
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and immunological effects of C14-linked 4,5-epoxymorphinan analogues as novel heroin vaccine haptens†
Eugene S.
Gutman
a,
Thomas C.
Irvin
a,
J. Brian
Morgan
a,
Rodell C.
Barrientos
 bc,
Oscar B.
Torres‡
bc,
Zoltan
Beck§
bc,
Gary R.
Matyas
b,
Arthur E.
Jacobson
*a and
Kenner C.
Rice
*a
bc,
Oscar B.
Torres‡
bc,
Zoltan
Beck§
bc,
Gary R.
Matyas
b,
Arthur E.
Jacobson
*a and
Kenner C.
Rice
*a
aDrug Design and Synthesis Section, Molecular Targets and Medications Discovery Branch, Intramural Research Program, National Institute on Drug Abuse and The National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Department of Health and Human Services, 9800 Medical Center Drive, Bethesda, MD 20892-3373, USA. E-mail: kennerr@nida.nih.gov; arthurj@nida.nih.gov; Tel: +1 301-451-4799 Tel: +1 301-451-5028
bLaboratory of Adjuvant and Antigen Research, U.S. Military HIV Research Program, Walter Reed Army Institute of Research, 503 Robert Grant Avenue, Silver Spring, MD 20910, USA
cHenry M. Jackson Foundation for the Advancement of Military Medicine, 6720A Rockledge Drive, Bethesda, MD 20817, USA
First published on 19th April 2021
Abstract
Active immunization is being explored as a potential therapeutic to combat accidental overdose and to mitigate the abuse potential of opioids. Hapten design is one of the crucial factors that determines the efficacy of a candidate vaccine to substance abuse and remains one of the most active areas of research in vaccine development. Herein we report for the first time the synthesis of three novel opiate surrogates with the linker attachment site at C14, 1 (6,14-AmidoHap), 2 (14-AmidoMorHap), and 3 (14-AmidoHerHap) as novel heroin haptens. The compounds 1, 2, and 3 are analogues with different substituents at C6: an acetamide, a hydroxyl moiety, and an acetate, respectively. All three haptens had a phenolic hydroxyl group at C3. The haptens were conjugated to the tetanus toxoid carrier protein, adjuvanted with liposomal monophosphoryl lipid A/aluminum hydroxide and were tested in mice in terms of immunogenicity and efficacy. Immunization of mice resulted in antibody endpoint titers of >105 against all the haptens. Neither of the conjugates of 1, 2, and 3 had induced antibodies with selectivity broad enough to recognize and bind heroin, 6-AM, and morphine resulting in little to no protection against the antinociceptive effects of heroin in vivo. Only the mice immunized with conjugate 3 were partially protected against heroin-induced antinociception. These results contribute to the growing body of knowledge that the linker position and the subtle structural differences in the hapten scaffold impact the selectivity of the induced antibodies. Together, these highlight the importance of rational hapten design for heroin vaccine development.
Introduction
Active immunization could serve as a potential therapeutic against opioid use disorders and unintentional overdose.1–4 Vaccines induce antibodies that function as a pharmacokinetic antagonist by sequestering the drugs in the blood, thus preventing their access to the brain and blunting their physiological effects.3 To induce an immune response, a surrogate of the target drug, also called a hapten, is synthesized and conjugated to an immunogenic carrier protein.3 The development of vaccines against opioids such as morphine,5 heroin,6–12 fentanyl,13–16 oxycodone,17 and related drugs have been reported.3Hapten design has been recognized as a crucial factor that impacts vaccine efficacy.3,18 It impacts the activation of hapten-specific B cells that can generate drug-specific humoral response through a T cell-mediated process.19,20 What remains unknown is the optimum hapten design that can induce polyclonal response to not only to a target drug but also its structural analogs. Heroin quickly hydrolyses to 6-acetylmorphine (6-AM) and morphine through the action of esterases (Fig. 1A).11,18 The hydrolysis product 6-AM, and to some extent, morphine, mediate the euphoric effects of heroin.21,22 As such, hapten design is particularly important in heroin vaccine development because not only heroin but also its bioactive metabolites 6-AM and morphine should be recognized and sequestered by the vaccine-induced antibodies. Importantly, antibodies should not cross-react to therapeutic drugs such as buprenorphine, methadone, naloxone, and naltrexone. Immunogens used to raise antibodies to opioids have the linker typically attached at the C2, C3, C6, or N17 positions of the morphinan scaffold (Fig. 1A).11 The facial recognition hypothesis by Matyas et al.,7 suggested that antibodies are generated against the hapten ‘face’ that is exposed during presentation to the immune system. The linker attachment site determines which part of the hapten can be effectively exposed. Early works have suggested that haptens with linker at N17 and carry specific functionality at the C3 and C6 elicit highly selective antibodies against drugs with matched substituents at the indicated positions.23,24 For example, by designing a hapten with appropriate functional groups at C3 and C6, e.g. hydroxyl groups, antibodies more selective to morphine with minimal cross-reactivity to 6-AM or heroin could be induced. Ideally, for a practical heroin vaccine, only one version of the hapten should suffice to induce antibodies effective against heroin and its metabolites. We have interrogated many heroin haptens with distinct functional groups and linker positions and demonstrated that haptens with different linker sites impacted vaccine efficacy.3,11,18 More recent works have shown that linker sites at C3 and C6 also yield antibodies selective to target drugs that translate to protection against heroin-induced physiological effects.3,8,17 With a few on-going efforts to translate experimental opioid vaccines to humans,25 there is a need to study many variants of a given hapten to fully understand how the molecular scaffold ultimately impacts the selectivity of the induced antibodies, which in turn dictate the vaccine efficacy in vivo.
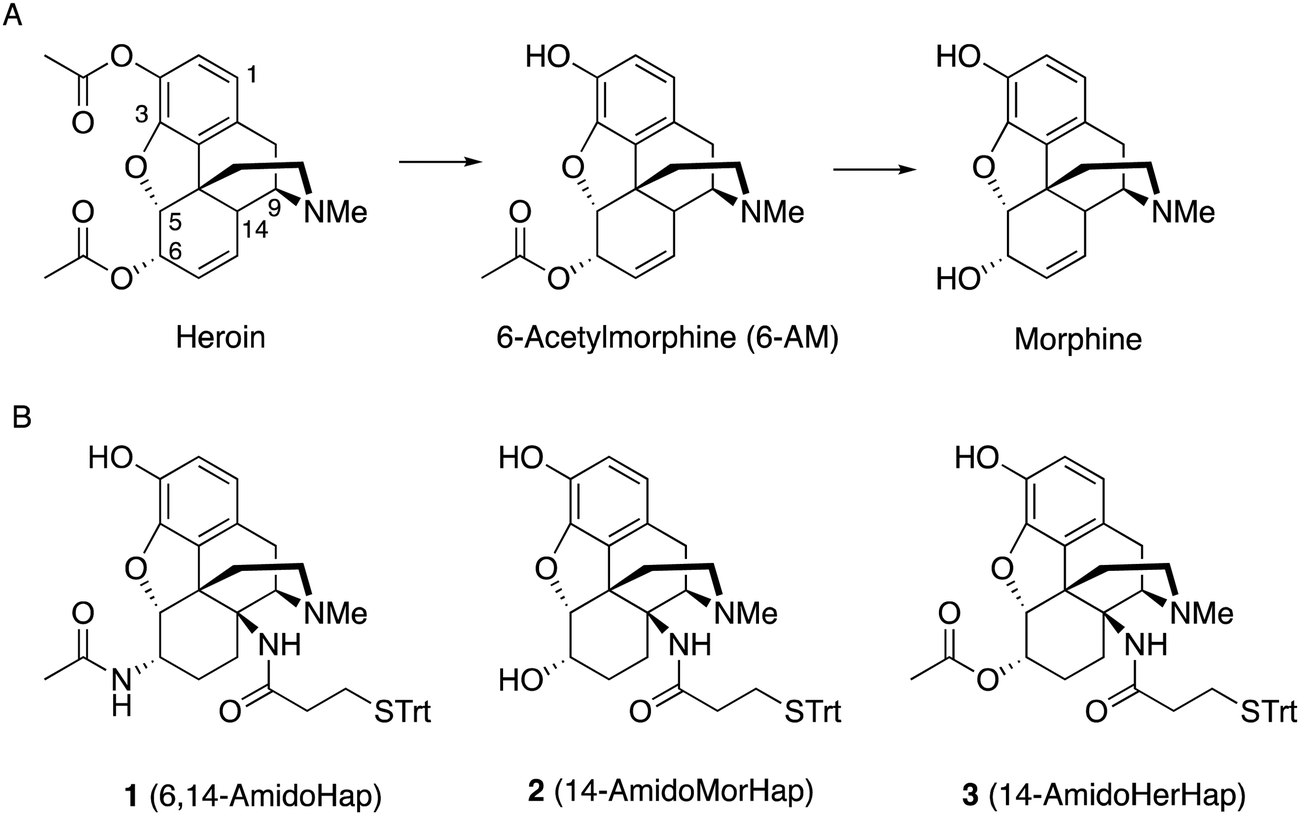 | ||
| Fig. 1 Heroin hydrolysis and structures of the haptens described in this study. (A) Hydrolysis of heroin. (B) Structures of haptens 1, 2, and 3. | ||
For the first time, we report in this study the synthesis of epoxymorphinan analogues with the linker attachment site at C14 (Fig. 1). Our rationale for choosing C14 as the attachment site for the linker was to continue our work examining the facial selectivity7 presented by different sites of attachment in terms of producing the optimum immune response. There are about 17 sites (depending on the compound) for possible linker attachment on the morphinan ring system and it was not obvious which site would be optimal. The C14 position was reasonably synthetically accessible in an area of the molecule not yet investigated by us or anyone else in terms of hapten design. We were able to design a relatively short synthesis that allowed us to obtain sufficient quantities for in vivo work.
These haptens differ in functionality at the C6 position, namely, an acetamide, a hydroxyl moiety, and an acetate for haptens 1, 2, and 3, respectively. We conjugated the haptens to tetanus toxoid (TT) carrier protein through the thiol-maleimide reaction using our established procedure.8,9 The conjugates were formulated into a vaccine adjuvanted with Army Liposome Formulation adsorbed to aluminum salt (ALF43A).26,27 The immunogenicity and efficacy of the vaccines were tested in mice. Results suggested that attachment of linker at the C14 position is plausible and that modifications at the C6 position result in the variation of the selectivity of the induced antibodies. In vivo data indicated that the C14-linked haptens did not provide vaccines that were an improvement over our previously reported metabolically stable hapten linked at C3.8
Results and discussion
Synthesis of hapten 1
Starting from thebaine, the first step was to introduce the C14 amine (Scheme 1). Using the method of Kirby and McLean,28 trichloroethyl N-hydroxycarbamate was oxidized with sodium periodate in the presence of thebaine. The in situ formed acyl nitroso intermediate efficiently underwent 4+2 cycloaddition with the diene moiety found in thebaine to give cycloadduct 4 and introduce the desired C–N bond at C14. Pd-catalyzed hydrogenation reduced the C7–C8 olefin and the carbamate was cleaved in the reaction to give desired 14β–aminodihydrocodeinone along with a small inseparable impurity resulting from the cleavage of the C5-oxide bridge. Previous work had suggested that performing the reduction in 10–20% acetic acid would prevent the C5 cleavage; however, when the reduction was repeated with either 10 or 20% acetic acid as the solvent, only the C5 cleavage product was obtained. The crude mixture was carried through to the Boc protection step and the desired product 5 and the C5 cleavage product were separated at this stage. Reductive amination with Ti(O–iPr)4, NH3 and NaBH4 cleanly afforded the C6-amine29 and conversion to acetamide 6 was straightforward. O-Demethylation was achieved upon treatment with BBr3 and the linker was attached with a TBTU-promoted coupling to give hapten 1.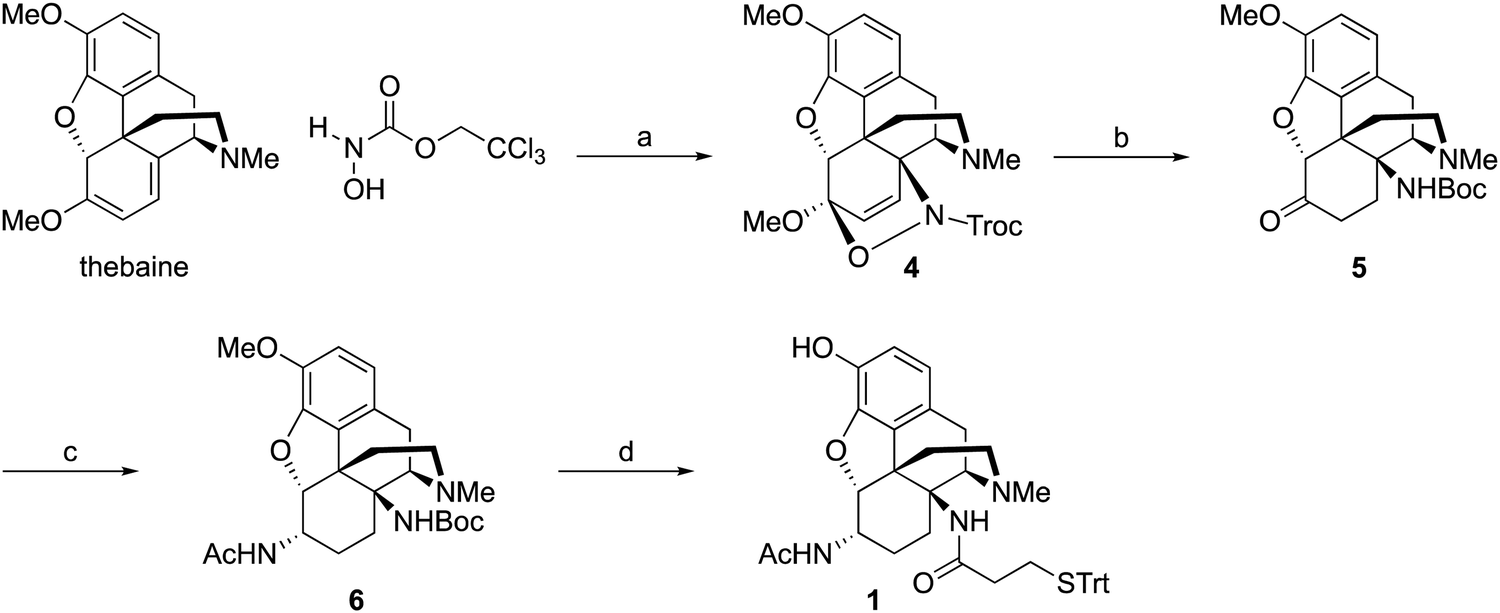 | ||
| Scheme 1 Synthesis of hapten 1. Reagent and conditions: (a) NalO4, aq. NaOAc/EtOAc, 23 °C, 96%. (b) (i) Pd/C, H2, AcOH/NaOAc/MeOH, 23 °C. (ii) Boc2O, NaHCO3, THF/H2O, 0 to 23 °C, 53%. (c) (i) NH3, Ti(OiPr)4, CH2Cl2, 0 to 23 °C. (ii) NaBH4, CH2Cl2, 23 °C. (iii) Ac2O, Et3N, CH2Cl2, 23 °C, 81%. (d) (i) BBr3, CH2Cl2, −78 to 23 °C. (ii) 3-(tritylthio)propanoic acid, TBTU, Et3N, 0 to 23 °C, 29%. | ||
As mentioned earlier, hydrogenolysis of 4 in the presence of 10% Pd/C resulted in significant amount C5 ring cleavage and was difficult to reproduce. Kirby and co-workers have observed similar ring opened side-products when attempting hydrogenation of 4.25 This issue was circumvented by first protecting the C6 position as the ketal followed by cleavage of the N–O bond and removal of the Troc group with zinc dust. Hydrolysis of the ketal group after reduction was accomplished utilizing concentrated HCl, affording 7 in 47% over three steps. 7 was selectively reduced with NaBH4 in methanol affording 8.27 Hydrogenation of 8 utilizing 10% Pd/C afforded 9 in good yield and no C5 ring cleavage was observed. Demethylation of 9 with BBr3 proceeded smoothly resulting in 10. TBTU mediated coupling of 10 to 3-(tritylthio)propanoic acid proved problematic. Sparse amounts of desired hapten 2 were initially isolated, the bulk of the material going towards over-coupled C3 phenolic ester. Fortunately, hydrolysis of the C3 phenolic ester with refluxing KOH in ethanol resulted in the formation of hapten 2 in moderate yield. Treatment of 2 with acetic anhydride and catalytic DMAP resulted in acylation at both C3 and C6 positions. Hydrolysis of the C3-acetyl with aqueous ammonium hydroxide gave desired hapten 3 in 39% yield (Scheme 2).
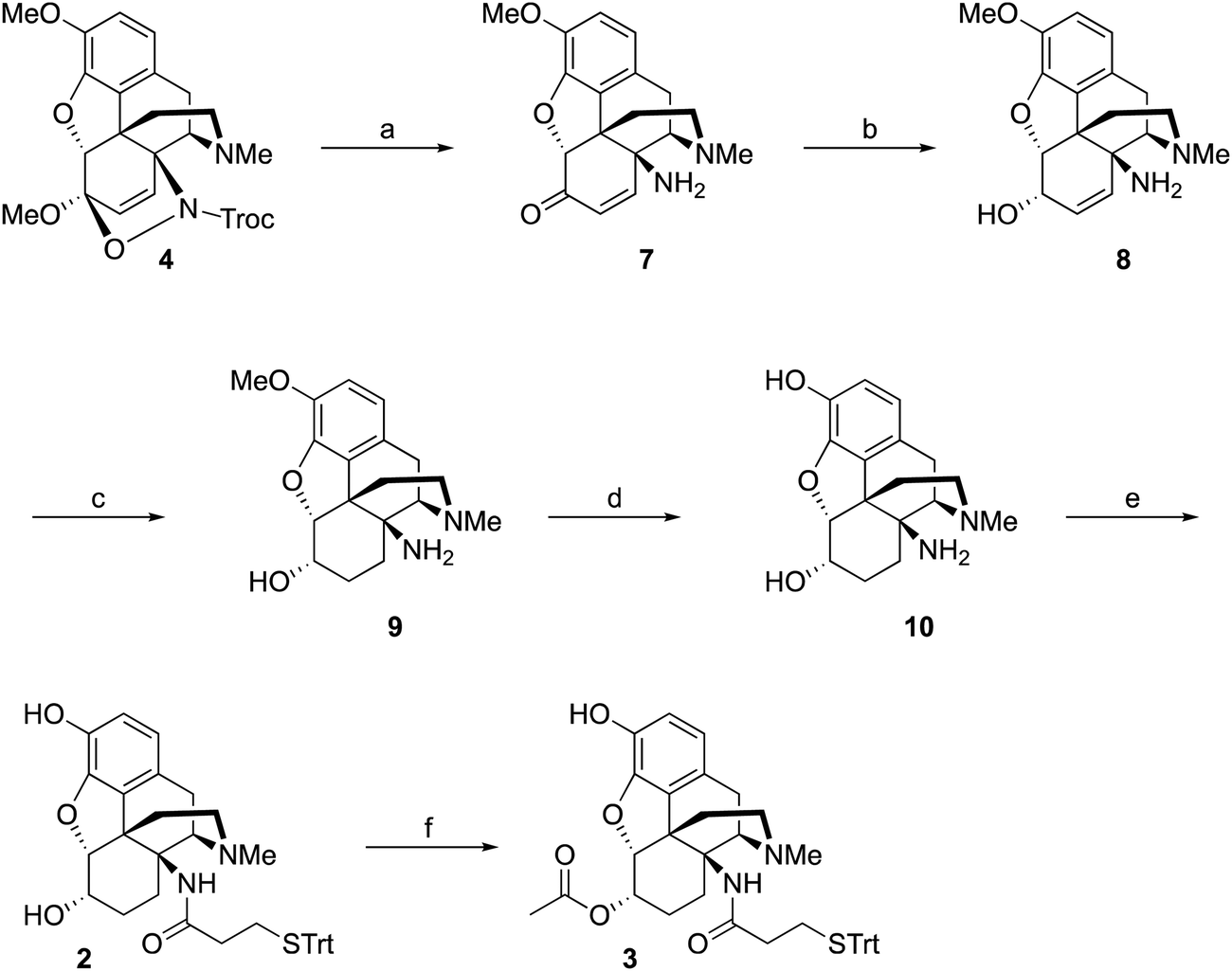 | ||
| Scheme 2 Synthesis of 2 and 3. Reagent and conditions: (a) (i) CSA, ethylene glycol, 23 °C. (ii) Zn, (NH4)2CO, 100 °C. (iii) HCl, MeOH/H2O, 80 °C, 47%. (b) NaBH4, MeOH, 23 °C, quant. (c) Pd/C, H2, MeOH, 23 °C, 81%. (d) BBr3, CH2Cl2, −78 °C, 84%. (e) 3-(tritylthio)propanoic acid, TBTU, Et3N, DMF, 0 °C. (ii) KOH, EtOH/THF, 80 °C, 45%. (f) Ac2O, DMAP, Et3N, CH2Cl2, 0°C, 39%. | ||
Biological results
Immune response to haptens
The haptens were conjugated to TT carrier protein to obtain the following respective vaccine immunoconjugates: TT-1, TT-2, and TT-3. Briefly, the carrier protein was reacted with SM(PEG)2, a heterobifunctional linker that carries an NHS ester on one end and a maleimide on the other. The primary surface amines reacted with the NHS end of the SM(PEG)2 to form the TT-maleimide. Finally, the hapten was conjugated to the TT-linker via the thiol–maleimide reaction (Fig. S1, ESI†).18 Successful conjugation was confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) (Fig. S2, ESI†). Hapten density was calculated by mass difference between unconjugated TT and conjugate. Hapten density ranged from ∼29–31 per carrier protein molecule for all the conjugates. Vaccines were formulated by mixing immunoconjugates (10 μg/50 μL dose) with unilamellar ALF43 and aluminum hydroxide (ALF43A).8,26 Immunization of mice resulted in a temporal increase in the anti-hapten IgG endpoint titers as measured by ELISA (Fig. 2). These results showed that these conjugates were immunogenic in mice. | ||
| Fig. 2 Time course of immune response to haptens 1, 2, and 3. Female BALB/c mice (n = 10 per group) were immunized at weeks 0, 3, 6, and 14 and sera from indicated weeks were collected for IgG endpoint titer measurements. The coating agents used for ELISA measurements were BSA conjugates of the appropriate hapten. | ||
Inhibition of heroin-induced antinociception
The vaccine efficacy was evaluated by heroin challenge experiments in mice using the tail immersion test. This test serves as one of the standard behavioral assays in evaluating the nociceptive response.13,30,31 On week 16, mice were challenged subcutaneously with 1.0 mg kg−1 heroin. Results indicated that mice immunized with TT-1 did not differ significantly with that of naïve, unimmunized mice (Fig. 3). Mice immunized with TT-1 had partial (∼60% MPE) but not significant protection (p = 0.1362). Mice immunized with TT-3 had modest (∼30% MPE) but significant (p = 0.0010) protection compared with unimmunized mice. Taken together, these results indicate that immunization using TT-3 could inhibit the antinociceptive effects of heroin in mice.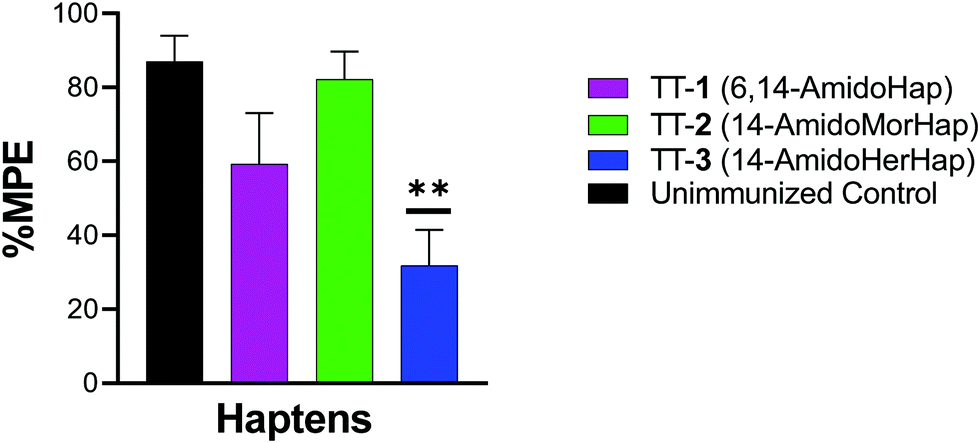 | ||
| Fig. 3 Inhibition of antinociceptive effects of heroin in vivo. Female BALB/c mice (n = 10 per group) were immunized on weeks 0, 3, 6, and 14, and challenged with heroin (1 mg kg−1) s.c. on week 16. Inhibition of antinociception was tested 15 min post-heroin injection using tail immersion and the results were reported as % MPE. Data shown are mean ± std error of the mean. Comparisons were made using ordinary one-way ANOVA and significance was defined as p < 0.05. ** p = 0.0010. | ||
Serum binding to heroin and metabolites
The overarching goal of a heroin vaccine is to induce antibodies that could potentially sequester heroin and its bioactive metabolites in the blood to prevent or slow down their access to the brain.1,3,4 To rationalize the observed in vivo data, we evaluated the potential of mice sera to sequester heroin, 6-acetyl-morphine (6-AM), and morphine. Sera collected at week 14 from immunized and unimmunized mice were used. Results showed that none of the sera from mice immunized with TT-1, TT-2, and TT-3 have significantly sequestered heroin in vitro (Fig. 4A). Sera from mice immunized with TT-1 and TT-3, but not TT-2 have significantly sequestered 6-AM (Fig. 4B). Only the sera from mice immunized with TT-2 have significantly sequestered morphine (Fig. 4C). Morphine is assumed to contribute little to the early effects of heroin.21 The inability of antibodies to block heroin and 6-AM could explain why TT-2 did not protect animals against the antinociceptive effects of heroin (Fig. 3). 6-AM has been found to drive behavioral effects in mice after heroin administration.21,22 Although TT-1 also bound 6-AM, the sequestration is weaker compared with TT-3, which may explain the partial but not significant protection observed in the antinociceptive assay (Fig. 2). These data suggest that antibodies induced by TT-1, TT-2, and TT-3 had distinct and different selectivity towards the target drugs. In contrast, sera from mice immunized with the TT-6-AmHap candidate vaccine effectively bound heroin, 6-AM and morphine.8,32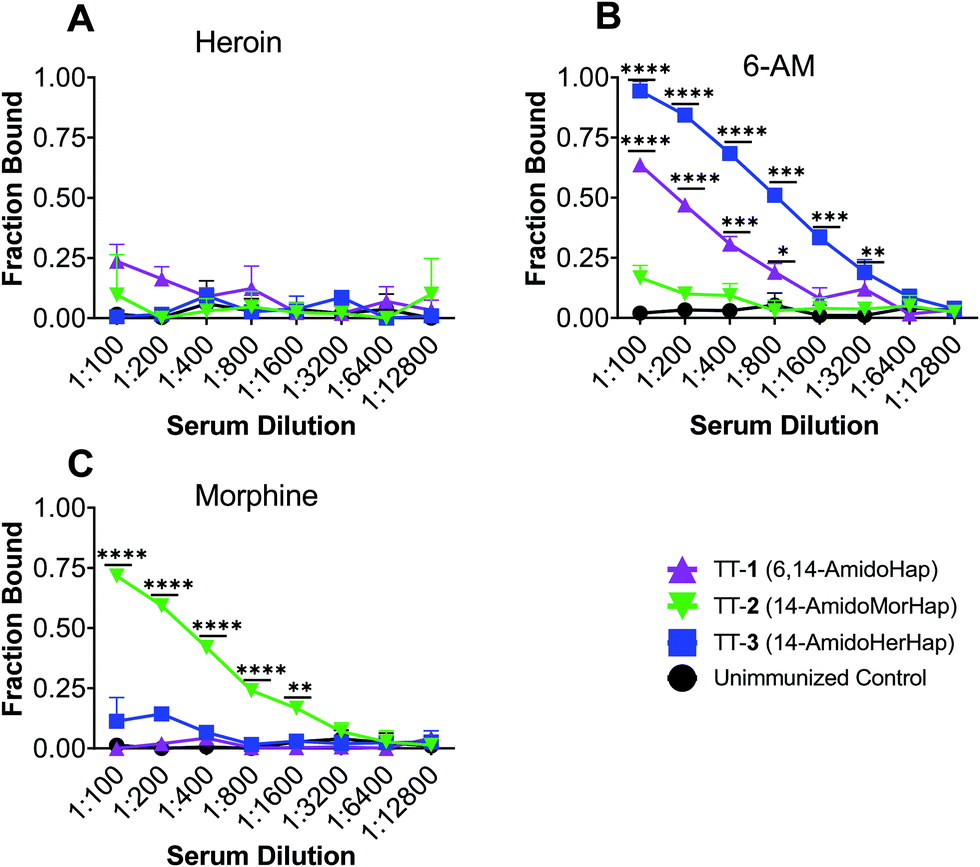 | ||
| Fig. 4 Heroin and metabolite sequestration of sera from mice immunized with TT-hapten conjugates. Female BALB/c mice (n = 10 per group) were immunized at week 0, 3, 6 and 14 and sera from week 14 were pooled for in vitro drug sequestration experiments. Pooled sera were diluted with equilibrium dialysis (ED) buffer containing the indicated drug at 5 nM, incubated in an ED plate against ED buffer. The concentration of the drug post-equilibrium was measured by LC-MS/MS, and fraction bound was computed. Data shown are mean ± std dev. of triplicate determinations. Comparison was made against unimmunized control sera using a two-tailed, unpaired t-test. Statistical significance was defined as p < 0.05 (****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05). (A) Heroin. (B) 6-Acetylmorphine (6-AM). (C) Morphine. | ||
Antibody affinity is an important determinant of vaccine efficacy.3 We measured the average antibody–drug dissociation constant (Kd) of the sera that bound a given drug. The sera from mice immunized with TT-1 and TT-3 had Kd values of 5.74 ± 0.52 nM and 3.12 ± 0.32 nM, respectively, against 6-AM. This apparent difference in Kd values is consistent with the in vivo data (vide supra). The sera from mice immunized with TT-2 had a Kd value of 2.89 ± 0.60 nM against morphine. The Kd values for heroin could not be calculated since none of the sera bound heroin. These nanomolar Kd suggest strong antibody–drug interaction comparable to antibodies induced by 6-AmHap conjugated to TT that ranged from 0.44 to 1.60 nM.33
Hapten design dictates the selectivity of the induced antibodies.7,11,23,24 Previous works have shown that subtle modifications at the C3 and C6 positions could have a large effect on the resulting vaccine efficacy. In this study, we showed that by changing the substituent at the C6 position, we effectively altered the selectivity of the induced antibodies. Specifically, TT-1 and TT-3, where C6 has been substituted by an acetate group and an amide group, respectively, induced antibodies that effectively sequestered 6-AM. On the other hand, when the C6 position was substituted with a hydroxy group, as in TT-2, no sequestration of 6-AM was observed. Importantly, none of these conjugates sequestered heroin. Although hapten 3 is susceptible to in vivo hydrolysis, to form hapten 2, antibodies induced by TT-3 did not bind morphine suggesting that hydrolysis at the C6 position to afford the formation of 2 is in accord with the slower rate of heroin hydrolysis at C6. With heroin, dissociation at C3 occurs more rapidly than the hydrolysis at C6.11
The observed serum binding can be rationalized in terms of the space-filling models of the drugs and the haptens (Fig. 5). Shown in Fig. 5 are the minimized structures of the opioids and haptens. At the molecular level, it can be envisioned that the portion of the molecule distal from the linker is the face that is effectively presented to the immune system for recognition. These faces have been marked by letters A and B and are emphasized by a blue arc. These structures suggest that heroin, having two acetyl groups at C3 and C6 positions, has a distinct structure compared to any of the haptens. Specifically, the haptens 1, 2, and 3 have a hydroxy substituent at the C3 position suggesting that these haptens might not induce antibodies that recognize heroin: this is consistent with the binding data (Fig. 4A). Similarly, the epitopes presented from TT-1 and TT-3 resemble that of 6-AM, as such, these conjugates induced antibodies that sequestered 6-AM (Fig. 4B). Of note, TT-1 had an acetamide group at the C6 position, while TT-3 had an acetate substituent. This single-atom difference (nitrogen vs. oxygen) of the C6 substituent resulted in significantly different binding to 6-AM (Fig. 4B). One possible explanation for this is that despite the overall similarity of the molecular geometry of 1 and 3, the oxygen atom is more electronegative than nitrogen, which may result in a stronger hydrogen bonding interaction during antigen presentation causing more effective recognition by the B cells; such a theory remains to be further investigated. A similar observation was noted before where a single atom difference in the heroin vaccine design yielded a significantly different affinity of the induced antibodies.34 On the other hand, because hapten 2 has a distinct substituent at C6, a hydroxy, sera from mice immunized with TT-2 could not recognize and bind 6-AM. Taken together, these observations agree with previous works3,11,24,34 that subtle differences in hapten structure could result in varying selectivity of the induced polyclonal sera.
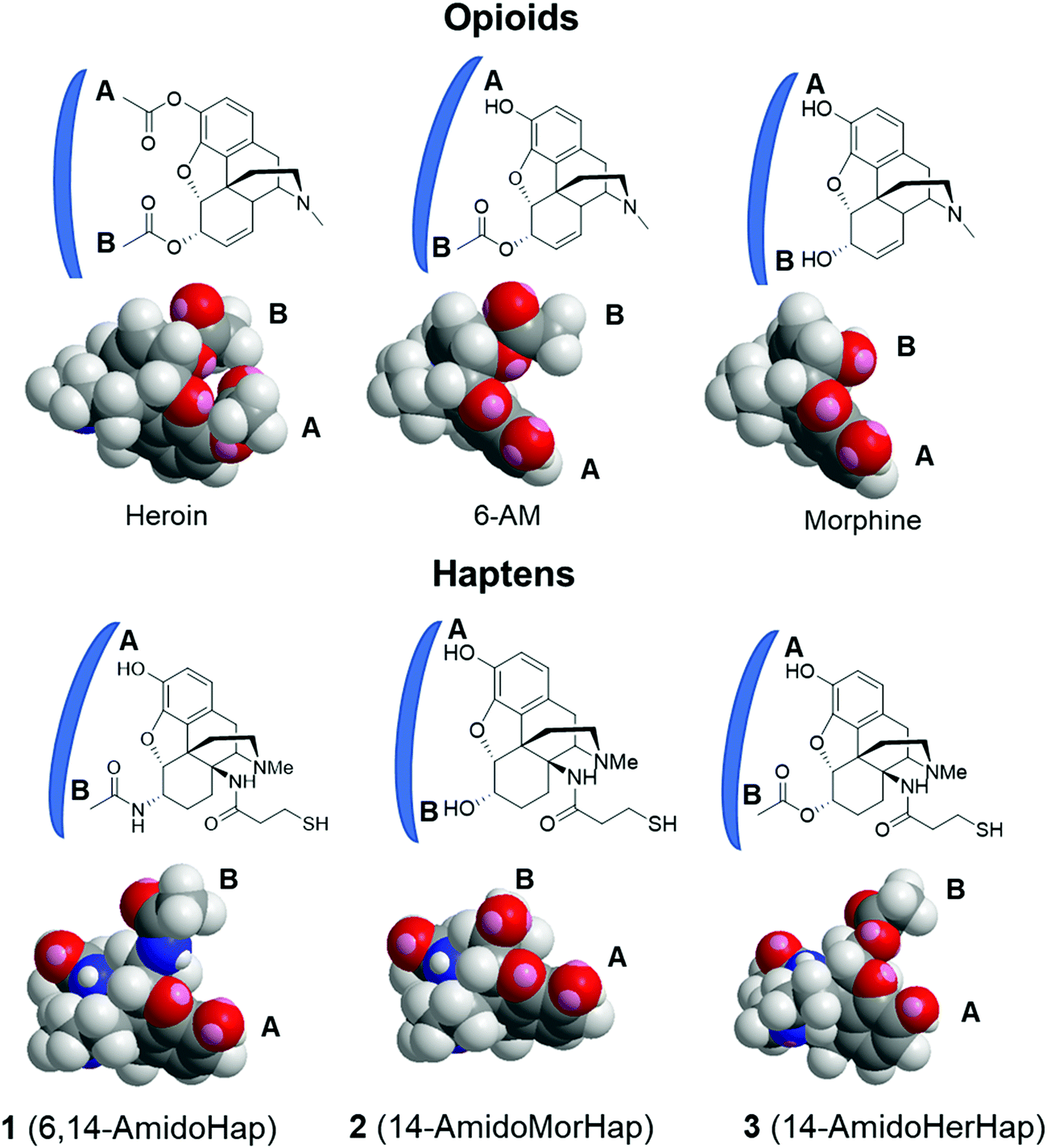 | ||
| Fig. 5 Space-filling models of opioids and haptens. Structures shown were minimized using the built-in MM2 method in ChemDraw 19.1. Postulated regions of the molecule recognized by the antibodies are labeled A and B and marked by a blue arc. | ||
Serum binding to therapeutic drugs
Antibodies induced by vaccines against opioids should not cross-react with drugs used for the management of opioid use disorders. The TT-3 conjugate attenuated the antinociceptive effects of heroin, which could be attributed to its ability to sequester 6-AM, suggesting the potential of this conjugate for further studies. We thus evaluated the cross-reactivity of sera from mice immunized with TT-3 against buprenorphine, methadone, naloxone, and naltrexone. Results of ED-LC-MS/MS revealed that no binding to any of the therapeutic drugs tested was observed in the week 14 sera at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 to 1:800 dilutions (Fig. 6). At these dilutions, significant binding was observed for 6-AM or morphine (Fig. 4). The space-filling models of these drugs (Fig. S3, ESI†) suggest that subtle differences in the C6 and N17 positions significantly change the 3D orientation these drugs in reference to 6-AM, effectively deviating from the predicted sites where the anti-3 could bind to. These results corroborate with previous findings using analogous but structurally distinct haptens that sera from immunized animals do not cross-react with therapeutic drugs.7,8
:100 to 1:800 dilutions (Fig. 6). At these dilutions, significant binding was observed for 6-AM or morphine (Fig. 4). The space-filling models of these drugs (Fig. S3, ESI†) suggest that subtle differences in the C6 and N17 positions significantly change the 3D orientation these drugs in reference to 6-AM, effectively deviating from the predicted sites where the anti-3 could bind to. These results corroborate with previous findings using analogous but structurally distinct haptens that sera from immunized animals do not cross-react with therapeutic drugs.7,8
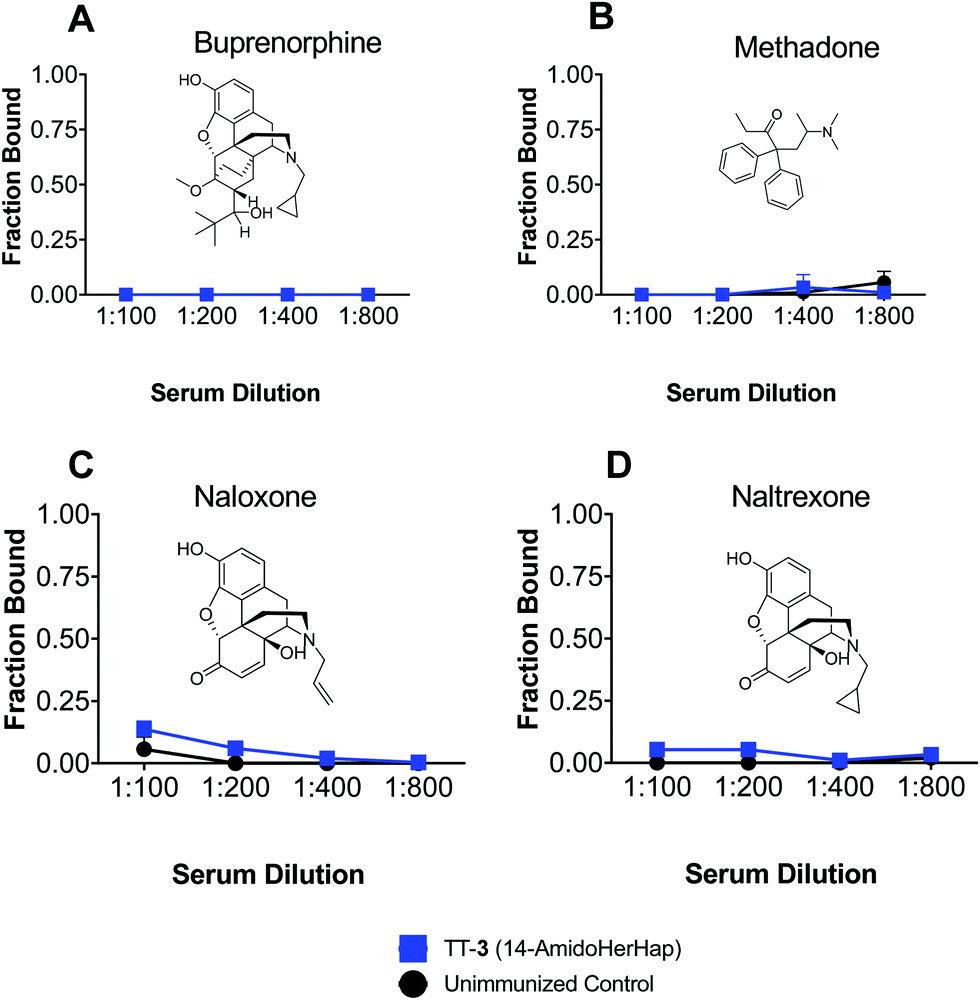 | ||
| Fig. 6 Therapeutic drug sequestration of sera from mice immunized with TT-3 conjugate. Female BALB/c mice (n = 10 per group) were immunized at weeks 0, 3, 6 and 14 and sera from week 14 were pooled for in vitro drug sequestration experiments. Pooled sera were diluted with ED buffer containing the indicated drug, incubated in an ED plate against ED buffer. The concentration of the drug post-equilibrium was measured by LC-MS/MS and fraction bound was computed. Data shown are mean ± std. dev. of triplicate determinations. (A) Buprenorphine. (B) Methadone. (C) Naloxone. (D) Naltrexone. | ||
Comparison with other heroin haptens
Many heroin surrogates have been reported and tested for immunogenicity and efficacy.3,11,18 Early works11,24 have suggested that the linker site at N17 could induce more selective antibodies. Janda's group has reported the use of a dynamic hapten with acetate groups at C3 and C6 with linker at N17 mimicking the intact heroin scaffold.11,35 This dynamic hapten degrades in vivo to generate a hydroxyl group at the C3 position that generated antibodies that could sequester heroin and 6-AM with no cross-reactivity to morphine.11,34–36 Pravetoni's group used a morphine analog with C6 as a linker attachment site5 analogous to the hapten design by Anton and Leff.12 Both Janda's (N17 linked) and Pravetoni's (C6 linked) haptens have been shown to elicit protection against heroin-induced effects.11,35,37 Our previous hapten designs had the linker attached at C3, C6, and N17.6–9,38 This is the first report of heroin haptens with linker attachment site at C14.The immunological effects, in terms of vaccine efficacy, of these C14-linked haptens are inferior to our previously reported haptens. The following haptens showed protective effects against heroin-induced effects: 6-PrOxyHap (C3-linked), MorHap (C6-linked), and DiAmHap (N17 linked).6,7,9,18 DiPrOxyHap (N17 linked) did not elicit protective effects in vivo.6 DiAmHap carries acetamide groups at C3 and C6, while DiProxyHap has oxopropyl groups at these sites. Despite both linked at N17, DiAmHap and DiPrOxyHap showed opposing effects, suggesting that the functional groups at C3 and C6 positions are also important. Serum binding and ELISA experiments performed on animal sera immunized with these protein-hapten conjugates revealed varying selectivity. Linker attachment at C14 did not induce antibodies with broad enough selectivity to simultaneously recognize and sequester heroin, 6-AM and morphine. It has been reported that a hydrolytically inactive acetamide at C3 induced high selectivity to heroin but not to 6-AM.34 Conversely, a hydrolytically active acetate group at C3 induced antibodies that are selective to both heroin and 6-AM.34 The haptens reported in this study had no hydrolyzable group at C3 position which may explain why no cross-reactivity to heroin was observed. These findings reinforced the importance of fine-tuning the hapten structure to elicit antibodies with the desired selectivity to the target drugs.
Author contributions
Dr Eugene S. Gutman: investigation, writing – original draft, writing – review and editing. Dr Thomas C. Irvin: investigation, writing – original draft. Dr J. Brian Morgan: investigation. Dr Rodell C. Barrientos: investigation, writing – original draft, writing – review and editing, project administration. Dr Oscar B. Torres: investigation, writing – review and editing. Dr Zoltan Beck: investigation, writing – review and editing. Dr Gary R. Matyas: conceptualization, data curation, project administration, funding acquisition, methodology, supervision. Dr Arthur E. Jacobson: conceptualization, writing – original draft, writing – review and editing, project administration. Dr Kenner C. Rice: conceptualization, writing – review and editing, supervision, funding acquisition, methodology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported in this publication was supported by the National Institute on Drug Abuse (NIDA) of the National Institutes of Health under Award Number UG3DA048351 and by an Avant-Garde award to GRM from NIDA (NIH grant no. 1DP1DA034787-01). The work of GRM, ZB, OBT, and RCB was supported through a Cooperative Agreement Award (no. W81XWH-07-2-067) between the Henry M. Jackson Foundation for the Advancement of Military Medicine and the U.S. Army Medical Research and Development Command (MRDC). The work of ESG, JBM, TCI, AEJ and KCR was supported by the NIH Intramural Research Program (IRP) of the National Institute on Drug Abuse and the National Institute of Alcohol Abuse and Alcoholism. This material has been reviewed by the Walter Reed Army Institute of Research and the NIDA. There is no objection to its presentation and/or publication. The views expressed are those of the authors and should not be construed to represent the positions of the U.S. Army, the Department of Defense, HJF, NIDA, NIH or the US government. Research was conducted under an approved animal use protocol in an AAALACi accredited facility in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, NRC Publication, 2011 edition. The authors thank Taeshia Washington and Alexander Duval for their outstanding technical assistance.References
- P. Skolnick, Annu. Rev. Pharmacol. Toxicol., 2018, 58, 143–159 CrossRef CAS PubMed.
- M. E. Olson and K. D. Janda, EMBO Rep., 2018, 19, 5–9 CrossRef CAS PubMed.
- P. T. Bremer and K. D. Janda, Pharmacol. Rev., 2017, 69, 298–315 CrossRef CAS PubMed.
- C. Baehr and M. Pravetoni, Neuropsychopharmacology, 2019, 44, 217–218 CrossRef PubMed.
- M. Pravetoni, M. D. Raleigh, M. Le Naour, A. M. Tucker, T. M. Harmon, J. M. Jones, A. K. Birnbaum, P. S. Portoghese and P. R. Pentel, Vaccine, 2012, 30, 4617–4624 CrossRef CAS PubMed.
- F. Li, K. Cheng, J. F. Antoline, M. R. Iyer, G. R. Matyas, O. B. Torres, R. Jalah, Z. Beck, C. R. Alving, D. A. Parrish, J. R. Deschamps, A. E. Jacobson and K. C. Rice, Org. Biomol. Chem., 2014, 12, 7211–7232 RSC.
- G. R. Matyas, K. C. Rice, K. Cheng, F. Li, J. F. Antoline, M. R. Iyer, A. E. Jacobson, A. V. Mayorov, Z. Beck, O. B. Torres and C. R. Alving, Vaccine, 2014, 32, 1473–1479 CrossRef CAS PubMed.
- A. Sulima, R. Jalah, J. F. G. Antoline, O. B. Torres, G. H. Imler, J. R. Deschamps, Z. Beck, C. R. Alving, A. E. Jacobson, K. C. Rice and G. R. Matyas, J. Med. Chem., 2018, 61, 329–343 CrossRef CAS PubMed.
- R. Jalah, O. B. Torres, A. V. Mayorov, F. Li, J. F. Antoline, A. E. Jacobson, K. C. Rice, J. R. Deschamps, Z. Beck, C. R. Alving and G. R. Matyas, Bioconjugate Chem., 2015, 26, 1041–1053 CrossRef CAS PubMed.
- C. S. Hwang, P. T. Bremer, C. J. Wenthur, S. O. Ho, S. Chiang, B. Ellis, B. Zhou, G. Fujii and K. D. Janda, Mol. Pharming, 2018, 15, 1062–1072 CrossRef CAS PubMed.
- G. N. Stowe, J. E. Schlosburg, L. F. Vendruscolo, S. Edwards, K. K. Misra, G. Schulteis, J. S. Zakhari, G. F. Koob and K. D. Janda, CNS Neurol. Disord.: Drug Targets, 2011, 10, 865–875 CrossRef CAS PubMed.
- B. Anton and P. Leff, Vaccine, 2006, 24, 3232–3240 CrossRef CAS PubMed.
- R. C. Barrientos, E. W. Bow, C. Whalen, O. B. Torres, A. Sulima, Z. Beck, A. E. Jacobson, K. C. Rice and G. R. Matyas, Mol. Pharmaceutics, 2020, 17, 3447–3460 CrossRef CAS PubMed.
- P. T. Bremer, A. Kimishima, J. E. Schlosburg, B. Zhou, K. C. Collins and K. D. Janda, Angew. Chem., Int. Ed., 2016, 55, 3772–3775 CrossRef CAS PubMed.
- M. D. Raleigh, F. Baruffaldi, S. J. Peterson, M. Le Naour, T. M. Harmon, J. R. Vigliaturo, P. R. Pentel and M. Pravetoni, J. Pharmacol. Exp. Ther., 2019, 368, 282–291 CrossRef CAS PubMed.
- C. Robinson, V. Gradinati, F. Hamid, C. Baehr, B. Crouse, S. Averick, M. Kovaliov, D. Harris, S. Runyon, F. Baruffaldi, M. LeSage, S. Comer and M. Pravetoni, J. Med. Chem., 2020, 63(23), 14647–14667 CrossRef CAS PubMed.
- M. D. Raleigh, M. Laudenbach, F. Baruffaldi, S. J. Peterson, M. J. Roslawski, A. K. Birnbaum, F. I. Carroll, S. P. Runyon, S. Winston, P. R. Pentel and M. Pravetoni, J. Pharmacol. Exp. Ther., 2018, 365, 346–353 CrossRef CAS PubMed.
- O. B. Torres, C. R. Alving, A. E. Jacobson, K. C. Rice and G. R. Matyas, in Biologics to Treat Substance Use Disorders: Vaccines, Monoclonal Antibodies, and Enzymes, ed. I. D. Montoya, Springer International Publishing, Cham, 2016, pp. 397–424 DOI:10.1007/978-3-319-23150-1_20.
- M. Laudenbach, F. Baruffaldi, J. S. Vervacke, M. D. Distefano, P. J. Titcombe, D. L. Mueller, N. J. Tubo, T. S. Griffith and M. Pravetoni, J. Immunol., 2015, 194, 5926–5936 CrossRef CAS PubMed.
- J. J. Taylor, M. Laudenbach, A. M. Tucker, M. K. Jenkins and M. Pravetoni, J. Immunol. Methods, 2014, 405, 74–86 CrossRef CAS PubMed.
- J. M. Andersen, A. Ripel, F. Boix, P. T. Normann and J. Mørland, J. Pharmacol. Exp. Ther., 2009, 331, 153–161 CrossRef CAS PubMed.
- E. L. Way, J. W. Kemp, J. M. Young and D. R. Grassetti, J. Pharmacol. Exp. Ther., 1960, 129, 144–154 CAS.
- S. J. Gross, J. D. Grant, S. R. Wong and R. Schuster, Immunochemistry, 1974, 11, 453–456 CrossRef CAS.
- J. W. Findlay, R. F. Butz and E. C. Jones, Clin. Chem., 1981, 27, 1524–1535 CrossRef CAS.
- E. A. Townsend and M. L. Banks, CNS Drugs, 2020, 34(5), 449–461 CrossRef CAS PubMed.
- C. R. Alving, K. K. Peachman, G. R. Matyas, M. Rao and Z. Beck, Expert Rev. Vaccines, 2020, 1–14, DOI:10.1080/14760584.2020.1745636.
- G. R. Matyas, A. V. Mayorov, K. C. Rice, A. E. Jacobson, K. Cheng, M. R. Iyer, F. Li, Z. Beck, K. D. Janda and C. R. Alving, Vaccine, 2013, 31, 2804–2810 CrossRef CAS PubMed.
- G. W. Kirby and D. McLean, J. Chem. Soc., Perkin Trans. 1, 1985, 1443–1445, 10.1039/P19850001443.
- B. Miriyala, S. Bhattacharyya and J. S. Williamson, Tetrahedron, 2004, 60, 1463–1471 CrossRef CAS.
- C. S. Hwang, L. C. Smith, C. J. Wenthur, B. Ellis, B. Zhou and K. D. Janda, Vaccine, 2019, 37, 4155–4163 CrossRef CAS PubMed.
- M. Barrot, Neuroscience, 2012, 211, 39–50 CrossRef CAS PubMed.
- O. B. Torres, A. J. Duval, A. Sulima, J. F. G. Antoline, A. E. Jacobson, K. C. Rice, C. R. Alving and G. R. Matyas, Anal. Bioanal. Chem., 2018, 410, 3885–3903 CrossRef CAS PubMed.
- O. B. Torres, J. F. Antoline, F. Li, R. Jalah, A. E. Jacobson, K. C. Rice, C. R. Alving and G. R. Matyas, Anal. Bioanal. Chem., 2016, 408, 1191–1204 CrossRef CAS PubMed.
- P. T. Bremer and K. D. Janda, J. Med. Chem., 2012, 55, 10776–10780 CrossRef CAS PubMed.
- G. N. Stowe, L. F. Vendruscolo, S. Edwards, J. E. Schlosburg, K. K. Misra, G. Schulteis, A. V. Mayorov, J. S. Zakhari, G. F. Koob and K. D. Janda, J. Med. Chem., 2011, 54, 5195–5204 CrossRef CAS PubMed.
- J. E. Schlosburg, L. F. Vendruscolo, P. T. Bremer, J. W. Lockner, C. L. Wade, A. A. K. Nunes, G. N. Stowe, S. Edwards, K. D. Janda and G. F. Koob, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9036–9041 CrossRef CAS PubMed.
- M. D. Raleigh, M. Pravetoni, A. C. Harris, A. K. Birnbaum and P. R. Pentel, J. Pharmacol. Exp. Ther., 2013, 344, 397–406 CrossRef CAS PubMed.
- O. B. Torres, C. R. Alving and G. R. Matyas, Methods Mol. Biol., 2016, 1403, 695–710 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00029b |
| ‡ Present address: Merck & Co., Inc., 126 E Lincoln Avenue, Rahway, NJ 07065-0900, USA. |
| § Present address: Pfizer, Inc., 401 N Middletown Rd, Pearl River, NY 10965, USA. |
| This journal is © The Royal Society of Chemistry 2021 |