
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfur modification in natural RNA and therapeutic oligonucleotides
Ya Ying
Zheng†
ab,
Ying
Wu†
ab,
Thomas J.
Begley
*bc and
Jia
Sheng
 *ab
*ab
aDepartment of Chemistry, University at Albany, State University of New York, 1400 Washington Ave. Albany, NY 12222, USA. E-mail: jsheng@albany.edu
bThe RNA Institute, University at Albany, State University of New York, 1400 Washington Ave. Albany, NY 12222, USA
cDepartment of Biological Science, University at Albany, State University of New York, 1400 Washington Ave. Albany, NY 12222, USA. E-mail: tbegley@albany.edu
First published on 27th April 2021
Abstract
Sulfur modifications have been discovered on both DNA and RNA. Sulfur substitution of oxygen atoms at nucleobase or backbone locations in the nucleic acid framework led to a wide variety of sulfur-modified nucleosides and nucleotides. While the discovery, regulation and functions of DNA phosphorothioate (PS) modification, where one of the non-bridging oxygen atoms is replaced by sulfur on the DNA backbone, are important topics, this review focuses on the sulfur modification in natural cellular RNAs and therapeutic nucleic acids. The sulfur modifications on RNAs exhibit diversity in terms of modification location and cellular function, but the various sulfur modifications share common biosynthetic strategies across RNA species, cell types and domains of life. The first section reviews the post-transcriptional sulfur modifications on nucleobases with an emphasis on thiouridine on tRNA and phosphorothioate modification on RNA backbones, as well as the functions of the sulfur modifications on different species of cellular RNAs. The second section reviews the biosynthesis of different types of sulfur modifications and summarizes the general strategy for the biosynthesis of sulfur-containing RNA residues. One of the main goals of investigating sulfur modifications is to aid the genomic drug development pipeline and enhance our understandings of the rapidly growing nucleic acid-based gene therapies. The last section of the review focuses on the current drug development strategies employing sulfur substitution of oxygen atoms in therapeutic RNAs.
 Ya Ying Zheng | Ya Ying Zheng graduated from College of Charleston with a dual degree in Biochemistry and Chemistry. She is currently a third year PhD student in the RNA institute at the SUNY Albany. Her research interest lies in understanding the roles of natural cellular RNA modifications play in biological processes such in gene regulation and protein expression. |
 Ying Wu | Ying Wu obtained her PhD in chemistry from Columbia University. Ying conducted postdoctoral research on oligonucleotide synthesis and RNA modifications with Dr Jia Sheng in The RNA Institute at SUNY-Albany, where she discovered the RNA phosphorothioate modification, the first natural modification reported on the backbone of RNA, in both prokaryote and eukaryotes. Ying is now pursuing a career in oligonucleotide-based genomic medicine. |
 Thomas J. Begley | Thomas J. Begley is Professor of Biological Sciences and Associate Director of the RNA Institute at the University at Albany, SUNY. His research interests include DNA damage and repair, RNA modifications and translation. The Begley lab works at the interfaces of nucleic acid biology, chemistry and computation. A major focus of current studies centers on how RNA modifications, which comprise the epitranscriptome, are collectively reprogrammed in response to stress to translationally regulate genes with specific codon usage patterns. In addition to science, he enjoys family, friends, tennis, hiking and sci-fi movies. |
 Jia Sheng | Jia Sheng is an associate professor in the Chemistry Department and The RNA Institute at University at Albany, State University of New York. The Sheng lab works at the interfaces of nucleic acid chemistry and biology, focusing on the chemical synthesis and structural studies of RNA natural modifications. The long-term goals of their research are to understand nature's strategies in diversifying the structures and functions of RNA, and to develop new molecular tools and therapeutics for biomedical applications. |
1. Introduction
Post-transcriptional modifications in cellular RNAs have gained significant attentions over the past few decades. To date, scientists have discovered over 170 chemical modifications in rRNA, tRNA and mRNA in all domains of life.1 The existence of these naturally occurring modifications added a new dimension beyond the basic building blocks of nucleic acids adenosine (A), uridine (U), cytidine (C) and guanosine (G). The presence of modified nucleosides on the base or ribose or backbone in specific sequence contexts confers unique features that allow for additional layers of gene expression and regulation.1–3 Although modifications can provide RNAs with expanded structural diversity, a majority of modifications can affect RNA functionality when working with their cellular targets. Such extraordinary chemical alterations have been observed playing roles in many biological processes such as transcription, translation, transportation, stability and protection of nucleic acids. These chemical modifications are catalytic products of specific writer enzymes that feature different reaction processes such as methylation, demethylation, amination, deamination, isomerization and thiolation, etc.4 The natural presence of these modifications affords more subtle functions of both DNA and RNA as evidenced by tRNA species with highly evolutionary conserved modifications at specific positions found in all the three domains of life.5 The development and advancement of new mass spectrometry-based methods serve as the dominant strategies to analyze and quantify the RNA epitranscriptome.6,7 Over 170 modifications across RNA species including ribosomal RNA (rRNA), messenger RNA (mRNA), transfer RNA (tRNA), microRNA (miRNA) and other non-coding RNA have been identified. However, the specific roles and the impacts of some modifications in RNA structures and functions have only been appreciated and thoroughly elucidated very recently.8 Overall speaking, the mapping and characterization for the majority of these modifications remain a big challenge considering the difficulty of studying the effects of a single modification at molecular and cellular levels.Sulfur based modifications are among the rapidly growing modifications being reported, with many important roles. Sulfur accounts for one of the most abundant and essential elements in our body and is found in the amino acids methionine and cysteine.9 Sulfur also plays important roles in the structure and regulation of proteins and coordinates many biomolecules in the cells via the formation of covalent disulfide bonds between cysteine residues in the proteins. Disulfide bond formation sometimes governs the enzymatic activity in many proteins, which points to the regulatory function of sulfur in cysteine.10 In addition to cysteine and methionine, sulfur is also found in glutathione, the vitamins biotin and thiamine, the cofactor S-adenosyl-Met, coenzyme A, the molybdenum cofactor (MoCo) and many other secondary metabolic products.10,11 Glutathione is a tripeptide containing cysteine and is known to detoxify metabolites and other stressors, making it an essential aspect of cellular homeostasis.12 These crucial sulfur-containing biomolecules and their biochemical functions render sulfur a good natural choice for nucleic acid modification. Several post-transcriptionally installed sulfur modifications in transfer ribonucleic acid (tRNA) have been discovered to maintain structural stability, promote effective codon recognition and enhance translation fidelity.13 tRNAs are by far the most modified nucleic acid species. More specifically, over 60 chemical modifications are found on uridine (U) in tRNA, of which 16 are thiolated at the C2 position of U.1,14 Nucleoside thiolation includes 2-thiouridine (s2U), 4-thiouridine (s4U), 2-thiocytidine (s2C), 2-thioribothymidine (s2T), 2-methythioadenosine (ms2A), and other derivatives such as 2-selenouridine (se2U) and 2-geranyl thiouridine (ges2U).5,15–17 The s2 and s4 thiolated nucleobases are derived by replacing the keto-oxygen with sulfur.18 Although the structural role of thio-modifications should promote functions in RNA species across all three domains of life, here we highlight the sulfur decorations on tRNA from well-studied organisms,19,20 and also integrate the newly discovered backbone phosphorothioate modification (Table 1).
| Structure | Symbol | Modification | Location | Function |
|---|---|---|---|---|
|
S2U | 2-thiouridine | tRNA U34 wobble position | Enhance the Accuracy and efficiency of protein translation |

|
S4U | 4-thiouridine | tRNA U8 | Serves as a photo-sensor for near-ultraviolet radiation |

|
Ges2U | Geranyl-2-thiouridine | tRNA U34 | Enhance codon recognition and reduce frameshift |

|
PS | Phosphorothioate | Backbone of RNAs | Unknown |
2. RNA sulfur modifications and their cellular functions
s2U and s4U modifications on tRNA
tRNAs possess numerous modifications on the bases and ribose moieties, with s2U and s4U among the most studied sulfur modifications.4 Sulfur modifications within the anticodon stem loop of tRNA are essential for proper decoding of mRNA codons in the ribosome and allow for accurate and efficient translation.21,22 tRNA acts as an adaptor molecule that bridges the nucleic acid blueprint of genomic information to amino acid peptide in the protein. In general, modified nucleosides alter the structure to regulate the affinity of anticodon-codon base pairing and the overall performance in protein synthesis. The central DNA theme governing all living organisms is the knowledge of how combinations of A, T, C and G form triplet codons that encode 20 amino acids. It is known that the codon degeneracy often occurs in the third position to allow for nonstandard pairing with wobble position 34 of the anticodon tRNA, while the first two base pairs are adhere to standard Watson–Crick rules for bonding with position 35 and 36 nucleotides in the anticodon stem loop.23 Modification to the wobble 34th position in tRNA, particularly s2U, is critical for achieving higher accuracy and efficiency in protein translation. Moreover, modifications located around the anticodon in positions 32 and 37 also have roles in improving reading frame maintenance.5,24,25 The thiolated nucleosides in the wobble position have also shown to be important as identity elements for certain aminoacyl-tRNA synthetase, in addition to ensuring correct codon binding in the ribosome.23,25,26Sulfur containing amino acids have also been shown to regulate translational capacity and metabolic homeostatic through modulation of tRNA thiolation. In yeast, the availability of sulfur containing amino acids methionine and cysteine can have direct effects on the thiolation status of wobble uridine U34. When those amino acids are limiting the sulfur source, s2U levels are downregulated, which serves as a cue to increase biosynthesis enzymes in methionine and cysteine production. Thus, tRNA thiolation is essential for achieving metabolic homeostasis and cell growth.27 Mutations in the s2U biosynthetic gene encoding the 2-thiouridylase TrmU are linked to acute infantile liver failure,28 and respiratory defects alongside with nonsyndromic deafness.29 Depletion of s2U caused by point mutations in mitochondrial tRNA genes can lead to a variety of human mitochondrial pathologies, including mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) and myoclonus epilepsy associated with ragged-red fibers (MERRF).30–33 Modifications occurring within the acceptor stem, the TψC-loop and the D-loop serve to govern structural stability, enhance recognition by aminoacyl-tRNA synthetases and fine-tune the overall performance.13,34 The s4U modification is a thionucleoside usually found at the position 8 in tRNA within the intersection of the acceptor stem and D-loop. This modification is responsible for sensing near-UV light in bacteria contributing to its unique λmax at 330 nm. When the cell is challenged with near-UV light, s4U crosslinks with cytidine 13, resulting in the destruction of the tRNA tertiary structure to cease translation.35 In some extreme thermophilic bacteria such as Thermus thermophilus and Pyrococcus furiosus, 5-methyl-2-thioridine (m5s2U) is found at position 54 on the TψC loop. The presence of the 2-thiolation content could strengthen the tRNA structure at elevated temperatures.36–38
It is worth mentioning that both thiolated nucleobases s2U and s4U have impacts on RNA structure, which includes base pairing stability, specificity and an alteration of thermodynamic parameters. As determined by UV-thermal melting and imino-proton NMR experiments, the order of duplex stability for a pentamer RNA sequence containing either s2U, s4U or U and 2′-O-methyl-ribonucleotide to a complementary strand supported that s2U is the most stable while s4U is the least stable when pairing with A.39 When comparing the base pairing affinity, s2U can stabilize base pairing with A more than G, with vice versa for s4U as it can stabilize base pairing with G more than A.40 Further studies also demonstrated that s2U containing oligonucleotides can stabilize both A and U base pairing in RNA duplexes. The thermodynamic stabilizing effect is possibly due to the tendency of s2U to pre-orient the RNA strand to single-stranded and facilitate the hybridization with its complementary target. In addition, the enhancement of the base stacking and highly polarizable nature of the sulfur atom could also contribute the stability enhancement.14 S2U has also shown to favor the C3′-endo conformation, which substantially stabilizes the nucleotides that likewise impart duplex thermostability.39,41
Geranyl-2-thiouridine (ges2U) and seleno-2-thiorudine (se2U) derivatives on tRNA
Wobble tRNA modifications can significantly alter anticodon-codon interactions, which points to their regulatory potential to speed up or slow down translation and affect fidelity.34,41 The 2-thiolation of U34 can increase the codon recognition efficiency of tRNAGluUUC,42 and plays a critical role in the ribosome binding of tRNALysUUU.43,44 In addition, the s2U can be further modified, forming a s2U based modification network that will be described in the next section. Particularly, the sulfur atom in s2U can be further replaced with selenium to form 2-selenouridine (se2U) by the tRNA modifying enzyme MnmH (also called SelU) using selenophosphate as the cofactor. MnmH has also been shown to use geranyl pyrophosphate as the cofactor to form geranylated 2-thiouridine (ges2U) in tRNAs specific for lysine (tRNALysUUU), glutamine (tRNAGlnUUG), and glutamic acid (tRNAGluUUC). Although the mechanisms of this dual function of MnmH remain elusive, MnmH has been known to contain a rhodanese domain, which is the key active working domain for both geranylation and selenation processes,45,46 and is ubiquitous protein that catalyzes the transfer of a sulfur atom from thiosulfate (rhodanese) or 3-mercaptopyruvate to e.g. cyanide in vitro.47,48 In addition, MnmH also contains a P-loop domain with a Walk-A motif, which is present in many ATP- or GTP-binding proteins and also involved in substrate binding.49,50It has also been suggested that ges2U is the intermediate product in the se2U formation process.46,51 The ges2U modification is lipid like with a 10-carbon hydrophobic chain on the wobble U. The ges2U modification can be further modified to 5-methylaminomethyl-2-gerenythioluridine (mnm5ges2U) or 5-carboxymethylaminomethyl-2-geranylthiouridine (cmnm5ges2U).17,52 This special lipid modification has been found in several bacteria including E. coli, E. aerogenes, P. aeruginosa, and S. typhimurium at a frequency of up to 6.7% in all the U34 residues of the three tRNAs (∼400 geranylated nucleotides per cell).17 Notably, the conjugation of the geranyl group has not been found in any eukaryotic cells, and the lipid modification is distinct in the bacterial kingdom. It has been known that both 2-thiouridine (s2U) and 2-selenouridine (se2U) enhance the discrimination of U:A pair over U:G pair (Fig. 1A–D).14,53 In contrast, the geranylated tRNAGluUUC was observed to increase the codon recognition efficiency to GAG over GAA,17 indicating that the geranylated uridine has a stronger base pairing preference with G over A at the end of codons. Indeed, by analyzing potential base pairing patterns (Fig. 1E–H), one direct consequence of the geranyl group is to switch the N3 of uridine from a hydrogen bond donor to an acceptor, which induces different base pairing patterns. As a result, only G that has two connected hydrogen bonding donors can form relatively stable two hydrogen bonds with the geranylated uridine. Based on this hydrogen bonding pattern and the fact that each of the two codons for glutamic acid, lysine, and glutamine ends in either A or G, it is speculated that geranylated uridine might be involved in translational regulation, by promoting base pairing with the G-ending codon for each amino acid, while restricting pairing with the A-ending codons.
 | ||
| Fig. 1 Base pairing patterns of 2-thiouridine (s2U), 2-selenouridine (Se2U) and geranylated 2-thiouridine (ges2U) with A, G, C, and U, respectively. G forms 2 H-bonds with ges2U, with A only forming 1 H-bond. | ||
Phosphorothioate (PS) on cellular RNAs
Post-transcriptional modifications of cellular RNAs are indispensable for cellular development and exert influence in biological processes in both normal and disease cells.54,55 The modifications on RNAs known earlier are either on nitrogenous base or ribose. The discovery of phosphorothioate (PS) modification on DNA has been documented in bacteria as well as in archaea.56,57 In a phosphorothioate internucleotide linkage, one of the non-bridging oxygen atoms is replaced with the sulfur, this renders the stereogenic phosphorus center. In both bacteria and archaea, the DNA phosphorothioate modifications have been elucidated to be in Rp configuration, with occurrence frequency of 4–31 PS modifications per 104 deoxy-nucleotides.58 More recently, the discovery of phosphorothioate modification in RNA was also reported in both prokaryotes and eukaryotes.59 The method of detection involved synthesizing RNA oligonucleotides containing PS modifications and subjecting it to nuclease enzymatic digestion to determine the optimal kinetics in which the normal phosphodiester bonds are hydrolyzed while the phosphorothioate bonds are intact. Different samples with prokaryotes and eukaryotes total RNAs were extracted and digested. The resulting mono-nucleosides and phosphorothioate dinucleotides together with 16 synthetic RNA PS dinucleotide standards were subjected to ultra-performance liquid chromatography coupled with mass spectrometry (UHPLC-MS) for analysis of RNA PS linkages. Quantification analysis estimated the number of phosphorothioate dinucleotide (PS) modification per 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nucleotides in total RNAs of HeLa, mouse liver and DM S2 as 4.7, 7.6, and 6.7 respectively. Additionally, CpsA, GpsC and GpsG are also found in the total RNA of L. lactis. The GpsG occurs at the highest frequency among all the samples tested and exists in stereo-specific Rp configuration. Further experiment showed that this GpsG modification locates on the rRNA in E. coli, L. lactis, and HeLa cells, and it is not detected in the rRNA-depleted total RNAs from these cells. Despite this big leap in the road of RNA modification, however, the cellular functions of RNA PS modification are still under investigation.
000 nucleotides in total RNAs of HeLa, mouse liver and DM S2 as 4.7, 7.6, and 6.7 respectively. Additionally, CpsA, GpsC and GpsG are also found in the total RNA of L. lactis. The GpsG occurs at the highest frequency among all the samples tested and exists in stereo-specific Rp configuration. Further experiment showed that this GpsG modification locates on the rRNA in E. coli, L. lactis, and HeLa cells, and it is not detected in the rRNA-depleted total RNAs from these cells. Despite this big leap in the road of RNA modification, however, the cellular functions of RNA PS modification are still under investigation.
3. Biosynthesis of RNA sulfur modifications
In this section, we summarize the biosynthesis of sulfur modifications located on nucleobases emphasising on 2-thiouridine, 4-thioruidine and geranyl derivatives on tRNA. Sulfur-modified nucleosides in eukaryotes and bacteria all utilize free L-cysteine in the cell as primary sulfur source. In general, the biosynthetic pathways for sulfur modifications in tRNA begin with the cysteine desulfurase protein, which activates L-cysteine residue to generate persulfide-SSH molecule. Enzyme linked persulfide is then delivered to downstream sulfur acceptor proteins and eventually incorporated into tRNA.60,61Biosynthesis of 2-Thiouridine (s2U)
The 2-thiouridine modification at the wobble position 34 of the anticodon stem loop in the tRNAs for glutamate acid, glutamine, and lysine is critical for enhancing translation due to its functional role in reducing frameshift and promoting codon recognition during protein synthesis.5 The biosynthesis of these thionucleosides has been well elucidated in eukaryote such as Saccharomyces cerevisiae and bacteria as in E. coli. The synthetic pathway can be categorized into two groups depending on the need for iron–sulfur [Fe–S] clusters. The overall pathway is summarized in Fig. 2. | ||
| Fig. 2 S2U (wobble position) biosynthesis pathway for both prokaryotes and eukaryotes. In E coli (black arrow), the biosynthesis S2U requires IscS, sulfur-relay system consists of Tus proteins and MnmA as the final thiolation enzyme. The thiolated U34 can be further converted into ges2U and se2U in the presence of selenouridine synthetase or SelU enzyme and geranyl pyrophosphate (GPP) or selenophosphate. In B. subtilis (green arrow), YrvO is an IscS like desulfurase and can directly transfer perfulfide sulfur to the thiolation enzyme MnmA. In S cerevisiae (red arrow), Nfs1 initiates the perfulfide transfer to rhodanese domain on Tum1 and Uba4. Urm1 serves as both sulfur acceptor and modifying enzyme on tRNA. | ||
In Gram-negative bacteria such as E. coli, the biosynthesis of 2-thiouridine requires two conserved enzymes, IscS and MnmA along with other enzymes: TusA, TusBCD heterohexameric complex and TusE enzymes, which serve as sulfur-relay system for the flow of persulfide sulfur in the pathway. IscS, the cysteine desulfurase, liberates sulfur from L-cysteine to form IscS-SSH enzyme bound persulfide intermediate. The binding of IscS with TusA stimulates the transfer of sulfur persulfide to TusA. The small sulfur-carrier protein TusA then transfers the sulfur to TusD aided by the interaction of TusE with the TusBCD complex. TusE serves as final sulfur persulfide intermediate between TusBCD complex and MnmA. It can interact with TusBCD complex and form a ternary complex with MnmA and tRNA before sulfur incorporation to U34 by MnmA. MnmA possesses a PP-loop moiety and belongs to the ATP-pyrophosphatase family. MnmA has dual functionalities, capable of adenylation by direct interaction with tRNA in the presence of ATP thereby activating U34 at C2 position as acyl-adenylated intermediate. MnmA is also capable of thiolation by performing a nucleophilic attack of terminal persulfide sulfur on Cys-199, which complete the generation of 2-thiouridine.5,62,63
In Gram-positive bacterium such as Bacillus subtilis, the biosynthesis of s2U is distinct from that of E. coli. Bacillus subtilis does not contain Tus proteins and cysteine desulfurase IscS. However, genomic analysis showed its genome interestingly encodes an active IscS-like cysteine desulfurase YrvO. It is worth noting that YrvO coding sequence is 31 bases upstream of MnmA gene and in the same genomic region as IscS gene. In vitro and in vivo studies showed that both of the YrvO and MnmA proteins are the sole requirement for s2U synthesis in Bacillus subtilis. In this two-component pathway, YrvO transfers persulfide sulfur to the cysteine residue within MnmA at the expense of ATP. Although the exact mechanism awaits further experimental investigation, the function of YrvO and MnmA in s2U tRNA formation and the lack of Tus sulfur relay protein system suggested the presence of a devoted cysteine desulfurase that can perform direct sulfur transfer from cysteine to MnmA.64
Bacteria contains at least two biosynthesis mechanisms of s2U tRNA: the 5-methoxycarbonylmethyl-2-thioruidne (mnm5s2U34) in cytoplasm and the 5-carboxymethylaminomethyl-2-thiouridine (cmnm5s2U34) in mitochondria. The 5-mnm and 5-cmnm groups are introduced through the MnmCDEG pathways65 and are also important for efficient codon recognition.42,66 The overall 2-thiouridine centered modification pathways are illustrated in Fig. 3 with several key enzymes still missed in this network. The differences in the biosynthesis pathways between bacteria and yeast is due to the involvement of [Fe-S] clusters and the usage of protein-thiocarboxylate as sulfur donor.67,68 The mechanistic pathway for the 2-thiolation of cy-tRNAs in S. cerevisiae begins with cysteine desulfurase Nfs1 catalyzing the first step, which is the transfer of persulfide to the rhodanese domain (Rhd) on Uba4 via Rhd of Tum1. Rhodanese serves as a sulfur carrier biomolecule and catalyzes the transfer reaction to downstream sulfur acceptor enzymes. Ubiquitin-related modifier 1 or Urm1 acts as a sulfur carrier while acyl-adenylate forms Urm1-COAMP intermediate and then thiocarboxylate Urm1-COSH at the C-terminus by receiving persulfide sulfur from the Rhd of Uba4. The sulfur from activated thiocarboxylate is then transferred to tRNA via Ncs6/Ncs2 heterodimer complex which catalyzes the final step of 2-thiolation U34 cy-tRNAs.5,68,69 This 2-thiouridine formation pathway is [Fe–S] clusters dependent and utilizes thiocarboxylated intermediate as the active sulfur donor. In addition to providing sulfur in s2U synthesis, the Nfs1 also serves as sulfur donor to [Fe–S] clusters by the assembly machineries ISC and CIA.70,71 In S. cerevisiae, S2U34 synthesis in the mitochondria required both NifS and Mtu1 proteins which are the homologs of IscS and MnmA in E.coli, and mt-tRNA thiolation process does not require [Fe–S] clusters.68,72 However, the exact mechanism and intermediate sulfur carriers are still unknown and require further investigation.
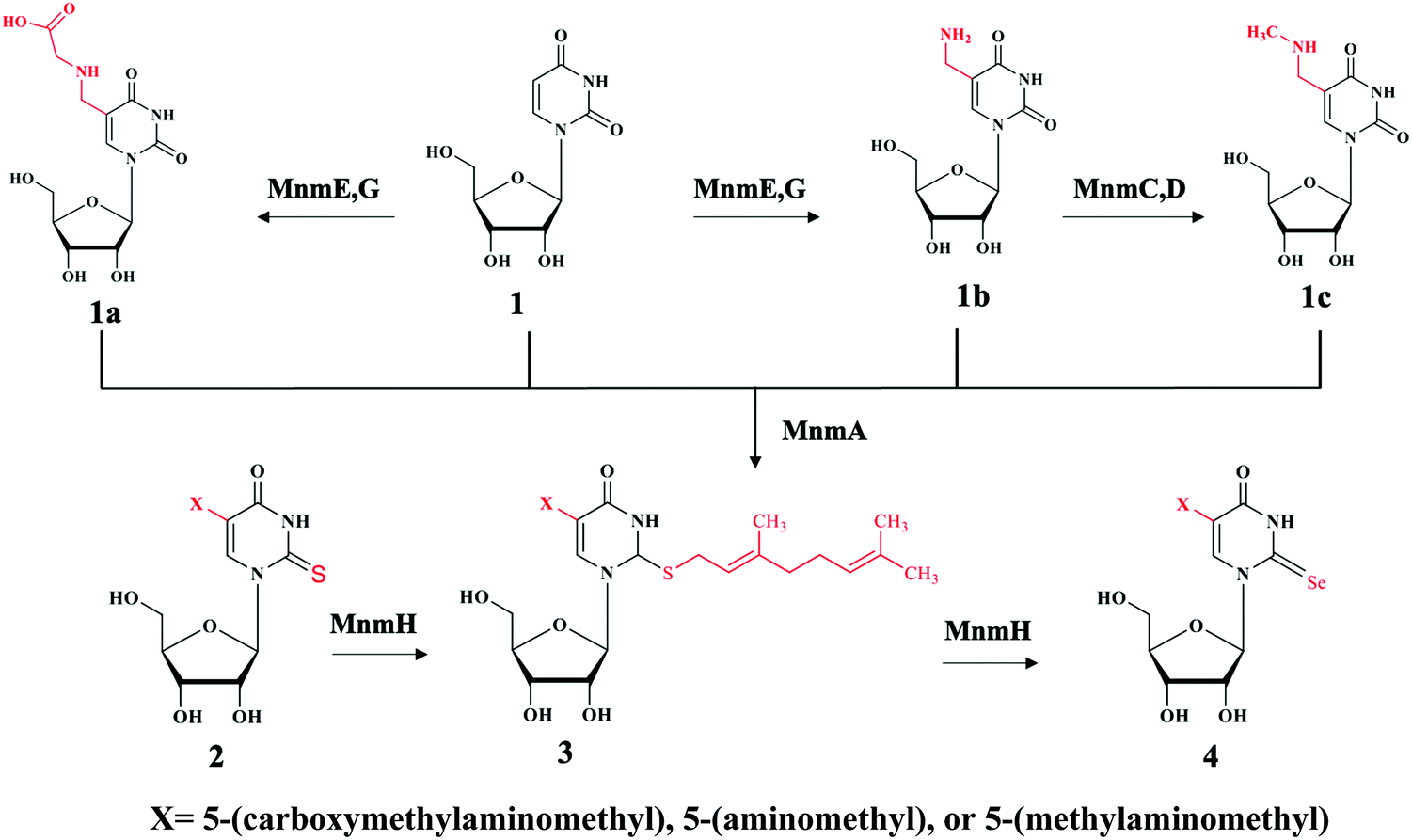 | ||
| Fig. 3 Chemical illustration of the biosynthetic pathway of s2U derivatives on both position 5 and 2. Compound 1 is uridine (U) with no substitution at the N5 position. When N5 is substituted with different moieties of X to produce compound 1a: 5-carboxymethylaminomethyluridine (cmnm5U), 1b: 5-aminomethyluridine (nm5U) both through enzyme MnmE, G, and 1c: 5-methylaminomethyluridine (mnm5U) through enzyme MnmC,D. These products can be further modified at the N2 position by the enzyme MnmA to generate compound 2 derivatives containing 2-thiouridine, with the products depending upon the X moieties. For example, U will be converted to s2U, cmnm5U to cmnm5s2U, nm5U to nm5s2U and mnm5U to mnm5s2U. Moreover, enzyme MnmH will further modified compound 2 derivatives eventually to compound 4 via common geranyl intermediate compound 3. The final products of compound 4 derivatives are Se2U via ges2U, cmnm5se2U via cmnm5ges2U, nm5se2U via nm5ges2U and mnm5se2U via mnm5ges2U intermediates. It is worth noting that enzyme MnmE, G can also use s2U and se2U as substrates and MnmC,D can also use nm5s2U and nm5se2U as substrates. | ||
In thermophilic bacterium such as T. thermophilus, the biosynthesis of m5s2U at position 54 in tRNA involves the TtuA/TtuB pathway. Cysteine desulfurase IscS or SufS begins the persulfide sulfur relay to the subsequent enzyme TtuB, a ubiquitin like sulfur carrier. TtuC activates TtuB at the C-terminal of Gly to form acyl-adenylated intermediate while being thiocarboxylated by accepting the sulfur from IscS/SufS. The final transfer of sulfur to tRNA in thiouridine formation is mediated by TtuA.5
Biosynthesis of 4-thiouridine (s4U)
4-Thiouridine at position 8 of tRNA serves as a photo-sensor for near-ultraviolet radiation found in bacteria. In Gram-negative bacterium such as E. coli, the synthesis of 4-thio modification required both IscS and ThiI enzymes. Earlier studies have reported that both enzymes also play roles in the synthesis of thiamine.73–75 IscS is a pyridoxal 5′-phosphate (PLP) dependent cysteine desulfurase that converts L-cysteine to L-alanine by catalyzing the cleavage of the carbon-sulfur bond in cysteine while liberating the sulfur atom to generate IscS linked persulfide intermediate IscS-SSH at its active site Cys-328.76,77 The subsequent step is the transfer of this activated persulfide sulfur to the acceptor molecule ThiI on Cys-456. ThiI contains the rhodanese-like sulfurtransferase domain which serves as the sulfur acceptor site. This persulfide on ThiI adenylates on U8 tRNA and donates sulfur for the synthesis of s4U.78 Characterization of ThiI reveals that this enzyme is common in the biosynthetic pathway for both thiamin and 4-thiouridine in tRNA. The overall synthesis pathway of s4U is shown in Fig. 4.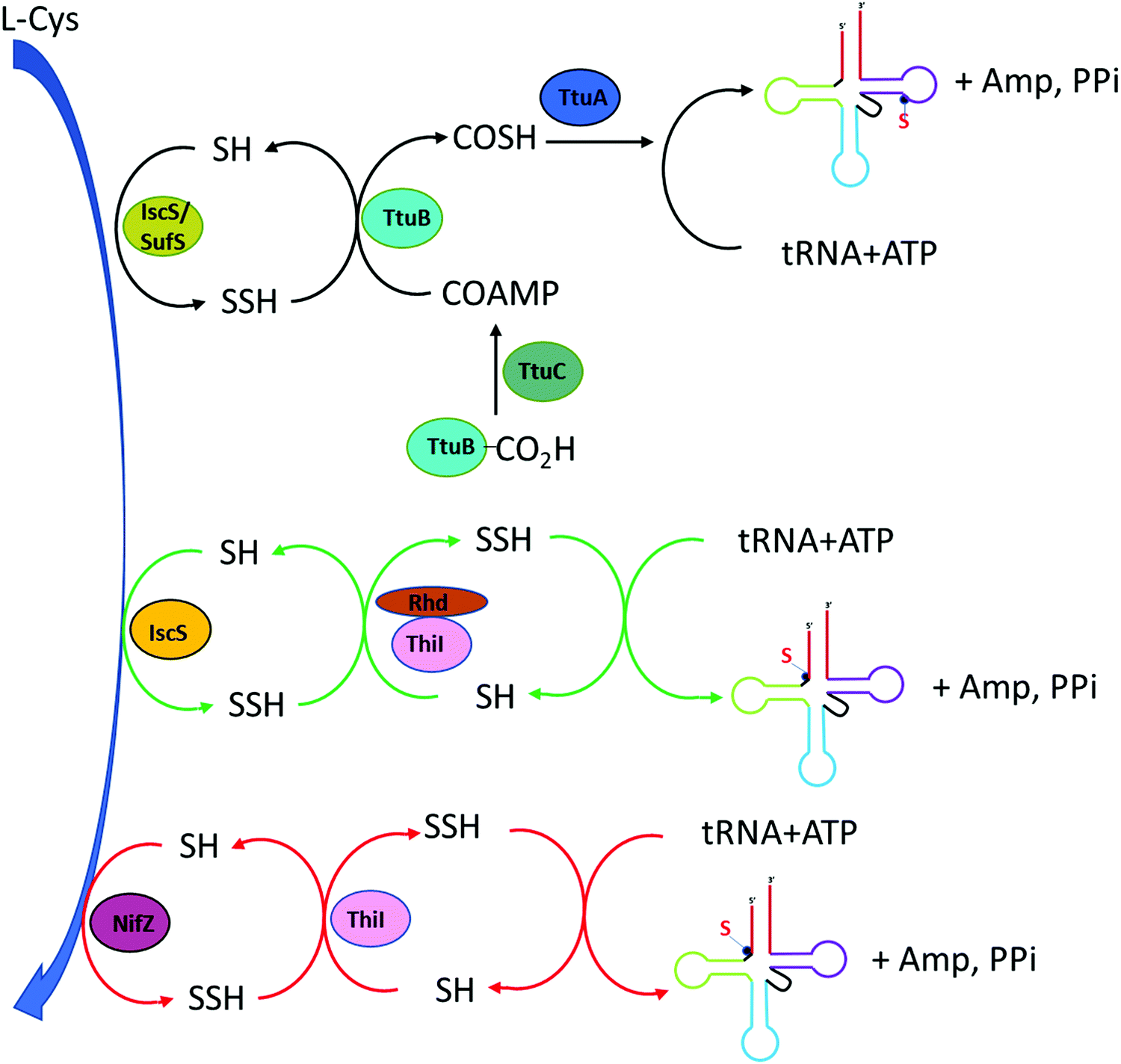 | ||
| Fig. 4 Biosynthesis pathway of s4U (position 8). In T. thermophilus (black arrow), the synthesis involves desulfurase IscS or SufS initiates the persulfide transfer to TtuB which is acyl-adenylated by TtuC. TtuA is the final modifying enzyme. In E coli (green arrow), IscS transfers perfulfide sulfur to ThiI in the Rhd domain as the sulfur acceptor site. In B. subtilis (red arrow), NifZ acts as desulfurase enzyme and ThiI can directly receive persulfide sulfur from NifZ despite lacking Rhd domain. | ||
The E. coli ThiI contains four domains: the C-terminal rhodanese like domain (RLD), the PP-loop pyrophosphatase domain (PP-loop domain), the N-terminal ferredoxin-like domain (NFLD), and the thiouridine synthases, RNA methyltransferase and pseudouridine synthases domain (THUMP domain).79 The NFLD and THUMP domains are involved in tRNA binding at the acceptor stem region. The proposed mechanism for the transfer of persulfide on the ThiI to form 4-thiouridine in tRNA involves the adenylation of 4-carbonyl group at the U8 tRNA in the PP-loop domain of ThiI utilizing the Mg-ATP. This activates U8 for the incorporation of sulfur atom. Sulfur is delivered from IscS to the Cys-456 in ThiI, forming ThiI persulfide. The transfer of terminal sulfur atom of persulfide on ThiI to U8 tRNA occurs in the rhodanese like domain (Rhd). Two mechanisms have been proposed for sulfur transfer mediated by ThiI.80 The nucleophilic attacking of persulfide sulfur on Cys-456 of ThiI active site to the C4 of uridine-8 in tRNA could initiate this process in concomitant with the release of AMP. In addition, this mechanism involves the enzymatic disulfide bond formation between Cys-456 and Cys-344 located in the PP-loop domain and generates s4U tRNA as the product. The assembly of disulfide bond serves to regenerate the enzyme for subsequent cycle which requires the reductive cleavage of the disulfide bond by a reductase through either a thiol group or another cysteine residue of ThiI. The regenerated cysteine active site is ready for another sulfur transfer from IscS and the cycle starts again.79 Alternatively, Cys-344 can also attack the bridging sulfur of Cys-456 followed by the release of sulfide that can feed directly into tRNA for the synthesis of s4U. The key feature of both mechanisms is the formation of disulfide bond between Cys-456 and Cys-344 as mark for the completion of the catalytic cycle.80,81
The s4U synthesis pathway in Gram-positive bacteria and archaea diverges from those of Gram-negative bacteria as in E. coli, although the exact mechanism is still unknown. Experimental data have revealed that majority of Gram-positive bacteria such as Bacillus subtilis do not contain IscS protein but actually encode another cysteine desulfurase enzyme NifZ. The sequence of NifZ exists four bases directly upstream of ThiI. The short sequence of ThiI produces protein that lacks the rhodanese domain but retains NFLD, THUMP and PP-loop domains. PP-loop domain reserves the Cys-344, the equivalent cysteine residue position in E. coli ThiI that is important for adenylation and thiolation of U8 tRNA.82 Despite lacking the rhodanese domain, B. subtilis ThiI and NifZ are sufficient to generate thiolation in tRNA as ThiI can directly receive persulfide sulfur from NifZ. In E. coli, Cys-344 involves in the final sulfur transferase step accompanied by the formation of disulfide bond with Cys-456 in the rhodanese domain. The missing of this important Cys-456 counterpart suggests the possibility of other unidentified ThiI cysteine for completing the thiolation.82 Similar to Gram-positive bacteria, the mechanism of s4U thiolation needs further elucidation since majority of archaea also lack cysteine desulfurase IscS and the rhodanese (Rhd) domain in ThiI protein. Study in Methanococcus maripaludis reveals that ThiI contains three conserved Cys residues (two come from a CXXC motif) in the putative catalytic region. A single mutation study showed that all three Cys residues are critical for persulfide transferase and replacing any Cys with Ala abolishes the function of ThiI as persulfide carrier. ThiI also contains [3Fe-4S] clusters which are essential for thiolation functionality in tRNA.83
Biosynthesis of RNA phosphorothioate (PS) backbone modification
RNA phosphorothioate (PS) modification was recently reported in both prokaryotes and eukaryotes.59 However, its corresponding regulatory enzymes and biological functions remain elusive. It has been demonstrated that the PS modification in RNA shares some common features with DNA counterpart such as the stereospecificity and relative abundance of the linked dinucleotides. For example, the GpsG, the most frequent RNA PS dinucleotide found so far, is only detected in the Rp stereo form. The abundance of RNA PS modification is also comparable to that of DNA PS modification found in bacteria. Therefore it is possible that this modification is regulated by the same set of enzymes or enzymes of similar functions. The DNA phosphorothioate backbone modification with one of the non-bridging oxygens replaced by the sulfur atom is the product of the DndA-E gene clusters together with DndFGH genes which constitute a defense mechanism that resemble restriction-modification system and protect bacteria from foreign invaders.84 The DndA-E gene clusters that regulate DNA PS modifications in both bacteria and archaeal are conserved in all domains of life. DndA actually shares significant nucleotide sequence identity and protein structural similarity with the pyridoxal 5-phosphate (PLP) dependent cysteine desulfurases IscS and NifS in eukaryotes.59,84 In addition, DndA was also reported as the sulfur source provider in the biosynthesis of DNA PS modification in bacteria. To test the possibility of DndA being involved in the biosynthesis of RNA PS modification as a writer, this gene was knocked out in E. coli BUN21/pML300 strain and the total RNAs were then extracted from both wild type and knock out strains. Quantification of GpsG with LC-MS/MS shows the modification level is reduced by half in DndA-knocked out strain comparing to the wild type BUN21. This result supports that DndA plays important roles in RNA PS biosynthesis pathway. Moreover, bacteria strains lacking the DndA gene were previously reported to still have detected DNA PS modification, which implies that there might be other devoted cysteine desulfurase such as IscS that serves as alternative sulfur source in this backbone thiolation process. Similar in DNA PS modification, there might be other enzymes in the Dnd gene clusters worthy of further investigation, probably guided by bioinformatic studies.594. Sulfur modifications in nucleic acid based therapeutics
Advances in biotechnology and genomics have led to rapid advances in nucleic acid based therapeutics, all of which have great potential in clinical applications for the treatment of cancer, infectious diseases, cardiovascular and neurological disorders. Currently, nucleic acids based therapeutic strategies such as antisense oligonucleotides (ASOs), RNA interference (RNAi) or small interfering RNA (siRNA) and aptamers all have been demonstrated to have promising results for biomedical applications since they are able to alter gene expression, thereby preventing or alleviating disease development.85These synthetic nucleic acids are designed to modulate gene expression with high specificity via target sequence recognition and hybridization. Antisense oligonucleotides (ASOs) can hybridize target mRNA by Watson–Crick base pairing and silence the gene expression by employing one of the three antisense mechanisms (Fig. 5): steric inhibition of mRNA translation; mRNA degradation caused by RNase H cleaving the RNA strand of the DNA–RNA duplex;86,87 and induction of splicing switch by exon skipping which restores the production of a protein.88 siRNA mediates mRNA degradation in the process of RNA interference (RNAi). Ago2, a RNase H like endonuclease can cleave targeted mRNA that is complementary to the guide RNA in siRNA bound to Ago2.89,90 Aptamers are single stranded nucleic acid that resemble monoclonal antibodies by self-assembling into 3D structure with the capability of binding to a variety of targets ranging from small molecules to proteins to whole cells with high affinity and specificity.91 It is also noteworthy the potential therapeutic applications of CRISPR-Cas9 technologies in genome editing.92 The clustered regularly interspaced short palindromic repeat (CRISPR) with CRISPR associated protein together constitute adaptive immunity possessed by prokaryotes, as a defense mechanism against foreign DNA or RNA-DNA interference. The single guide RNA (sgRNA) is versatile, its sequence can be programmed to guide Cas protein to cause precise gene editing at any target DNA loci.93 To improve in vivo activity and specificity, the engineered sgRNA with chemical modification can be obtained by synthesis.94 One of the main restrictions or challenges in the development of nucleic acid based drugs is the nuclease degradation before reaching the target sites. To solve this problem, different chemical modifications have been used to increase the biostability of these oligonucleotides such as base methylation, 2′-substitutions like 2′-fluoro (2′-F), 2′-methoxy (2′-OMe), 2′-O-methoxyethyl (2′-MOE), locked nucleic acid (LNA), phosphorodiamidate morpholino (PMO), N-acetylgalactosamine (GalNAc) and phosphorothioate (PS), etc. The incorporation of these modifications have proven to drastically stabilize the synthetic agents.95 In the case of CRISPR-Cas9 system, sgRNA with PS along or in conjunction with 2′-modifications have reported to reduce off target and significantly enhanced the editing ability by improving target binding of Cas9 protein.94,96Table 2 shows the FDA approved oligo therapies.97–106 These modified RNA-based drugs have shown resistance toward nuclease degradation, improving target selectivity, increasing binding affinity, as well as enhancing metabolic uptake. In particular, nusinersen, mipomersen, inotersen and volanesorsen are all modified with 2′-methoxy and phosphorothioate backbone linkages and fomivirsen, a DNA based ASO, have all PS modifications.106
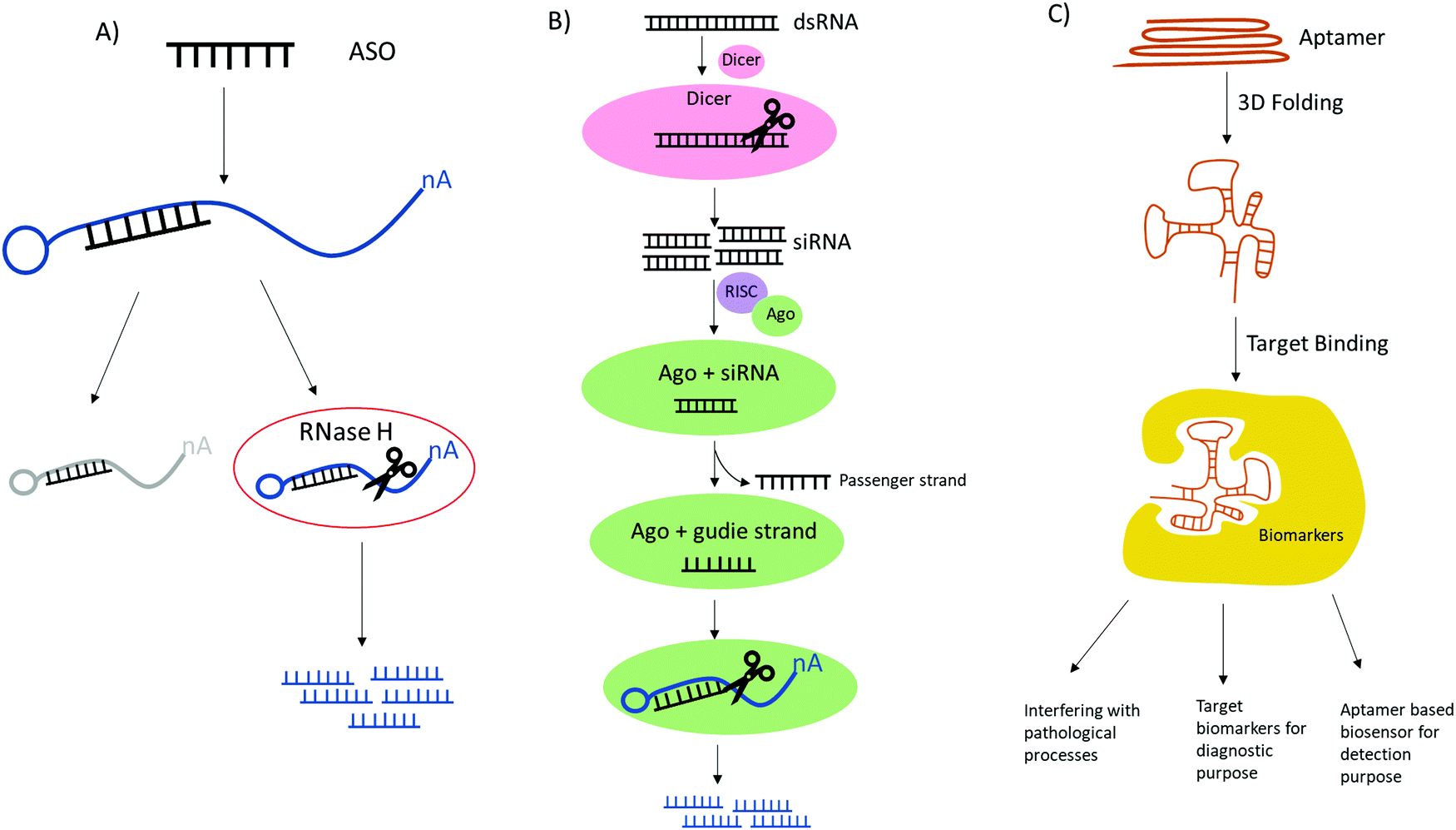 | ||
| Fig. 5 Comparison of the mechanistic action of ASO, siRNA and aptamer. (A) ASO has two antisense mechanisms by obstruction of target mRNA leading to cease translation and RNase mediated degradation of target mRNA. (B) siRNA mediates mRNA degradation involving Dicer and RISC complex containing Ago, RNase H like endonuclease which cleaves target mRNAs that are complementary to guide RNA. (C) Aptamer can be activated by folding into 3D structures and can bind to a variety of targets therefore achieving broad applications. | ||
| Drug name | Target disease | Oligo type | Mechanism | Year of approval |
|---|---|---|---|---|
| FDA, Food and Drug Administration; ASO, antisense oligonucleotides; siRNA, Small interfering RNA. | ||||
| Nusinersen | Spinal Muscular Atrophy (SMA) | ASO | Splicing switch exon 7 inclusion | 2016 |
| Macugen | Neovascular age-related Macular Dgeneration (AMD) | Aptamer | Steric block | 2004 |
| Fomivirsen | Cytomegalovirus Rhinitis | ASO | Steric block | 1998 |
| Mipomersen | Familial hypercholesterolemia (FH) | ASO | RNase H-mediated mRNA degradation | 2013 |
| Eteplirsen | Duchenne muscular dystrophy | ASO | Splicing switch exon 51 Skipping | 2016 |
| Inotersen | Hereditary transthyretin amyloidosis | ASO | RNase H-mediated mRNA degradation | 2018 |
| Patisiran | Hereditary TTR-mediated amyloidosis (hATTR) | siRNA | Ago 2 | 2018 |
| Volanesorsen | Familial Chylomicronemia | ASO | RNase H-mediated mRNA degradation | 2019 |
| Givosiran | Acute hepatic porphyria (AHP) | siRNA | Ago 2 | 2019 |
| Golodirsen | Duchenne muscular dystrophy (DMD) | ASO | Splicing switch exon 53 Skipping | 2019 |
| Lumasiran | Primary Hyperoxaluria type 1 (PH1) | siRNA | Reduce glycolate oxidase (GO) enzyme | 2020 |
PS modifications, where a sulfur atom replaces one of the non-bridging oxygens on the oligonucleotide strand, are one of the most important and widely used strategies to improve the drug-like properties of nucleic acid based therapeutics to enhance both cellular uptake and biological stability towards nuclease degradation.107 This characteristic is essential for the intended therapeutic effect as the molecules will remain in circulation until reaching to the target location for gene silencing.108 Out of a series of chemical modifications either on the backbone, nucleobase, or ribose sugar, the internucleotide PS linkage exhibits most resistance to RNase H cleavage.109 The chemical differences between the phosphorothioate and phosphate group arise from a larger van der Waals radius of 1.85 Å of sulfur atom versus 1.44 Å of oxygen, which resulted in the lengthened P-S bond. Moreover, phosphorothioate occurs as the mixture of stereo-isomers (Rp and Sp) since its phosphate center is connected with four different ligands. This chirality renders the isomers with different binding affinity and interaction modes with enzymes. In general, the Rp stereo-isomers have a stronger binding to the target mRNA and form a more stable complex with higher melting temperatures.110 However, Rp configuration has shown to be more susceptible to RNase H degradation compared to the Sp linkage that has shown to possess a higher resistance to nuclease cleavage activity.111 For example, Sp isomer confers greater stability for 3′ exonuclease by RNase A and snake venom phosphodiesterase than Rp isomer.112,113 Therefore, stereo-control synthesis of oligonucleotides must be attainable to examine the fundamental properties of individual isomers. Instead of using the regular nucleoside phosphoramidites for solid phase synthesis, using the bicyclic oxazaphospholidine derivatives as monomer building block has demonstrated the success of obtaining a high quality of stereo-pure Rp or Sp diastereomers. The 3′-O-bicyclic oxazaphospholidine derivatives were generated by the reaction between the 3′-OH of the corresponding protected nucleosides and 2-chloro-1,3,2-oxazaphospholidine derivatives, and the resultant trans-oxazaphospholidine isomers were configurationally very stable without any loss of diastereopurity even under acidic condition.114
Thio-cap mRNA analogs
It is also worth mentioning the thio-containing methyl-7-guanosine triphosphate cap at the 5′-end of mRNA (5′m7G) connected via a 5′,5′-triphosphate linkage with the first nucleotide, which is an evolutionarily conserved across eukaryotic species. This modification confers unique functions such as protection against degradation, pre-mRNA processing, nuclear exportation and modulating protein synthesis.115 In cancer immunotherapy, phosphorothioate modified cap analogs have been shown to profoundly increase the biostability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo.116 In another study where the mRNA bears the phosphorothioate modification at either the α, β, or γ position of the 5′,5′-triphosphate chain, it has been demonstrated that the thio-modified cap generally binds tighter to eIF4E, the eukaryotic translation initiation factor 4E, and are more resistant to the decapping pyrophosphatase DcpS. Particularly, the β-substituted analog m7GPPsPG has the strongest binding affinity toward eIF4E, and g-substituted analog shows strong resistant to hydrolysis by DcpS.117 The 5′-m7G cap analog containing 6-thioguanosine can be applied to study nucleic acid and cap dependent protein interaction through photo-inducible crosslinking experiments. 2′-O-methylation within the m7G moiety ensures correct cap incorporation during mRNA synthesis, which is known as anti-reverse cap analogs (ARCAs).118 The 6-thioguanosine serves as a photo-activable nucleoside which absorbs higher wavelength light compared with the natural nucleic acids and proteins. This allows for identification of the binding site between nucleic acid and protein when the system is selectively activated with certain wavelength.119 Cap analogs modified with 1,2-dithiodiodiphosphate containing a sulfur substitute at the two neighboring phosphate moieties have shown to protect mRNA from decapping and enhance its overall translational efficiency.120 The thio-modified cap has also been tried in formulating COVID-19 mRNA vaccines to increase their biostability and overall efficiency, which represents a very useful strategy for future vaccine development.1215. Summary
Overall, the biosynthetic pathways of nucleosides containing sulfur are quite complicated since these processes usually involve different sulfur-transporting enzymes rather than a direct single reaction. In addition, these pathways could vary significantly in different species and organisms. More detailed functions and working mechanisms in these biological reactions are still largely unknown. In general, the unique functions of sulfur modifications are based on the biochemical properties of sulfur-containing motifs in the nucleic acid structures. Sulfur atoms are significantly less electronegative (EN = 2.58) than oxygen (EN = 3.44), leading to changes in non-covalent interactions such as hydrogen bonding properties when oxygen atoms are replaced by sulfurs. These reduced or enhanced non-covalent interactions rendered by sulfur substitution alter a wide range of cellular RNAs’ functions from base pairing specificity to three dimensional structures and RNA stability toward nuclease degradation. The sulfur residues can also respond to cellular stress and environmental stimuli to induce a cascade of biological processes. The phosphorothioate bond containing a chiral center with two stereo configurations does not alter the RNA structure but substantially reduce their susceptibility towards nuclease degradation. Advancement in chemical synthesis and the huge progress in understanding genetic mutations and molecular drivers of diseases have led to rapid advancement of modified oligonucleotide-based therapeutics and provided solutions to solve the RNA degradation issues. As we have seen in COVID-19 vaccines, mRNA holds great promise in future drug development and the thio-modifications including the thio-caps on mRNA will play important roles in achieving more effective therapeutics for targeted diseases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to NSF (CHE-1845486 and MCB-1715234) and the NERF grant from the University at Albany, State University of New York, for the financial support.References
- P. Boccaletto, M. A. Machnicka, E. Purta, P. Piatkowski, B. Baginski, T. K. Wirecki, V. de Crecy-Lagard, R. Ross, P. A. Limbach, A. Kotter, M. Helm and J. M. Bujnicki, Nucleic Acids Res., 2018, 46, D303–D307 Search PubMed.
- R. J. Ontiveros, J. Stoute and K. F. Liu, Biochem. J., 2019, 476, 1227–1245 Search PubMed.
- H. Grosjean, Fine-tuning of RNA functions by modification and editing, Springer, Berlin, New York, 2005 Search PubMed.
- J. E. Jackman and J. D. Alfonzo, Wiley Interdiscip. Rev.: RNA, 2013, 4, 35–48 Search PubMed.
- N. Shigi, Front. Genet., 2014, 5, 67 Search PubMed.
- C. T. Chan, M. Dyavaiah, M. S. DeMott, K. Taghizadeh, P. C. Dedon and T. J. Begley, PLoS Genet., 2010, 6, e1001247 Search PubMed.
- M. Basanta-Sanchez, S. Temple, S. A. Ansari, A. D'Amico and P. F. Agris, Nucleic Acids Res., 2016, 44, e26 Search PubMed.
- S. Li and C. E. Mason, Annu. Rev. Genomics Hum. Genet., 2014, 15, 127–150 Search PubMed.
- M. E. Nimni, B. Han and F. Cordoba, Nutr. Metab., 2007, 4, 24 Search PubMed.
- K. Saito, Plant Physiol., 2004, 136, 2443–2450 Search PubMed.
- T. Leustek, The arabidopsis book, 2002, 1, e0017 Search PubMed.
- V. A. Pompella, A. Paolicchi, V. De Tata and A. F. Casini, Biochem. Pharmacol., 2003, 66(8), 1499–1503 Search PubMed.
- B. El Yacoubi, M. Bailly and V. de Crecy-Lagard, Annu. Rev. Genet., 2012, 46, 69–95 Search PubMed.
- A. T. Larsen, A. C. Fahrenbach, J. Sheng, J. Pian and J. W. Szostak, Nucleic Acids Res., 2015, 43, 7675–7687 Search PubMed.
- N. Shigi, Y. Sakaguchi, S. Asai, T. Suzuki and K. Watanabe, EMBO J., 2008, 27, 3267–3278 Search PubMed.
- A. J. Wittwer, L. Tsai, W. M. Ching and T. C. Stadtman, Biochemistry, 1984, 23, 4650–4655 Search PubMed.
- C. E. Dumelin, Y. Chen, A. M. Leconte, Y. G. Chen and D. R. Liu, Nat. Chem. Biol., 2012, 8, 913–919 Search PubMed.
- E. G. Mueller, Nat. Chem. Biol., 2006, 2, 185–194 Search PubMed.
- O. Bimai, S. Arragain and B. Golinelli-Pimpaneau, Curr. Opin. Struct. Biol., 2020, 65, 69–78 Search PubMed.
- M. Cavuzic and Y. Liu, Biomolecules, 2017, 7, 27 Search PubMed.
- P. F. Agris, EMBO Rep., 2008, 9, 629–635 Search PubMed.
- F. V. T. Murphy, V. Ramakrishnan, A. Malkiewicz and P. F. Agris, Nat. Struct. Mol. Biol., 2004, 11, 1186–1191 Search PubMed.
- P. F. Agris, Biochimie, 1991, 73, 1345–1349 Search PubMed.
- H. Tukenmez, H. Xu, A. Esberg and A. S. Bystrom, Nucleic Acids Res., 2015, 43, 9489–9499 Search PubMed.
- J. Urbonavicius, Q. Qian, J. M. Durand, T. G. Hagervall and G. R. Bjork, EMBO J., 2001, 20, 4863–4873 Search PubMed.
- U. L. RajBhandary, Nature, 1988, 336, 112–113 Search PubMed.
- S. Laxman, B. M. Sutter, X. Wu, S. Kumar, X. Guo, D. C. Trudgian, H. Mirzaei and B. P. Tu, Cell, 2013, 154, 416–429 Search PubMed.
- A. Zeharia, A. Shaag, O. Pappo, A. M. Mager-Heckel, A. Saada, M. Beinat, O. Karicheva, H. Mandel, N. Ofek, R. Segel, D. Marom, A. Rotig, I. Tarassov and O. Elpeleg, Am. J. Hum. Genet., 2009, 85, 401–407 Search PubMed.
- M. X. Guan, Q. Yan, X. Li, Y. Bykhovskaya, J. Gallo-Teran, P. Hajek, N. Umeda, H. Zhao, G. Garrido, E. Mengesha, T. Suzuki, I. del Castillo, J. L. Peters, R. Li, Y. Qian, X. Wang, E. Ballana, M. Shohat, J. Lu, X. Estivill, K. Watanabe and N. Fischel-Ghodsian, Am. J. Hum. Genet., 2006, 79, 291–302 Search PubMed.
- Y. Kirino, T. Yasukawa, S. Ohta, S. Akira, K. Ishihara, K. Watanabe and T. Suzuki, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15070–15075 Search PubMed.
- Y. Kirino and T. Suzuki, RNA Biol., 2005, 2, 41–44 Search PubMed.
- T. Yasukawa, T. Suzuki, N. Ishii, S. Ohta and K. Watanabe, EMBO J., 2001, 20, 4794–4802 Search PubMed.
- T. Yasukawa, Y. Kirino, N. Ishii, I. J. Holt, H. T. Jacobs, T. Makifuchi, N. Fukuhara, S. Ohta, T. Suzuki and K. Watanabe, FEBS Lett., 2005, 579, 2948–2952 Search PubMed.
- Y. Motorin and M. Helm, Biochemistry, 2010, 49, 4934–4944 Search PubMed.
- A. Favre, A. M. Michelson and M. Yaniv, J. Mol. Biol., 1971, 58, 367–379 Search PubMed.
- K. Watanabe, T. Oshima, M. Saneyoshi and S. Nishimura, FEBS Lett., 1974, 43, 59–63 Search PubMed.
- J. A. Kowalak, J. J. Dalluge, J. A. McCloskey and K. O. Stetter, Biochemistry, 1994, 33, 7869–7876 Search PubMed.
- N. Horie, M. Hara-Yokoyama, S. Yokoyama, K. Watanabe, Y. Kuchino, S. Nishimura and T. Miyazawa, Biochemistry, 1985, 24, 5711–5715 Search PubMed.
- R. K. Kumar and D. R. Davis, Nucleic Acids Res., 1997, 25, 1272–1280 Search PubMed.
- S. M. Testa, M. D. Disney, D. H. Turner and R. Kierzek, Biochemistry, 1999, 38, 16655–16662 Search PubMed.
- P. F. Agris, Nucleic Acids Res., 2004, 32, 223–238 Search PubMed.
- M. K. Kruger, S. Pedersen, T. G. Hagervall and M. A. Sorensen, J. Mol. Biol., 1998, 284, 621–631 Search PubMed.
- S. S. Ashraf, G. Ansari, R. Guenther, E. Sochacka, A. Malkiewicz and P. F. Agris, RNA, 1999, 5, 503–511 Search PubMed.
- C. Yarian, M. Marszalek, E. Sochacka, A. Malkiewicz, R. Guenther, A. Miskiewicz and P. F. Agris, Biochemistry, 2000, 39, 13390–13395 Search PubMed.
- M. D. Wolfe, F. Ahmed, G. M. Lacourciere, C. T. Lauhon, T. C. Stadtman and T. J. Larson, J. Biol. Chem., 2004, 279, 1801–1809 Search PubMed.
- G. Jager, P. Chen and G. R. Bjork, PLoS One, 2016, 11, e0153488 Search PubMed.
- D. Bordo and P. Bork, EMBO Rep., 2002, 3, 741–746 Search PubMed.
- R. Cipollone, P. Ascenzi and P. Visca, IUBMB Life, 2007, 59, 51–59 Search PubMed.
- J. E. Walker, M. Saraste, M. J. Runswick and N. J. Gay, EMBO J., 1982, 1, 945–951 Search PubMed.
- E. V. Koonin, J. Mol. Biol., 1993, 229, 1165–1174 Search PubMed.
- P. Bartos, A. Maciaszek, A. Rosinska, E. Sochacka and B. Nawrot, Bioorg. Chem., 2014, 56, 49–53 Search PubMed.
- P. Chen, P. F. Crain, S. J. Nasvall, S. C. Pomerantz and G. R. Bjork, EMBO J., 2005, 24, 1842–1851 Search PubMed.
- H. Sun, J. Sheng, A. E. Hassan, S. Jiang, J. Gan and Z. Huang, Nucleic Acids Res., 2012, 40, 5171–5179 Search PubMed.
- S. Nachtergaele and C. He, RNA Biol., 2017, 14, 156–163 Search PubMed.
- N. Jonkhout, J. Tran, M. A. Smith, N. Schonrock, J. S. Mattick and E. M. Novoa, RNA, 2017, 23, 1754–1769 Search PubMed.
- T. Tong, S. Chen, L. Wang, Y. Tang, J. Y. Ryu, S. Jiang, X. Wu, C. Chen, J. Luo, Z. Deng, Z. Li, S. Y. Lee and S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E2988–E2996 Search PubMed.
- L. Xiong, S. Liu, S. Chen, Y. Xiao, B. Zhu, Y. Gao, Y. Zhang, B. Chen, J. Luo, Z. Deng, X. Chen, L. Wang and S. Chen, Nat. Commun., 2019, 10, 1688 Search PubMed.
- L. Wang, S. Chen, K. L. Vergin, S. J. Giovannoni, S. W. Chan, M. S. DeMott, K. Taghizadeh, O. X. Cordero, M. Cutler, S. Timberlake, E. J. Alm, M. F. Polz, J. Pinhassi, Z. Deng and P. C. Dedon, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2963–2968 Search PubMed.
- Y. Wu, Y. Tang, X. Dong, Y. Y. Zheng, P. Haruehanroengra, S. Mao, Q. Lin and J. Sheng, ACS Chem. Biol., 2020, 15, 1301–1305 Search PubMed.
- S. Leimkuhler, M. Buhning and L. Beilschmidt, Biomolecules, 2017, 7, 5 Search PubMed.
- H. K. Lundgren, K. Nilsson, T. G. Hagervall and G. R. Björk, J. Bacteriol., 2002, 184, 6830–6835 Search PubMed.
- T. Numata, S. Fukai, Y. Ikeuchi, T. Suzuki and O. Nureki, Structure, 2006, 14, 357–366 Search PubMed.
- T. Numata, Y. Ikeuchi, S. Fukai, T. Suzuki and O. Nureki, Nature, 2006, 442, 419–424 Search PubMed.
- K. A. Black and P. C. Dos Santos, J. Bacteriol., 2015, 197, 1952–1962 Search PubMed.
- M. E. Armengod, I. Moukadiri, S. Prado, R. Ruiz-Partida, A. Benitez-Paez, M. Villarroya, R. Lomas, M. J. Garzon, A. Martinez-Zamora, S. Meseguer and C. Navarro-Gonzalez, Biochimie, 2012, 94, 1510–1520 Search PubMed.
- C. Yarian, H. Townsend, W. Czestkowski, E. Sochacka, A. J. Malkiewicz, R. Guenther, A. Miskiewicz and P. F. Agris, J. Biol. Chem., 2002, 277, 16391–16395 Search PubMed.
- H. V. Miranda, N. Nembhard, D. Su, N. Hepowit, D. J. Krause, J. R. Pritz, C. Phillips, D. Soll and J. A. Maupin-Furlow, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4417–4422 Search PubMed.
- Y. Nakai, M. Nakai, R. Lill, T. Suzuki and H. Hayashi, Mol. Cell. Biol., 2007, 27, 2841–2847 Search PubMed.
- A. Noma, Y. Sakaguchi and T. Suzuki, Nucleic Acids Res., 2009, 37, 1335–1352 Search PubMed.
- P. Yuvaniyama, J. N. Agar, V. L. Cash, M. K. Johnson and D. R. Dean, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 599–604 Search PubMed.
- C. J. Schwartz, O. Djaman, J. A. Imlay and P. J. Kiley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9009–9014 Search PubMed.
- N. Umeda, T. Suzuki, M. Yukawa, Y. Ohya, H. Shindo, K. Watanabe and T. Suzuki, J. Biol. Chem., 2005, 280, 1613–1624 Search PubMed.
- R. Kambampati and C. T. Lauhon, Biochemistry, 1999, 38, 16561–16568 Search PubMed.
- E. G. Mueller, C. J. Buck, P. M. Palenchar, L. E. Barnhart and J. L. Paulson, Nucleic Acids Res., 1998, 26, 2606–2610 Search PubMed.
- C. T. Lauhon and R. Kambampati, J. Biol. Chem., 2000, 275, 20096–20103 Search PubMed.
- D. H. Flint, Biol. Chem., 1996, 5, 16068–16074 Search PubMed.
- H. Mihara, T. Kurihara, T. Yoshimura and N. Esaki, J. Biochem., 2000, 127, 559–567 Search PubMed.
- R. Kambampati and C. T. Lauhon, Biochemistry, 2003, 42, 1109–1117 Search PubMed.
- Y. Liu, X. Zhu, A. Nakamura, R. Orlando, D. Soll and W. B. Whitman, J. Biol. Chem., 2012, 287, 36683–36692 Search PubMed.
- P. M. Palenchar, C. J. Buck, H. Cheng, T. J. Larson and E. G. Mueller, J. Biol. Chem., 2000, 275, 8283–8286 Search PubMed.
- C. Zheng, K. A. Black and P. C. Dos Santos, Biomolecules, 2017, 7, 33 Search PubMed.
- L. J. Rajakovich, J. Tomlinson and P. C. Dos Santos, J. Bacteriol., 2012, 194, 4933–4940 Search PubMed.
- Y. Liu, D. J. Vinyard, M. E. Reesbeck, T. Suzuki, K. Manakongtreecheep, P. L. Holland, G. W. Brudvig and D. Soll, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12703–12708 Search PubMed.
- L. Wang, S. Jiang, Z. Deng, P. C. Dedon and S. Chen, FEMS Microbiol. Rev., 2019, 43, 109–122 Search PubMed.
- H. Xu, Z. Li and J. Si, J. Biomed. Nanotechnol., 2014, 10, 3483–3507 Search PubMed.
- H. Wu, W. F. Lima and S. T. Crooke, J. Biol. Chem., 1999, 274, 28270–28278 Search PubMed.
- M. H. Ho, M. De Vivo, M. Dal Peraro and M. L. Klein, J. Am. Chem. Soc., 2010, 132, 13702–13712 Search PubMed.
- R. Kole, A. R. Krainer and S. Altman, Nat. Rev. Drug Discovery, 2012, 11, 125–140 Search PubMed.
- K. Kim, Y. S. Lee and R. W. Carthew, RNA, 2007, 13, 22–29 Search PubMed.
- J. Liu, M. A. Carmell, F. V. Rivas, C. G. Marsden, J. M. Thomson, J. J. Song, S. M. Hammond, L. Joshua-Tor and G. J. Hannon, Science, 2004, 305, 1437–1441 Search PubMed.
- S. Ni, Z. Zhuo, Y. Pan, Y. Yu, F. Li, J. Liu, L. Wang, X. Wu, D. Li, Y. Wan, L. Zhang, Z. Yang, B. T. Zhang, A. Lu and G. Zhang, ACS Appl. Mater. Interfaces, 2021, 13(8), 9500–9519 Search PubMed.
- A. A. Dominguez, W. A. Lim and L. S. Qi, Nat. Rev. Mol. Cell Biol., 2016, 17, 5–15 Search PubMed.
- R. Barrangou and L. A. Marraffini, Mol. Cell, 2014, 54, 234–244 Search PubMed.
- S. B. Moon, D. Y. Kim, J. H. Ko, J. S. Kim and Y. S. Kim, Trends Biotechnol., 2019, 37, 870–881 Search PubMed.
- J. K. Watts and D. R. Corey, J. Pathol., 2012, 226, 365–379 Search PubMed.
- M. Rahdar, M. A. McMahon, T. P. Prakash, E. E. Swayze, C. F. Bennett and D. W. Cleveland, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E7110–7117 Search PubMed.
- Q. Li, Yonsei Med. J., 2020, 61, 273–283 Search PubMed.
- V. Mathew and A. K. Wang, Drug Des., Dev. Ther., 2019, 13, 1515–1525 Search PubMed.
- E. W. Ng, D. T. Shima, P. Calias, E. T. Cunningham, Jr., D. R. Guyer and A. P. Adamis, Nat. Rev. Drug Discovery, 2006, 5, 123–132 Search PubMed.
- C. M. Perry and J. A. Balfour, Drugs, 1999, 57, 375–380 Search PubMed ; discussion 381.
- E. W. A. T. Goldberg, P & T, 2014, 39, 119–122 Search PubMed.
- K. R. Lim, R. Maruyama and T. Yokota, Drug Des., Dev. Ther., 2017, 11, 533–545 Search PubMed.
- S. M. Hoy, Drugs, 2018, 78, 1625–1631 Search PubMed.
- J. L. Witztum, D. Gaudet, S. D. Freedman, V. J. Alexander, A. Digenio, K. R. Williams, Q. Yang, S. G. Hughes, R. S. Geary, M. Arca, E. S. G. Stroes, J. Bergeron, H. Soran, F. Civeira, L. Hemphill, S. Tsimikas, D. J. Blom, L. O'Dea and E. Bruckert, N. Engl. J. Med., 2019, 381, 531–542 Search PubMed.
- Y. A. Heo, Drugs, 2020, 80, 329–333 Search PubMed.
- S. T. Crooke, P. P. Seth, T. A. Vickers and X. H. Liang, J. Am. Chem. Soc., 2020, 142, 14754–14771 Search PubMed.
- F. Eckstein, Nucleic Acid Ther., 2014, 24, 374–387 Search PubMed.
- C. M. Miller and E. N. Harris, RNA & disease, 2016, 3, e1393 Search PubMed.
- D. R. Scoles, E. V. Minikel and S. M. Pulst, Neurol. Genet., 2019, 5, e323 Search PubMed.
- M. Boczkowska, P. Guga and W. J. Stec, Biochemistry, 2002, 41, 12483–12487 Search PubMed.
- M. Koziolkiewicz, A. Krakowiak, M. Kwinkowski, M. Boczkowska and W. J. Stec, Nucleic Acids Res., 1995, 23, 5000–5005 Search PubMed.
- F. Eckstein, P. M. Burgers and D. H. Hunneman, J. Biol. Chem., 1979, 25, 7476–7478 Search PubMed.
- K. A. Lennox, R. Owczarzy, D. M. Thomas, J. A. Walder and M. A. Behlke, Mol. Ther.--Nucleic Acids, 2013, 2, e117 Search PubMed.
- N. Oka, M. Yamamoto, T. Sato and T. Wada, J. Am. Chem. Soc., 2008, 130, 16031–16037 Search PubMed.
- A. Ramanathan, G. B. Robb and S. H. Chan, Nucleic Acids Res., 2016, 44, 7511–7526 Search PubMed.
- A. N. Kuhn, M. Diken, S. Kreiter, A. Selmi, J. Kowalska, J. Jemielity, E. Darzynkiewicz, C. Huber, O. Tureci and U. Sahin, Gene Ther., 2010, 17, 961–971 Search PubMed.
- J. Kowalska, M. Lewdorowicz, J. Zuberek, E. Grudzien-Nogalska, E. Bojarska, J. Stepinski, R. E. Rhoads, E. Darzynkiewicz, R. E. Davis and J. Jemielity, RNA, 2008, 14, 1119–1131 Search PubMed.
- E. Grudzien-Nogalska, J. Jemielity, J. Kowalska, E. Darzynkiewicz and R. E. Rhoads, RNA, 2007, 13, 1745–1755 Search PubMed.
- M. Nowakowska, J. Kowalska, F. Martin, A. d'Orchymont, J. Zuberek, M. Lukaszewicz, E. Darzynkiewicz and J. Jemielity, Org. Biomol. Chem., 2014, 12, 4841–4847 Search PubMed.
- M. Strenkowska, R. Grzela, M. Majewski, K. Wnek, J. Kowalska, M. Lukaszewicz, J. Zuberek, E. Darzynkiewicz, A. N. Kuhn, U. Sahin and J. Jemielity, Nucleic Acids Res., 2016, 44, 9578–9590 Search PubMed.
- R. Rhoads, ASBMBTODAY, 2020.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |