lawrence Berkeley National Laboratory in mass spectrometry with Dr. Trent Northen. He is currently a Scientist within the Environmental Genomics and Systems Biology Division (EGSB) in the Northen Lab. His research is focused on developing and applying mass spectrometry based high-throughput screening for unraveling microbial interactions and studying microbial gene function in microbiomes. La Zhen Han obtained a BA from Sarah Lawrence College and is currently a Research Assistant at the Northen Lab, where she contributes to projects within the Environmental Genomics and Systems Biology Division (EGSB) and Joint Genome Institute (JGI). Her work includes liquid chromatography-mass spectrometry (LCMS) sample processing and instrumentation. Amber Golini earned her BS in Environmental Management and Protection with a minor in Biology from California Polytechnic State University, San Luis Obispo. She is currently a Research Assistant within the Environmental Genomics and Systems Biology Division (EGSB) and Joint Genome Institute (JGI) in the Northen Lab. She assists with the preparation, processing, and analysis of samples for metabolomics projects using liquid chromatography-mass spectrometry (LCMS) based methods, as well as operating and maintaining LCMS instrumentation systems. Dr. Petzold obtained his BS in Chemistry and BA in Physics at Taylor University followed by a PhD in Analytical Chemistry from Purdue University with Prof. Hilkka Kenttämaa. He then did post-doctoral research at the University of California, Berkeley in bioanalytical mass spectrometry with Prof. Julie Leary. Dr. Petzold joined Prof. Jay Keasling's lab at the University of California, Berkeley in 2005 and joined Berkeley Lab in 2008. He is currently a Staff Scientist within the Biological Systems and Engineering Division (BSE). Dr. Petzold's research is focused on the development of high-throughput analytical methods and quantitative proteomics. Dr. Northen obtained his BS in Chemical Engineering at the University of California Santa Barbara followed by a PhD in Chemistry and Biochemistry from Arizona State University with Prof. Neal Woodbury. He then did a Post-Doctoral Fellow at the Scripps Research Institute in mass spectrometry and metabolomics with Prof. Gary Siuzdak. Dr. Northen started his lab at Berkeley Lab in 2008 and is currently Deputy Division Director and a Senior Scientist within the Environmental Genomics and Systems Biology Division (EGSB). The Northen group's research is focused on using exometabolomics to link genomes with environments to understand how webs of microbes cycle carbon and sustain biomes. |
1. Introduction
High-throughput screening (HTS) is a critical step in drug discovery and an important tool for elucidating gene and protein function.1,2 It is widely used across pharmaceutical and biotechnological applications with key applications including protein characterization, disease and health monitoring, synthetic biology, and drug development.3 High-throughput screening is traditionally defined as the rapid analysis of samples, exceeding 103 samples per day.4–6 As technology advances, the screening of a large population of biological entities for a particular metabolite, enzyme, protein, nucleic acid, phenotype, or mutation is becoming a significant challenge. Primary difficulties in accomplishing this include the high costs and amount of time required to refine large libraries to a limited number of candidates for further characterization. Therefore, there are three important goals to achieve in HTS: (1) high speed analysis with low-cost operations, (2) high specificity, and (3) high sensitivity for precise measurements.Mass spectrometry (MS) is a label-free detection technique and has become a method of choice for high-throughput assays.7,8 Currently, mass spectrometry is heavily utilized in many research areas due to its high selectivity based on analysis of a characteristic mass to charge ratio (m/z) of analytes and their fragments using tandem mass spectrometry (MS/MS). The range of platforms available for mass spectrometry, based on a diverse set of ionization techniques, enables the analysis of a broad range of sample types.9–12 However, traditional liquid chromatography-mass spectrometry (LC/MS) approaches for screening samples are time consuming due to chromatographic separation and require relatively large sample volumes, which often makes them cost prohibitive for HTS efforts. While this review paper is focused on integration of MS with microfluidics, it is important to note that there are a range of other label-free analytical methods that can also be considered, including surface-based sensing techniques such as surface-plasmon resonance (SPR)13,14 and surface enhanced Raman spectroscopy (SERS).15,16 The reader is referred to several excellent review papers on these topics.17–20
Microfluidics is widely used in biotechnology and is central to next-generation sequencing technologies.21–23 Within the field of proteomics, there is substantial interest in the integration of microfluidic technologies to process proteins to peptides and carry out the analysis of low samples of low abundance, including those associated with single cells.24 A range of approaches are used to enable manipulation of droplets containing picoliter to nanoliter volumes.25–29 Together, such methods provide the capability to rapidly process millions of samples, multiplex numerous processes in parallel, and improve sensitivity even for a low abundance sample by increasing the concentration. These properties of microfluidics, when coupled with mass spectrometry systems, provide potential for high sensitivity and specificity, as well as throughput analysis.30,31 Thanks to recent substantial developments in microfluidic technologies, miniaturized microfluidic ion sources have been a very important development for mass spectrometry analyses.32,33 The further integration of droplet microfluidic approaches with mass spectrometry has tremendous potential to increase the throughput of the system, which is important for a wide range of important applications. In this review, we introduce the key microfluidic and mass spectrometry approaches and describe the latest applications of coupling microfluidics with mass spectrometry.
2. Introduction to relevant microfluidic approaches
Microfluidics is a rapidly developing field and is currently regarded as a critical component to various life sciences.34 For instance, it has been well established that microfluidic devices provide numerous advantages for biochemical assays as well as the synthesis of pharmaceuticals.30,31,35 In particular, the scale of microfluidic devices enables improved control of reaction conditions, leading to faster, higher-yield production and reduced reagent consumption and system cost.36,37 Various types of microfluidic devices have been coupled to mass spectrometry platforms to manipulate samples (including processes such as cell lysis, separation, purification, and step-wise chemical and biological reactions),32,38–40 and here we will discuss the most frequently reported types of coupling that have emerged over the past four years.Continuous-flow system
The most common type of microfluidic device is a continuous-flow system where the flow through the microchannel is driven by pressure. A variety of pressure sources can be used to control the flow in this type of system. The Quake group pioneered this type of system and developed high-density microfluidic chips that contain plumbing networks with thousands of micromechanical valves and hundreds of individual chambers41,42 (Fig. 1A). This setup is capable of automating hundreds of thousands of experiments in parallel in a single device. Continuous-flow microfluidic devices have been extensively coupled with mass spectrometry as it is particularly useful to automate the multiple steps of sample pretreatment (e.g., cell lysis, protein extraction, purification, desalting, etc.) prior to mass spectrometry analysis.43–45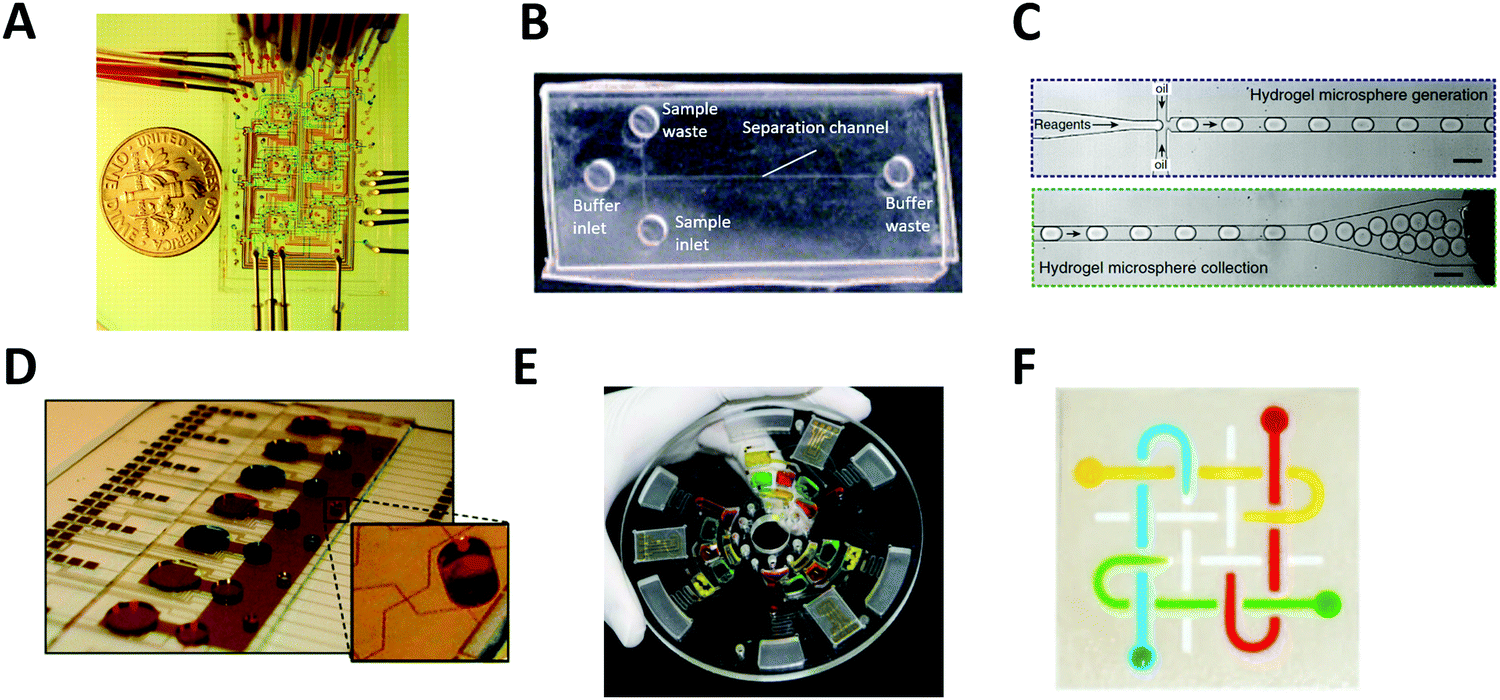 | ||
| Fig. 1 Major types of microfluidic systems. (A) Continuous-flow microfluidic chip (by the Quake group). Flow is driven by pressure through the microchannel. Adapted from ref. 42 with permission from the American Association for the Advancement of Science, copyright © 2005. (B) Microchip electrophoresis system (by the Woolley group). Electroosmotic flow is driven by high voltage between buffer inlet and buffer waste. Adapted from ref. 46 with permission from the Royal Society of Chemistry, copyright © 2009. (C) Droplet microfluidics (by the Weitz group). Reactants are compartmentalized into droplets that function as individual reaction vessel. Adapted from ref. 190 with permission from Elsevier, copyright © 2015. (D) Digital microfluidic system (by the Wheeler group). Droplet movement is controlled by electrodes. Adapted from ref. 71 with permission from the Royal Society of Chemistry, copyright © 2010. (E) Centrifugal microfluidic chip (by the Madou & Cho groups). Centrifugal force drives liquid samples radially outwards from the center of a disc-shaped device. Adapted from ref. 77 with permission from the Royal Society of Chemistry, copyright © 2013. (F) Paper microfluidic chip (by the Whitesides group). A small volume of fluid is placed and moved by capillary force along patterned microchannels. Adapted from ref. 79 with permission from the National Academy of Sciences, copyright © 2008. | ||
Microchip electrophoresis
Another type of continuous-flow system is driven by an electric field (rather than pressure) via capillary electrophoresis (CE) (Fig. 1B).46 CE is a separation technique with high separation efficiency and low consumption of sample and reagents.47,48 CE is employed in diverse applications including DNA and protein separation,49–51 detection of disease biomarkers,52,53 environment monitoring,54,55 and pharmaceutical analysis.48,56–59 Furthermore, it can readily be miniaturized using microfluidic chip technology, called microchip electrophoresis (MCE). MCE has been used for high-resolution separations where portability or small sample volumes are especially important and it has been commonly coupled with electrospray ionization mass spectrometry (ESI-MS).35,60,61 The MCE system requires the use of high voltage for operation, which makes parallel sample processing challenging without the use of multiple devices simultaneously.Droplet microfluidics
Droplet microfluidic (DMF) systems compartmentalize reactants through the use of an inert carrier fluid (typically oil) to encapsulate aqueous samples in droplets. (Fig. 1C).62 These systems can produce droplets in the volume range of 0.05 pL to 1 nL (i.e., 5–120 μm in diam.) instead of the microliter volumes commonly used in conventional methods.26 Droplets can encapsulate cells, DNA, or other molecules inside the aqueous phase without risking cross-contamination, and manipulation and measurement of droplets at kHz speeds enable up to 108 samples to be screened in one day.26,63 This method of droplet microfluidics can increase assay sensitivity by raising the effective concentration of low abundance species and decreasing the time required to reach reaction equilibrium and detection thresholds. When coupled with next-generation sequencing or MS, droplet microfluidics enables high-throughput screening applications such as single-cell and single-molecule assays that are currently unfeasible or impossible with conventional methods.64,65 However, controlled droplet manipulation for downstream mass spectrometry analysis can be challenging, and droplet stability over a longer period at an elevated temperature can become an issue. When coupled with mass spectrometry, the carrier oil phase used in the droplet microfluidics can degrade the stability, efficiency, and accuracy of mass spectrometry detection and the sample transportation rates might not sync with the MS data acquisition rate for transient signal acquisition.66 Optionally, the droplet contents can be extracted from the oil phase to an aqueous channel for subsequent ionization.67–69Digital microfluidics
Digital microfluidics is a developing liquid-handling technology that utilizes electrodes to individually control droplets.70 These microliter-sized droplets (typically larger than ones used in aforementioned droplet microfluidics) can be made to move, merge, and split across the array (Fig. 1D).71 Because of its unique advantages (such as programmable control of sample handling and flexible device geometry) digital microfluidics has been applied to a wide range of fields.72–74 Digital microfluidics is a particularly useful option for step-wise sample pretreatments in chemical and biological reactions (such as cell lysis and protein extraction) prior to mass spectrometry analysis.75 However, device fabrication is costly and complicated. In addition, the device is prone to failure at high reaction temperature or high voltage.Centrifugal microfluidics
In centrifugal microfluidics, centrifugal forces induced on the sample drive the liquids radially outwards from the center of a disc-shaped device (Fig. 1E),76,77 and the flow of fluids in a centrifugal platform has been well characterized.78 In comparison to other systems, the instrumentation demands of centrifugal microfluidics are much lower. The only required component is a simple, compact motor to create the forces needed for fluid manipulation, thereby eliminating any complex tubing or external pressure systems. Centrifugal microfluidic chips can also be mass-produced using inexpensive materials, making them low-cost and suitable for disposable devices. However, large-scale integration is challenging, as throughput is often limited to a single centrifuge. Furthermore, contact-free on-line interface with downstream mass spectrometry analysis is not currently available.Paper microfluidics
Microfluidic paper-based analytical devices (μPADs) or lateral-flow microfluidic systems are made of patterned paper on which a small volume of fluid is placed and moved by capillary force for subsequent chemical or biological reactions (Fig. 1F).79 The typical readout can be obtained from a colorimetric assay, but other sophisticated detection modalities such as electrochemical and mass spectral detection have been also explored. Paper is not only inexpensive, but it is also a very light substrate that can be stored and transported easily. The detailed outstanding features of paper-based microfluidic devices were summarized elsewhere.80 Many paper microfluidic devices have been developed in an attempt to make diagnostic devices environmentally friendly and affordable. μPADs are also the subject of development for rapid testing (for example, diagnosis of the SARS-CoV-2 virus) in urgent situations or remote areas where more complex and expensive technologies are unavailable.81 However, μPADs have limitations related to the material properties of paper, the fabrication techniques used, and the detection methods connected to the devices.80 Specifically, the sample retention within paper-fluidic channels and the sample evaporation during transport can result in a low efficiency of sample delivery within the device, which in turn can negatively impact downstream mass spectrometry analysis. Furthermore, some hydrophobic agents used to pattern devices fail to build barriers strong enough to repel low surface tension samples.3. Coupling of microfluidics with mass spectrometry
Microfluidic chips have been coupled with mass spectrometry via various interfaces and ionization methods. Electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI) are currently the most popular techniques coupled with microfluidics. More recently, other ionization techniques have also been coupled with mass spectrometry, including surface-assisted laser desorption ionization mass spectrometry (SALDI), desorption electrospray ionization (DESI), inductively coupled plasma (ICP), and surface acoustic wave nebulization (SAWN).3A. Electrospray ionization
Electrospray ionization (ESI) is the most common ionization method coupled with microfluidic systems. In ESI, a fluid is pumped through a capillary or channel and subjected to an electric field, producing a Taylor cone, which ejects charged droplets that are desolvated and introduced into the mass spectrometry system. The range of microfluidic devices integrated with ESI-MS is discussed below.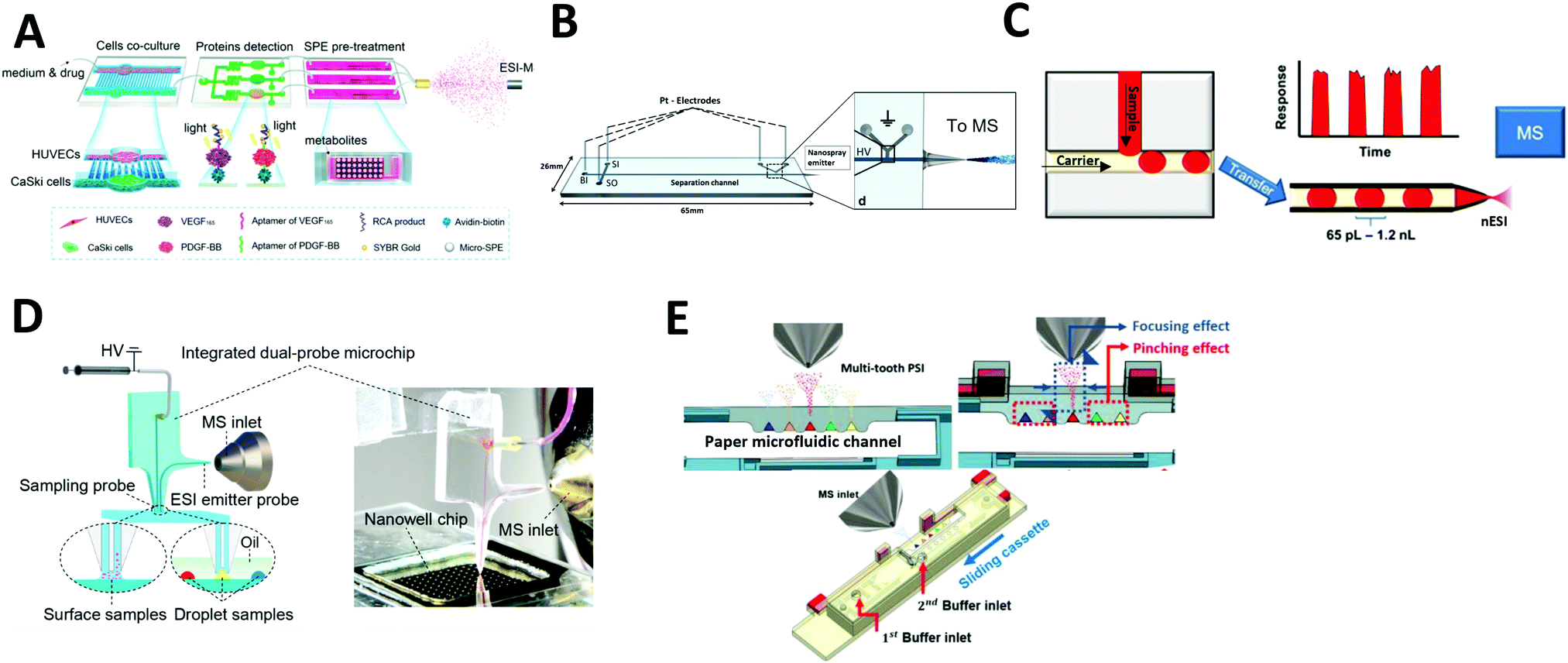 | ||
| Fig. 2 Coupling of microfluidics with electrospray ionization mass spectrometry (ESI-MS). (A) Continuous flow microfluidic chip with multiple functions coupled with ESI-MS. Adapted from ref. 44 with permission from the American Chemical Society, copyright © 2017. (B) Microchip electrophoresis chip coupled with ESI-MS. Adapted from ref. 87 with permission from Springer Nature, copyright © 2018. (C) Droplet microfluidics coupled with nano-ESI-MS. Adapted from ref. 93 with permission from the American Chemical Society, copyright © 2019 (D) direct surface and droplet micro-sampling probe for downstream ESI-MS. Adapted from ref. 95 with permission from the American Chemical Society, copyright © 2017. (E) Paper microfluidic device equipped with paper spray ionization. Adapted from ref. 98 with permission from the IEEE, copyright © 2020. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 samples (Fig. 2C). The signal was stable for droplets as small as 65 pL and for throughputs up to 10 droplets per s. Wink et al. developed a more advanced droplet microfluidic system for the analysis of secondary metabolites produced inside the droplets through simultaneous fluorescence imaging and ESI-MS analysis. For this method, microbes were encapsulated in ∼200 pL droplets; long-term incubation was performed on one chip before the droplets were transferred to a second microfluidic chip for MS analysis. Fluorescent markers were monitored before the droplets were ionized, which enabled analysis of metabolites and fluorescent labels in a complex biological matrix. They also demonstrated the detection of streptomycin produced in situ by ESI-MS using Streptomyces griseus hyphae. Recently, the Belder group has even explored the integration of droplet microfluidics with on-chip liquid chromatography for the separation of analytes for the downstream analysis by ESI-MS.83,94 The sample flows through the microfluidic channel packed with chromatographic beads, followed by the droplet generation.
000 samples (Fig. 2C). The signal was stable for droplets as small as 65 pL and for throughputs up to 10 droplets per s. Wink et al. developed a more advanced droplet microfluidic system for the analysis of secondary metabolites produced inside the droplets through simultaneous fluorescence imaging and ESI-MS analysis. For this method, microbes were encapsulated in ∼200 pL droplets; long-term incubation was performed on one chip before the droplets were transferred to a second microfluidic chip for MS analysis. Fluorescent markers were monitored before the droplets were ionized, which enabled analysis of metabolites and fluorescent labels in a complex biological matrix. They also demonstrated the detection of streptomycin produced in situ by ESI-MS using Streptomyces griseus hyphae. Recently, the Belder group has even explored the integration of droplet microfluidics with on-chip liquid chromatography for the separation of analytes for the downstream analysis by ESI-MS.83,94 The sample flows through the microfluidic channel packed with chromatographic beads, followed by the droplet generation.
Another method, developed by Huang et al., involves using a microfluidic device for direct surface or droplet micro-sampling followed by ESI.95 A single glass microfluidic chip integrates a sampling probe, an electrospray emitter probe, and an on-line mixer (Fig. 2D). Furthermore, two types of sampling probes were developed: a parallel-channel probe for dry spot droplets and a U-shaped channel probe for liquid-phase droplets. The system was demonstrated to be capable of MS analysis of nanoliter-scale chemical reactions. The assay throughput was ∼13 s per droplet, hundreds of times faster than those of conventional LC–MS systems.
When coupling droplet microfluidics with ESI-MS, carryover between droplets that are ionized consecutively needs to be addressed. Other droplet microfluidic systems coupled with ESI-MS have been previously reported.40 However, most of them require off-line sample injection for ionization.
When analyzing samples in complex biological matrices by ESI-MS, among other mass spectrometry techniques, ion suppression is a critical issue especially in the analysis of miniscule amounts of metabolites originating from single cells.99 Ion suppression is a process where the detection of a given analyte is reduced by the presence of another—for example, salty samples or those containing polyethylene glycol (PEG) surfactants often suffer from this effect. Another challenge for ESI-coupled technologies is the loss of sensitivity over consecutive measurements due to accumulation of materials on the source. When dealing with droplets containing the sample in the respective matrix, the proper choice of surfactants is also crucial as it might reduce the ionization during the electrospray process.100
3B. Desorption electrospray ionization
Desorption electrospray ionization (DESI) is an ambient ionization technique which combines features of electrospray and desorption ionization methods.101–103 In DESI, charged solvent droplets are directed onto the surface of the sample to be analyzed. The impact of the charged droplets produces ions from the surface that are directed towards the inlet of a mass spectrometer. DESI is the most used technique for ambient mass spectrometry-based bioanalysis since it allows analysis of both solid and liquid samples with little or no sample preparation. This spray-based technique benefits from increased throughput and the ability for spatially resolved molecular features to be probed under ambient conditions.103 de Freitas et al. reported the use of DESI mass spectrometry imaging for dried sample on the microfluidic paper-based analytical devices (μPADs).104 In paper microfluidics with colorimetric readouts, the formation of color gradients or lack of color uniformity on the detection zone can compromise the readout reliability. L-DESI measurements revealed a heterogeneous distribution of chromogenic agent (iodide and triiodide ions) at the zone edge (Fig. 3A). However, compared to vacuum desorption mass spectrometry, a disadvantage of DESI is lower spatial resolution (approximately 1800–200 μm, compared to 30–50 μm for MALDI).105 Recently, a nano-DESI probe was developed to improve the spatial resolution to approximately 12 μm. With the nano-DESI probe fabricated using two capillaries, controlled desorption of analytes present in a specific region of specimen has been demonstrated using a small amount of solvent (∼0.5 μL for each spectrum acquisition).106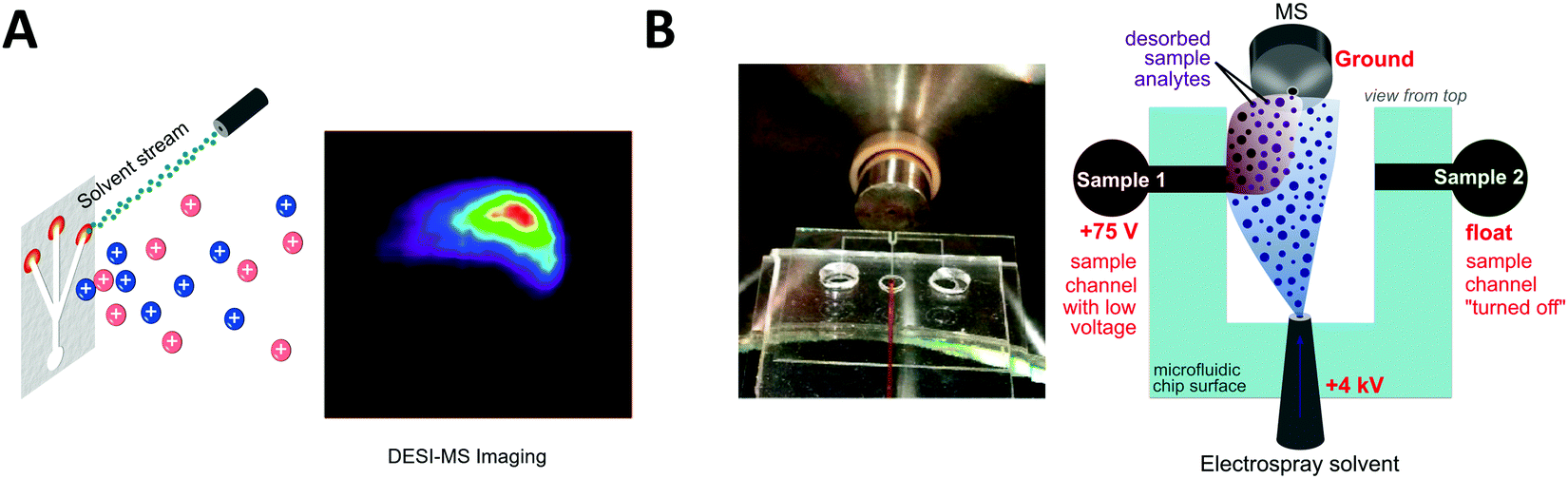 | ||
| Fig. 3 Coupling of microfluidics with desorption electrospray ionization mass spectrometry (DESI-MS). (A) Microfluidic paper-based analytical device with DESI-MS. Adapted from ref. 104 with permission from the American Chemical Society, copyright © 2018. (B) Image of microfluidic voltage-assisted liquid-DESI (left) where the liquid sample is desorbed by the stream of solvent, and a schematic illustration of the working principle (right). Adapted from ref. 107 with permission from the American Chemical Society, copyright © 2017. | ||
3C. Surface-based laser desorption ionization mass spectrometry
Laser desorption ionization mass spectrometry (LDI-MS) has been widely used in organic and biological sample analysis.110 Of the many LDI-MS methods, matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) is the most frequently used and has been used to analyze biological samples for protein and bacteria identification, as well as biomarker and metabolite detection.111–113 Since LDI is very rapid (nanosecond timescale) these techniques can be extremely high-throughput.MALDI has frequently been coupled with microfluidics, as it allows for automated and high-throughput sample preparation. Thus, microfluidic devices with MALDI-MS can be useful tools to investigate samples in real-time without compromising sample integrity. Filla et al. described a continuous-flow microfluidic device capable of automated cell lysis, metabolite extraction, and quenching of enzymatic activity.114 Quenching efficiency was measured with an off-line MALDI-MS assay of exogenous isotopic adenosine triphosphate (ATP) hydrolysis to isotopic adenosine diphosphate (ADP), which was used as a marker of metabolite degradation. The samples processed on the chip were manually transferred onto a MALDI plate for mass spectrometry analysis.
Grant et al. developed a more advanced continuous-flow microfluidic system to directly prepare samples over a MALDI-MS surface. An elastic polydimethylsiloxane (PDMS) microfluidic chip was reversibly clamped against a chemically functionalized gold layer to create a MALDI surface (Fig. 4A). The sample flows through the microfluidic channel while conducting an enzyme reaction. Then, the reaction product is immobilized onto the floor of the microfluidic channel (gold layer) to form a self-assembled monolayer which is later scanned to visualize reaction progress via mass spectrometry analysis.
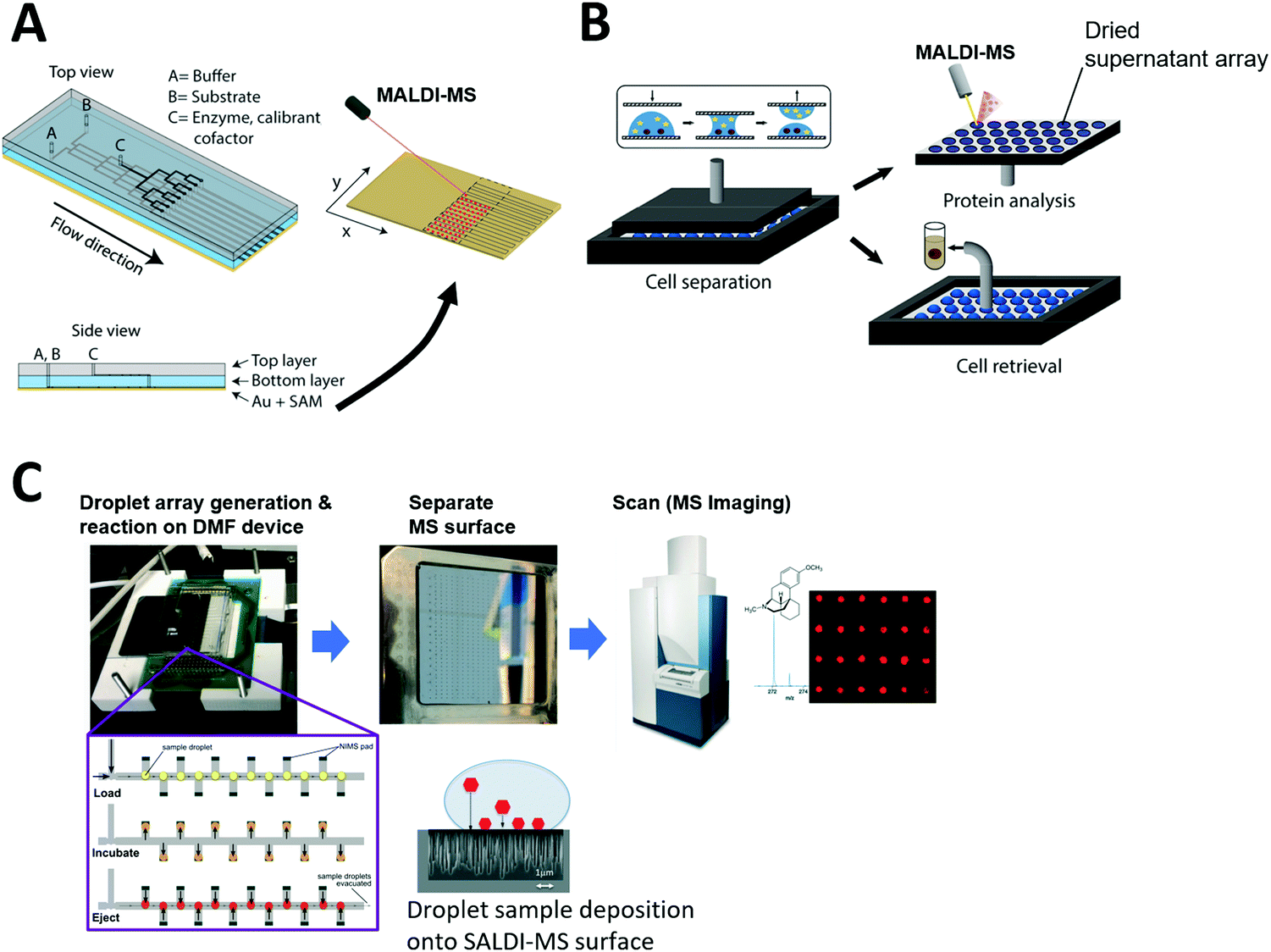 | ||
| Fig. 4 Coupling of microfluidics with laser desorption ionization mass spectrometry (LDI-MS). (A) Continuous-flow microfluidic chip with a functionalized bottom layer as Matrix Assisted Laser Desorption Ionization (MALDI) surface for enzyme reaction (left) and the MALDI-MS scanning of the separated MS surface. Adapted from ref. 191 with permission from the American Chemical Society, copyright © 2018. (B) Nanoliter droplet arrays for protein analysis in single yeast cells by MALDI-MS; samples of cell supernatant are taken by parallel plate-based droplet splitting method. Adapted from ref. 116 with permission from the American Chemical Society, copyright © 2020. (C) Digital microfluidic (DMF) device interfaced with SALDI-MS surface (nanostructure initiator mass spectrometry, NIMS). Enzyme reactions are performed inside the arrayed droplets, and the mass spectrometry surface with the dried sample array can be separated from the DMF device and scanned for mass spectrometry imaging. Adapted from ref. 133 with permission from the Royal Society of Chemistry, copyright © 2017. | ||
The Dittrich group previously developed a droplet spotting and analysis platform for MALDI-MS analysis of secreted proteins from single cells.115 Droplets containing single cells are spotted onto a custom-made indium tin oxide (ITO)-glass plate and the hydrophilic/hydrophobic pattern on the plate guides droplets to a predefined hydrophilic position. Recently, they developed a new way to split arrayed droplet to extract cell supernatant.116 A second plate was placed above the droplet array plate and brought in contact with the droplets (Fig. 4B). All 200 droplets were sampled in parallel by this plate-based droplet splitting and analyzed via MALDI-MS upon drying to detect the protein brazzein secreted from ∼500 cells in each droplet.
Unfortunately, most reported microfluidic systems require manual sample transfer to MALDI plate prior to mass spectrometry analysis. Even in cases where sample arrays are prepared using microfluidic devices, the mass spectrometry surface needs to be manually separated from the microfluidic device before transfer to a mass spectrometry system Thus, a drawback of using MALDI-MS for microfluidic coupling is the difficulty of complete on-line analysis without human intervention. Significant advancements in the microfluidic interface are required for automated on-line analysis.
However, there have been only a few reports where microfluidic devices were coupled with SALDI-MS, including where a DMF device was used for automated sample handling and followed by SALDI-MS at very low throughput.132 Recently, Heinemann et al. developed a higher throughput system that integrates DMF with NIMS.133 Briefly, NIMS uses liquid initiator-coated silicon nanostructures to generate gas phase ions from surface-adsorbed molecules upon laser irradiation. The microfluidic device constructed by Heinemann et al. contains electrodes for droplet manipulation and is patterned with the NIMS array for sample deposition for downstream MS analysis (Fig. 4C). In the study, enzyme reactions were carried out inside droplets using a premixed enzyme reaction solution, and arrayed in discrete locations that had local MS surfaces where sample could be deposited. Once the reaction was completed over the NIMS array, the droplets were removed and the NIMS array was scanned in a commercial MALDI-MS system. The system proved capable of directly measuring the substrates and products of enzymatic reactions and is broadly applicable to many molecular classes including metabolites, drugs, and peptides. This proof-of-principle study reveals SALDI surfaces can be fabricated into an array format for high-throughput microfluidic sample processing. This system can be potentially adapted for diverse array-based compound screening in the future. However, to overcome low sensitivity observed for certain sample types, other sample loading techniques such as complete drying of sample droplets on the MS surface can be considered in the future. Also, complete on-chip sample manipulation, rather than manipulation via premixed solutions, would be desired to automatically screen large compound libraries and possible reaction combinations.
Recently, a centrifugal microfluidic device was coupled with SALDI-MS. Zhao et al. developed a centrifugal microfluidic disc that performs sample cleanup on human serum samples (∼5 μL) for subsequent metabolite analysis by SALDI-MS.38 The disc device consists of six layers of polyester film with a 10 cm diameter and 100 μm thickness per layer. This system demonstrated sufficient removal of proteins, lipids, and other biomolecules for effective downstream MS analysis of multiple small-ion metabolites in the human serum samples. After cleanup in the rotating centrifugal disc, the sample was manually transferred to the SALDI-chip (silicon nanoposts deposited on a silicon wafer) for SALDI-MS analysis. This two-chip system could potentially be integrated into a single device by incorporating the SALDI surface within the chamber in the centrifugal disc for on-line MS analysis.
3D. Inductively coupled plasma
Inductively coupled plasma mass spectrometry (ICP-MS) is a powerful analytical instrument for trace elements detection.134 In particular, ICP-MS enables accurate identification and quantification of trace elements and their species in cells. When on-line coupled with IPS-MS, widely used water-in-oil droplet microfluidics for single cell analysis encounters several problems. Frequently used carrier oil is not suitable for ICP-MS measurement due to relatively high carbon content. Wang et al. developed a droplet microfluidic chip on-line coupled with ICP-MS that uses a high-viscosity alcohol as a carrier phase to generate single-cell droplets (Fig. 5A).135 Droplets were generated at a frequency of 3–6 × 106 droplets min−1, and 2500 single-cell droplets were injected and analyzed per minute. Recently, they used this system to investigate cellular uptake of AuNPs by single HeLa cells as a tool to study intracellular drug delivery and cell/tissue imaging.136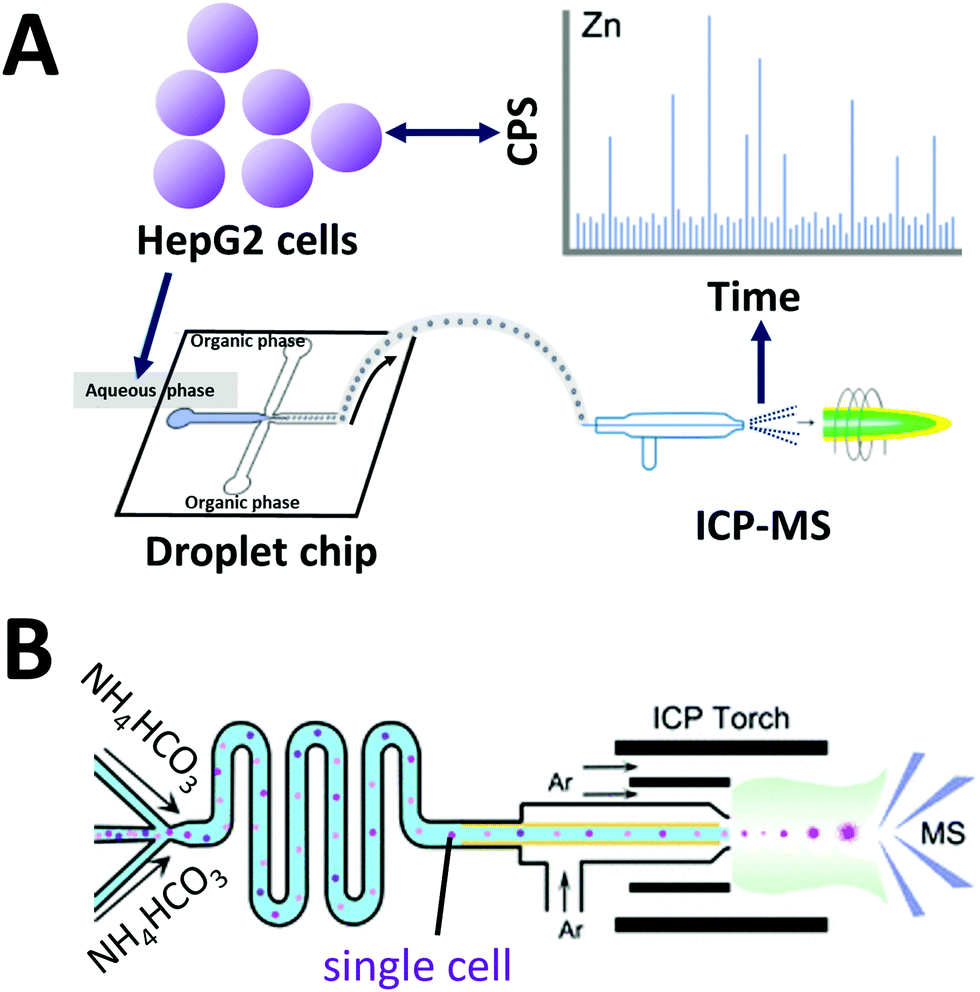 | ||
| Fig. 5 Coupling of microfluidics with inductively coupled plasma mass spectrometry (ICP). (A) Droplet chip-ICP-MS single-cell analysis system using high viscosity alcohol (alternative to oil phase) to generate single-cell droplets Adapted from ref. 135 with permission from the American Chemical Society, copyright © 2017. (B) Direct infusion of single cells with thermo-decomposable buffer as carrier phase into a mass spectrometer via ICP torch. Adapted from ref. 66 with permission from the American Chemical Society, copyright © 2020. | ||
Considering cells are already specialized “droplets” with a hydrophilic surface and an elastic hydrophobic membrane, Zhou et al. developed an oil-free passive microfluidic system (OFPMS) for the direct infusion of single cells (∼10 μm in diam., pL-range) into a micronebulizer for ICP-MS66 (Fig. 5B). This system enables single cell isolation through the use of a thermo-decomposable buffer that eliminates the use of any oil and incompatible polymer carriers. The quantitative single-cell transportation of endogenous zinc (Zn) and exogenous AuNPs in HeLa cells and RAW264.7 macrophages were measured with adjustable throughput ranging from 400 to 25000 cells per min.
3E. Acoustic wave nebulization/ionization
Acoustic-based microfluidics has been developed for use with micro and nanoscale samples and appears to be an effective method of manipulating small volumes.24,137,138 With an acoustic-based approach, researchers utilize gentle pressure waves to control a minute amount of sample in a contactless and precise manner for numerous research and industrial applications.139 A technique called surface acoustic wave nebulization (SAWN) utilizes standing waves to ionize droplet samples on a surface in an ambient environment at atmospheric pressure.24,137,140,141 SAWN was pioneered by Goodlett et al. as a microfluidic interface for mass spectrometry.24,142,143 In SAWN, the surface acoustic wave is generated through electrodes embedded on a dielectric chip, onto which a liquid sample droplet is placed. The acoustic energy transferred to the droplet overcomes its surface tension leading to atomization. When the samples are atomized, fine particles are produced, a small fraction of which then become charged due to microscopic fluctuations in the initial droplet. Recently, SAWN has been combined with other ambient MS ionization techniques such as atmospheric-Pressure chemical Ionization (APCI)140 and low-temperature plasma ionization (LTPI)144 to enhance the sensitivity. Unlike other microfluidic methods for sample manipulation, SAWN chip does not require pressure driven pumps or interconnects, nor the integration of microchannels. Monkkonen et al. previously combined a DMF device with the SAWN-MS to implement both controllable droplet movement and nebulization processes to perform hydrogen/deuterium exchange (HDX) of peptides.143,145 Recently, the same group integrated anisotropic ratchet conveyors (ARCs) on the SAWN transducer surfaces to automate the sample preparation and droplet delivery process in addition to nebulization.145,146 The system did not require the complex control circuitry needed in DMF devices, and the droplet could be manipulated on top of the SAWN chip in an open environment, without an enclosed flow channel or top cover (see Fig. 6). It is important to note that the ion signal from SAWN is typically 2–3 orders of magnitude lower than ESI signal; while SAWN can produce mass spectra similar to ESI, its applications are more limited due to this issue. However, the simple geometry of SAWN devices allows potentially attractive combinations with other ionization techniques to potentially improve overall ionization efficiency.140,144 | ||
| Fig. 6 On-chip surface acoustic wave nebulization (SAWN). (A) Droplet movements (moving, merging, mixing) on the SAWN chip surface integrated with anisotropic ratchet conveyors (ARCs) pattern. ARCs utilize hydrophilic patterns on a hydrophobic background to control the droplet transport by imbalanced hysteresis force. (B) Droplets nebulization. Adapted from ref. 145 with permission from the Royal Society of Chemistry, copyright © 2020. | ||
All these recent endeavors of coupling microfluidics with mass spectrometry are summarized in Table 1.
| Ionization technique | Type of microfluidic system | Recent applications [ref.] | Microfluidic coupling | |
|---|---|---|---|---|
| Strengths | Challenges | |||
| Electrospray ionization (ESI) | Continuous flow | • Single-cell study: in situ lipid extraction and detection160 | • Ideal for multi-step sample preparation/pretreatment (e.g. Cell lysis, protein extraction, purification, separation, concentration) | • Loss of sensitivity due to accumulation of materials on the ionization source |
| • Extraction and quantification of quinolones in milk192 | • Coupling with MS is simple | • Microfluidic chip may require numerous physical valve structures to process and ionize multiple samples in a single device | ||
| • Tumor–endothelial cell interaction and drug screening44 | • Flexible adjustment of injection volume | |||
| Microchip electrophoresis | • Charge variant profiling of therapeutic proteins164 | • Great for sample separation prior to MS analysis | • Use of high voltage during sample separation/migration | |
| • Glycoprotein characterization62 | • Minimal sample required (as low as ∼1 nL) | • Sample dilution/loss prior to MS due to the sheath fluid used in a common structure | ||
| • Low flow rate of MCE is compatible with ESI | • Difficult parallel sample processing in a single device | |||
| • Great for charge variant profiling | ||||
| Droplet microfluidics | • Analysis of secondary metabolites from Actinobacteria99 | • Rapid generation and processing of a large number of sample droplets | • Loss of sensitivity due to oil contamination | |
| • Enzyme activity screening93,163 | • Discrete reaction in each droplet | • Alternating sample droplet and carrier oil can interfere with the formation of a stable ESI plume | ||
| • Detection of pesticide residue on fruit surfaces95 | • Minimal sample (pico-to-nanoliter range) in each droplet | • Potential carryover or cross-contamination between droplets | ||
| • Moving droplets can be influenced by high ESI potentials (electrowetting) | ||||
| • Precise droplet manipulation can be challenging | ||||
| Paper microfluidics | • Urine and hair sample analysis (Drug abuse screening test)98 | • Low-cost microfluidic device | • Risk of sample retention and evaporation during sample delivery and ionization | |
| • Simple device operation (e.g., simple pipetting) | • Multi-step sample processing is challenging | |||
| • Rapid sample preparation | ||||
| Desorption electrospray ionization (DESI) | Continuous flow | • Analysis of biofluids (e.g., urine, serum, and cell lysate)107 | • Little or no sample preparation | • Potential cross-contamination of sample from sprayed solvents |
| • Quantification of free amino acids in food109 | • Can measure liquid, solid, or dried sample | • Challenging to measure multiple liquid samples in a single device | ||
| • Quantification of targeted miRNAs109 | ||||
| Paper microfluidics | • Enzyme activity screening104 | • Rapid and simple sample preparation | • Potentially suffer from low efficiency of sample delivery (evaporation, undesired retention) prior to MS analysis | |
| • Low-cost and portable microfluidic device | • Lower spatial resolution for MS imaging compared to MALDI-MS | |||
| Matrix-assisted laser desorption ionization (MALDI) | Continuous flow | • Analysis of intracellular metabolites114 | • Fast sample preparation using enclosed flow channel over the MS surface | • Mostly off-line coupling, requiring manual sample transfer prior to MS analysis |
| • Separation & identification of bacteria in food39 | • May require manual separation of MS surface from the microfluidic flow channel | |||
| • Enzyme kinetic assay191 | • Potential sample cross-contamination | |||
| • Single-cell metabolic analysis159 | ||||
| Droplet microfluidics | • Protein analysis of single yeast cells116 | • Rapid generation of sample in an array format | • Array generation over the MALDI surface can be challenging | |
| • Screening enzyme reaction in yeast cells (secretion/metabolism)115 | • A large number of isolated samples can be processed in parallel | • Matrix coating can compromise sample integrity and cause cross-contamination | ||
| • No separation prior to MS lead to high noise if the sample droplet contains a complex biological matrix | ||||
| Surface-assisted laser desorption ionization (SALDI) | Digital microfluidics | • Enzyme activity screening for biomass deconstruction133 | • Programable sample handling, ideal for automated multi-step sample processing/pretreatment | • DMF device fabrication is complex and prone to mechanical failure at high temperatures, pressures, and voltage |
| • Can be scaled up for parallel sample processing in an array format | • Difficult to manipulate concentrated samples | |||
| • May require manual separation of MS surface from the DMF device prior to MS analysis | ||||
| • SALDI requires custom MS surface/substrate | ||||
| Centrifugal microfluidics | • Sample cleanup and analysis of human serum38 | • Rapid sample processing | • Large-scale integration is challenging | |
| • Requires a minimal amount of instrumentation | • Requires off-line analysis (manual transport prior to MS analysis) | |||
| • Disposable device and low-cost microfluidic device | • SALDI requires custom MS surface/substrate | |||
| Inductively coupled plasma (ICP) | Droplet microfluidics | • Single-cell detection of metal ions66,135 | • A powerful method to detect trace elements | • Commonly used carrier oil is not ideal for ICP-MS due to relatively high carbon content |
| • Rapid injection and analysis of the discrete sample in droplets | • Limited types of sample can be analyzed | |||
| • Lighter elements (e.g., chromium and iron) are prone to more interferences | ||||
| • Challenging to manipulate droplets and potential cross-contamination of consecutive droplets | ||||
| Acoustic wave nebulization | Surface acoustic wave nebulization (SAWN) chip | • Sample droplet manipulation and peptide detection145,146 | • Compound desorption/ionization from the surface without heating the sample | • Device fabrication is complex and costly |
| • Higher survival yield of fragmentation-prone ions | • Lower ionization efficiency; the ion signal is typically 2–3 orders of magnitude lower than ESI-MS signal | |||
| • Works for both polar and non-polar analytes | • Limited to liquid sample (in most cases) | |||
| • Difficult to accurately control particle size | ||||
4. Recent applications
Given the dramatic technical advances in integrating mass spectrometry and microfluidics, a broadening range of applications have been reported in proteomics, metabolomics, cell analysis, and clinical diagnosis. Here we discuss recent applications that have received substantial attention.4A. Single-cell mass spectrometry analysis
The direct characterization of biomolecules from single cells is an important technical advance.147 Technologies that enable the direct measurement of genome, transcriptome, proteome, and metabolome components from individual cells can lead to new insights that would be otherwise unattainable in studies of a bulk population.148 Analyzing heterogeneity in cellular responses to chemical and physical perturbations requires miniaturized systems that can perform tens of thousands of experiments on single cells or small communities, and microfluidic approaches have proven to be rapid and cost-effective tools for such research.149 Specialized microfluidics systems have been designed for single-cell analysis based on their HTS capabilities, including direct manipulation of cells, controlled cell lysis, and controlled chemical reactions.25,150–152 Microfluidics coupled with mass spectrometry has also proven to be a key technology for understanding cellular states, improving upon genome amplification and sequencing approaches, which only yield indirect measurements. Proteomic and metabolomic analyses via mass spectrometry improve detection of cellular states by providing more direct characterization of phenotypes, which is crucial for understanding cellular functions and regulatory networks.147Metabolites are challenging molecules to measure due to their chemical diversity, which encompasses a vast range of concentrations. In order for a mass spectrometry system to reach adequate sensitivity for single-cell metabolomic studies, each step must be optimized. Furthermore, the correct analyte sampling method must be used, and since cell metabolomes rapidly respond to changes in the environment, perturbations upon sampling must be minimized. Over the past few years, different mass spectrometry-based approaches have been developed to profile metabolites in single cells and address these issues. The Sweedler group has pioneered the use of MALDI-MS imaging for studying single-cells by measuring chemical variations that can also be used to classify cellular subpopulations.153,154 Briefly, cells were dispersed onto a microscope slide before coordinates were assigned via optical imaging; the coordinates were then used to automate MALDI-MS measurements of targeted cells. Such optically guided MALDI-MS works well to assess lipid and peptide content for large populations of cells. Recently, they combined this MALDI-MS imaging with capillary electrophoresis electrospray ionization (CE-ESI-MS) to quantify low-mass metabolites which are difficult to measure because of MALDI matrix interferences.155
One strategy developed to increase the throughput of single-cell MALDI-MS measurement involves microarrays for mass spectrometry (MAMS), which feature hydrophilic reservoirs in an omniphobic surface.156,157 Applying a cell suspension onto the MAMS surface (which also serves as the MALDI target) enables the rapid and efficient singularization of large numbers of cells in an array format. Such high-density single-cell arrays have been implemented for metabolic analyses of single yeast cells.158 More recently, Guillaume-Gentil et al. utilized a high-throughput MAMS surface combined with a new sample collection technique to analyze cytoplasmic metabolites from single live cells.159 Unlike previous studies, the intracellular metabolites were analyzed without needing to remove the cell from its environment and cell viability was maintained. Using fluidic force microscopy, quantitative withdrawal of intracellular fluid at sub-picoliter resolution from individually arrayed cells was performed, and the fluids were then automatically transferred to another surface to create an array for MALDI-MS analysis (Fig. 7A). The extraction of 1 pL sample from each cell took around 2–3 min. They demonstrated the detection and identification of 20 metabolites recovered from the cytoplasm of individual HeLa cells while overcoming some of the obstacles often associated with working with live cells. In addition, their approach enabled the analysis of intracellular metabolites at multiple time points and demonstrated possibilities for further investigations of different types of molecules from an individual cell (e.g. combined transcriptional readouts and metabolite profiling).
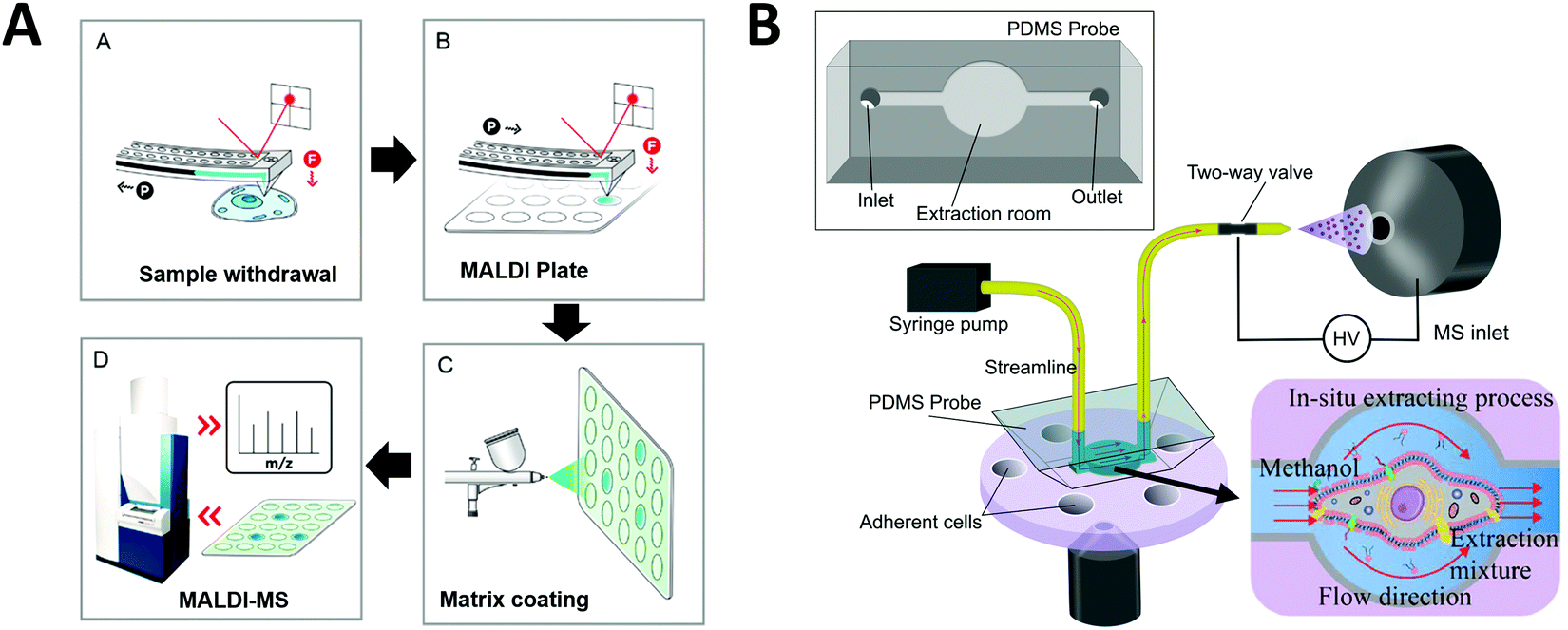 | ||
| Fig. 7 Applications of microfluidic-mass spectrometry in single-cell analysis. (A) Single-cell metabolite analysis by withdrawing intracellular fluid using fluidic force microscopy, followed by MALDI-MS analysis. Adapted from ref. 159 with permission from the American Chemical Society, copyright © 2017. (B) Microfluidic in situ single-cell entrapment and extraction for online ESI-MS analysis for single-cell identification and classification. Adapted from ref. 160 with permission from the Royal Society of Chemistry, copyright © 2019. | ||
Another single-cell MS approach was developed to uncover cellular heterogeneity in a human cell line. Huang et al. developed a flow-based microfluidic system for identification and classification of cells, including in-situ extraction of single cells with lipid analysis via an on-line ESI-MS.160 Their microfluidics-based in situ single-cell recognition system (ISCRS) can physically isolate single cells using PDMS valve structures and extract phosphatidylcholine for downstream MS analysis (Fig. 7B). The extraction from an isolated cell took ∼12 min. However, the throughput could be increased by adding multiple chambers for parallel sample processing within a single device. This method can be useful for automated multi-step pretreatment of cells.
4B. Compound screening
Compound screening is important in many areas such as drug discovery and bioproduct developments. In particular, by re-engineering an amino acid sequence through directed evolution, enzymes can be developed to explore new substrate and reaction conditions.161,162 New compounds produced through this process can be of major biological and chemical importance, but screening enzyme libraries is often the rate-limiting step in drug and biomarker discovery programs. Hence, the creation of technologies for carrying out rapid analysis of enzyme performance is an area of active research interest. Microfluidics-based HTS technology can be an effective option to dramatically decrease the assay volume required and increase the rate of analysis.Droplet microfluidics interfaced with ESI-MS provides a label-free HTS platform. Diefenbach et al. developed a droplet-MS method in a pharmaceutical setting for industrial enzyme screening, in addition to exploring methods to improve overall throughput of the system.163 The enzymatic assays were carried out in a multi-well plate and sample droplets were generated from each well using a custom Teflon fluid path. The droplets were then infused into ESI-MS (Fig. 8A). They demonstrated droplet enzyme reactions using two different transaminase libraries and analyzed the intact reaction mixture droplets by ESI-MS. Throughput was improved to 3 Hz compared to their previous system (1.7 Hz), with a wide range of droplet sizes (10–50 nL) achieved by tuning the sheath flow within the CE-MS source. These results suggest that mass spectrometry analysis of microfluidic droplets could significantly accelerate processes that require fast throughput, such as the screening of enzyme evolution libraries.
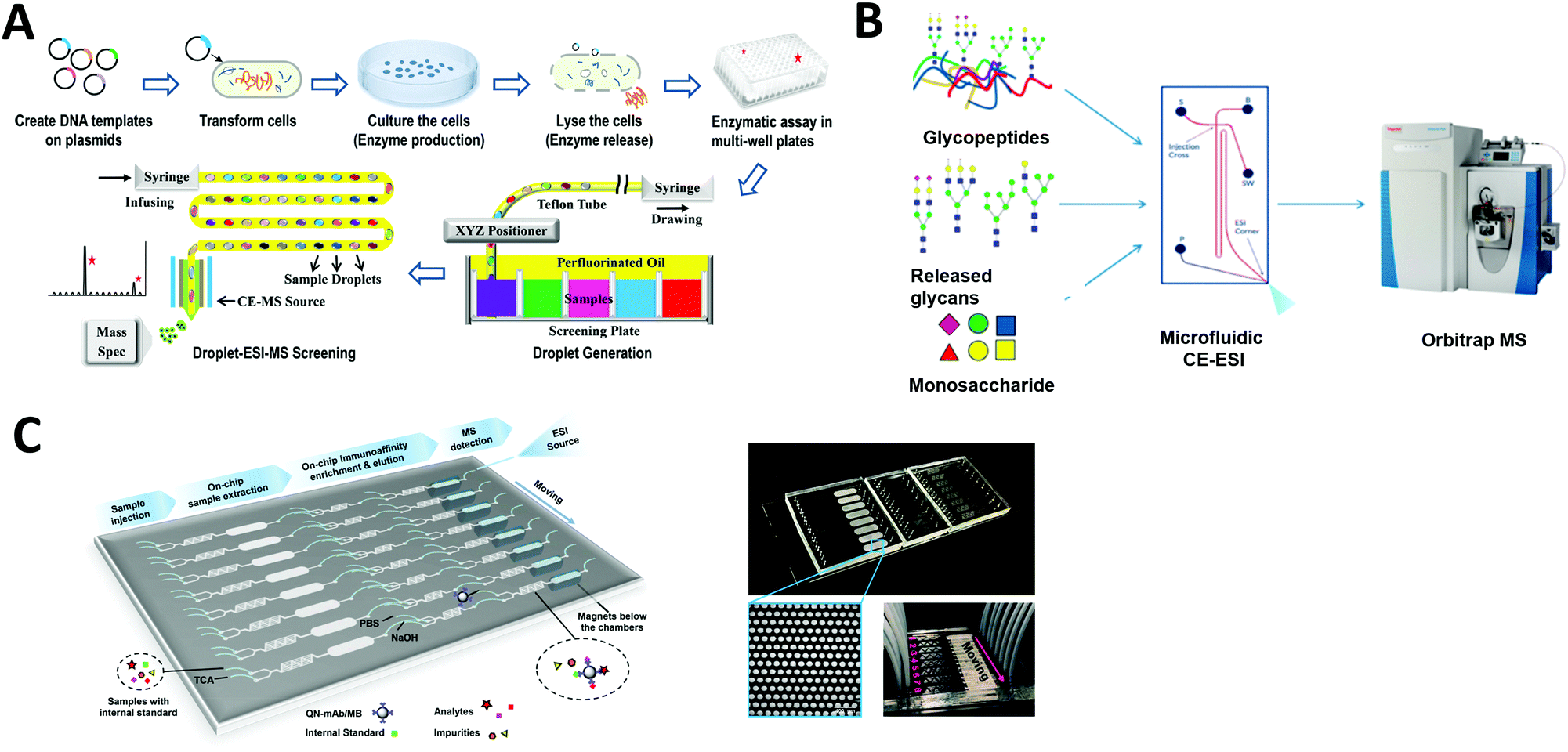 | ||
| Fig. 8 Applications of microfluidic-mass spectrometry in compound screening. (A) Screening of enzyme libraries using droplet microfluidics interfaced with ESI-MS. Adapted from ref. 163 with permission from the American Chemical Society, copyright © 2018. (B) Microfluidic CE-ESI-MS system for analysis of released glycans, glycopeptides and monosaccharides. Adapted from ref. 62 with permission from the American Chemical Society, copyright © 2017. (C) Continuous microfluidic device for an automated multi-step sample processing for an online MS analysis of seven different regulated quinolones in milk samples. Adapted from ref. 192 with permission from the American Chemical Society, copyright © 2019. | ||
Enzyme reactions carried out in droplets can also be arrayed on discrete SALDI plates. As mentioned in the previous section ‘SALDI-MS’, Heinemann et al. developed a DMF device patterned with an array of local SALDI-MS surface for HTS of enzyme kinetics at defined time intervals. As a proof-of-concept, a glycoside hydrolase enzyme (CelECC_CBM3a) was screened against a glycan substrate (1,4-b-D-cellotetraose-probe as a model plant biomass) for biofuels and bioenergy applications.133 This research demonstrates that such array-based SALDI-MS approaches have potential for screening large compound libraries and we expect to see developments for other existing SALDI surfaces in the future.
Charge variant profiling of therapeutic proteins is required by the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) and is traditionally performed by CE or ion exchange chromatography. Mass spectrometric determination of charge variant profiles acquired from electrophoretic separation is now possible through improvements in coupling microfluidic CE with MS, as well as the introduction of MS-compatible background electrolytes. Carillo et al. developed an MCE system coupled with ESI-MS using a custom “Zipchip” platform.164 With their technology, therapeutic monoclonal antibodies rituximab, trastuzumab, and bevacizumab drug products were separated and analyzed for proteoform identification, with an average mass accuracy of <15 ppm. 52 proteoforms were identified for trastuzumab, while rituximab samples indicated the presence of fragments and sialylated N-glycans. Khatri et al. also utilized an MCE-MS system for analysis of glycans, glycopeptides and monosaccharides, which require efficient separation methods for characterization of heterogeneous glycoform populations (Fig. 8B).62 Thus they demonstrated glycoproteomics analyses (a challenge if using conventional LC-MS) by improving electrophoretic separation followed by MS analysis. The separation of each sample in this MCE-MS system took around 10–20 min.
Affinity purification (AP) is another powerful technology to elucidate protein–protein interactions in cells and tissues. Zhao et al. developed a multifunction microfluidic system including AP coupled to ESI-MS for on-line analysis of seven different quinolones (QNs) in milk samples. Sample extraction, immunoaffinity enrichment, magnetic separation, elution, and ESI-MS were performed sequentially in a single device (Fig. 8C). This system permits automated on-chip immunoaffinity enrichment and accurate MS detection without additional off-line cleanup procedures.
4C. Study of microorganisms
Currently, microorganisms are often identified using 16S rRNA or 18S rRNA gene sequencing. However, because of the incredible complexity of microbial systems, it requires detailed scientific evaluations that yield both chemical and spatial information.165 Mass spectrometry analysis can provide chemical information relevant to genomic and transcriptomic data. In recent years, MALDI-MS has emerged as a promising tool for microbial detection and identification.166–168 The Sweedler group, which has pioneered the use of MALDI-MS imaging for studying single-cells,153–155 has recently used this approach for high-throughput label-free screening of multistep enzymatic reactions in bacterial colonies.165,169 During the MALDI-MS process, microbes are identified using either intact cells or cell extracts and the process is rapid, sensitive, and low cost in terms of labor and reagents. This technology has been widely adapted by microbiologists for bacterial strain typing, including the identification of water- and food-borne pathogens and antibiotic resistance.111 However, it is important to note that this technology is limited by peptide mass fingerprint data, as identification of new isolates is dependent on existing database information for a taxonomic rank. For instance, a fingerprint is required when attempting to identify an unknown isolate from a given genera, species, subspecies, or strain.Condina et al. reported a microfluidic device for separation of microbes, followed by off-line identification of beer spoilage microbes using MALDI-MS.39 Their system combines inertial microfluidics with secondary flows in a spiral microchannel for high-throughput separation of yeasts (Saccharomyces pastorianus and Saccharomyces cerevisiae, ∼5 μm) from beer spoilage microorganisms (Lactobacillus brevis and Pediococcus damnosus, ∼0.3–3 μm). The microorganisms were then identified at the species level using the MALDI-MS off-chip. Even though the MS analysis was performed separately off-chip, this could be modified for on-line MS analysis in the future with a sample transfer system.
Microfluidic cultivation systems coupled with high resolution MS provide single-cell product analysis and quantification capabilities, which can be very useful for microbial studies. Dusney et al. reported an analytical framework that interfaces microfluidic trapping and cultivation of a few bacterial cells (using their previously developed negative dielectrophoresis (nDEP)-based device) with the analysis of their catalytic products by Fourier-transform ion cyclotron resonance mass spectrometry (FT-ICR-MS).170 Using the biocatalytic model system of Corynebacterium glutamicum DM 1919 pSenLys cells, they analyzed cells that synthesized L-lysine from D-glucose. Cell trapping (with as few as 19 cells per experiment) was performed on chip for cultivating bacterial cells under continuous perfusion via negative dielectrophoresis (Fig. 9A). Quantification of catalytic products from only a few living cells was also demonstrated via microfluidics coupled with MS; 1.5 μL of cell supernatant was sampled into microcapillaries, which was then analyzed using a nanoESI ion source coupled to a FT-ICR-MS. Wink et al. explored a chip that combined MS with an epifluorescence read-out for on-line monitoring of bioactive metabolites produced by incubated Actinobacteria. This was conducted with Streptomyces griseus hyphae encapsulated in droplets of ∼200 pL. Detection of streptomycin produced in situ via ESI-MS was demonstrated, in addition to highlighting the feasibility of detecting fluorophores inside droplets just before they are electrosprayed (Fig. 9B).99 Recently, Terekhov et al. introduced a droplet-generating system equipped with fluorescence-activated cell sorting (FACS) followed by off-chip next-generation sequencing and LC-MS analysis to analyze the secretomes of encapsulated bacteria.23
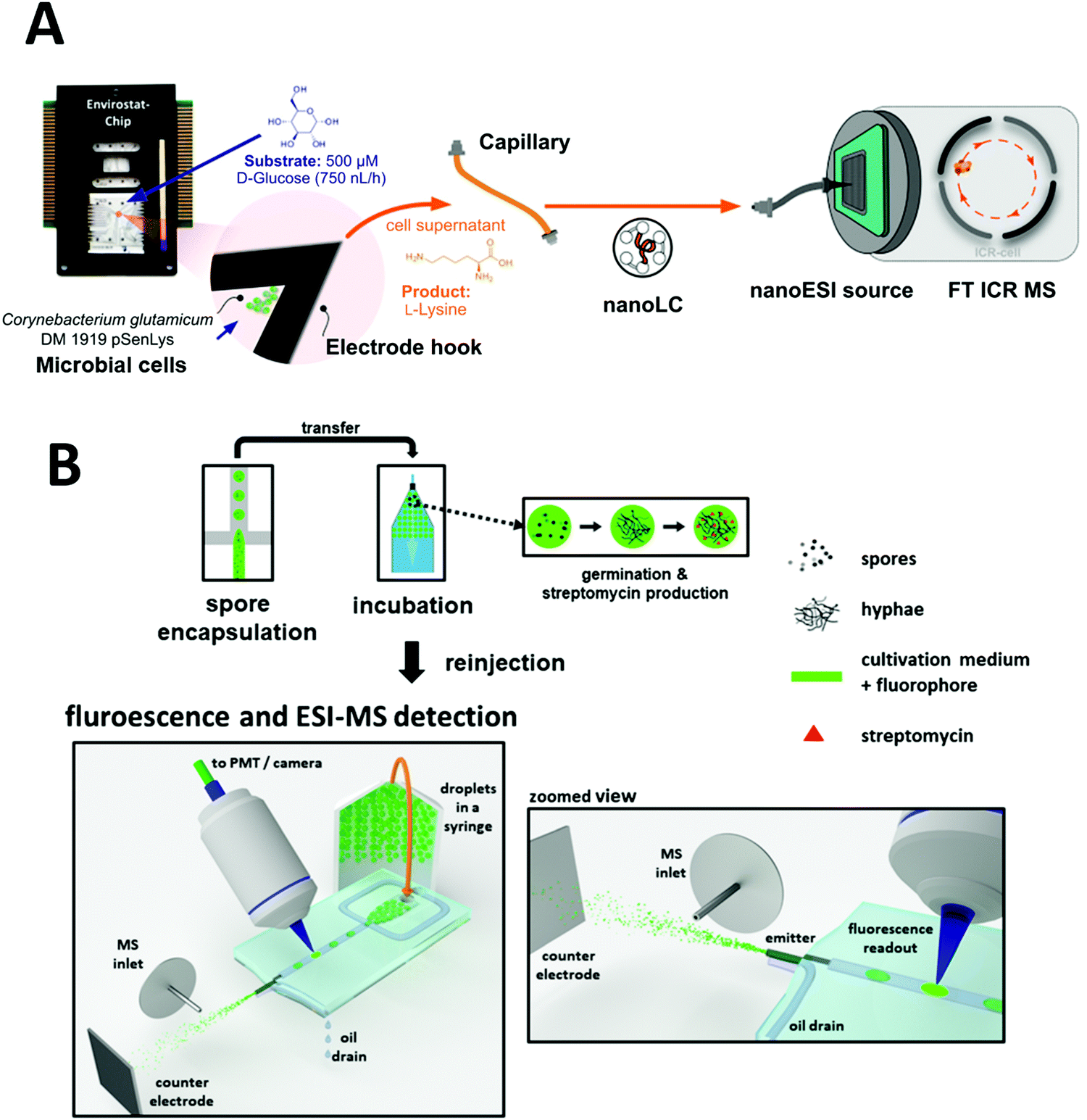 | ||
| Fig. 9 Applications of microfluidic-mass spectrometry in microbial study. (A) Microfluidic trapping and cultivation of a few microbial cells for the analysis of their catalytic products by Fourier-transform ion cyclotron resonance mass spectrometry. Adapted from ref. 170 with permission from the American Chemical Society, copyright © 2019. (B) Encapsulation of spores of Streptomyces griseus in droplets followed by incubation and ESI-MS analysis of produced Streptomycin. Adapted from ref. 99 with permission from Springer Nature, copyright © 2018. | ||
5. Outlook
Although enormous strides have been made in microfluidics and their integration with mass spectrometry, further improvements are needed to unlock its full potential as a powerful HTS platform. Here we list several areas that we think are key in this path forward.Towards on-line chip-to-mass spectrometry coupling
Real-time in situ measurement of samples by mass spectrometry without any additional handling steps after on-chip operation is ideal. However, the majority of reported approaches still involve off-line sample handling prior MS analysis. Manual steps often include sampling, sample preparation, and sample measurements. These off-chip approaches can compromise sample integrity and lower analysis throughput. Therefore, it will be important to move towards on-line coupling of microfluidic devices with mass spectrometry analysis for optimal throughput and minimal variation. Additionally, many microfluidic devices will benefit from these developments. For instance, the Abate group has developed a novel technology called ‘printed droplet microfluidics’, which encapsulates reagents and single cells in picoliter droplets, then actively selects and deposits desired droplets in an arrayed format on a printing substrate.27,64 Their systems have been used for various single-cell assays including protein profiling mainly using fluorescence detection and DNA sequencing (and off-chip mass spectrometry analysis in some cases) as readouts. Such an advanced microfluidic platform can take advantage of on-line coupling with mass spectrometry to further expand their applications.Universal sample processing system
A major advantage of microfluidic chips is the ability to automate and multiplex multiple processes with various functionalities. It can include cell culture, sample extraction, purification, desalting, concentration, drying, and droplet encapsulation.44,45,171–174 More efforts can be made to integrate multiple sample processing/pretreatment steps for biological samples into a single microfluidic system prior to ionization. And this universal sample processing system could be coupled with various types of ionization technique (e.g., ESI, MALDI) for downstream mass analysis. We anticipate, for example, microfluidic devices that can transform cells, culture cells and extract metabolites from the cells to study the gene function at large scale. Such universal sample processing system can be very powerful, introducing the needed experimental flexibility to perform.Coupling emerging ionization techniques with microfluidics
In addition to aforementioned ionization techniques, other emerging ionization approaches have been recently coupled with microfluidic devices. For example, Sathyanarayanan et al. developed a digital microfluidic devices equipped with desorption atmospheric pressure photoionization (DAPPI) for the ambient ionization of samples.175 This technology uses a microfabricated nebulizer chip with an internal heater to localize the gas jet (250–350 °C) onto the sample droplet, resulting in thermal desorption via gas-phase chemical reactions.Recently, Sinclair et al. reported an acoustic mist ionization (AMI) for direct injection of charged femto-liter sample droplets into the MS system.176 This platform uses acoustic energy (thereby eliminating the need for physical contact) to load ionized liquid samples from a microtiter plate to a mass spectrometer. In AMI-MS, sample ionization is achieved by applying high voltage (0.5–4 kV) above the test well, which leads to charge separation in the sample prior to droplet generation. Then, ultrasonic pulses are delivered from an acoustic transducer to produce a fluid cone and a spray of charged droplets. AMI-MS systems can deliver quantitative results up to 50 times faster than conventional LC–MS, with the system capable of analyzing up to 3 samples per second and 100000 samples per day on a single mass spectrometer.176,177 A few commercial AMI-MS (or Echo-MS) platforms have been developed (e.g., XEVO G2-XS QTOF-MS with AMI by Labcyte Inc. & Waters Corp., Echo® MS by AB SCIEX Ltd).177,178 To the best of our knowledge, there have been no publications on microfluidic devices integrated with the AMI-MS. Due to the potential of such technology, the development of microfluidic systems with direct acoustic sample droplet injection methods would be of considerable significance.
Coupling microfluidics with a miniaturized mass spectrometer
Even with the coupling of mass spectrometer with various microfluidic chips, conventional mass spectrometry system still suffers from portability issue due to its bulky equipment size.179,180 Cooks et al. pioneered the miniaturization of mass spectrometry system, called miniature mass spectrometer (MMS). The group has developed multiple generations of MMS (including the 25 kg “backpack” version) equipped with the miniaturized ion trap and ambient ionization source181–183 that can perform direct mass spectrometry analysis on complex samples without sample preparation or chromatographic separation (including sample types such as whole blood, untreated food, and environmental samples). Kirby et al. coupled digital microfluidics with the MMS for quantitation of drugs in urine.75 A custom digital microfluidic system was used to deliver droplets of solvent to dried urine samples, which were analyzed on MMS after the analytes were sent through an array of nanoelectrospray emitters. Cocaine, benzoylecgonine, and codeine were quantified in less than 15 min from four dried urine samples.To make microfluidic-mass spectrometry systems truly portable, more coupling of various microfluidic systems with improved MMS can be explored.
Higher specificity
Separation of samples prior to mass spectrometry analysis is important to improve sensitivity and specificity, especially if the analytes of interests are in complex biological matrices. In addition to on-chip purification or separation capabilities via microchip electrophoresis or microchip liquid chromatography (separation column embedded on chip),82,83 other off-chip separation techniques such as ion-mobility spectrometry (IMS) can be coupled with mass spectrometry to further enhance detection specificity of the microfluidic-mass spectrometry system. IMS separates ions based on the difference in mobility in an electric field in the gas phase, caused by their mass, shape/size and charge and integration with mass spectrometry can result in rapid analyte separation for mass spectrometry-based measurements.184 An increasing number of commercial IMS-MS platforms from several different vendors are available.185 This combined with microfluidic sample preparation will allow for further improvement in specificity. Another approach to improve MS detection specificity is tandem mass spectrometry (MS/MS). MS/MS involves two stages, where in the first stage ions of a desired m/z are isolated from the rest of the ions emanating from the ion source. These precursor ions are then induced to undergo a process to increase the internal energy of the ions leading to fragmentation. The ions resulting from the reactions are termed product ions, and these are analyzed with the second stage of MS/MS. The resulting MS/MS spectra provide additional specificity and can be compared with reference MS/MS spectra to further support chemical identifications.Multi-omics platforms
Genomics and transcriptomics though DNA and RNA sequencing have emerged as powerful tools for characterizing cells, including single cells. However, not all phenotypes of interest can be observed through changes in gene expression. For example, traditional sequencing approaches cannot capture the epigenetic state, protein expression, enzyme activity, and morphology of a cell or set of cells.65 Thus, combining genomic/transcriptomic analyses with proteomic and metabolomic analyses via mass spectrometry into a single microfluidic system could reveal genotype-to-phenotype relationships23 and provide insight into the molecular basis of cellular function.23 As a recent example, Zhang et al. reported a droplet microfluidic platform to connect optical imaging with gene expression profiling of single cells.65 In the future, such system could be also coupled with MS for more comprehensive characterization of single cells.A range of other analytical techniques can also be coupled with microfluidics and mass spectrometry such as surface-plasmon resonance (SPR) and surface enhanced Raman spectroscopy (SERS). SPR sensors can provide quantitative real-time binding data particularly useful to investigate the protein–protein interactions.19 However, as the monitoring of an interaction between a protein or small molecular ligands and a receptor molecule provides ambiguous information on the identity of the bound material due to lack of selectivity, a second technique is necessary for identification.186 The combination of SPR and mass spectrometry is emerging as a sensitive tool for the elucidation of novel protein–protein interactions as recently reported.14,186,187 Another powerful analytical technique, SERS detects Raman signal enhancement of analytes located close to or directly adsorbed onto the metal (nanoparticle) surface.17,20 There have been efforts of integrating SERS with mass spectrometry including the recent example of coupling SERS with paper spray ionizatio.15,16,188 These coupling approaches can, for example, enable analysis of cell biomass which could facilitate normalization of mass spectrometry data.
Detection of pathogenic microorganisms
The recent appearance of the novel corona virus (SARS-CoV-2 or COVID-19) and its alarming spread highlighted the importance of rapid diagnostic systems.189 Currently, laboratory-based reverse transcription-polymerase chain reaction (RT-PCR) is the primary method of confirming COVID-19 infection. However, RT-PCR tests demand well-equipped laboratories and skilled personnel. To address this challenging situation, rapid testing is under development, with a large component based on later-flow microfluidics (or μPADs) to make diagnostic devices faster and more affordable.81 In addition to common colorimetric readouts, paper microfluidics could accurately identify and quantify analytes of interest via downstream MS analysis, such as LDI-MS or L-DESI. Recently, Nachtigall et al. has proposed to use MALDI-MS pathogen identification combined with machine learning analysis for SARS-CoV-2 testing.112 They obtained mass spectra from 362 samples and, through the use of machine learning, were able to analyze and select the peaks that could distinguish a positive SARS-CoV-2 sample from a negative one. Such approaches can potentially become powerful diagnostic tools if sample processing can be performed via microfluidics. Based on recent advances in microfluidics and its coupling with MS techniques, microfluidic-MS would enable rapid and accurate detection of various pathogenic microorganisms. If further developed and properly validated such new approaches would enable point-of-care testing, leading to more efficient and decentralized screening among suspected cases.6. Conclusions
Advanced microfluidics enables the translation of chemical and biological assays to scales and rates unachievable in conventional laboratory workflows. The successful integration of microfluidic systems with mass spectrometry analysis provides a powerful approach to increase the sensitivity, specificity, and throughput of many conventional assays. It also allows for the integration and automation of sample processing to minimize sample and reagent consumption and to further streamline experimental workflows to make faster, better, and cheaper assays. Currently, a wide variety of mass spectrometry ionization techniques have been effectively coupled to different microfluidic systems and used for diverse applications. We expect that the integration of additional mass spectrometry capabilities outside the microfluidic chips such as the Ion-mobility spectrometry-mass spectrometry (IMS-MS) or tandem MS/MS configuration can also further enhance the specificity of these assays. Despite substantial advancements in microfluidic sample manipulation and analysis, there are still challenges for many microfluidic devices to be widely adapted for high-throughput screening applications. Although we believe that all systems reviewed here have the potential for increased throughput via various methods, some technologies have not yet been demonstrated for high-throughput assays. Broad adoption of these techniques will require robust implementation of strategies for the stable sample storage, containment, and sample tracking. Overall, we are optimistic that microfluidic-mass spectrometry systems will provide faster analysis while offering more sensitive and comprehensive analyses that eliminate off-line or manual sample handling steps to enable massive-scale screening capabilities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Contributions from NH and CP were funded by the DOE Joint BioEnergy Institute (http://www.jbei.org) and those of MdR, LZH, and AG were funded by the ENIGMA-Ecosystems and Networks Integrated with Genes and Molecular Assemblies (http://enigma.lbl.gov) Science Focus Area Program, and TN was funded by both. Both JBEI and ENIGMA are supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes.References
- S. Fox, S. Farr-Jones, L. Sopchak, A. Boggs, H. W. Nicely, R. Khoury and M. Biros, J. Biomol. Screening, 2006, 11(7), 864–869 CrossRef PubMed.
- R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. S. Green, R. P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer and G. S. Sittampalam, Nat. Rev. Drug Discovery, 2011, 10, 188–195 CrossRef CAS PubMed.
- E. E. Kempa, K. A. Hollywood, C. A. Smith and P. E. Barran, Analyst, 2019, 144, 872–891 RSC.
- L. M. Mayr and D. Bojanic, Curr. Opin. Pharmacol., 2009, 9, 580–588 CrossRef CAS PubMed.
- S. A. Sundberg, Curr. Opin. Biotechnol, 2000, 11, 47–53 CrossRef CAS PubMed.
- E. M. Payne, D. A. Holland-Moritz, S. Sun and R. T. Kennedy, Lab Chip, 2020, 20, 2247–2262 RSC.
- A. El-Aneed, A. Cohen and J. Banoub, Appl. Spectrosc. Rev., 2009, 44, 210–230 CrossRef CAS.
- T. P. Roddy, C. R. Horvath, S. J. Stout, K. L. Kenney, P.-I. Ho, J.-H. Zhang, C. Vickers, V. Kaushik, B. Hubbard and Y. K. Wang, Anal. Chem., 2007, 79, 8207–8213 CrossRef CAS PubMed.
- M. Bantscheff, M. Schirle, G. Sweetman, J. Rick and B. Kuster, Anal. Bioanal. Chem., 2007, 389, 1017–1031 CrossRef CAS PubMed.
- M. de Raad, C. R. Fischer and T. R. Northen, Curr. Opin. Chem. Biol., 2016, 30, 7–13 CrossRef CAS PubMed.
- D. B. Liesenfeld, N. Habermann, R. W. Owen, A. Scalbert and C. M. Ulrich, Cancer Epidemiol., Biomarkers Prev., 2013, 22, 2182–2201 CrossRef CAS PubMed.
- K. Deng, J. M. Guenther, J. Gao, B. P. Bowen, H. Tran, V. Reyes-Ortiz, X. Cheng, N. Sathitsuksanoh, R. Heins, T. E. Takasuka, L. F. Bergeman, H. Geertz-Hansen, S. Deutsch, D. Loqué, K. L. Sale, B. A. Simmons, P. D. Adams, A. K. Singh, B. G. Fox and T. R. Northen, Front. Bioeng. Biotechnol., 2015, 3, 153 Search PubMed.
- N. F. C. Visser, A. Scholten, R. H. H. van den Heuvel and A. J. R. Heck, ChemBioChem, 2007, 8, 298–305 CrossRef CAS PubMed.
- A. Madeira, E. Öhman, A. Nilsson, B. Sjögren, P. E. Andrén and P. Svenningsson, Nat. Protoc., 2009, 4, 1023–1037 CrossRef CAS PubMed.
- B. Nie, R. N. Masyuko and P. W. Bohn, Analyst, 2012, 137, 1421–1427 RSC.
- G. Grasso, L. D’Urso, E. Messina, F. Cataldo, O. Puglisi, G. Spoto and G. Compagnini, Carbon, 2009, 47, 2611–2619 CrossRef CAS.
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. García de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J.-F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J.-M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L.-L. Tay, K. G. Thomas, Z.-Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- I. Abdulhalim, M. Zourob and A. Lakhtakia, Electromagnetics, 2008, 28, 214–242 CrossRef.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Maragò and M. A. Iatì, J. Phys.: Condens. Matter, 2017, 29, 203002 CrossRef PubMed.
- S. Fornasaro, F. Alsamad, M. Baia, L. A. E. Batista de Carvalho, C. Beleites, H. J. Byrne, A. Chiadò, M. Chis, M. Chisanga, A. Daniel, J. Dybas, G. Eppe, G. Falgayrac, K. Faulds, H. Gebavi, F. Giorgis, R. Goodacre, D. Graham, P. La Manna, S. Laing, L. Litti, F. M. Lyng, K. Malek, C. Malherbe, M. P. M. Marques, M. Meneghetti, E. Mitri, V. Mohaček-Grošev, C. Morasso, H. Muhamadali, P. Musto, C. Novara, M. Pannico, G. Penel, O. Piot, T. Rindzevicius, E. A. Rusu, M. S. Schmidt, V. Sergo, G. D. Sockalingum, V. Untereiner, R. Vanna, E. Wiercigroch and A. Bonifacio, Anal. Chem., 2020, 92, 4053–4064 CrossRef CAS PubMed.
- H. Kim, M. J. Jebrail, A. Sinha, Z. W. Bent, O. D. Solberg, K. P. Williams, S. A. Langevin, R. F. Renzi, J. L. Van De Vreugde, R. J. Meagher, J. S. Schoeniger, T. W. Lane, S. S. Branda, M. S. Bartsch and K. D. Patel, PLoS One, 2013, 8, e68988 CrossRef CAS PubMed.
- T. W. Murphy, Y.-P. Hsieh, B. Zhu, L. B. Naler and C. Lu, Anal. Chem., 2020, 92, 2519–2526 CrossRef CAS PubMed.
- S. S. Terekhov, I. V. Smirnov, A. V. Stepanova, T. V. Bobik, Y. A. Mokrushina, N. A. Ponomarenko, A. A. Belogurov, M. P. Rubtsova, O. V. Kartseva, M. O. Gomzikova, A. A. Moskovtsev, A. S. Bukatin, M. V. Dubina, E. S. Kostryukova, V. V. Babenko, M. T. Vakhitova, A. I. Manolov, M. V. Malakhova, M. A. Kornienko, A. V. Tyakht, A. A. Vanyushkina, E. N. Ilina, P. Masson, A. G. Gabibov and S. Altman, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2550–2555 CrossRef CAS PubMed.
- S. R. Heron, R. Wilson, S. A. Shaffer, D. R. Goodlett and J. M. Cooper, Anal. Chem., 2010, 82, 3985–3989 CrossRef CAS PubMed.
- H. N. Joensson and H. Andersson Svahn, Angew. Chem., Int. Ed., 2018, 12176–12192 Search PubMed.
- M. T. Guo, A. Rotem, J. A. Heyman and D. A. Weitz, Lab Chip, 2012, 12, 2146–2155 RSC.
- R. H. Cole, S.-Y. Tang, C. A. Siltanen, P. Shahi, J. Q. Zhang, S. Poust, Z. J. Gartner and A. R. Abate, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8728–8733 CrossRef CAS PubMed.
- R. Seemann, M. Brinkmann, T. Pfohl and S. Herminghaus, Rep. Prog. Phys., 2012, 75, 016601 CrossRef PubMed.
- A. Kulesa, J. Kehe, J. E. Hurtado, P. Tawde and P. C. Blainey, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6685–6690 CrossRef PubMed.
- N. S. Ha, S. Sadeghi and R. M. Van Dam, Micromachines, 2017, 8, 337 CrossRef PubMed.
- D. B. Weibel and G. M. Whitesides, Curr. Opin. Chem. Biol., 2006, 10, 584–591 CrossRef CAS PubMed.
- D. Gao, C. Song and J.-M. Lin, in Microfluidics for Single-Cell Analysis, ed. J.-M. Lin, Springer, Singapore, 2019, pp. 163–195 Search PubMed.
- X. Wang, L. Yi, N. Mukhitov, A. M. Schrell, R. Dhumpa and M. G. Roper, J. Chromatogr. A, 2015, 1382, 98–116 CrossRef CAS PubMed.
- P. S. Dittrich, K. Tachikawa and A. Manz, Anal. Chem., 2006, 78, 3887–3908 CrossRef CAS PubMed.
- E. A. Redman, J. S. Mellors, J. A. Starkey and J. M. Ramsey, Anal. Chem., 2016, 88, 2220–2226 CrossRef CAS PubMed.
- T. M. Squires and S. R. Quake, Rev. Mod. Phys., 2005, 77, 977–1026 CrossRef CAS.
- J. Melin and S. R. Quake, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 213–231 CrossRef CAS PubMed.
- Y. Zhao, Y. Hou, J. Ji, F. Khan, T. Thundat and D. J. Harrison, Anal. Chem., 2019, 91, 7570–7577 CrossRef CAS PubMed.
- M. R. Condina, B. A. Dilmetz, S. R. Bazaz, J. Meneses, M. E. Warkiani and P. Hoffmann, Lab Chip, 2019, 19, 1961–1970 RSC.
- T. Ngernsutivorakul, D. J. Steyer, A. C. Valenta and R. T. Kennedy, Anal. Chem., 2018, 90, 10943–10950 CrossRef CAS PubMed.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580–584 CrossRef CAS PubMed.
- F. K. Balagaddé, L. You, C. L. Hansen, F. H. Arnold and S. R. Quake, Science, 2005, 309, 137–140 CrossRef PubMed.
- N. Gasilova, K. Srzentić, L. Qiao, B. Liu, A. Beck, Y. O. Tsybin and H. H. Girault, Anal. Chem., 2016, 88, 1775–1784 CrossRef CAS PubMed.
- L. Lin, X. Lin, L. Lin, Q. Feng, T. Kitamori, J.-M. Lin and J. Sun, Anal. Chem., 2017, 89, 10037–10044 CrossRef CAS PubMed.
- S. Kumar, V. Sahore, C. I. Rogers and A. T. Woolley, Analyst, 2016, 141, 1660–1668 RSC.
- X. Sun, W. Yang, Y. Geng and A. T. Woolley, Lab Chip, 2009, 9, 949–953 RSC.
- J. A. Jankowski, S. Tracht and J. V. Sweedler, TrAC, Trends Anal. Chem., 1995, 14, 170–176 CAS.
- N. S. Ha, J. Ly, J. Jones, S. Cheung and R. M. van Dam, Anal. Chim. Acta, 2017, 985, 129–140 CrossRef CAS PubMed.
- G. G. Mironov, C. M. Clouthier, A. Akbar, J. W. Keillor and M. V. Berezovski, Nat. Chem. Biol., 2016, 12, 918–922 CrossRef CAS PubMed.
- G. G. Morbioli, T. Mazzu-Nascimento, A. Aquino, C. Cervantes and E. Carrilho, Anal. Chim. Acta, 2016, 935, 44–57 CrossRef CAS PubMed.
- B. C. Durney, C. L. Crihfield and L. A. Holland, Anal. Bioanal. Chem., 2015, 407, 6923–6938 CrossRef CAS PubMed.
- Z. Yang and J. V. Sweedler, Anal. Bioanal. Chem., 2014, 406, 4013–4031 CrossRef CAS PubMed.
- A. N. de Macedo, M. Irfan Yasin Jiwa, J. Macri, V. Belostotsky, S. Hill and P. Britz-McKibbin, Anal. Chem., 2013, 85, 11112–11120 CrossRef CAS PubMed.
- S.-K. Ruokonen, F. Duša, J. Lokajová, I. Kilpeläinen, A. W. T. King and S. K. Wiedmer, J. Chromatogr. A, 2015, 1405, 178–187 CrossRef CAS PubMed.
- A. M. Skelley, J. R. Scherer, A. D. Aubrey, W. H. Grover, R. H. C. Ivester, P. Ehrenfreund, F. J. Grunthaner, J. L. Bada and R. A. Mathies, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1041–1046 CrossRef CAS PubMed.
- E. R. Castro and A. Manz, J. Chromatogr. A, 2015, 1382, 66–85 CrossRef CAS PubMed.
- D. Janasek, J. Franzke and A. Manz, Nature, 2006, 442, 374–380 CrossRef CAS PubMed.
- J. Jones, N. S. Ha, A. G. Barajas, A. F. Chatziioannou and R. M. van Dam, Anal. Chem., 2020, 92(4), 3483–3491 CrossRef CAS PubMed.
- J. Ly, N. S. Ha, S. Cheung and R. M. van Dam, Anal. Bioanal. Chem., 2018, 410, 2423–2436 CrossRef CAS PubMed.
- N. G. Batz, J. S. Mellors, J. P. Alarie and J. M. Ramsey, Anal. Chem., 2014, 86, 3493–3500 CrossRef CAS PubMed.
- A. Pettersson Dahlin, M. Wetterhall, G. Liljegren, S. K. Bergström, P. Andrén, L. Nyholm, K. E. Markides and J. Bergquist, Analyst, 2005, 130, 193–199 RSC.
- K. Khatri, J. A. Klein, J. R. Haserick, D. R. Leon, C. E. Costello, M. E. McComb and J. Zaia, Anal. Chem., 2017, 89, 6645–6655 CrossRef CAS PubMed.
- D. T. Chiu, R. M. Lorenz and G. D. M. Jeffries, Anal. Chem., 2009, 81, 5111–5118 CrossRef CAS PubMed.
- P. Shahi, S. C. Kim, J. R. Haliburton, Z. J. Gartner and A. R. Abate, Sci. Rep., 2017, 7, 1–12 CrossRef CAS PubMed.
- J. Q. Zhang, C. A. Siltanen, L. Liu, K.-C. Chang, Z. J. Gartner and A. R. Abate, Genome Biol., 2020, 21, 49 CrossRef CAS PubMed.
- Y. Zhou, Z. Chen, J. Zeng, J. Zhang, D. Yu, B. Zhang, X. Yan, L. Yang and Q. Wang, Anal. Chem., 2020, 92, 5286–5293 CrossRef CAS PubMed.
- R. T. Kelly, J. S. Page, I. Marginean, K. Tang and R. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 6832–6835 CrossRef CAS PubMed.
- X. Sun, K. Tang, R. D. Smith and R. T. Kelly, Microfluid. Nanofluid., 2013, 15, 117–126 CrossRef CAS PubMed.
- Y. Zhu and Q. Fang, Anal. Chem., 2010, 82, 8361–8366 CrossRef CAS PubMed.
- K. Choi, A. H. C. Ng, R. Fobel and A. R. Wheeler, Annu. Rev. Anal. Chem., 2012, 5, 413–440 CrossRef CAS PubMed.
- I. Barbulovic-Nad, S. H. Au and A. R. Wheeler, Lab Chip, 2010, 10, 1536–1542 RSC.
- P. Y. Keng, S. Chen, H. Ding, S. Sadeghi, G. J. Shah, A. Dooraghi, M. E. Phelps, N. Satyamurthy, A. F. Chatziioannou, C.-J. ‘CJ’ Kim and R. M. van Dam, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 690–695 CrossRef CAS PubMed.
- J. Li, N. S. Ha, T. Leo’ Liu, R. M. van Dam and C.-J. ‘CJ’ Kim, Nature, 2019, 572, 507–510 CrossRef CAS PubMed.
- A. R. Wheeler, H. Moon, C.-J. Kim, J. A. Loo and R. L. Garrell, Anal. Chem., 2004, 76, 4833–4838 CrossRef CAS PubMed.
- A. E. Kirby and A. R. Wheeler, Anal. Chem., 2013, 85, 6178–6184 CrossRef CAS PubMed.
- M. Tang, G. Wang, S.-K. Kong and H.-P. Ho, Micromachines, 2016, 7, 26 CrossRef PubMed.
- T.-H. Kim, K. Abi-Samra, V. Sunkara, D.-K. Park, M. Amasia, N. Kim, J. Kim, H. Kim, M. Madou and Y.-K. Cho, Lab Chip, 2013, 13, 3747–3754 RSC.
- R. Gorkin, J. Park, J. Siegrist, M. Amasia, B. Seok Lee, J.-M. Park, J. Kim, H. Kim, M. Madou and Y.-K. Cho, Lab Chip, 2010, 10, 1758–1773 RSC.
- A. W. Martinez, S. T. Phillips and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19606–19611 CrossRef CAS PubMed.
- X. Li, D. R. Ballerini and W. Shen, Biomicrofluidics, 2012, 6, 011301 CrossRef PubMed.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Anal. Chem., 2010, 82, 3–10 CrossRef CAS PubMed.
- I. M. Lazar, P. Trisiripisal and H. A. Sarvaiya, Anal. Chem., 2006, 78, 5513–5524 CrossRef CAS PubMed.
- R. F. Gerhardt, A. J. Peretzki, S. K. Piendl and D. Belder, Anal. Chem., 2017, 89, 13030–13037 CrossRef CAS PubMed.
- J. Stadlmann, O. Hudecz, G. Krššáková, C. Ctortecka, G. Van Raemdonck, J. Op De Beeck, G. Desmet, J. M. Penninger, P. Jacobs and K. Mechtler, Anal. Chem., 2019, 91, 14203–14207 CrossRef CAS PubMed.
- J. O. de Beeck and F. Impens, A Novel Nanoflow LC—MS Approach for Bottom-up Proteomics Using Micro Pillar Array Columns, PharmaFluidics, 2018 Search PubMed , https://www.pharmafluidics.com/products/%c2%b5pac-caplc-column/.
- C. A. Croushore, S. Supharoek, C. Y. Lee, J. Jakmunee and J. V. Sweedler, Anal. Chem., 2012, 84, 9446–9452 CrossRef CAS PubMed.
- T. Scholl, C. Dietze, M. Schmidt, S. Ohla and D. Belder, Anal. Bioanal. Chem., 2018, 410, 5741–5750 CrossRef CAS PubMed.
- R. T. Kelly, K. Tang, D. Irimia, M. Toner and R. D. Smith, Anal. Chem., 2008, 80, 3824–3831 CrossRef CAS PubMed.
- X. Sun, R. T. Kelly, K. Tang and R. D. Smith, Analyst, 2010, 135, 2296–2302 RSC.
- R. T. Kelly, C. Wang, S. J. Rausch, C. S. Lee and K. Tang, Anal. Chem., 2014, 86, 6723–6729 CrossRef CAS PubMed.
- Y. Cong, S. Katipamula, C. D. Trader, D. J. Orton, T. Geng, E. S. Baker and R. T. Kelly, Lab Chip, 2016, 16, 1544–1548 RSC.
- S. Sun and R. T. Kennedy, Anal. Chem., 2014, 86, 9309–9314 CrossRef CAS PubMed.
- D. J. Steyer and R. T. Kennedy, Anal. Chem., 2019, 91, 6645–6651 CrossRef CAS PubMed.
- A. J. Peretzki, S. Schmidt, E. Flachowsky, A. Das, R. F. Gerhardt and D. Belder, Lab Chip, 2020, 20, 4456–4465 RSC.
- C.-M. Huang, Y. Zhu, D.-Q. Jin, R. T. Kelly and Q. Fang, Anal. Chem., 2017, 89, 9009–9016 CrossRef CAS PubMed.
- J. Liu, H. Wang, N. E. Manicke, J.-M. Lin, R. G. Cooks and Z. Ouyang, Anal. Chem., 2010, 82, 2463–2471 CrossRef CAS PubMed.
- G. I. J. Salentijn, H. P. Permentier and E. Verpoorte, Anal. Chem., 2014, 86, 11657–11665 CrossRef CAS PubMed.
- Y.-C. Li, M.-H. Cheng and C.-H. Lin, in 2020 IEEE 33rd International Conference on Micro Electro Mechanical Systems (MEMS), 2020, pp. 98–101.
- K. Wink, L. Mahler, J. R. Beulig, S. K. Piendl, M. Roth and D. Belder, Anal. Bioanal. Chem., 2018, 410, 7679–7687 CrossRef CAS PubMed.
- C. A. Smith, X. Li, T. H. Mize, T. D. Sharpe, E. I. Graziani, C. Abell and W. T. S. Huck, Anal. Chem., 2013, 85, 3812–3816 CrossRef CAS PubMed.
- C. S. Clendinen, M. E. Monge and F. M. Fernández, Analyst, 2017, 142, 3101–3117 RSC.
- G. A. Harris, A. S. Galhena and F. M. Fernández, Anal. Chem., 2011, 83, 4508–4538 CrossRef CAS PubMed.
- M. Wleklinski, B. P. Loren, C. R. Ferreira, Z. Jaman, L. Avramova, T. J. P. Sobreira, D. H. Thompson and R. G. Cooks, Chem. Sci., 2018, 9, 1647–1653 RSC.
- S. V. de Freitas, F. R. de Souza, J. C. R. Neto, G. A. Vasconcelos, P. V. Abdelnur, B. G. Vaz, C. S. Henry and W. K. T. Coltro, Anal. Chem., 2018, 90(20), 11949–11954 CrossRef CAS PubMed.
- E. Gemperline, B. Chen and L. Li, Bioanalysis, 2014, 6, 525–540 CrossRef CAS PubMed.
- J. Laskin, B. S. Heath, P. J. Roach, L. Cazares and O. J. Semmes, Anal. Chem., 2012, 84, 141–148 CrossRef CAS PubMed.
- X. Li, R. Xu, X. Wei, H. Hu, S. Zhao and Y.-M. Liu, Anal. Chem., 2017, 89, 12014–12022 CrossRef CAS PubMed.
- X. Li, P. Rout, R. Xu, L. Pan, P. B. Tchounwou, Y. Ma and Y.-M. Liu, Anal. Chem., 2018, 90, 13663–13669 CrossRef CAS PubMed.
- H. Hu, S. Smith, X. Li, Z. Qian, Y. Su, M. Lin, J. Tu and Y.-M. Liu, Anal. Bioanal. Chem., 2020, 412, 1947–1954 CrossRef CAS PubMed.
- K. Song and Q. Cheng, Appl. Spectrosc. Rev., 2020, 55, 220–242 CrossRef CAS.
- N. Singhal, M. Kumar, P. K. Kanaujia and J. S. Virdi, Front. Microbiol., 2015, 6, 791 Search PubMed.
- F. M. Nachtigall, A. Pereira, O. S. Trofymchuk and L. S. Santos, Nat. Biotechnol., 2020, 1–6 Search PubMed.
- D. S. Cornett, M. L. Reyzer, P. Chaurand and R. M. Caprioli, Nat. Methods, 2007, 4, 828–833 CrossRef CAS PubMed.
- L. A. Filla, K. L. Sanders, J. B. Coulton, R. T. Filla and J. L. Edwards, Anal. Bioanal. Chem., 2019, 411, 6399–6407 CrossRef CAS PubMed.
- D. Haidas, S. Bachler, M. Köhler, L. M. Blank, R. Zenobi and P. S. Dittrich, Anal. Chem., 2019, 91, 2066–2073 CrossRef CAS PubMed.
- D. Haidas, M. Napiorkowska, S. Schmitt and P. S. Dittrich, Anal. Chem., 2020, 92, 3810–3818 CrossRef CAS PubMed.
- K. P. Law and J. R. Larkin, Anal. Bioanal. Chem., 2011, 399, 2597–2622 CrossRef CAS PubMed.
- T. R. Northen, J.-C. Lee, L. Hoang, J. Raymond, D.-R. Hwang, S. M. Yannone, C.-H. Wong and G. Siuzdak, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3678–3683 CrossRef CAS PubMed.
- T. de Rond, J. Gao, A. Zargar, M. de Raad, J. Cunha, T. R. Northen and J. D. Keasling, Angew. Chem., 2019, 58(30), 10114–10119 CrossRef CAS PubMed.
- E. Al-Hetlani, M. O. Amin, M. Madkour and B. D’Cruz, Talanta, 2021, 221, 121556 CrossRef CAS PubMed.
- K. Deng, J. Zeng, G. Cheng, J. Gao, K. L. Sale, B. A. Simmons, A. K. Singh, P. D. Adams and T. R. Northen, Biotechnol. Biofuels, 2018, 11, 266 CrossRef CAS PubMed.
- J. Wang, M. Jie, H. Li, L. Lin, Z. He, S. Wang and J.-M. Lin, Talanta, 2017, 168, 222–229 CrossRef CAS PubMed.
- Q. Zhu, F. Teng, Z. Wang, Y. Wang and N. Lu, Anal. Bioanal. Chem., 2019, 411, 1135–1142 CrossRef CAS PubMed.
- N. Li, S. Dou, L. Feng, X. Wang and N. Lu, Talanta, 2019, 205, 120085 CrossRef CAS PubMed.
- T. R. Northen, O. Yanes, M. T. Northen, D. Marrinucci, W. Uritboonthai, J. Apon, S. L. Golledge, A. Nordström and G. Siuzdak, Nature, 2007, 449, 1033–1036 CrossRef CAS PubMed.
- J. Gao, M. de Raad, B. P. Bowen, R. N. Zuckermann and T. R. Northen, Anal. Chem., 2016, 88, 1625–1630 CrossRef CAS PubMed.
- J. A. Fincher, D. R. Jones, A. R. Korte, J. E. Dyer, P. Parlanti, A. Popratiloff, C. A. Brantner, N. J. Morris, R. K. Pirlo, V. K. Shanmugam and A. Vertes, Sci. Rep., 2019, 9, 17508 CrossRef PubMed.
- A. R. Korte, N. J. Morris and A. Vertes, Anal. Chem., 2019, 91, 3951–3958 CrossRef CAS PubMed.
- A. R. Korte, S. A. Stopka, N. Morris, T. Razunguzwa and A. Vertes, Anal. Chem., 2016, 88, 8989–8996 CrossRef CAS PubMed.
- B. N. Walker, J. A. Stolee, D. L. Pickel, S. T. Retterer and A. Vertes, J. Phys. Chem. C, 2010, 114, 4835–4840 CrossRef CAS.
- H. N. Abdelhamid, Microchim. Acta, 2019, 186, 682 CrossRef PubMed.
- F. Lapierre, G. Piret, H. Drobecq, O. Melnyk, Y. Coffinier, V. Thomy and R. Boukherroub, Lab Chip, 2011, 11, 1620–1628 RSC.
- J. Heinemann, K. Deng, S. C. C. Shih, J. Gao, P. D. Adams, A. K. Singh and T. R. Northen, Lab Chip, 2017, 17, 323–331 RSC.
- M. He, B. Chen, H. Wang and B. Hu, Appl. Spectrosc. Rev., 2019, 54, 250–263 CrossRef CAS.
- H. Wang, B. Chen, M. He and B. Hu, Anal. Chem., 2017, 89, 4931–4938 CrossRef CAS PubMed.
- H. Wang, B. Chen, M. He, X. Li, P. Chen and B. Hu, Talanta, 2019, 200, 398–407 CrossRef CAS PubMed.
- X. Ding, P. Li, S.-C. Steven Lin, Z. S. Stratton, N. Nama, F. Guo, D. Slotcavage, X. Mao, J. Shi, F. Costanzo and T. Jun Huang, Lab Chip, 2013, 13, 3626–3649 RSC.
- T. Franke, A. R. Abate, D. A. Weitz and A. Wixforth, Lab Chip, 2009, 9, 2625–2627 RSC.
- P. Zhang, H. Bachman, A. Ozcelik and T. J. Huang, Annu. Rev. Anal. Chem., 2020, 13, 17–43 CrossRef CAS PubMed.
- L. Song, Y. You and T. Evans-Nguyen, Anal. Chem., 2019, 91, 912–918 CrossRef CAS PubMed.
- L. Pintabona, A. Astefanei, G. L. Corthals and A. C. van Asten, J. Am. Soc. Mass Spectrom., 2019, 30, 2655–2669 CrossRef CAS PubMed.
- S. H. Yoon, Y. Huang, J. S. Edgar, Y. S. Ting, S. R. Heron, Y. Kao, Y. Li, C. D. Masselon, R. K. Ernst and D. R. Goodlett, Anal. Chem., 2012, 84, 6530–6537 CrossRef CAS PubMed.
- L. Monkkonen, J. S. Edgar, D. Winters, S. R. Heron, C. L. Mackay, C. D. Masselon, A. A. Stokes, P. R. R. Langridge-Smith and D. R. Goodlett, J. Chromatogr. A, 2016, 1439, 161–166 CrossRef CAS PubMed.
- A. Kiontke, M. Roudini, S. Billig, A. Fakhfouri, A. Winkler and C. Birkemeyer, Sci. Rep., 2021, 11, 2948 CrossRef CAS PubMed.
- D. Sun, K. F. Böhringer, M. Sorensen, E. Nilsson, J. S. Edgar and D. R. Goodlett, Lab Chip, 2020, 20, 3269–3277 RSC.
- D. Sun, K. F. Böhringer, M. Sorensen, E. Nilsson and D. R. Goodlett, in 2019 20th International Conference on Solid-State Sensors, Actuators and Microsystems Eurosensors XXXIII (Transducers Eurosensors XXXIII), 2019, pp. 49–52.
- Y. Zhu, P. D. Piehowski, R. Zhao, J. Chen, Y. Shen, R. J. Moore, A. K. Shukla, V. A. Petyuk, M. Campbell-Thompson, C. E. Mathews, R. D. Smith, W.-J. Qian and R. T. Kelly, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed.
- K. Rosenthal, V. Oehling, C. Dusny and A. Schmid, FEMS Microbiol. Rev., 2017, 41, 751–780 CrossRef CAS PubMed.
- Y. Li, J. D. Motschman, S. T. Kelly and B. B. Yellen, Anal. Chem., 2020, 92, 2794–2801 CrossRef CAS PubMed.
- A. R. Wheeler, W. R. Throndset, R. J. Whelan, A. M. Leach, R. N. Zare, Y. H. Liao, K. Farrell, I. D. Manger and A. Daridon, Anal. Chem., 2003, 75, 3581–3586 CrossRef CAS PubMed.
- H. Yin and D. Marshall, Curr. Opin. Biotechnol, 2012, 23, 110–119 CrossRef CAS PubMed.
- R. N. Zare and S. Kim, Annu. Rev. Biomed. Eng., 2010, 12, 187–201 CrossRef CAS PubMed.
- E. T. Jansson, T. J. Comi, S. S. Rubakhin and J. V. Sweedler, ACS Chem. Biol., 2016, 11, 2588–2595 CrossRef CAS PubMed.
- T.-H. Ong, D. J. Kissick, E. T. Jansson, T. J. Comi, E. V. Romanova, S. S. Rubakhin and J. V. Sweedler, Anal. Chem., 2015, 87, 7036–7042 CrossRef CAS PubMed.
- T. J. Comi, M. A. Makurath, M. C. Philip, S. S. Rubakhin and J. V. Sweedler, Anal. Chem., 2017, 89, 7765–7772 CrossRef CAS PubMed.
- S. K. Küster, M. Pabst, R. Zenobi and P. S. Dittrich, Angew. Chem., Int. Ed., 2015, 54, 1671–1675 CrossRef PubMed.
- S. K. Küster, S. R. Fagerer, P. E. Verboket, K. Eyer, K. Jefimovs, R. Zenobi and P. S. Dittrich, Anal. Chem., 2013, 85, 1285–1289 CrossRef PubMed.
- A. J. Ibáñez, S. R. Fagerer, A. M. Schmidt, P. L. Urban, K. Jefimovs, P. Geiger, R. Dechant, M. Heinemann and R. Zenobi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8790–8794 CrossRef PubMed.
- O. Guillaume-Gentil, T. Rey, P. Kiefer, A. J. Ibáñez, R. Steinhoff, R. Brönnimann, L. Dorwling-Carter, T. Zambelli, R. Zenobi and J. A. Vorholt, Anal. Chem., 2017, 89, 5017–5023 CrossRef CAS PubMed.
- Q. Huang, S. Mao, M. Khan, W. Li, Q. Zhang and J.-M. Lin, Chem. Sci., 2019, 11, 253–256 RSC.
- F. H. Arnold, Acc. Chem. Res., 1998, 31, 125–131 CrossRef CAS.
- R. Baran, W. Reindl and T. R. Northen, Curr. Opin. Microbiol., 2009, 12, 547–552 CrossRef CAS PubMed.
- X. W. Diefenbach, I. Farasat, E. D. Guetschow, C. J. Welch, R. T. Kennedy, S. Sun and J. C. Moore, ACS Omega, 2018, 3, 1498–1508 CrossRef CAS PubMed.
- S. Carillo, C. Jakes and J. Bones, J. Pharm. Biomed. Anal., 2020, 185, 113218 CrossRef CAS PubMed.
- S. J. B. Dunham, J. F. Ellis, B. Li and J. V. Sweedler, Acc. Chem. Res., 2017, 50, 96–104 CrossRef CAS PubMed.
- R. D. Holland, J. G. Wilkes, F. Rafii, J. B. Sutherland, C. C. Persons, K. J. Voorhees and J. O. Lay, Rapid Commun. Mass Spectrom., 1996, 10, 1227–1232 CrossRef CAS PubMed.
- J. O. Lay and R. D. Holland, in Mass Spectrometry of Proteins and Peptides: Mass Spectrometry of Proteins and Peptides, ed. J. R. Chapman, Humana Press, Totowa, NJ, 2000, pp. 461–487 Search PubMed.
- T. L. Simmons, R. C. Coates, B. R. Clark, N. Engene, D. Gonzalez, E. Esquenazi, P. C. Dorrestein and W. H. Gerwick, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4587–4594 CrossRef CAS PubMed.
- T. Si, B. Li, T. J. Comi, Y. Wu, P. Hu, Y. Wu, Y. Min, D. A. Mitchell, H. Zhao and J. V. Sweedler, J. Am. Chem. Soc., 2017, 139, 12466–12473 CrossRef CAS PubMed.
- C. Dusny, M. Lohse, T. Reemtsma, A. Schmid and O. J. Lechtenfeld, Anal. Chem., 2019, 91, 7012–7018 CrossRef CAS PubMed.
- X. Feng, B.-F. Liu, J. Li and X. Liu, Mass Spectrom. Rev., 2015, 34, 535–557 CrossRef CAS PubMed.
- W. Wu, D. Zhang, K. Chen, P. Zhou, M. Zhao, L. Qiao and B. Su, Anal. Chem., 2018, 90, 14395–14401 CrossRef CAS PubMed.
- J. Wang, P. H. Chao and R. Michael van Dam, Proceedings of the 23rd International Conference on Miniaturized Systems for Chemistry and Life Sciences (MicroTAS), Basel, Switzerland, 2019, pp. 546–547 Search PubMed.
- J. Kehe, A. Kulesa, A. Ortiz, C. M. Ackerman, S. G. Thakku, D. Sellers, S. Kuehn, J. Gore, J. Friedman and P. C. Blainey, Proc. Natl. Acad. Sci. U. S. A., 2019, 201900102 Search PubMed.
- G. Sathyanarayanan, M. Haapala and T. Sikanen, Micromachines, 2018, 9, 649 CrossRef PubMed.
- I. Sinclair, M. Bachman, D. Addison, M. Rohman, D. C. Murray, G. Davies, E. Mouchet, M. E. Tonge, R. G. Stearns, L. Ghislain, S. S. Datwani, L. Majlof, E. Hall, G. R. Jones, E. Hoyes, J. Olechno, R. N. Ellson, P. E. Barran, S. D. Pringle, M. R. Morris and J. Wingfield, Anal. Chem., 2019, 91, 3790–3794 CrossRef CAS PubMed.
- SCIEX Ltd, Echo MS | SCIEX, https://sciex.com/products/integrated-solutions/echo-ms?utm_source=adwords+search&utm_medium=cpc&utm_campaign=2020+product+launches&utm_term=g-b-h-a-echo+ms+search+ads-may-20&utm_content=echo+ms&gclid=EAIaIQobChMI8NCQ0vm56gIVmx-tBh0wTAqwEAAYASAAEgKZtvD_BwE, accessed 15 October 2020.
- J. Rhys, E. Hoyes, E. Sprake, P. DeLand, L. Ghislain, E. Hall, S. Datwani, I. Sinclair, M. Bachmann and J. Wingfield, Waters corporation, https://www.labcyte.com/media/pdf/POS-Waters-AZ-Labcyte-Acoustic-Mist-Ionisation.pdf.
- W. Xu, N. E. Manicke, G. R. Cooks and Z. Ouyang, JALA, 2010, 15, 433–439 CAS.
- D. T. Snyder, C. J. Pulliam, Z. Ouyang and R. G. Cooks, Anal. Chem., 2016, 88, 2–29 CrossRef CAS PubMed.
- L. Li, T.-C. Chen, Y. Ren, P. I. Hendricks, R. G. Cooks and Z. Ouyang, Anal. Chem., 2014, 86, 2909–2916 CrossRef CAS PubMed.
- Z. Ouyang and R. G. Cooks, Annu. Rev. Anal. Chem., 2009, 2, 187–214 CrossRef CAS PubMed.
- P. I. Hendricks, J. K. Dalgleish, J. T. Shelley, M. A. Kirleis, M. T. McNicholas, L. Li, T.-C. Chen, C.-H. Chen, J. S. Duncan, F. Boudreau, R. J. Noll, J. P. Denton, T. A. Roach, Z. Ouyang and R. G. Cooks, Anal. Chem., 2014, 86, 2900–2908 CrossRef CAS PubMed.
- G. A. Eiceman, TrAC, Trends Anal. Chem., 2002, 21, 259–275 CrossRef CAS.
- E. J. Davis, P. Dwivedi, M. Tam, W. F. Siems and H. H. Hill, Anal. Chem., 2009, 81, 3270–3275 CrossRef CAS PubMed.
- E. C. A. Stigter, G. J. de Jong and W. P. van Bennekom, TrAC, Trends Anal. Chem., 2013, 45, 107–120 CrossRef CAS.
- S. Joshi, H. Zuilhof, T. A. van Beek and M. W. F. Nielen, Anal. Chem., 2017, 89, 1427–1432 CrossRef CAS PubMed.
- P. W. Fedick, F. Pu, N. M. Morato and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2020, 31, 735–741 CrossRef CAS PubMed.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N.-H. Wu, A. Nitsche, M. A. Müller, C. Drosten and S. Pöhlmann, Cell, 2020, 181, 271–280.e8 CrossRef CAS PubMed.
- A. M. Klein, L. Mazutis, I. Akartuna, N. Tallapragada, A. Veres, V. Li, L. Peshkin, D. A. Weitz and M. W. Kirschner, Cell, 2015, 161, 1187–1201 CrossRef CAS PubMed.
- J. Grant, S. H. Goudarzi and M. Mrksich, Anal. Chem., 2018, 90, 13096–13103 CrossRef CAS PubMed.
- Y. Zhao, M. Tang, F. Liu, H. Li, H. Wang and D. Xu, Anal. Chem., 2019, 91, 13418–13426 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |