
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemogenomics for drug discovery: clinical molecules from open access chemical probes
Robert B. A.
Quinlan
 a and
Paul E.
Brennan
*ab
a and
Paul E.
Brennan
*ab
aTarget Discovery Institute, Nuffield Department of Medicine, University of Oxford, Old Road Campus, Oxford OX3 7FZ, UK. E-mail: paul.brennan@cmd.ox.ac.uk
bAlzheimer's Research (UK) Oxford Drug Discovery Institute, Nuffield Department of Medicine, University of Oxford, Oxford, OX3 7FZ, UK
First published on 26th March 2021
Abstract
In recent years chemical probes have proved valuable tools for the validation of disease-modifying targets, facilitating investigation of target function, safety, and translation. Whilst probes and drugs often differ in their properties, there is a belief that chemical probes are useful for translational studies and can accelerate the drug discovery process by providing a starting point for small molecule drugs. This review seeks to describe clinical candidates that have been inspired by, or derived from, chemical probes, and the process behind their development. By focusing primarily on examples of probes developed by the Structural Genomics Consortium, we examine a variety of epigenetic modulators along with other classes of probe.
 Robert B. A. Quinlan | Robert Quinlan completed his MChem in Medicinal and Biological Chemistry (2016) at the University of Edinburgh, during which he spent time in the labs of Dr Nicolas Blanchard (Université de Strasbourg) and Professor Guy Lloyd-Jones FRS FRSE. He then joined the Synthesis for Biology and Medicine Centre for Doctoral Training (SBM CDT) at the University of Oxford. He began his DPhil studies under the supervision of Professor Paul Brennan in 2017, working on the development of small molecule epigenetic modulators for the treatment of neurodegenerative diseases. |
 Paul E. Brennan | Paul Brennan received his PhD in organic chemistry from the University of California, Berkeley under the mentorship of Paul Bartlett working on synthetic methodology for combinatorial chemistry and synthesizing inhibitors for new anti-bacterial targets. Following post-doctoral research with Steve Ley at Cambridge University, Paul returned to California to take a position at Amgen. After two years at Amgen, Paul accepted a position as medicinal chemistry design lead at Pfizer in Sandwich, UK. In 2011 Paul joined the Structural Genomics Consortium at the University of Oxford, before becoming Professor of Medicinal Chemistry for the Centre for Medicines Discovery and head of chemistry of the Alzheimer's Research UK Oxford Drug Discovery Institute. His research is focused on drug discovery for dementia and on finding small molecule chemical probes for unexplored protein families. |
Introduction
Progress in the understanding of cellular and disease biology has advanced our knowledge of protein function beyond solely catalysis.1–5 We have begun to better characterise the extra-catalytic roles of proteins and how these contribute to disease pathology, whether as readers of epigenetic marks, as molecular chaperones, or scaffolding proteins. To that end, non-destructive means of target validation and investigation are useful to examine the precise role of a protein in a cellular process. Where the use of RNA inhibition (RNAi) or CRISPR gene editing precludes this, chemical probes are an invaluable tool for rapid onset, reversible, and domain specific protein inhibition.6–8 They permit the evaluation of a particular role a protein plays, whilst leaving other interactions intact. In combination with techniques such as RNAi, they paint a complete picture of a protein's role within a cell.7Chemical probes are defined by four main criteria:6,9,10
1. A minimal in vitro potency of less than 100 nM.
2. Greater than 30-fold selectivity over sequence-related proteins.
3. Profiled against an industry standard selection of pharmacologically relevant targets.
4. On-target cellular effects at greater than 1 μM.
Their utility in interrogating the function of a protein and thus its relevance11 as a drug target has led a consortium of industrial and academic researchers to establish a collaboration for the development of probes for the entire proteome.12 Target 2035 aims to translate the advances made in genomics13 to advances in the clinic, specifically new small-molecules for the treatment of disease. One advantage of chemical probes often touted is that they are more likely to mimic the pharmacology of a drug.7 Whilst not designed with drug-like characteristics in mind (for example absorption, distribution, metabolism, and excretion (ADME) properties),7 there is nevertheless a belief that probes can provide a small-molecule starting point to accelerate the drug discovery process. In this review, we highlight some small-molecule chemical probes that have both inspired and mirrored clinical candidates, with a focus on epigenetic modulators. We describe the target and development of the probe itself, the medicinal chemistry optimisation that led to the eventual clinical candidate. We also include an estimate of the relative drug-likeness of each compound calculated using the Molsoft drug-likness and molecular property prediction tool. A higher score indicates greater drug-likeness with drugs distributed around a score of one, and non-drugs at zero.14
BET bromodomains
Bromodomains (BRDs) are a class of epigenetic reader domains that recognise acetylated lysine (KAc) residues,15 a reversible post-translational modification with a key role in regulating transcription.16 A subset of BRDs have also been shown to recognise propionylated, butyrylated, and crotonylated lysines,17,18 demonstrating the importance of these domains in interpreting the chromatin landscape. Evolutionarily conserved, they form part of diverse nuclear proteins and complexes.19 The bromo- and extra C-terminal (BET) subfamily of bromodomains is perhaps the best studied and validated in a disease context.4,20,21 Its members (BRD2, BRD3, BRD4, and BRDT) have been implicated in a variety of disease processes, including cancer, viral infection, and inflammation.22–25The probe: (+)-JQ1
Disclosed just months apart, the first pan-BET inhibitors (+)-JQ126 and I-BET76227 (Fig. 1) represented key milestones in the targeting of BET proteins. Inspired by a triazolothienodiazepine scaffold patented in 2009,28(+)-JQ1 was developed following molecular modelling of potential ligands against the bromodomain of BRD4. It was a potent inhibitor of both bromodomains of BRD4 (KD(BRD4(1)) = 50 nM, KD(BRD4(2)) = 90 nM by isothermal titration calorimetry (ITC)), with similar potency against both bromodomains of BRD3. It showed approximately three-fold weaker binding against BRD2 and BRDT. Pan-BET inhibition using (+)-JQ1 has shown anti-proliferative effects against a multitude of haematological and solid malignancies, including breast,29,30 colorectal,31 and brain cancers,32,33 as well as multiple myeloma (MM),34,35 leukaemia,36–38 and lymphoma.35,38,39(+)-JQ1 was key to establishing the mechanistic significance of BET inhibition, however was unsuitable for clinical progression due to its short half-life26 and thus its required dose concentrations being above tolerable levels in vivo.40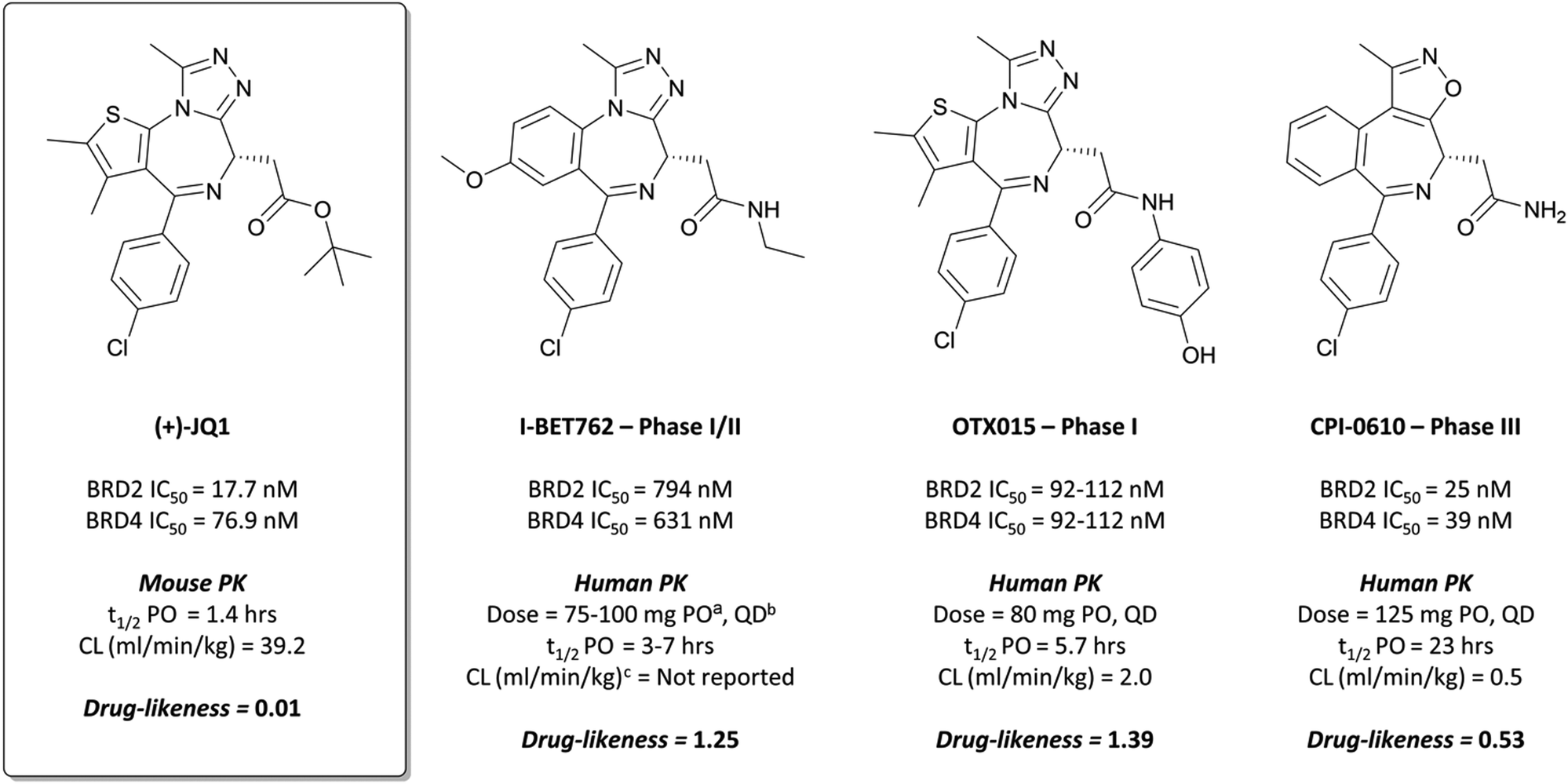 | ||
Fig. 1 Chemical probe (+)-JQ1 and structurally derived clinical candidates: I-BET762, OTX015, and CPI-0610. Their potency against relevant BET bromodomains is presented, along with pharmacokinetic (PK) data where available, and their clinical status. Also included is a relative drug-likeness score to highlight the changes in drug-likeness moving from probe to drug. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PO = per os, by mouth. bQD = quaque die, once daily. cAssume 70 kg bodyweight for humans. PO = per os, by mouth. bQD = quaque die, once daily. cAssume 70 kg bodyweight for humans. | ||
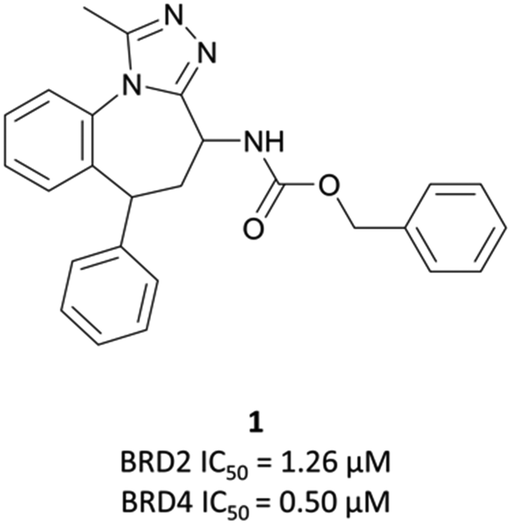 | ||
| Fig. 2 Initial hit 1 in a screen for compounds that upregulated ApoA1 expression. | ||
Optimisation efforts focused on improving potency, target selectivity, physiochemical properties, and compound stability.41 Previous studies on the stability of triazolobenzodiazepines had identified problems with ring-opening under acidic conditions, precluding oral dosing.43 The authors observed this with some analogues of 1, measuring very short half-lives (<1 h) when the compounds were suspended in buffer at pH = 2.0. They confronted this by eliminating the nitrogen at the 3-position of the benzodiazepine ring of 1, replacing the amide with the acetamide of I-BET762. This had the added benefit of simplifying the enantioselective synthesis of further analogues, as aspartic acid could be used as a chiral precursor. The ethylacetamide of I-BET762 in lieu of the phenylcarbamate of 3 lowered both the logP and molecular weight (MW) to further improve the oral profile. Other improvements came from SAR that introduced the methoxy- and chloro-substituents on their respective phenyl rings to yield I-BET762 (IC50 (FP) = 794 nM, 398 nM, 631 nM for BRD2, -3, and -4 respectively).
Whilst structurally very similar to (+)-JQ1, I-BET762 displayed more favourable pharmacokinetic (PK) properties, with good solubility and improved half-life41,44 reflected in an improved drug-likeness. I-BET762 showed no appreciable activity in safety assays and was selective against other representative bromodomains. It induced growth inhibition in in vivo models of nuclear protein in testis (NUT) midline carcinoma,44 MM,45 and prostate cancer.46 In 2012, I-BET762 was advanced to the clinic for the treatment of NUT carcinoma and other solid tumours.44 Target engagement was observed with once-daily dosing, with several patients experiencing clinical benefit. Despite that, I-BET762 showed rapid elimination (t1/2 = 3–7 hours) and transient responses, thought to be due to the activation of resistance mechanisms to BET inhibition. Nevertheless, it is currently under clinical investigation for the treatment of acute myeloid leukaemia (AML) (NCT01943851), with manageable adverse events and some objective responses observed.47 It is also being evaluated as part of a combination therapy for breast cancer (NCT02964507, expected completion August 2021)48 and prostate cancer (NCT03150056, expected completion August 2021).49
After preclinical models demonstrated good oral bioavailability and PK properties,53,56OTX015 entered the clinic as a treatment for a variety of haematological malignancies. It showed dose-limiting toxicities (DLTs) in patients with lymphoma and MM,57 and in a study for patients with leukaemia.58 These findings informed a once-daily dosing regimen that led to some partial responses, however it was rapidly eliminated (t1/2 = 5.7 h) and there was no evidence of clinical activity that met objective response criteria the majority of patients. It was also evaluated against some solid tumours (prostate cancer, NUT midline carcinoma, and non-small-cell lung cancer (NSCLC)),59 however displayed DLTs alongside a lack of efficacy. The same lack of efficacy was observed in a phase I trial of OTX015 against glioblastoma (NCT02698176). As a result of these collective failures, Merck terminated the OTX015 program (NCT02698189).
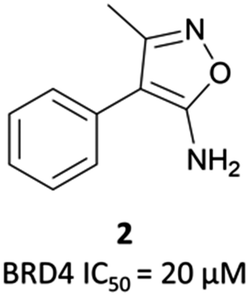 | ||
| Fig. 3 Aminoisoxazole fragment hit 2 identified from a screen against the first bromodomain of BRD4. | ||
2 binds to BRD4(BD1) with an IC50 (FRET) of 20 μM, which improved to 440 nM following the introduction of an azepine ring. Interestingly, this was more potent than the corresponding triazole compound, thought to be because it better engaged in hydrogen bonding interactions with Asn140 and Tyr97 in the binding site. Substitution on the methylene bridge of the azepine ring found that the (S)-enantiomer of an N-unsubstituted acetamide gave the most potent compound (CPI-0610) with a BRD4(1) IC50 (FRET) of 39 nM, binding in identical fashion to (+)-JQ1 (Fig. 4b) and with slight improvement in drug-likeness. Modulation of the chlorophenyl group gave no advantage, and whilst more potent compounds were synthesised through substitution on the benzo ring, these improvements in potency were not observed in cells. CPI-0610 was selective against non-BET bromodomains, as well as against targets in a CEREP express panel of 50 GPCRs, ion channels and transporters, and showed negligible inhibition of CYP450.
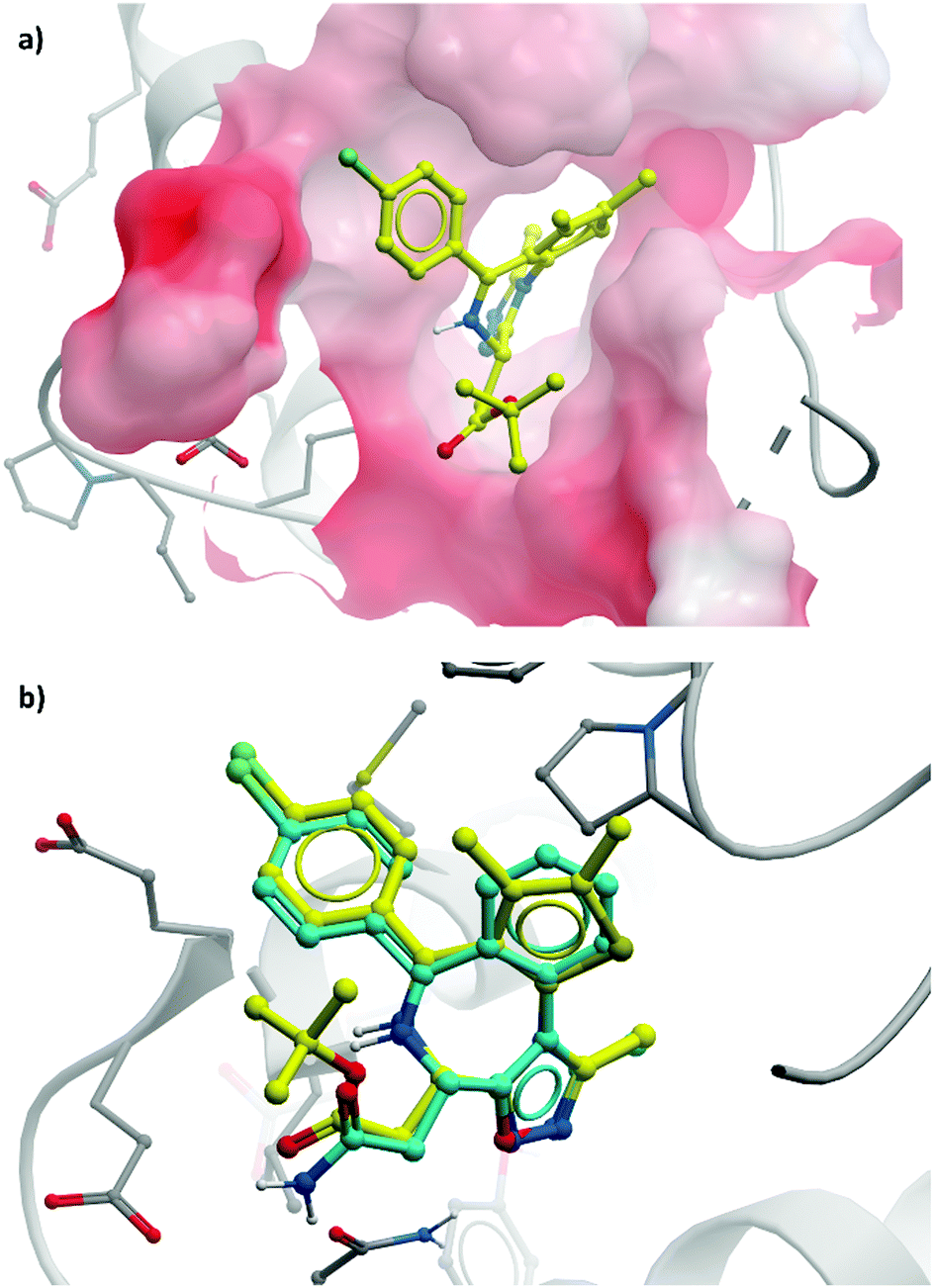 | ||
| Fig. 4 (a) (+)-JQ1 bound to the first bromodomain of BRD4, extending into the acetyllysine pocket (PDB: 3MXF); (b) the binding mode of CPI-0610 (cyan) overlaid with (+)-JQ1 (yellow), showing significant overlap (PDB:5HLS). | ||
CPI-0610 displayed favourable PK in various species, with low clearance, moderate half-life, and good oral bioavailability.60 In a mouse xenograft model of AML, it induced a dose-dependent reduction in MYC mRNA levels, as well as significant tumour growth inhibition, with no associated weight loss observed. It also induced growth inhibition and improved survival in a mouse xenograft model of MM.61 These findings led Constellation to advance CPI-0610 to early phase clinical studies for the treatment of haematological malignancies. Phase 1 evaluation in patients with relapsed or refractory lymphoma62–64 showed that once-daily oral dosing was well tolerated and induced antitumour responses, as well as dose-dependent decreases in the expression of BET target genes. CPI-0610 demonstrated good oral bioavailability and stability (t1/2 = 23 h).62
Following these results, CPI-0610 was initiated in a phase II trial for the treatment of myelofibrosis.65,66 The trial had two arms, with CPI-0610 as a monotherapy for myelofibrosis (NCT04603495, expected completion September 2023), or in combination with the Janus kinase (JAK) 1/2 inhibitor ruxolitinib (NCT02158858, expected completion December 2021).67 Its use as part of a combination therapy derives from the results of a previous study that showed combining JAK and BET inhibition (with ruxolitinib and (+)-JQ1) in mice models of myelofibrosis led to a marked reduction in disease burden and the serum levels of inflammatory cytokines in vivo.68 Thus far, the combination has been well tolerated and objective responses have been observed in all evaluable patients,66 indicating promise for the use of CPI-0610 in combination with ruxolitinib for the treatment of myelofibrosis.
The probe: PFI-1
In the search for life beyond triazolodiazepine BET inhibitors, several groups described orthogonal chemotypes that also bound to BET proteins.69–72 In particular, 3,5-dimethylisoxazole- and quinazolinone-based fragments were found to mimic the N-acetyllysine binding mode of the histone peptide, forming a productive hydrogen bond with Asn140 in the BRD4(BD1) binding pocket (vide supra the effective use of an isoxazole fragment in the development of CPI-0610). Inspired by these findings, a collaboration between scientists at Pfizer and the SGC used a fragment-led approach to discover PFI-1 (Fig. 7),73 a structurally novel pan-BET inhibitor.Taking inspiration from previous work by groups from GSK71 and the SGC,72 the authors chose fragments based on a 4-dihydro-3-methyl-2(1H)-quinazolinone scaffold as their starting point, identifying a brominated derivative (Fig. 5, 3) as a promising initial hit.73 A crystal structure of 3 in complex with BRD4(BD1) showed that the cyclic urea motif was indeed engaging in a hydrogen bonding interaction with Asn140, and that the rest of the scaffold was interacting with a variety of lipophilic residues in the protein. Interrogation of the crystal structure suggested the introduction of a kink in the molecule, using the 6-bromo moiety as a synthetic handle, would enable engagement of the lipophilic WPF shelf at the opening of the binding pocket. This was effectively achieved through the introduction of an arylsulfonamide group off this position, resulting in PFI-1 that had an IC50 (alphascreen) of 220 nM 98 nM for BRD2(BD1) and (BD2) respectively.74 Thermal shift assays showed that PFI-1 was selective for the BET bromodomains over other representative bromodomains, and assessment against other pharmacologically relevant targets selectivity over a panel of 65 GPCRs, ion channels, and kinases.73
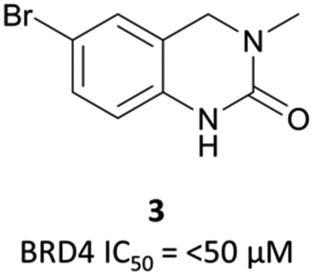 | ||
| Fig. 5 Quinazolinone fragment 3. | ||
PFI-1 induced growth inhibition and apoptosis in a dose-dependent manner in leukaemia cells,74 and also downregulated Aurora B kinase expression, which is implicated in a number of cancers, including leukaemia.75 It displayed poor PK properties in rats, with poor oral bioavailability, high clearance, and a short half-life. Nevertheless, its selectivity, along with its on-target and cellular potency, identified it as an effective chemical probe for interrogation of the BET bromodomains.
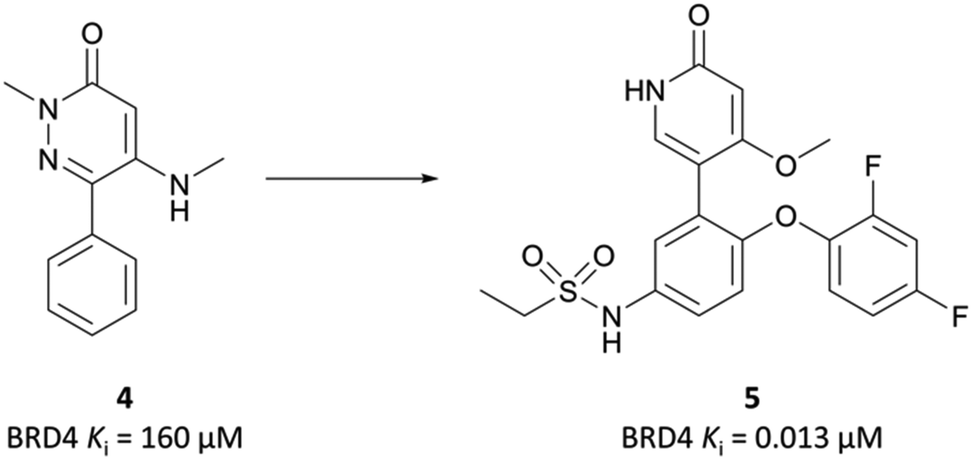 | ||
| Fig. 6 Phenylpyridazinone fragment 4 around which SAR exploration led to 5. | ||
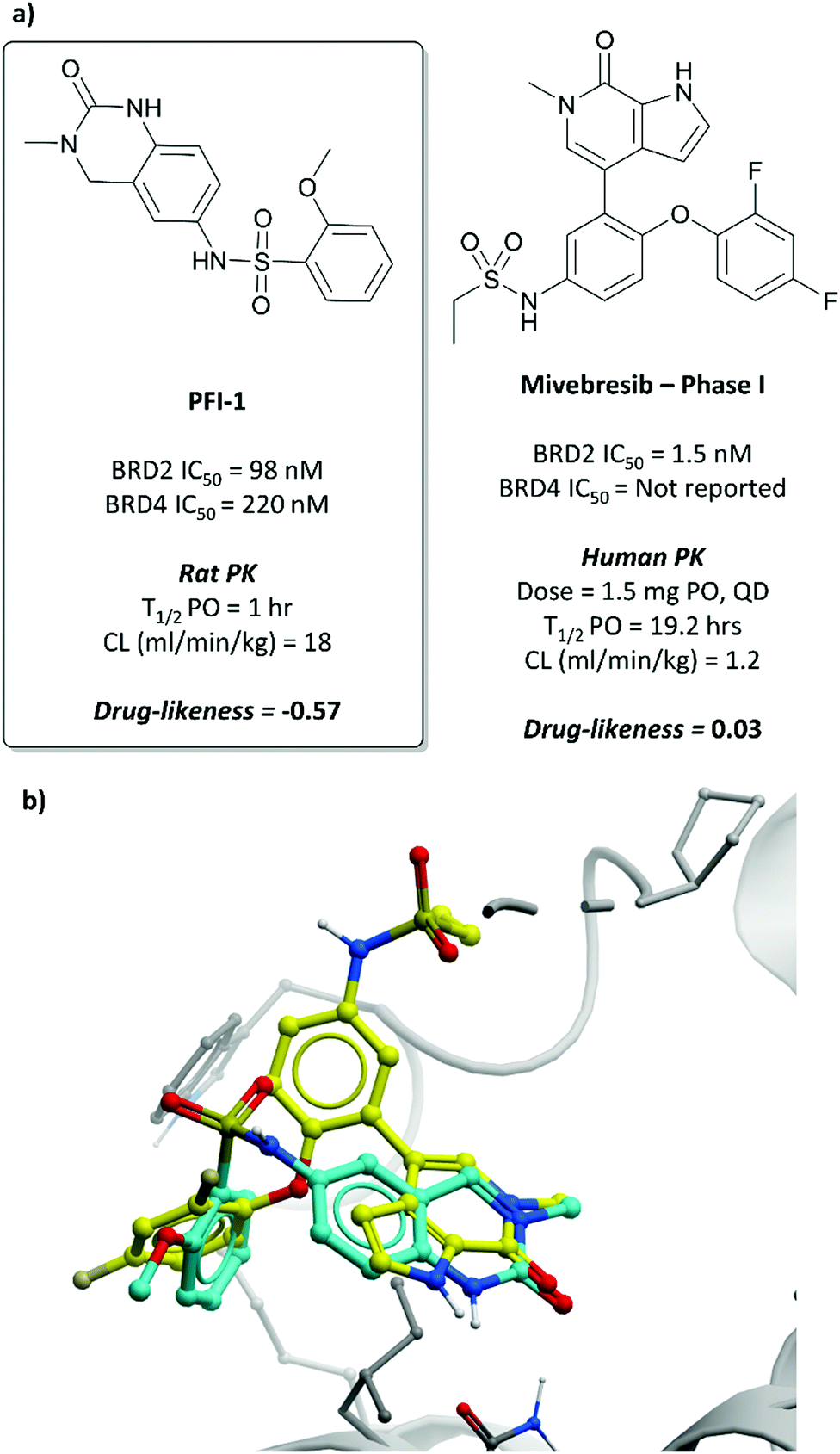 | ||
| Fig. 7 (a) Probe PFI-1 and clinical molecule mivebresib; (b) overlaid crystal structures of PFI-1 (cyan) and mivebresib (yellow) bound to the first bromodomain of BRD4. The quinazolinone of PFI-1 and pyrrolopyridone of mivebresib align, mimicking an acetylated lysine. The phenylether of mivebresib also occupies the same vector as the arylsulfonamide of PFI-1 (PDB: 4E96 and 5UVW). | ||
Because of its favourable PK properties, namely good oral bioavailability (F% (mouse) = 63), 5 was chosen for further evaluation in a mouse xenograft model of MM.76 Oral dosing led to significant dose-dependent tumour growth inhibition, with acceptable weight loss observed. Enthused by these results, the authors decided to evaluate whether further gains in potency could be achieved through a bidentate interaction that would engage the NH2 group of Asn433 in the BRD4(BD2) binding pocket.77 By transforming the pyridone core to a bicyclic pyrrolopyridone (mivebresib), they were able to form productive hydrogen bonds to both the carbonyl and the NH2 of Asn433. Mivebresib was a potent binder, with a Ki (FRET) against BRD4(BD2) of 1.5 nM and remained potent in cellular assays (EC50 (MX-1 proliferation) = 13 nM). Whilst displaying poor drug-likeness, it was substantially improved relative to PFI-1. It was selective against a panel of 79 pharmacologically relevant targets and exhibited favourable PK and drug-like properties, including low clearance in human liver microsomes.78,79
In a mouse xenograft model of AML, mivebresib was dosed orally at 1 mg kg−1 for 25 days and achieved 99% tumour growth inhibition, with acceptable weight loss observed. Further PK characterisation across species identified this compound as a clinical candidate, leading to its evaluation as a treatment for patients with relapsed/refractory solid tumours, including breast and prostate cancer, as well as melanoma.80Mivebresib was well tolerated, with some DLTs and a half-life of 16.1–19.9 h. Despite these favourable characteristics, very little efficacy was observed beyond stable disease for a modest subset of patients. Preclinical studies demonstrated that this lack of efficacy may be improved by its use as a combination therapy,81 and so a phase I trial evaluating mivebresib in combination with the BCL2 inhibitor navitoclax is ongoing (NCT04480086, expected completion July 2024).
Whilst involved in the suppression of tumour formation, p53 has also been shown to mediate excessive apoptotic death in normal cells following cancer therapy.94 As well as this, its overactivity has been implicated in a variety of neurodegenerative and cardiovascular diseases,95–100 suggesting its inhibition may be of therapeutic value.
The probe: CBP-30
The potential benefit of blocking the interaction of CBP with p53 and thus preventing the transcription of p53-mediated genes101,102 led researchers at the SGC to embark on the development of CBP-30 (Fig. 9),93 a potent and selective inhibitor of the CBP/p300 bromodomains.The starting point was a non-selective isoxazole fragment (Fig. 8, 6) that inhibited CBP/p300 and BRD4 with similar micromolar potency.103 Subsequent derivatives would be designed to achieve selectivity for CBP/p300 over the BET bromodomains. Synthesis of a variety of analogues of 6 led to the discovery that N1- and C2-substitution increased the potency to sub-micromolar levels against CBP, inducing some selectivity over BRD4(BD1). Optimisation of both the N1-amine substituent and the C2-aryl component through introduction of morpholine and 3-chloro-4-methoxyphenyl moieties respectively, led to compound 7 (Fig. 8). 7 had a KD (ITC) of 28 nM and 480 nM against CBP and BRD4(BD1) respectively, a selectivity of 17-fold.
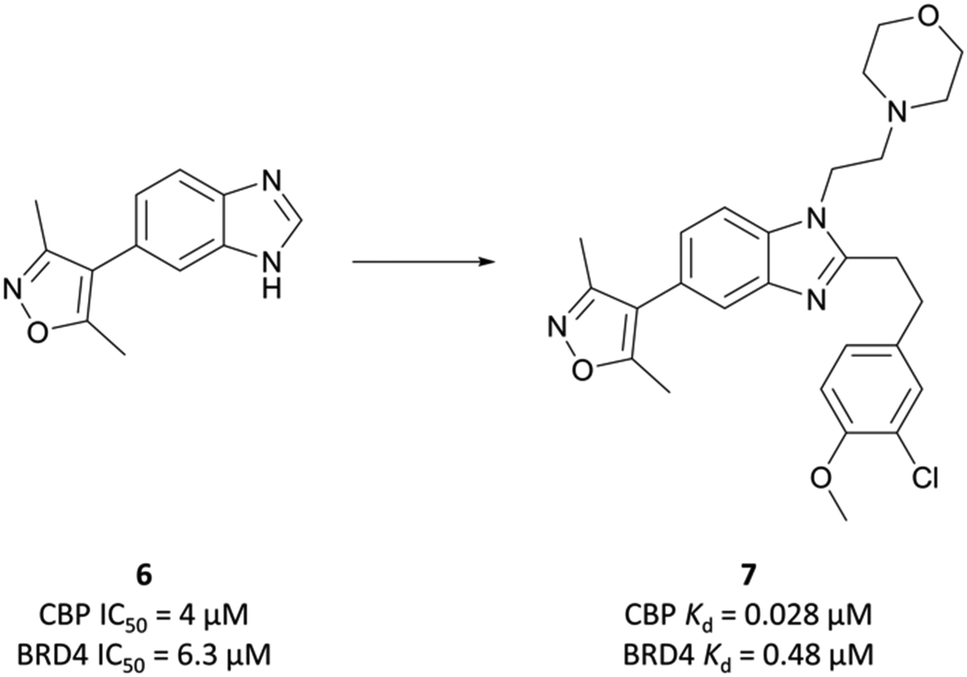 | ||
| Fig. 8 Starting isoxazole fragment 6 and the subsequently derived 7, with improved potency for CBP and selectivity over BRD4. | ||
 | ||
| Fig. 9 Chemical probe for CBP/p300 CBP30, and clinical candidate CCS1477. The selectivity for CBP over BRD4 is highlighted, along with available PK data. | ||
To build on this, the authors evaluated the binding mode of 7 in complex with CBP and BRD4(BD1) and found that the C2-substituent sits in a different conformation in each.93 They reasoned that constraining the molecule in the CBP binding conformation may lead to improved potency and selectivity. By introducing stereochemistry in the N1-linker, the (S)-enantiomer specifically (CBP-30), they were able to impart approximately 40-fold selectivity for CBP over BRD4(BD1) (KD = 21 nM vs. 850 nM, respectively). CBP-30 was also selective by thermal shift assay for CBP/p300 against other representative bromodomains, with only minor ΔTm observed against the other BET proteins.104
In cells, CBP-30 effectively displaced CBP from acetylated lysines at 100 nM, with no effect on BRD4 activity.93 It inhibited doxorubicin-induced p53 activity in a dose-dependent manner (IC50 = 1.54 μM), although it was unclear if this was due to CBP inhibition or its BRD4 inhibition at these concentrations. In addition, CBP-30 was shown to inhibit IL-17A production at 2 μM in human cells derived from patients with ankylosing spondylitis and psoriatic arthritis, with a far more limited effect on gene expression observed than that associated with pan-BET inhibitor treatment.104CBP-30 displayed moderate cytotoxicity (CC50 = 80 μM), which was above efficacious on-target concentrations. Despite good drug-likeness, in vitro ADME evaluation showed very rapid metabolism in human liver microsomes (HLMs), with no compound remaining after 60 minutes. These results precluded its use as an oral in vivo probe, but it remains a useful, selective in vitro probe for the function of CBP/p300.
CCS1477 has a KD of 1.3/1.7 nM for CBP/p300, compared to a KD of 222 nM for BRD4, with approximately 170-fold selectivity. It was selective over 32 representative bromodomains (<50% activity at 1 μM), as well as 97 kinases at 10 μM,111 showing no activity in a safety panel of 44 targets. CCS1477 potently inhibited the growth of androgen receptor (AR) driven prostate cancer cell lines (IC50 = 49–230 nM),112,113 and induced significant growth inhibition in AML and MM cell lines at 100 nM.114,115 In mouse xenograft models of prostate cancer, AML, and MM, CCS1477 induced tumour growth inhibition at 20 mg kg−1 that persisted after dose cessation. It was orally bioavailable and displayed reasonable PK properties in various species, predicting good human PK and informing a dosing regimen for clinical trials.
CCS1477 entered the clinic in July 2018, in a phase I trial for castration resistant prostate cancer (NCT03568656).116 In August 2019 it began further evaluation in a phase I trial for haematological malignancies (NCT04068597) and is due to begin analysis and dissemination of findings for both by December 2021.117
Di- (me2) and tri- (me3) methylation of H3K79 is associated with active gene transcription and euchromatin formation121,123 thought to be due to its ability to prevent the formation of other transcriptionally repressive lysine methylation marks.124 DOT1L activity has been implicated in a subset of aggressive leukaemias that involve a chromosomal translocation of the mixed-lineage leukaemia (MLL) gene on chromosome 11q23.125,126 Typically, the MLL gene encodes a KMT responsible for H3K4 methylation,127,128 which is lost during the translocation.125,126 The remaining MLL protein is fused to a variety of partners that then bind and recruit DOT1L.129–131 The recruitment of DOT1L and its transcriptionally activating methyltransferase activity leads to the increased expression of pro-leukaemogenic genes.131–133 Knockout of DOT1L has been shown to reduce the viability of MLL cell lines,134,135 suggesting that DOT1L inhibition may be a useful therapeutic strategy for the treatment of these leukaemias.
The probes: EPZ004777 and SGC0946
Epizyme approached the development of its DOT1L inhibitor, EPZ004777 (Fig. 12), through targeting the SAM cofactor binding domain.136 They began with a SAM analogue (Fig. 10, 8), replacing the methionine moiety of SAM with a dimethylamino group. 8 bound in the same conformation as SAM with reasonable potency (Ki = 38 μM), validating its use as a starting point for derivatisation. They began to diversify the amine substituents in an attempt to extend into the lysine binding pocket of DOT1L and were surprised to find that an Fmoc-protected intermediate (Fig. 10, 9) displayed modest potency (Ki = 20 μM).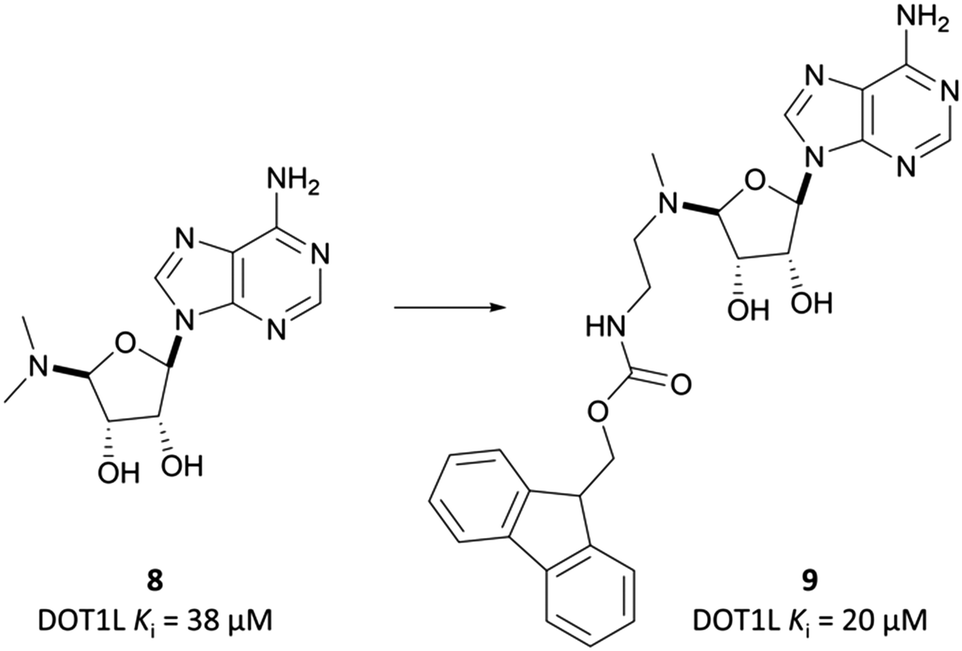 | ||
| Fig. 10 SAM-analogue 8 that was iteratively developed to the Fmoc-protected intermediate 9. | ||
This finding led them to explore substituting the amine with a large hydrophobic group, linked by a short tether.136 Introduction of a tert-butyl phenyl urea and a propyl tether led to dramatic increases in potency. Modification of the nucleoside scaffold showed that deazapurine analogues were also potent inhibitors and combining the two led to EPZ004777, which had a Ki of 0.3 nM for DOT1L.136 The root of this potency was found to be the engagement of EPZ004777 with a previously unknown binding pocket adjacent to the SAM-binding site, which wasn’t engaged by either SAM or its reaction product, S-adenosylhomocysteine (SAH). The urea of EPZ004777 recapitulates the binding site interactions of the amino acid portion of SAM, whilst the bulky tert-butyl phenyl group induces a conformational change in a variety of hydrophobic residues, opening the pocket to accommodate it (Fig. 11b).136 As a result, and despite the universality of SAM as a cofactor, EPZ004777 was highly selective for DOT1L over other KMTs, with >1000-fold selectivity.129
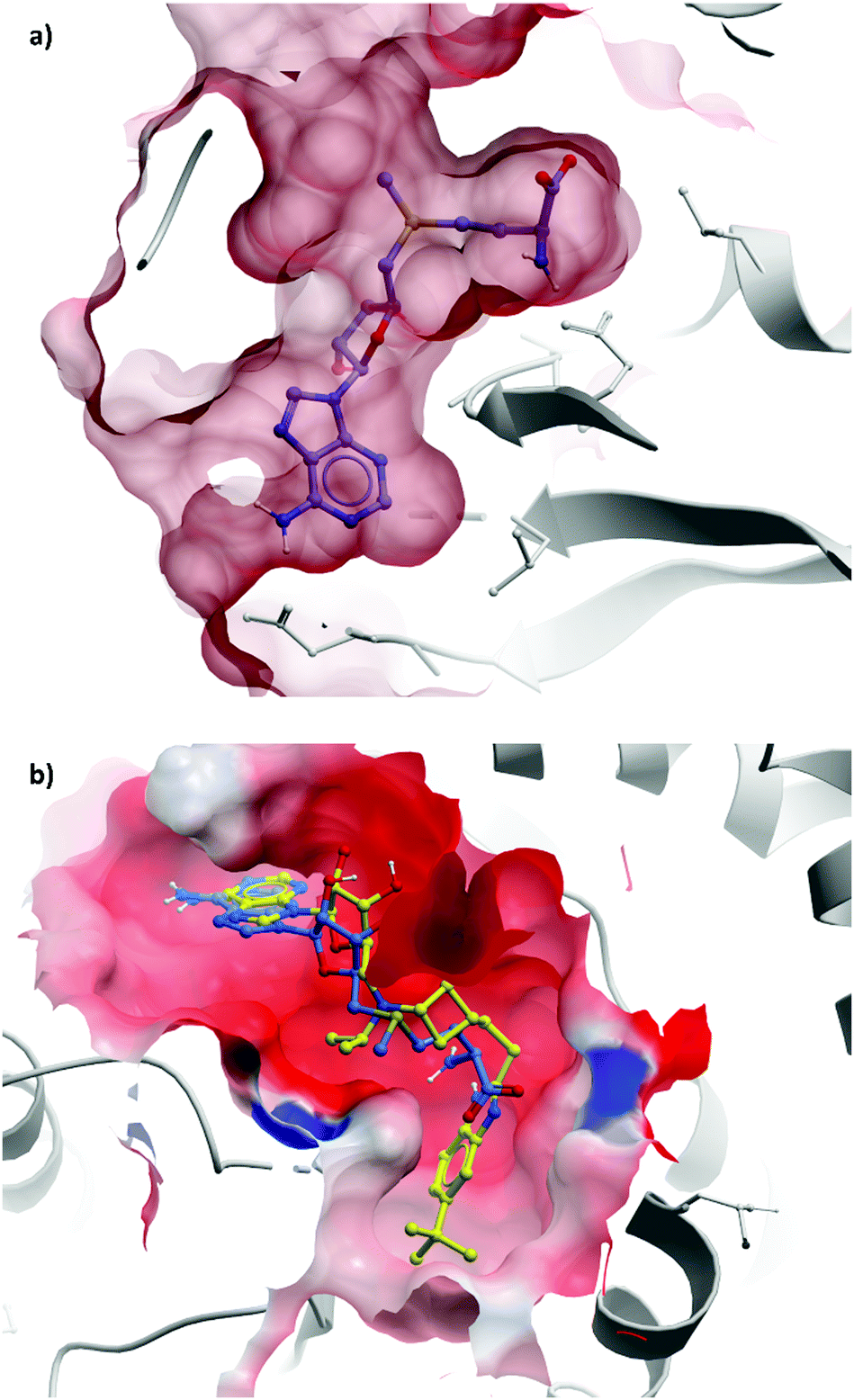 | ||
| Fig. 11 (a) Partially occluded view of SAM bound to DOT1L, showing the depth of the pocket occupied by the amino acid moiety (PDB: 3QOW); (b) overlaid crystal structures of SAM (blue) and pinometostat (yellow), showing the new hydrophobic cleft opened by the bulky tert-butyl benzimidazole group (PDB: 4HRA). | ||
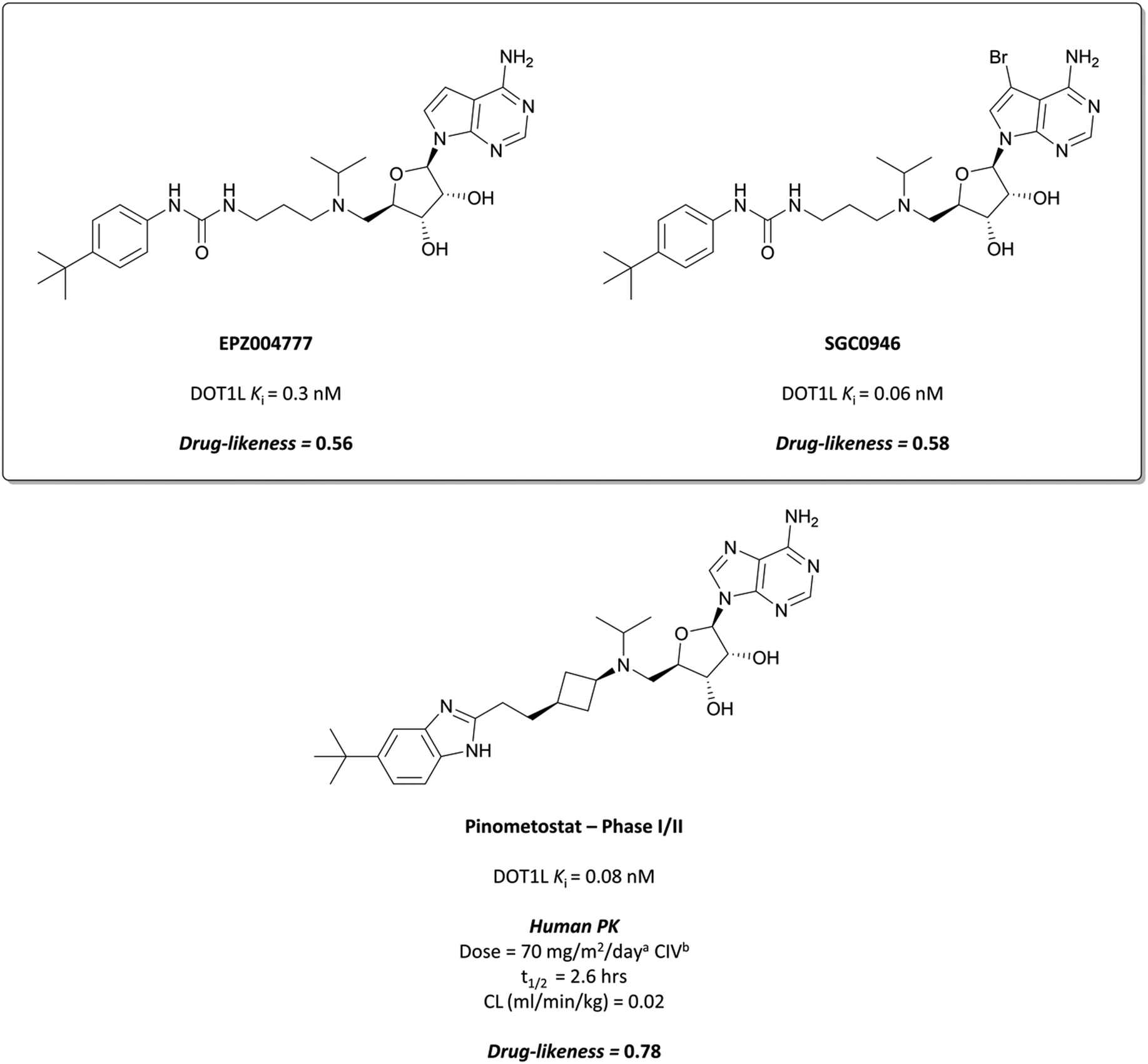 | ||
| Fig. 12 Probe EPZ004777 and its brominated derivative with improved permeability, SGC0946. Clinical candidate pinometostat retains several structural features and pharmacophores from both molecules, as well as potency against DOT1L. amg m−2 day−1 dosing based on body surface area. bCIV = continuous intravenous infusion. | ||
Treatment of patient derived MLL-rearranged AML cells with EPZ004777 resulted in a concentration-dependent, selective reduction in global H3K79me1 and H3K79me2 levels.129 It also reduced the expression of key MLL fusion target genes (IC50 (scintillation proximity assay (SPA)) = 700 nM), and induced apoptosis-mediated cell death in MLL-rearranged cells. In vivo testing in mouse xenograft models showed that H3K79me2 levels were reduced, efficacious doses were well-tolerated, and survival was extended.
EPZ004777 displayed very poor permeability which precluded its clinical development,129 exemplified by the fact that it required administration via subcutaneous osmotic pumps to permit in vivo assessment. The scaffold was latterly improved following assessment of the binding pose of EPZ004777 in the SAM binding pocket, which identified a hydrophobic cleft around C7 of the deazapurine ring.137 Bromination of this position yielded SGC0946 (Fig. 12) and a five-fold increase in potency (Ki = 0.06 nM), along with an almost ten-fold increase in potency for abrogating H3K79me2 in MCF10A cells. SGC0946 was also more active relative to EPZ004777 in cells transformed with an MLL fusion oncogene, effectively reducing the expression of MLL target genes. SGC0946 has subsequently been employed in a variety of settings to further elucidate the role of DOT1L in cancer and the mechanism of MLL fusion target gene expression.138–141
000-fold selectivity observed over related protein methyltransferases, and displays improved drug-likeness as well.
Continuous intravenous infusion (CIV) at 70 mg kg−1 of pinometostat in a rat xenograft model of MLL-rearranged leukaemia caused complete tumour regression after 14 days with no observed weight loss or toxicity.142 This route of administration was necessary for maintaining plasma levels of pinometostat at efficacious concentrations, due to its poor oral bioavailability and short half-life. Pinometostat showed moderate to high clearance in mice, rats, and dogs, and was predicted to be a similarly moderate to high clearance compound in humans.
Pinometostat was progressed into the clinic for the treatment of adult and paediatric MLL-rearranged acute leukaemias.144,145 Interestingly, the observed clearance (0.08 L h−1) was far lower than predicted. This ‘vertical allometry’, as is observed with drugs like diazepam and warfarin,146 was found to be a result of binding to alpha1-acid glycoprotein (AAG), which is present in higher levels in human plasma than in mouse, rat, or dog plasma.143Pinometostat had a short half-life (2.6 h) and so was given by CIV over 28 days, displaying an acceptable safety profile however no clinical activity.147 Preclinical studies have suggested pinometostat may be more effective as a combination therapy,148 and a phase Ib/II study is currently ongoing to investigate the use of pinometostat in combination with the DNA-hypomethylating agent azacitidine149 as a treatment for MLL-rearranged leukaemias (NCT03701295, completed with results not yet reported).
The probes: UNC0638 and A-366
The first reported inhibitor for G9a/GLP was BIX-01294 (Fig. 13), a peptide-competitive inhibitor discovered after a high throughput screen (HTS) of compounds with potential KMT inhibitory activity.160 Whilst selective for G9a/GLP over other KMTs, it displayed only moderate potency (IC50 = 1.9 μM (G9a), 0.7 μM (GLP)).161BIX-01294 reduced promoter-proximal H3K9me2 marks, although was found to only modestly increase transcriptional upregulation of G9a target genes.160 It has found use as a probe for the role G9a in cellular reprogramming162,163 and HIV-1 latency,164 however the concentrations required for cellular efficacy were cytotoxic.165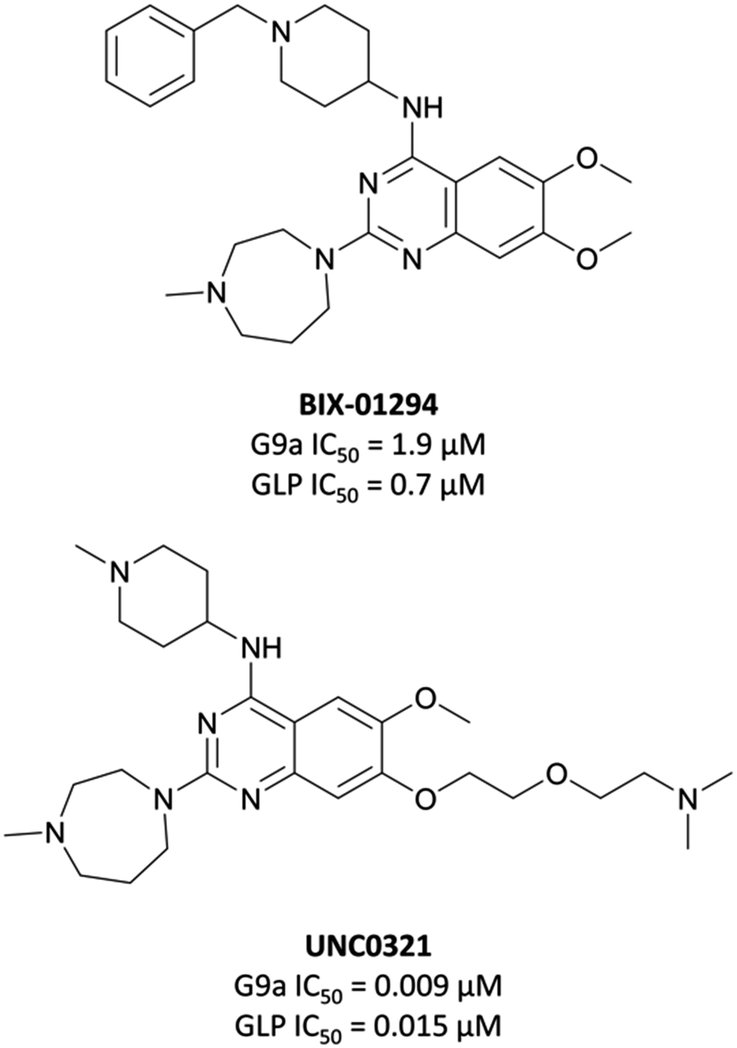 | ||
| Fig. 13 The first reported G9a/GLP inhibitor, BIX-01294, and the first iteration of more potent chemical probes developed following SAR, UNC0321. | ||
The lack of reported SAR around the scaffold of BIX-01294 led researchers at the University of North Carolina at Chapel Hill, in collaboration with the SGC, to investigate the quinazoline template as a means of improving potency.159,166–168 The crystal structure of BIX-01294 bound to GLP161 showed that the benzyl group on the piperidine ring lay outside the binding pocket, so its replacement should not lead to a loss in potency, whilst beneficially reducing MW and lipophilicity. It was postulated that modulation of the 7-methoxy moiety of BIX-01294 would allow for penetration into the histone lysine binding channel of G9a/GLP, leading to further gains in potency. Diverse analogues of BIX-01294 were synthesised to probe this SAR, resulting in UNC0321 (Fig. 13), which had an IC50 (enzyme-coupled SAH detection (ECSD)) of 9 nM against G9a and 15 nM against GLP. The key structural difference between BIX-01294 and UNC0321 was the introduction of an ethoxyethyl-linked dimethylamino group that occupied the lysine binding channel of G9a/GLP. UNC0321 was highly selective against other protein methyltransferases, however displayed reduced potency in cellular assays compared to BIX-01294, inhibiting H3K9me2 accumulation in MDA-MB-231 cells with an IC50 of 11 μM.159
This lack of cellular potency was thought to be due to insufficient lipophilicity, leading to reduced membrane permeability.159,168 Increasing the lipophilic bulk of the 4-(piperidin-4-yl)amino capping group, substitution of the methylhomopiperazinyl group with a cyclohexyl group, and introducing a pyrrolidine in place of the dimethylamino group at the 7-position, yielded UNC0638 (Fig. 14).168UNC0638 was a potent G9a/GLP inhibitor (IC50 (ECSD) = <15 nM/19 nM), which reduced H3K9me2 levels in MDA-MB-231 cells with an IC50 of 81 nM, displaying vastly improved cellular potency compared to UNC0321. In all lines tested it reduced H3K9me2 by 60–80% at 250 nM, a comparable level to G9a/GLP knockdown.159UNC0638 showed an improved toxicity/function ratio across several cell lines compared to BIX-01294 at functional doses, as well as excellent selectivity against a range of epigenetic and non-epigenetic targets.159
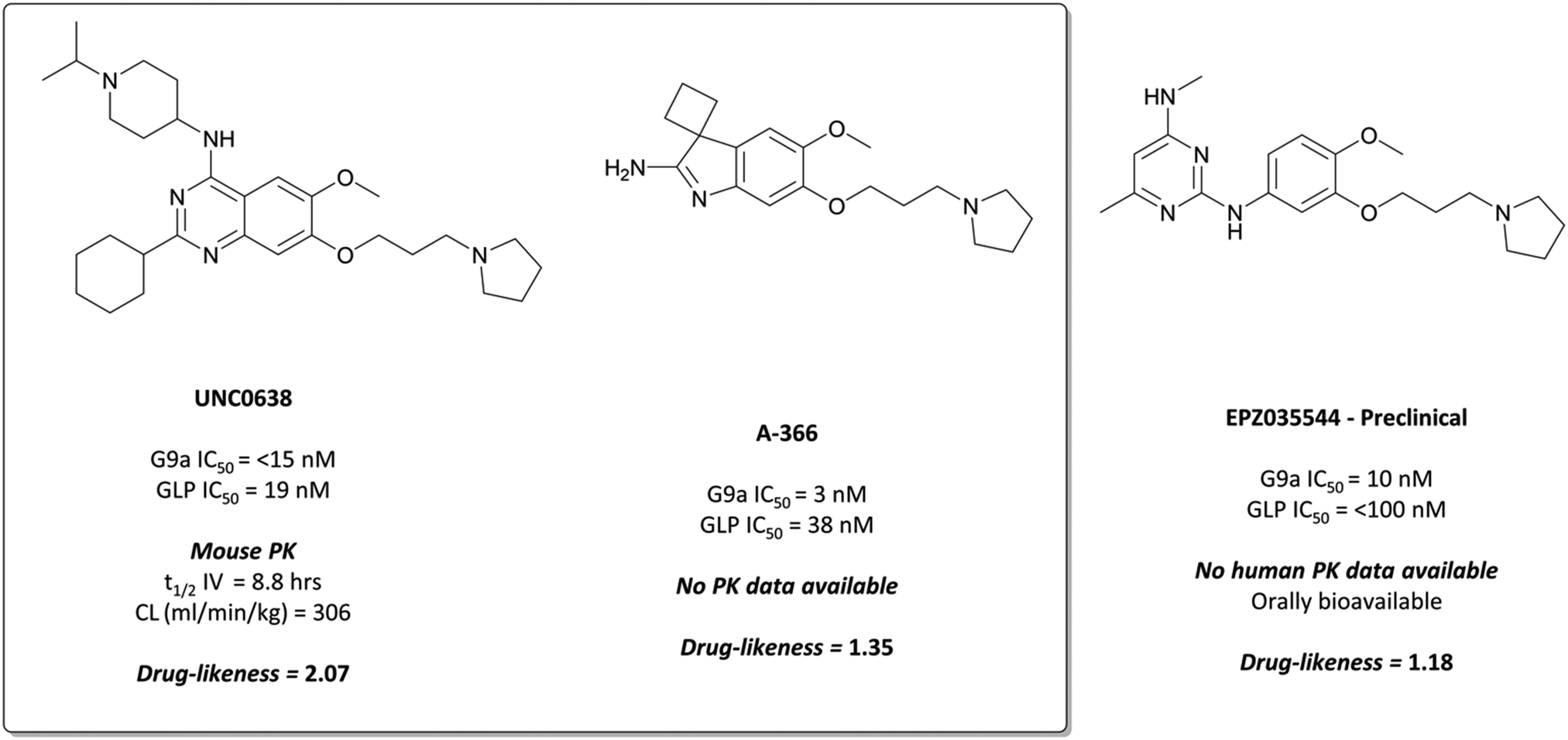 | ||
| Fig. 14 G9a/GLP chemical probe UNC0638 and the structurally similar clinical candidate EPZ035544, which displays improved potency for G9a/GLP. | ||
In an effort to discover G9a inhibitors chemically distinct from the quinazoline based BIX-01294 and UNC0638, researchers at the SGC Toronto in collaboration with Abbvie disclosed the development of A-366 (Fig. 14).169 An initial spiro[cyclobutane-1,3′-indol]-2′-amine hit was combined with the tethered pyrrolidine of UNC0638 to give A-366, which had an IC50 (SPA) of 3 nM against G9a and 38 nM against GLP. It was selective up to 50 μM against 20 HMTs and DNA (cytosine-5)-methyltransferase 1 (DNMT1), and reduced H3K9me2 levels in PC3 cells by 50% after dosing at 3 μM for 72 hours.169 It was also shown to induce modest (45%) tumour growth inhibition in a mouse xenograft model of AML, after 30 mg kg−1 dosing for two weeks.170
Despite primarily being used in investigations against cancer cells,159,165,171,172 G9a/GLP inhibition using UNC0638 was also shown to induce the expression of foetal haemoglobin.173,174 In human red blood cells, it decreased repressive H3K9me2 levels and increased activating H3K9Ac levels at the locus of the foetal γ-globin gene. Inhibition of G9a also reduced recruitment of RNA polymerase II to the β-globin locus, leading to reduced expression of the β-globin gene. Together, this led to increased production of foetal haemoglobin and reduced production of the problematic β-globin chains of adult haemoglobin, suggesting inhibition of G9a may be of therapeutic benefit for haematological disorders such as sickle cell disease.
EPZ035544 is a potent inhibitor of G9a (IC50 = 10 nM), with a cellular IC50 of 55 nM against H3K9me2.176 It is highly selective against physiologically relevant targets (>2000-fold by Ki against KMTs, >1000-fold by Ki against kinases), and induces γ-globin expression in a dose dependent manner.177 The favourable PK properties and oral bioavailability of EPZ03544 permitted extensive in vivo investigation of G9a inhibition in mice over 90 days. Doses of 50 and 75 mg kg−1 led to a 100-fold increase in the levels of embryonic haemoglobin mRNA observed in peripheral blood mononuclear cells (PBMCs), with quantitative mass spectrometry confirming concurrent increases at the protein level. The compound was well tolerated with no significant adverse effects reported in mice, leading Epizyme to advance EPZ03544 further towards the clinic as a treatment for sickle cell anaemia. Their next iteration of compounds led to EZM8266 (structure undisclosed);178 however preclinical toxicity concerns led to a discontinuation of the program.179
Aberrant expression of EZH2 and hypertrimethylation of H3K27 have been implicated in a variety of cancers,186 including myeloma,187 lymphoma,188,189 prostate,190,191 and breast.192,193 As well as this, loss-of-function mutations of one of the lysine demethylases (KDM) responsible for demethylation of H3K27me3 (UTX) are associated with various renal and throat cancers, as well as with myeloma.194 Alongside its role in these cancers, PRC2-EZH2 is also integral in regulating cellular differentiation.195 As such, its evaluation as a safe and effective therapeutic target would require a chemical probe with favourable PK properties that would permit regular dosing to examine the effects of chronic inhibition.
The probe: EPZ005687
To this end, groups at Epizyme and GSK disclosed almost concurrently (within ten days of each other) small-molecule inhibitors of EZH2 with similar structures, EPZ005687 and GSK126 (Fig. 17).196–198 Scientists at GSK had published details of an assay for the discovery of EZH2 inhibitors some months earlier, identifying GSK-A (Fig. 15) as a SAM competitive EZH2 inhibitor, with an IC50 (SPA) of 210 nM and a Ki of 700 nM.199 A breast cancer cell line was exposed to GSK-A for three days, resulting in a dose-dependent reduction in H3K27me3 levels (∼50% reduction at 8 μM) and identifying GSK-A as a cell permeable, specific EZH2 inhibitor.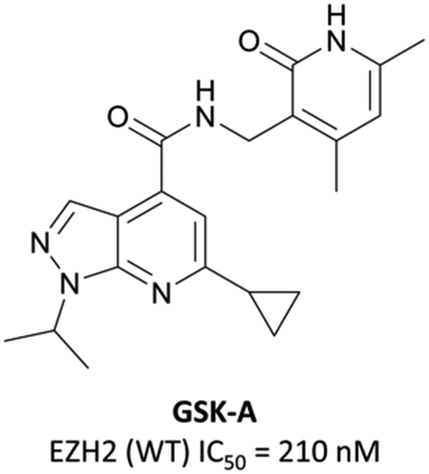 | ||
| Fig. 15 GSK-A. | ||
An initial screen of 180000 compounds carried out by Epizyme discovered a pyridone pharmacophore.196 Hit expansion led to a compound with a remarkably similar structure to GSK-A and an IC50 of 620 nM against EZH2. Replacement of a pyrazolopyridine with an indazole, the introduction of a 4-benzylmorpholine, and increased lipophilicity at the 1-position of the indazole eventually yielded EPZ005687, with improved solubility and potency over the initial hit. It inhibited EZH2 with an IC50 of 54 nM, acting as a SAM-competitive inhibitor, and was selective (>500-fold) against a variety of methyltransferases, with the exception of EZH1 (50-fold). It showed less than 50% inhibition against 73/77 GPCRs and ion channels at 10 μM, with the lowest IC50 for the remaining four 1.5 μM. EPZ005687 remained potent against a variety of mutant enzymes associated with certain cancers,200–202 and also shows good drug-likeness.
EPZ005687 displayed potent, selective reduction of H3K27me2/3 (IC50 = 80 nM) in cells196 and led to cell death in lymphoma lines bearing EZH2 Tyr641 and Ala677 mutations, with an IC50s of 300–400 nM across Tyr641 mutant cell lines. It was particularly potent against the Ala677Gly mutant line (IC50 = 36 nM), rendering it an incredibly valuable tool for the interrogation of the role of wild type (WT)- and mutant-EZH2 in the pathogenesis of disease.
GSK126 displayed a similar Ki for both WT and mutant EZH2 (0.5–3 nM), a 1400-fold improvement in potency over GSK-A.197 It was greater than 1000-fold selective for EZH2 over 20 other human methyltransferases and displayed a selectivity of 150-fold over EZH1, despite a large degree of sequence homology. It induced a loss of H3K27me3 in mutant and WT EZH2 diffuse large B-cell lymphoma (DLBCL) lines at concentrations from 7–252 nM. Across a variety of lymphoma cell lines including Hodgkin's, non-Hodgkin's, and Burkitt lymphoma, DLBCL cells remained the most sensitive to EZH2 inhibition by GSK126, with seven out of 18 displaying sub-micromolar growth IC50s (28–861 nM) including both cytostatic and cytotoxic responses. A variety of transcriptional changes were observed, with chromatin immunoprecipitation sequencing (ChIP-seq) showing that the up-regulated genes were enriched for H3K27me3 prior to treatment, suggesting them as targets for EZH2.
These results also translated to mouse xenograft models of DLBCL.197 Complete tumour growth inhibition was observed at 50 mg kg−1, with regression observed at higher doses. Post-dosing at 50 mg kg−1, tumour stasis was observed, correlating with increased survival against vehicle-treated animals. As well as this, the compound was well tolerated, with no significant adverse effects reported.
GSK126 entered a phase I trial in 2014 in patients with haematologic and solid tumours.203 There were adverse events reported in all patients, with one-third experiencing a serious adverse event. Eventually, dose-limiting toxicities and poor anticancer activity, along with a sub-optimal half-life, precluded further investigation of GSK126 as a candidate for targeting EZH2 in patients.
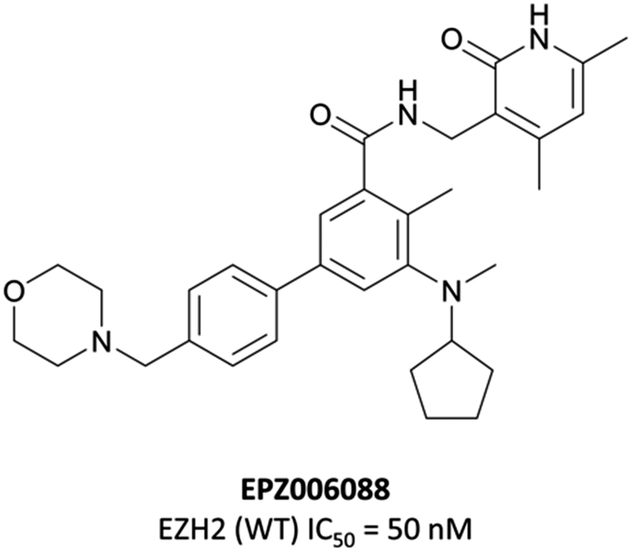 | ||
| Fig. 16 EPZ006088, a derivative of EPZ005687 with a monocyclic core. | ||
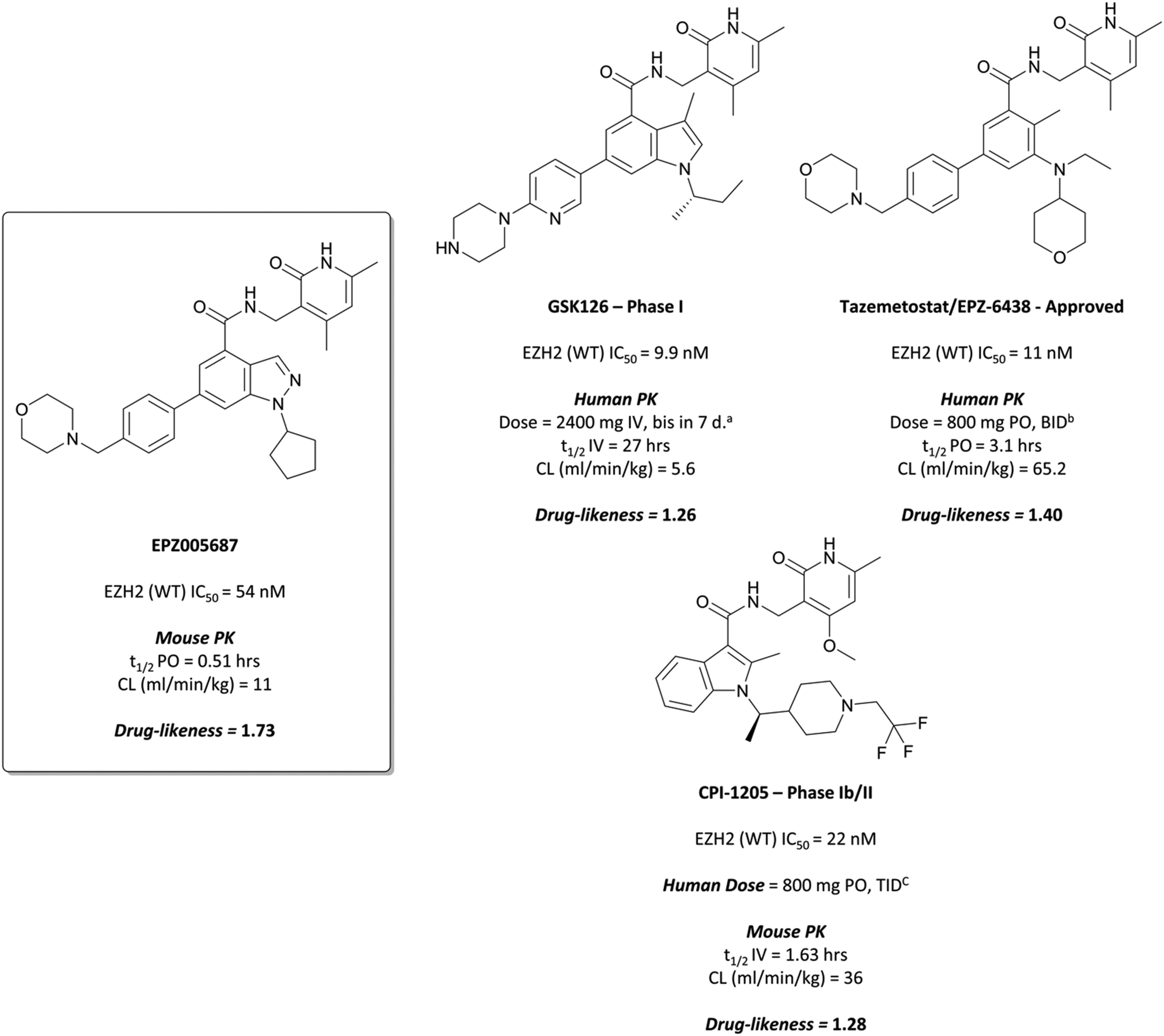 | ||
Fig. 17 EZH2 chemical probe EPZ005687, containing the crucial pyridone and arylamide pharmacophores. The structurally related clinical molecules GSK126 and CPI-1205 are presented, along with the recently approved drug Tazemetostat. aBis in 7 d. = twice weekly. b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) BID = bis in die, twice daily. cTID = ter in die, three times daily. BID = bis in die, twice daily. cTID = ter in die, three times daily. | ||
Further elaboration found that N-ethylation resulted in a modest increase in potency.204 Replacing the cyclopentane with a tetrahydropyran led to no improvement in potency but crucially lowered the logD, resulting in lower clearance. These modifications preserved drug-likeness and led to EPZ-6438 (Fig. 17), otherwise known as tazemetostat, which had a Ki of 2.5 nM against EZH2 but importantly displayed higher cellular potency (EC50 = 0.2 μM) along with reduced clearance and good oral bioavailability. Tazemetostat remained selective against EZH1 (35-fold) and 14 other HMTs tested (>4500-fold).205
Tazemetostat showed promise in a number of preclinical models of various tumour types.205,206 14-day, 1 μM treatment of SMARCB1-deleted malignant rhabdoid tumour (an aggressive childhood cancer)207 cells led to a reduction in H3K27me3 marks with concurrent growth inhibition. In a mouse xenograft model, tumours were almost entirely eliminated following twice-daily 250 mg kg−1 dosing, with no re-growth observed 32 days post-dose.205 In an EZH2-mutant lymphoma models, tazemetostat potently induced a reduction in H3K27me3 marks (IC50 = 9 nM),206 and in mouse xenografts, dose-dependent growth inhibition was observed along with complete and sustained tumour regression after 28 days. EZH2A682G mutant cells were particularly sensitive, with a dose of 114 mg kg−1 leading to tumour eradication after 28 days.
The success of the preclinical studies prompted evaluation of tazemetostat in a number of clinical studies.208–210 A phase I trial in non-Hodgkin lymphoma patients showed a favourable safety profile for chronic dosing, as well as antitumour activity in patients with refractory B-cell non-Hodgkin lymphoma. It was subsequently advanced to phase II for patients with advanced solid tumours or with B-cell lymphomas.211 In patients with follicular lymphoma, it showed an objective response rate in 69% of patients with EZH2 mutant tumours and 35% with EZH2 WT tumours, suggesting its use as a treatment for follicular lymphoma. A phase II trial for advanced epithelioid sarcoma resulted in 15% of patients showing objective responses to treatment.212 As a result, it was approved by the FDA in January 2020 for the treatment of advanced epithelioid sarcoma213 and in June 2020 for the treatment of follicular lymphoma.214Tazemetostat is also currently being evaluated in a phase II trial for paediatric patients with relapsed or refractory tumours with EZH2, SMARCB1 or SMARCA4 mutations (NCT03213665, expected completion September 2024).
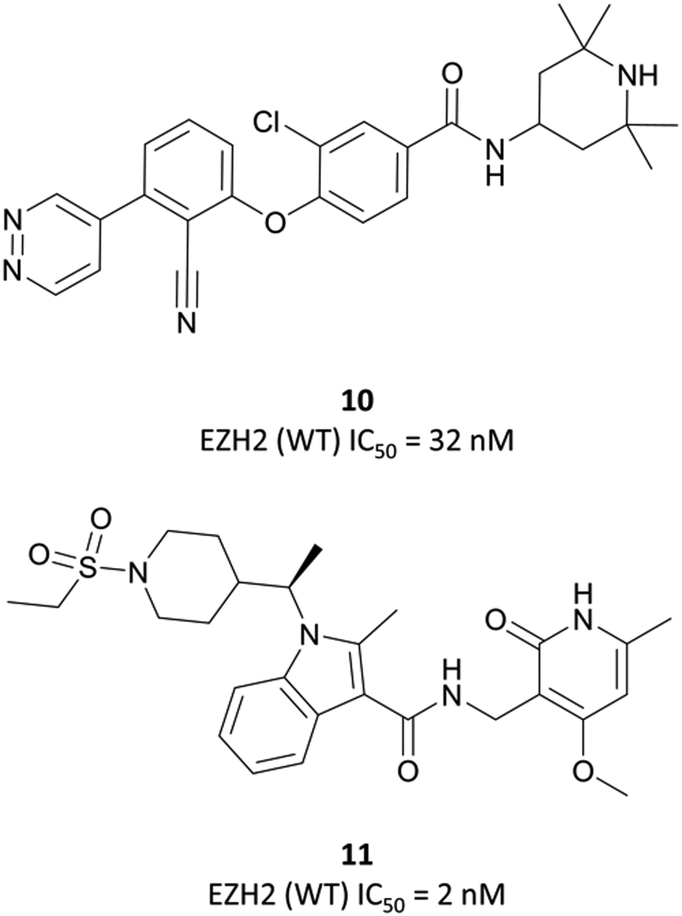 | ||
| Fig. 18 10, a non-pyridone-based EZH2 inhibitor. Poor cellular permeability led to the development of 11 following a second HTS. | ||
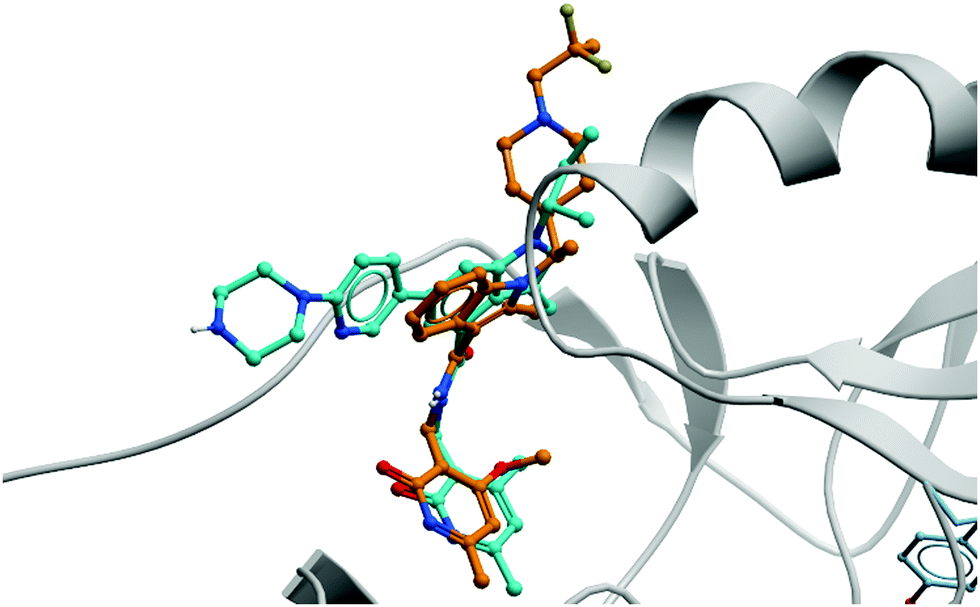 | ||
| Fig. 19 Crystal structure of GSK126 (cyan) bound to EZH2 overlaid with the structure of a defluorinated analogue of CPI-1205 (orange). The pyridone group is forced out of plane by the bicyclic core, occupying the same pocket in both structures (PDB: 5LS6 and 5WG6). | ||
As the poor PK properties of 10 precluded its use in vivo,217 researchers revisited the initial HTS and identified a pyridone-based hit from which to begin optimisation studies. Attempts to replace the tetramethylpiperidinyl group in 10 with the pyridone proved fruitless, and so it was abandoned. Analogous to GSK126, they found that a central indole scaffold led to sub-100 nM potency, and as with EPZ006088, that forcing the amide bond out of plane with the central ring system was essential. This led to a series of N-substituted, 2-methyl indoles, the most potent of which was a 1-(ethylsulfonyl)piperidine derivative. A methoxy group was introduced on the pyridone scaffold to improve metabolic stability, yielding 11 (Fig. 18), which had an IC50 of 2 nM against EZH2 (WT) and an EC50 of 80 nM against H3K27me3 in cells. Subcutaneous dosing in a mouse xenograft model of lymphoma led to dose-dependent tumour growth inhibition, along with regression at higher doses.
Despite the improved profile of 11, it was poorly bioavailable, and suffered from high clearance and a short half-life.217,218 Replacement of the sulfonamide with an N-trifluoroethyl group maintained potency and drug-likeness with improvements in oral bioavailability and half-life. The resultant compound, CPI-1205 (Fig. 17), had an IC50 of 22 nM against EZH2 (WT) and a cellular EC50 of 32 nM against H3K27me3. In a mouse xenograft model of B-cell lymphoma, oral dosing at 160 mg kg−1 over 25 days led to significant tumour growth inhibition, with no significant weight loss observed. CPI-1205 was selective against 30 other DNA and protein methyltransferases (>250-fold) and displayed no significant preclinical toxicity. As a result, it was advanced to the clinic in 2015 for the treatment of B-cell lymphoma.219 It was found to have an acceptable safety profile with some evidence of anti-tumour activity, leading to an expansion phase for lymphoma. It is also being investigated in phase Ib trials as part of a combination treatment for castration-resistant prostate cancer (NCT03480646, estimated completion date May 2021).220,221
Type I PRMTs
Amongst the diverse protein posttranslational modifications, arginine methylation plays a key role in myriad cellular processes, including transcriptional regulation, cell signalling, mRNA translation, and cell-fate decision.222–224 Methylation does not affect the charge of the residue, but rather imparts greater bulkiness and hydrophobicity, which is important for its recognition by reader domains.223,225 The increased steric bulk as a result of methylation directs the position of the cation within the binding pocket, facilitating favourable cation-π interactions, and permitting discrimination between differing arginine methylation states (mono-methylation, symmetric dimethylation, and asymmetric dimethylation).Type I protein arginine methyltransferases (PRMTs) are responsible for the mono- and asymmetric dimethylation of arginine residues, including PRMT1, -3, -4 (CARM1), -6, and -8.222–224 This is in contrast to type II PRMTs (PRMT5 and PRMT9) which catalyse mono- and symmetric dimethylation, and type III (PRMT7) which catalyse monomethylation. Type I PRMTs are implicated in a variety of human cancers:226 PRMT1 and PRMT6 dysregulation is associated with bladder and lung cancer,227 and PRMT4 has been found to be overexpressed in breast,228 colorectal,229 and prostate cancers.229–231
The probes: EPZ020411 and MS-023
The relevance of PRMT6 to human disease prompted Epizyme to pursue the development of a selective inhibitor for target validation studies.232 An initial HTS identified an aryl pyrazole hit (Fig. 20, 12) as a potent inhibitor of PRMT1, PRMT6 and PRMT8. A crystal structure of 12 in complex with PRMT6 showed that the diamine motif occupied the arginine side-chain pocket, with the terminal amine engaging in favourable hydrogen bonding interactions with various residues and water molecules in the pocket. The pyrazole also engaged in hydrogen bonding, with the aryl ring forming favourable π–π interactions with aromatic residues. Coupled with general hydrophobic interactions, these contributed to the low nanomolar potency of the hit against the three PRMTs (IC50 (SPA) = 11 nM, 67 nM, and 18 nM against PRMT6, PRMT8, PRMT1 respectively).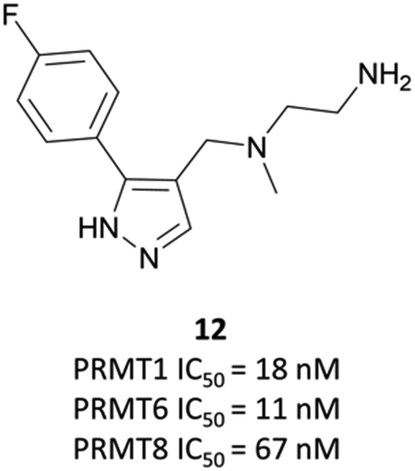 | ||
| Fig. 20 Type I PRMT inhibitor, and aryl pyrazole hit 12. | ||
Regioisomers of the pyrazole were found to have no detrimental effect on binding,232 and selectivity for PRMT6 over PRMT1 and PRMT8 could be achieved through extension of the vector off the para-position of the aryl group. Introduction of an oxygen-linked alkyl group yielded EPZ020411 (Fig. 21), with high drug-likeness and an IC50 of 10 nM against PRMT6 compared to 223 nM for PRMT8 and 119 nM for PRMT1. EPZ020411 was 100-fold selective for PRMT6, -1, and -8 compared to other histone methyltransferases, including four other PRMTs. It induced a dose-dependent decrease in the levels of PRMT6-mediated H3R2 methylation (IC50 = 637 nM) and was found to be 10-fold less potent against a PRMT1-specific monomethyl arginine mark. PK evaluation showed moderate clearance and reasonable half-life, however poor permeability which translated to poor oral bioavailability (<5%). Subcutaneous dosing resulted in good bioavailability (65.6%), with the unbound concentration remaining above the observed PRMT6 IC50 for longer than 12 hours.232 It was thus recommended as useful tool compound for in vivo PRMT6 target validation studies.
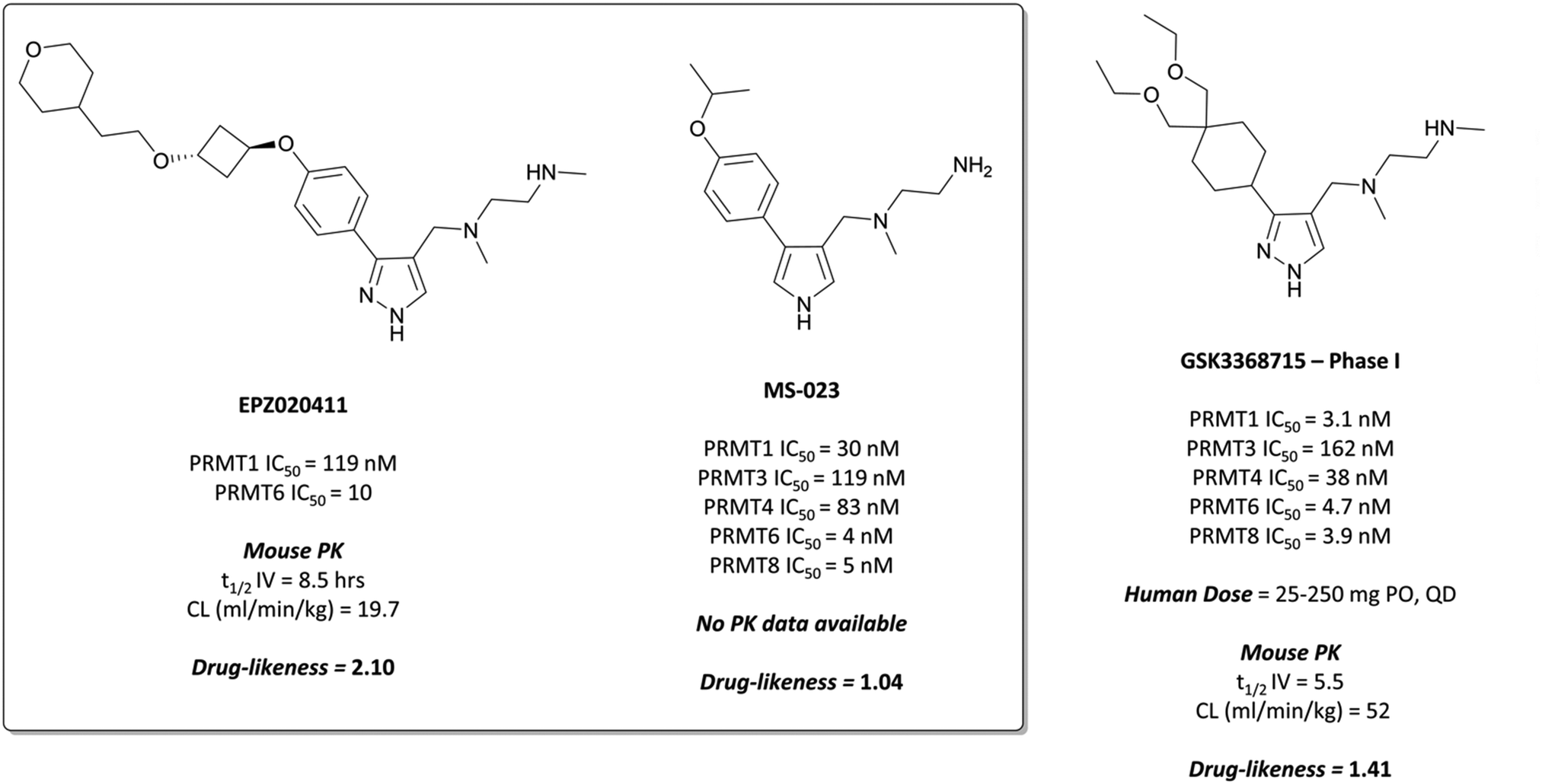 | ||
| Fig. 21 PRMT6 chemical probe EPZ020411 and type I PRMT probe MS-023, alongside clinical candidate GSK3368715. | ||
Inspired by the discovery of EPZ020411, researchers at Mt Sinai and the SGC Toronto began their own investigations into the development of a type I PRMT selective chemical probe.233 They recognised the ethylenediamino moiety as an arginine mimetic and thus essential component of any potential probe, but also that the bulky aryl substituent of EPZ020411 was crucial for PRMT6 selectivity, so opted for smaller substituents to enable targeting of other type I PRMTs.
Evaluation of 1,2,3-triazole- and pyrrole-based probes led to better understanding of the contribution of ring electronics to binding.233 The para-vector off the aryl ring remained important for activity against type I PRMTs, with meta-substituted aryl rings displaying far lower potency. Iterative compound design resulted in MS-023 (Fig. 21), which contained a pyrrole core, para-isopropoxy group, and a terminal primary amine, in contrast to the terminal secondary amine of EPZ020411. MS-023 was highly potent against the type I PRMTs, with IC50s ranging from 4 to 119 nM across them (Fig. 21), with relatively good drug-likeness.
MS-023 showed no inhibition of type II/III PRMTs up to 10 μM, nor of 25 KMTs and DNA methyltransferases (DNMTs), or of three KDMs.233 It was shown to be non-competitive with either SAM or peptide substrates, thought to be because affinity for the peptide derives from interactions removed from the arginine binding pocket, and thus inhibition has no effect on binding. Crystal structure analysis demonstrated that the ethylenediamine moiety did indeed occupy the arginine binding pocket, with the terminal amine forming both direct and water-mediated hydrogen bonds with residues in the pocket. Much like the interactions observed with EPZ020411, the pyrrole engaged in hydrogen bonding and the aryl ring in π–π interactions with a binding site tyrosine residue.
Cell assays showed that MS-023 could reduce both PRMT1- and PRMT6-dependent arginine methylation marks in a concentration dependent manner (IC50 = 9 nM and 56 nM for PRMT1, -6 respectively). When evaluated as a pan-type I PRMT inhibitor, a global decrease in arginine asymmetric dimethylation was observed, along with a concurrent increase in monomethylation, an effect consistent with PRMT1 knockout.234 Cell growth arrest was observed at concentrations as low as 100 nM over 10 days in a breast cancer line. These results cemented EPZ020411 and MS-023 as first-in-class probes for the investigation of type I PRMT biology.
GSK3368715 was selective over the remaining PRMTs (>100-fold) and several other methyltransferases with less than 20% inhibition observed at 10 μM.235 Crystal structures of the compound in complex with PRMT1, along with kinetic studies, suggested a similar mode of binding to EPZ020411 and MS-023. GSK3368715 induced a global loss of asymmetric dimethylarginine (ADMA) in a panel of cancer cell lines, with an IC50 of 13.6 nM, and a concurrent increase in monomethyl- and symmetric dimethylarginine.
Anti-proliferative effects were observed across a variety of cell lines derived from both haematological and solid tumours.235 The most sensitive lines to type I PRMT inhibition were lymphoma and AML lines, with some subsets of NSCLC and pancreatic cancer lines also displaying sensitivity. Favourable PK properties, particularly oral bioavailability, allowed for further evaluation of these effects in mouse xenograft models of solid and haematological malignancies. Once-daily oral dosing at 150 and 300 mg kg−1 led to significant tumour growth inhibition in models of DLBCL, pancreatic adenocarcinoma, clear cell renal carcinoma and triple-negative breast cancer.
As a result of the promising effect of pan-type I PRMT inhibition, and following further safety and PK profile evaluation, GSK3368715 entered phase I trials for the treatment of solid tumours and DLBCL in 2018 (NCT03666988, expected completion date 2022).235 In the design of MS-023, it was noted that reducing bulk at the para-position of the aryl ring would enable the targeting of other type I PRMTs beyond PRMT6, a strategy that has yielded dividends. Indeed, overlaying the crystal structures of both compounds shows the same binding mode, occupying the pocket in almost exactly the same fashion (Fig. 22).
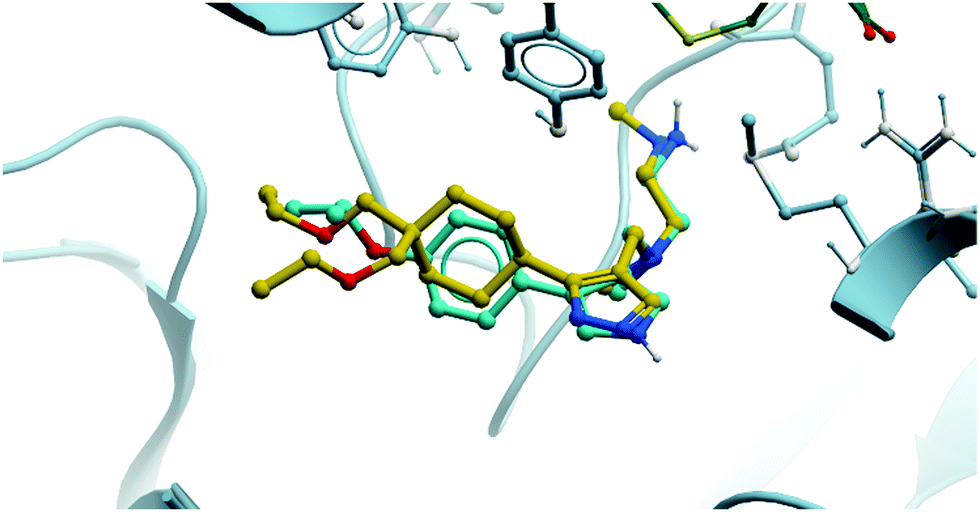 | ||
| Fig. 22 Comparison of the binding modes of MS-023 (cyan) and GSK3368715 (yellow), showing almost exactly the same pocket occupancy by both molecules. | ||
The probes: EPZ015666 and EPZ015866
In order to validate its clinical relevance, researchers from Epizyme and GSK began development of a selective probe for PRMT5.252,253 An initial, peptide-competitive, tetrahydroisoquinoline (THIQ) hit (Fig. 23, 13) from an HTS showed inhibition of PRMT5, with reasonable physiochemical properties. SAR around this scaffold demonstrated that the THIQ motif was essential, forming a cation-π interaction with the partial positive charge of the SAM methyl group. It was also involved in a π-stacking interaction with Phe327, and removal of the THIQ phenyl ring led to a complete loss of activity.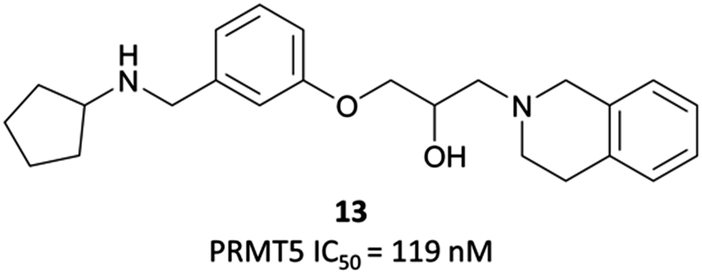 | ||
| Fig. 23 Tetrahydroisoquinoline hit 13. | ||
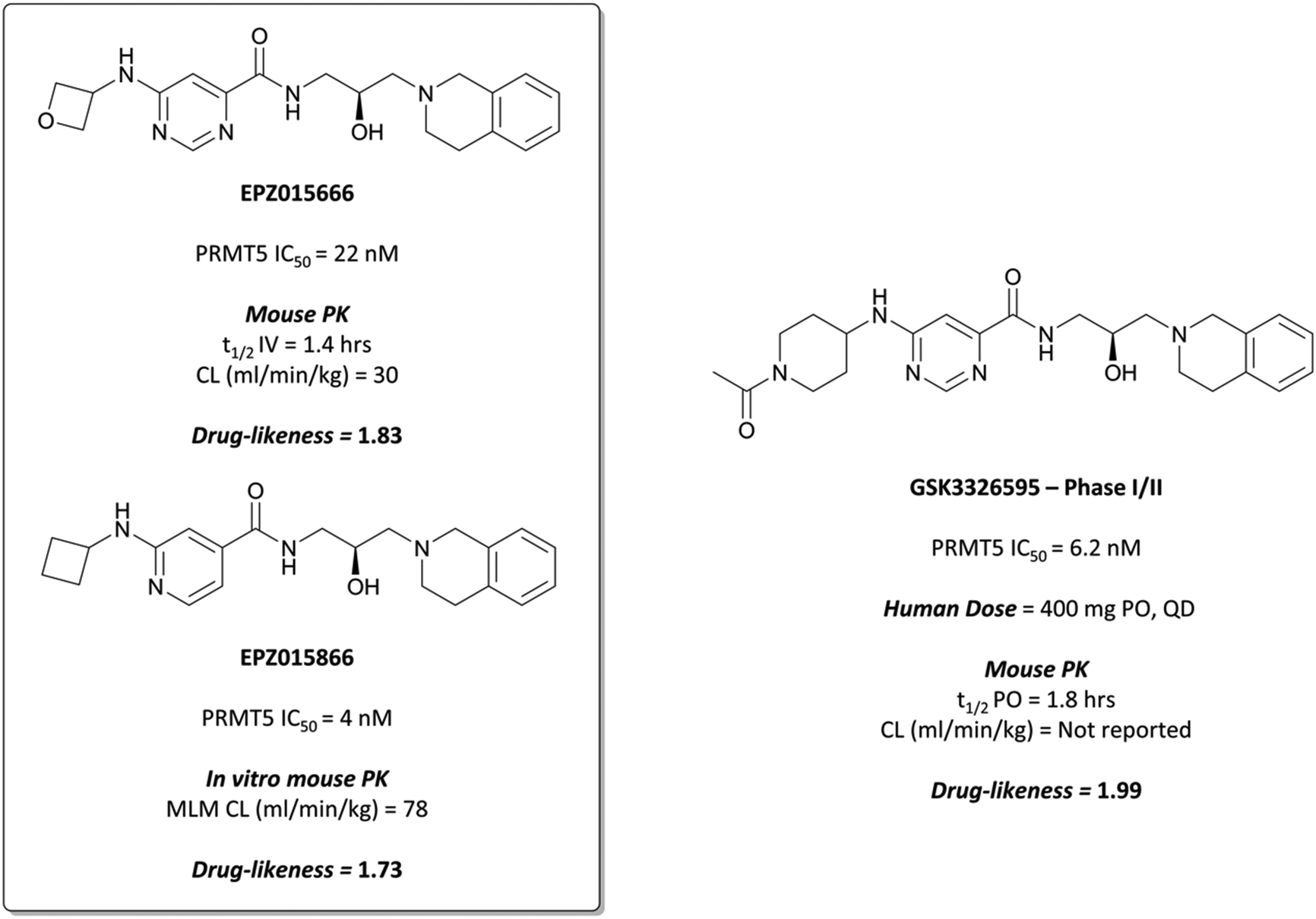
Although their initial derivatives showed excellent potency, this was accompanied by significant instability in microsomal assays, with clearances approaching hepatic blood flow rates.253 To address this issue, they sought to reduce the clogD by incorporating less lipophilic amide analogues. In doing so, they identified EPZ015666 (Fig. 24) and EPZ015866 (Fig. 24), with IC50s of 22 nM and 4 nM respectively against PRMT5. Both retained potency but EPZ015666 showed markedly improved clearance in both human and mouse liver microsomes. Both were highly selective over other protein methyltransferases, with no inhibition observed at 50 μM. EPZ015666 showed a dose-dependent reduction in cellular symmetrical dimethylarginine marks, with IC50s (western blot) ranging between 4–347 nM in mantle cell lymphoma lines.252 Both EPZ015666 and EPZ015866 potently inhibited the proliferation of a lymphoma cell line, with IC50s of 351 nM and 62 nM respectively.
 | ||
| Fig. 24 In vivo probe EPZ015666 and in vitro probe EPZ015866, alongside the structurally derived clinical molecule GSK3326595. | ||
Despite good drug-likeness, the poor PK properties of EPZ015866 precluded its use in vivo,253 but EPZ015666 was evaluated against mouse xenograft models of mantle cell lymphoma.252 Twice-daily oral dosing led to dose-dependent tumour growth inhibition with no significant weight loss observed at the highest dose. Both compounds represented the first PRMT tool compounds, with EPZ015666 orally bioavailable and suitable for in vivo studies.
GSK3326595 induced a reduction in global symmetric dimethylarginine in various cancer cell lines, with EC50s ranging from 2.5–180 nM, and was selective (>4000-fold) over 20 other methyltransferases. It inhibited growth and induced cell death in various cancer lines with gIC50s ranging from 2.5 nM to over 10 μM, and breast, AML, and MM lines displaying the highest degree of sensitivity. Subsequently, it was evaluated in a mouse xenograft model of lymphoma, resulting in dose-dependent tumour growth inhibition and regression following 21 days oral dosing, with no significant weight loss observed.
A phase I trial to evaluate the effect of GSK3326595 in patients with solid tumours and non-Hodgkin's lymphoma began in August 2016 (NCT02783300).255,256 Thus far, there has been a strong pharmacodynamic effect observed, accompanied by clinical activity in several tumour types, with an expected completion date of April 2025. A second phase I trial of GSK3326595 began in October 2018 for patients with myelodysplastic syndrome and AML (NCT03614728, expected completion date March 2023).257
The probe: LLY-283
In collaboration with Eli Lilly, researchers at SGC Toronto identified LLY-283 (Fig. 25) as a potent, SAM-pocket binding inhibitor of PRMT5,258 in contrast to EPZ015666 and associated peptide-competitive inhibitors. LLY-283 is deazadenosine-based and adopts a similar binding pose in the pocket to SAM. The phenyl moiety causes a conformational change, acting to displace the Phe237 side chain which adopts an alternate structure compared to when bound to the peptide substrate.259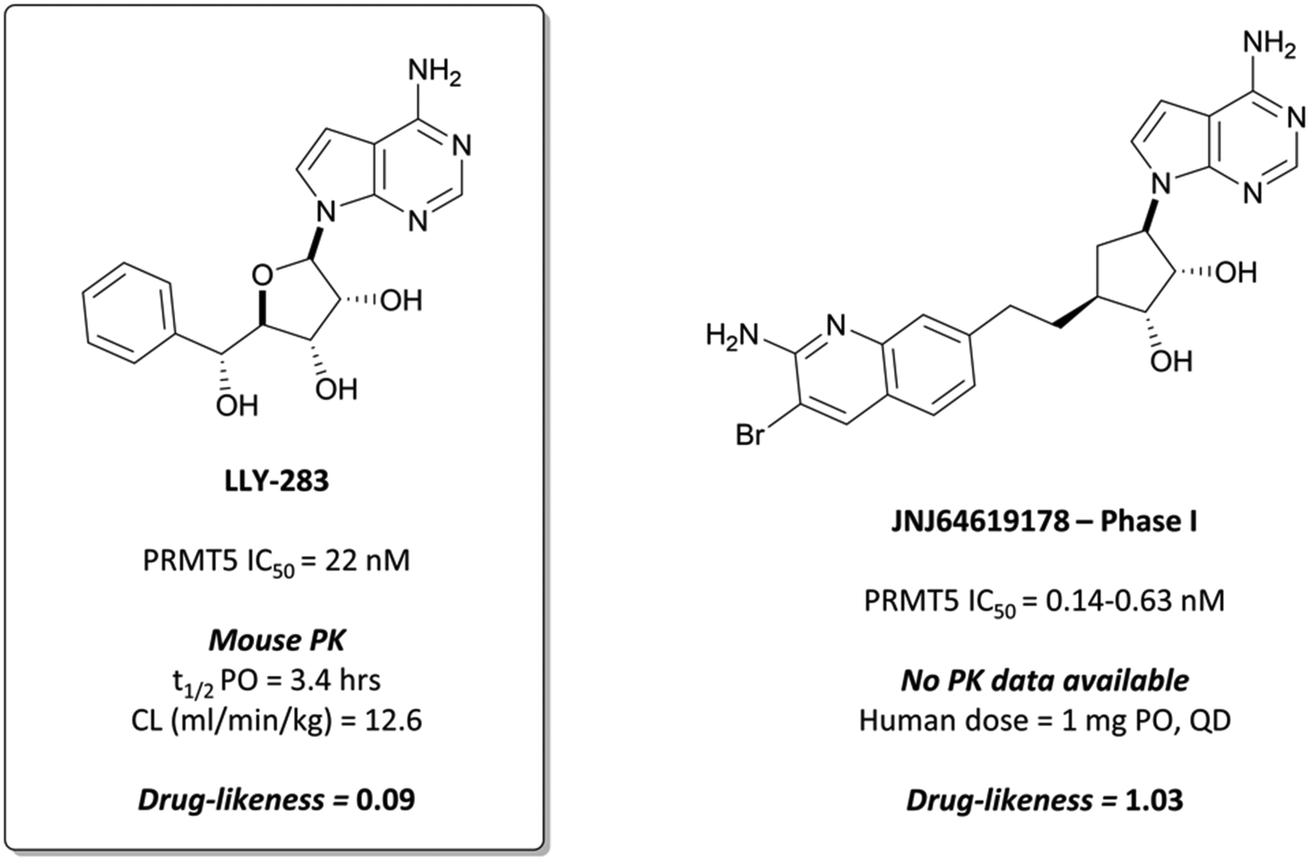 | ||
| Fig. 25 SAM competitive PRMT5 probe LLY-283, and the structurally related clinical candidate JNJ64619178. | ||
LLY-283 was a potent inhibitor of PRMT5, with an IC50 (SPA) of 22 nM.258 Despite clear binding to the SAM pocket being observed, the compound wasn’t competitive with either SAM or the peptide substrate when evaluated against differing concentrations of each, with no clear explanation available. This had been previously observed for other compounds and methyltransferases (for example MS-023),233 however not when binding was occurring in the SAM pocket. LLY-283 was selective against a panel of 32 other methyltransferases, with no loss of activity observed for any of the panel at 10 μM. It inhibited the symmetric dimethylation of SmBB′ protein with an IC50 of 25 nM in a breast cancer line and was effective at inducing antiproliferative effects in various cancer cell lines, with haematological tumours the most sensitive (IC50s (SPA) = 3–85 nM).
Favourable PK properties enabled oral dosing of LLY-283 in a mouse xenograft model of melanoma. Once-daily, 20 mg kg−1 dosing over 28 days led to significant tumour growth inhibition, with no significant weight loss observed. Alongside the peptide-competitive inhibitors, LLY-283 provides a useful addition to the toolbox of PRMT5 inhibitors.
These results led Janssen to advance JNJ64619178 to phase I trials in 2018 for the treatment of relapsed/refractory B cell non-Hodgkin lymphoma or advanced solid tumours (NCT03573310).268 Thus far, any observed adverse events have been manageable and there has been evidence of clinical activity. Full results are expected to be reported in July 2022.
Beyond epigenetic probes
Whilst the focus of the review has thus far been on probes for epigenetic targets, there are other examples of chemical probes leading to drugs for other target classes.7 We shall examine a select few, along with the role they have played in inspiring clinical molecules.RIPK1
Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) plays a key role in deciding the fate of the cell in response to various pro-death and inflammatory stimuli.269 It is involved in the regulation of necroptosis, a form of non-apoptotic cell death implicated in various inflammatory and neurodegenerative diseases such as multiple sclerosis (MS),270 amyotrophic lateral sclerosis (ALS),271 and Alzheimer's disease (AD).272 RIPK1 activity is regulated by tumour necrosis factor (TNF) binding to TNF receptor 1 (TNRF1), which leads to the RIPK1-kinase activity-dependent formation of a protein complex that induces necroptosis.273–275 Dependent on the context, RIPK1 activation can also cause apoptosis and neuroinflammation.269The probe: GSK′481
As a kinase, RIPK1 presents an attractive target for the development of small-molecule inhibitors of pro-necroptotic and pro-inflammatory pathways. The first identified RIPK1 inhibitors were the necrostatins, which displayed remarkable selectivity for RIPK1 over other kinases but suffered from relatively poor potency and metabolic stability.276–278 In an attempt to identify novel and improved RIPK1 inhibitors, scientists at GSK optimised an HTS hit and developed GSK′963 (Fig. 26), which had an IC50 (FP) of 29 nM and was selective (>10000-fold) for RIPK1 over 339 other kinases.279 Despite this, GSK′963 had poor oral exposure in rodents, which precluded its development as an in vivo probe.
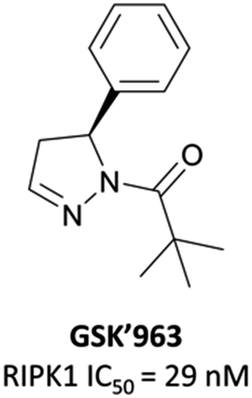 | ||
| Fig. 26 RIPK1 inhibitor GSK′963. | ||
Undeterred, the scientists at GSK screened a DNA-encoded small-molecule library against RIPK1 in the search for compounds that combined high potency and selectivity for RIPK1 with good PK properties.280 The screen identified a set of amino acid building blocks that were structurally distinct from more commonly known kinase hinge-binding motifs, combinations of which were synthesised and evaluated for potency against RIPK1. One such combination was the benzoxazepinone-based GSK′481 (Fig. 27), which had an IC50 (FP) of 10 nM against RIPK1 and was selective (>7500-fold) over 456 other kinases.280 In the human monocytic U937 cellular assay, which measures the inhibition of necroptosis induced by TNF and a caspase inhibitor, GSK′481 maintained its on-target potency with an IC50 of 10 nM. Despite this, a favourable PK profile remained elusive; GSK′481 was very lipophilic with high clearance and poor oral exposure in rats.281 Nevertheless, its favourable in vitro profile makes it an attractive tool for probing RIPK1 biology in cells.
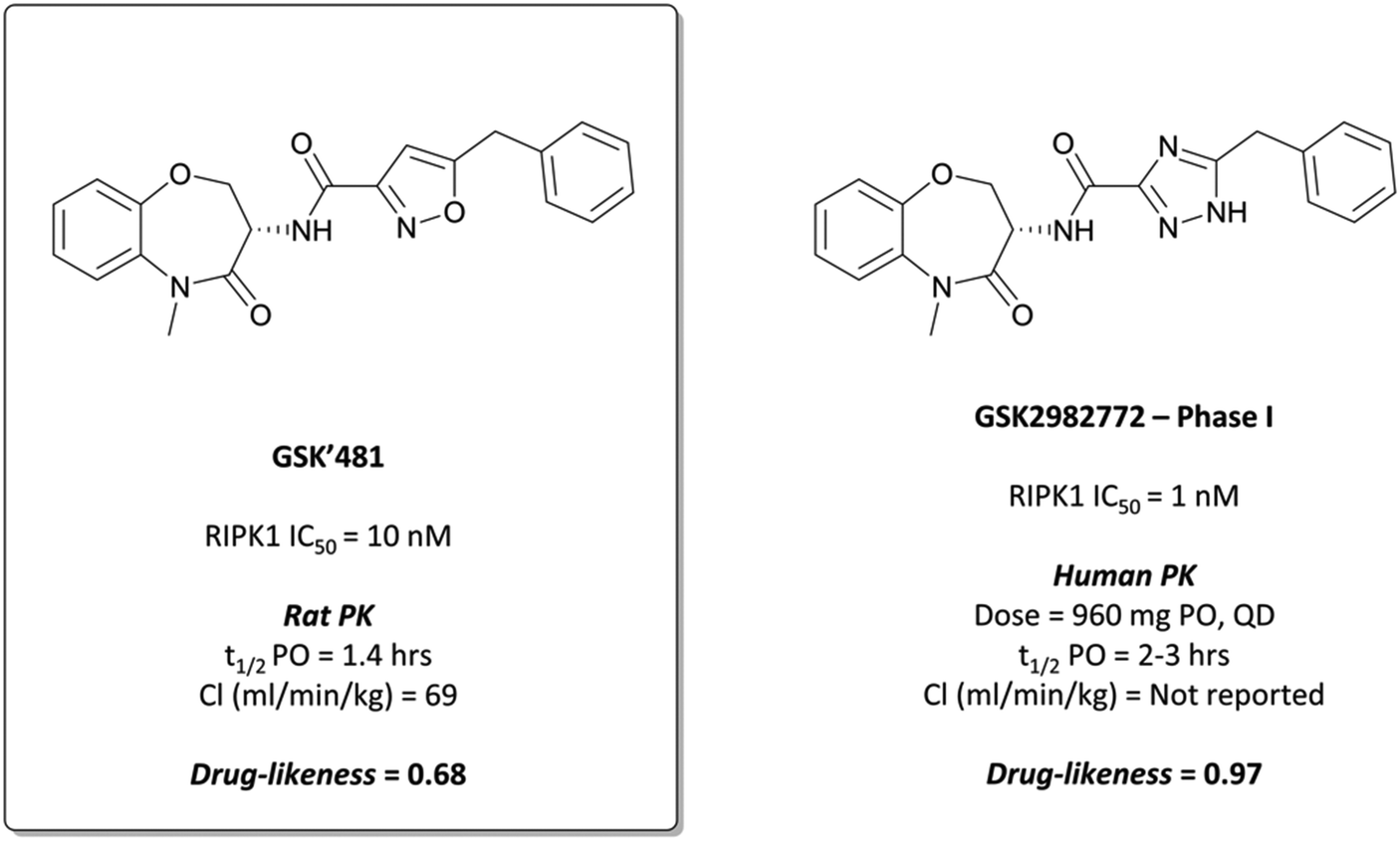 | ||
| Fig. 27 RIPK1 chemical probe GSK′481, from whose benzoxazepinone scaffold is derived clinical candidate GSK2982772. | ||
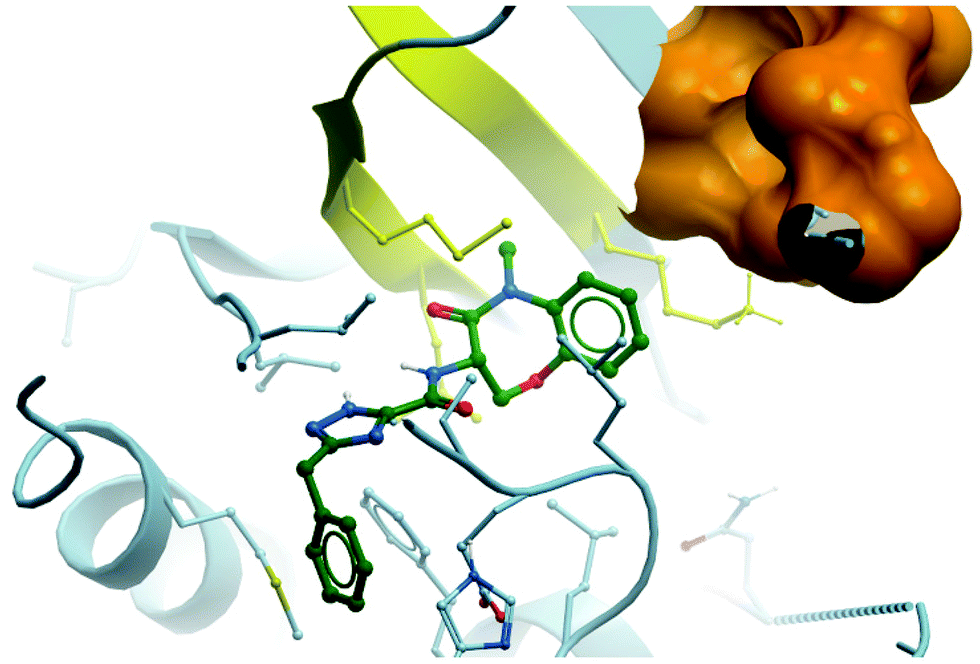 | ||
| Fig. 28 Binding mode of GSK2982772 (green) to RIPK1, showing the tight pocket formed by two β-strands (yellow) around the benzoxazepinone moiety, with the ATP binding region shown in orange (PDB: 5TX5). | ||
Eventually, modification of the central isoxazole led to the greatest improvements in lipophilicity, oral exposure, and solubility.281 The introduction of a 1,2,4-triazole resulted in GSK2982772 (Fig. 27), which had an IC50 (FP) of 1 nM against RIPK1. Crucially, GSK2982772 had a clogD of 3.8, which was a two-log improvement over GSK′481 (clogD = 5.9), and displayed a seven-fold improvement in both solubility and oral exposure in rats, echoed in an improvement in drug-likeness. A crystal structure of GSK2982772 in complex with RIPK1 showed it didn’t interact with any of the hinge residues and was instead binding more deeply within the ATP binding pocket (Fig. 28).281 The benzoxazepinone moiety sits in a pocket tightly flanked by two β-strands, explaining why any structural modification that resulted in a change in its conformation was not tolerated. This unexpected binding mode resulted in exquisite selectivity (>10000-fold) for RIPK1 over 339 other kinases evaluated.
GSK2982772 potently inhibited (as measured by the inhibition of RIPK1-dependent inflammatory cytokine production) necroptosis induced by caspase and apoptosis inhibitors in human primary neutrophils and whole blood (IC50 = 1.6 and 2 nM respectively).281 It reduced cytokine production from ulcerative colitis explant tissue in a dose-dependent fashion and was 93% protective in a mouse model of TNF-induced lethal shock after dosing at 50 mg kg−1. GSK2982772 displayed favourable PK properties, including good clearance, half-life, and oral bioavailability.281,283
GSK2982772 was advanced to the clinic for the treatment of peripheral autoimmune diseases in 2015.283 Excellent target engagement was achieved at well-tolerated doses, with no serious adverse events reported. Following this, it entered phase II trials in rheumatoid arthritis (NCT02858492), ulcerative colitis (NCT02903966), and plaque-type psoriasis (NCT02776033) in 2017.283,284 The drug was well tolerated however efficacy for these indications has not yet been demonstrated.284,285 It was re-entered into phase 1b trials in 2020 for psoriasis at a much higher dose (960 mg vs. 60 mg QD, NCT04316585), with results expected in October 2021.
FXR
Bile acids are amphipathic molecules that act as regulators of a diverse array of processes, such as lipid and glucose metabolism.286 They undergo enterohepatic circulation which acts as an important means of regulating their synthesis and transport, along with exerting a wider effect on whole-body lipid metabolism. Bile acids are the natural ligands for the farnesoid X receptor (FXR), a nuclear receptor that plays a key role in this process.287,288 Activation of FXR initiates a cascade that reduces the conversion of cholesterol to bile acids by repressing the transcription of the cholesterol 7α-hydroxylase gene (CYP7A1), a critical enzyme for bile acid synthesis.286,289 As well as this, FXR regulates the expression of several lipid-modifying proteins, modulating lipogenesis through the reduction of triglyceride levels.289,290Loss-of-function mutations in FXR and reduced FXR expression are associated with cholestasis, a condition resulting from the accumulation of bile salts due to impaired or obstructed bile flow.291 This build-up leads to the destruction of intrahepatic bile ducts and causes primary biliary cholangitis (PBC), a chronic and potentially fatal disease.292 FXR has also been implicated in nonalcoholic steatohepatitis (NASH), a condition caused by the accumulation of fat in the liver that leads to inflammation and fibrosis.293 As a result, FXR agonists that can suppress lipogenesis and the production of bile acids are of great interest as potential therapeutics for these diseases.
The probe: GW4064
Following the identification of FXR as a receptor for bile acids,287,288 scientists at GSK began a combinatorial library screen to discover a chemical tool to probe the pharmacology of the receptor.294 Initially, isoxazole 15 (Fig. 29) showed partial agonist activity (EC50 (FRET) = 70 nM) and was the basis for further exploration of SAR, with a focus on modulation of the three aromatic rings. Isoxazoles with bulky, lipophilic substituents were favoured, leading to the discovery of GW4064 (Fig. 31), which had an EC50 (FRET) of 15 nM against the FXR. In cells transfected with human and mouse FXR expression vectors, GW4064 had EC50s (FRET) of 90 and 80 nM respectively and was selective (>100-fold) against other nuclear receptors.294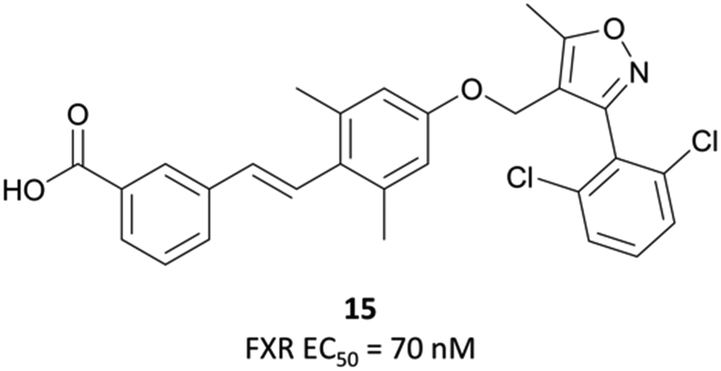 | ||
| Fig. 29 Initial isoxazole hit 15 from a combinatorial library screen against FXR. | ||
GW4064 induced a dose-dependent decrease in serum triglycerides in rats, with an ED50 of 20 mg kg−1, verifying a role for FXR in the regulation of lipid metabolism. GW4064 had a modest half-life (3.5 hours) but was poorly orally bioavailable (10%), precluding further in vivo evaluation. Despite this, it was the first nonsteroidal FXR agonist identified and remains a useful tool for the probing of FXR biology.
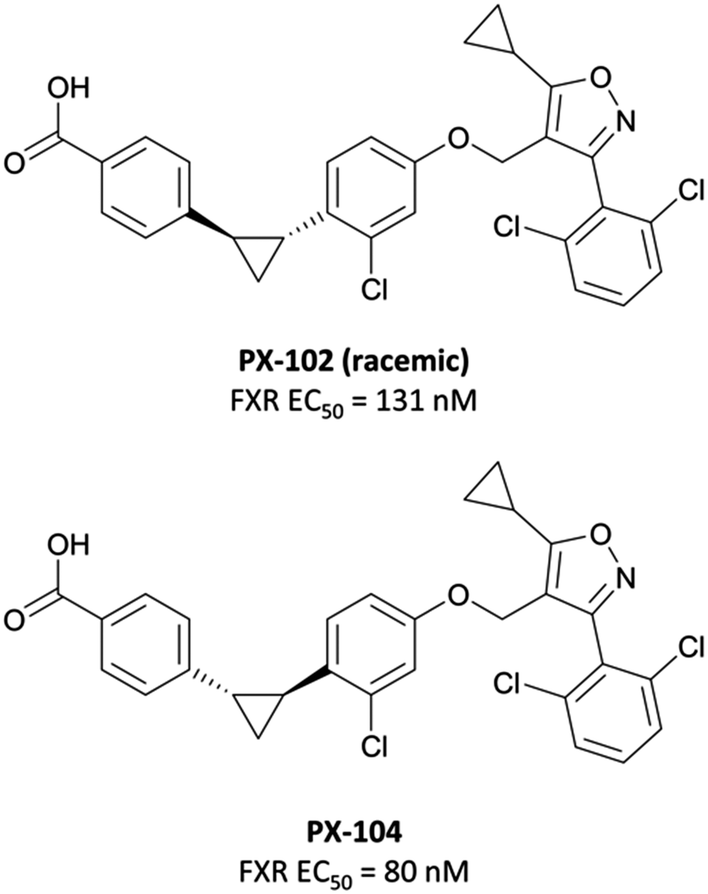 | ||
| Fig. 30 Racemic, cyclopropyl-based FXR agonist PX-102 and its eutomer PX-104. | ||
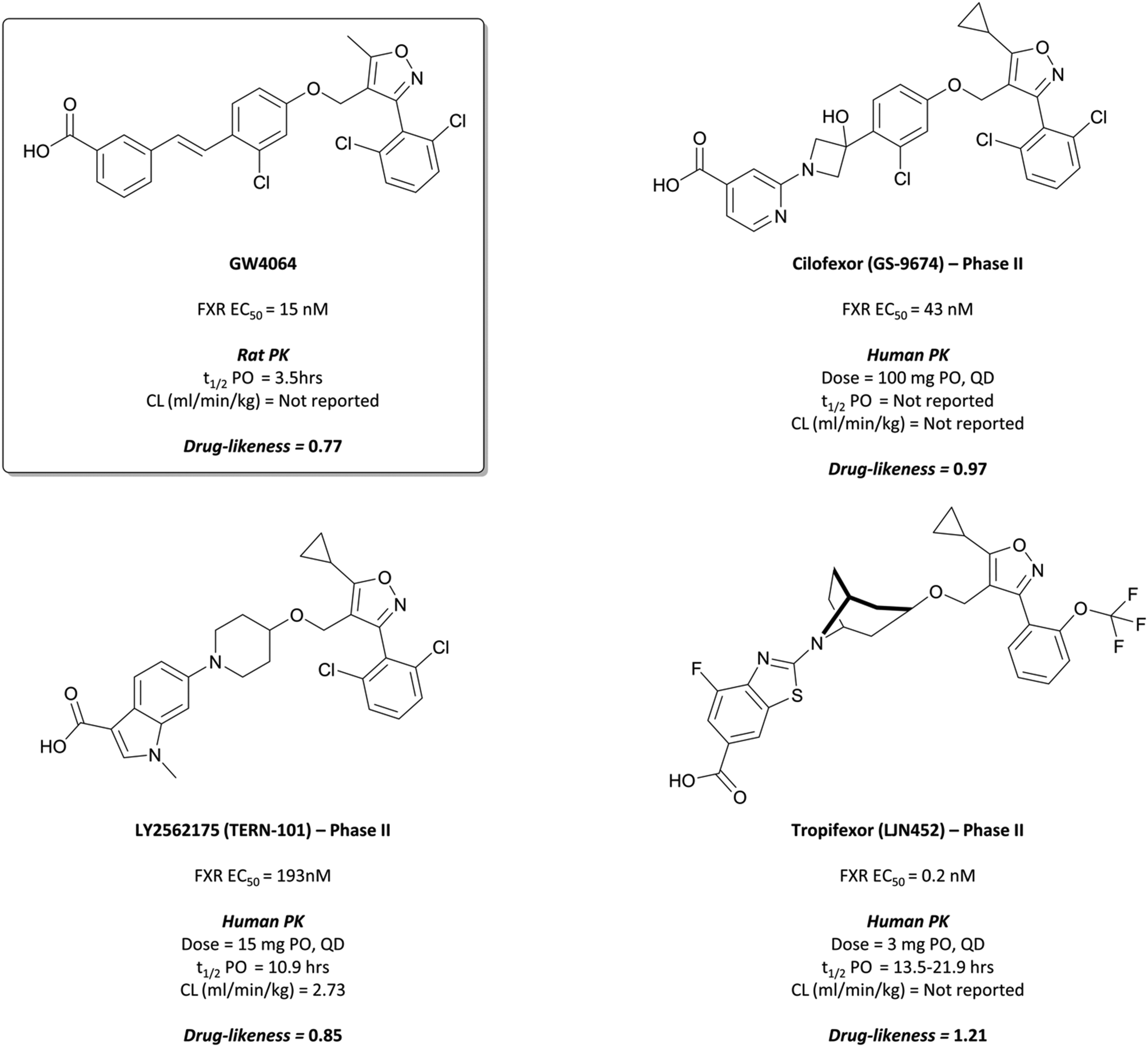 | ||
| Fig. 31 Pioneering early FXR agonist GW4064 and the isoxazole-containing drugs it inspired: Cilofexor, LY2562175, and Tropifexor. | ||
The results of these trials have not been published, however scientists at Phenex reported that the chiral cyclopropyl linker was synthetically inaccessible in a cost-efficient manner, leading to the need for appropriate achiral alternatives.300 This was achieved with the introduction of an hydroxy-azetidinyl linker, with further modification to include a 4-carboxylpyridine leading to the more drug-like GS-9674, or cilofexor (Fig. 31).300,303Cilofexor has an EC50 (FRET) of 43 nM for the FXR and demonstrated anti-inflammatory and anti-fibrotic effects in preclinical models of liver fibrosis.304 It is currently in phase III trials for primary sclerosing cholangitis (NCT03890120, expected completion August 2023). Thus far, it has shown significant reductions in bile acids and improvements in markers of cholestasis at tolerable doses, with no severe adverse events reported.305 As well as this, a phase 2 trial in patients with NASH (NCT02854605) demonstrated that at tolerable doses cilofexor induced significant reductions in hepatic steatosis (lipid retention) as well as serum bile acid levels.306
LY2562175 was orally bioavailable and showed favourable PK properties across species (mouse, dog, and monkey), with low clearance and a good half-life.308 It was progressed to phase I for evaluation of safety and PK and was well-tolerated at doses as high as 600 mg, with a half-life of 16–24 hours supporting a once-daily dosing regimen. LY2562175 was subsequently licensed to Terns Pharmaceuticals,309 where it was rebranded as TERN-101 and evaluated in models of NASH310 (Terns poster ref). It was shown to reduce liver steatosis and inflammation, thus inspiring Terns to evaluate novel formulations of the drug in a phase I trial, investigating their absorption and PK311 (pharmacokinetics of two oral formulations poster). The tablet formulation gave faster absorption and better systemic exposure and is currently being investigated in phase II trials for the treatment of NASH (NCT04328077, results expected May 2021).
000-fold) for FXR over other enzymes, ion channels, nuclear receptors, and GPCRs.
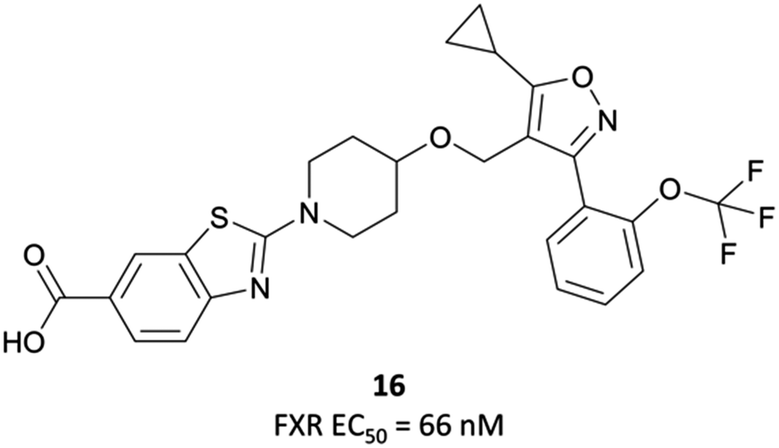 | ||
| Fig. 32 LY2562175 analogue with improved potency and metabolic stability. | ||
In rats, tropifexor was shown to potently induce the expression of FXR-dependent genes at doses as low as 0.01 mg kg−1, and to lower the levels of serum triglyceride following oral administration.291Tropifexor displayed favourable PK properties across species, prompting its evaluation in a phase I trial for safety and tolerability. Doses up to 3 mg were well-tolerated, with a moderate rate of absorption and half-life of 13.5–21.9 hours, supporting a once-daily oral dosing regimen.312 It was subsequently entered into phase II trials for PBC, where it has been shown to reduce the amount of γ-glutamyl transferase (GGT), a liver enzyme that acts as a biomarker for liver disease(novartis reference).313 A once-daily dose of 0.09 mg for 28 days reduced GGT levels by 72% compared to a placebo, with no significant adverse effects reported. In a phase II trial for NASH, 0.2 mg tropifexor induced a 64% reduction of hepatic fat fraction with no serious adverse events reported.314
BCL-2
Finally, an analysis of the value of high-quality chemical probes for drug discovery would be remiss without discussion of the development of the BCL-2 selective chemical probe ABT-737 (Fig. 34),315,316 and how it was essential to the discovery of the highly effective chemotherapeutic venetoclax (Fig. 34).The BCL-2 family of proteins are critical regulators of programmed cell death, functioning within a complex network of protein–protein interactions.316,317 The first identified, BCL-2 was found to inhibit apoptosis and prolong survival in cancer lines,318,319 and further anti-apoptotic members such as BCL-XL and MCL1 were subsequently identified.317,320,321 These anti-apoptotic proteins are often highly expressed in cancer cells, contributing to tumour initiation and progression, as well as resistance to therapy.322,323 Targeting the BCL-2 family of proteins is challenging: extended, hydrophobic interactions with endogenous ligands324 and a high degree of sequence similarity with related proteins makes the design of selective and potent inhibitors incredibly difficult.316
The probe: ABT-737
In the pursuit of a potent inhibitor of the BCL-2 family of proteins, researchers from Abbott carried out a high-throughput NMR screen of a chemical library against BCL-XL.315 They identified two compounds (Fig. 33, 17 and 18) that bound to two distinct sites in the BH3 binding cleft, observing similar binding modes to the endogenous peptide substrate.324 Substitution of the carboxyl of 17 with a bioisosteric acylsulfonamide provided an optimal vector through which to link the two sites, whilst maintaining the position of the acidic proton within the pocket. Further SAR led to compound 19 (Fig. 33), with 18 effectively replaced by a 3-nitro-4-(2-phenylthioethyl)aminophenyl group that spanned both initial binding sites. 19 had a Ki (FP) of 36 nM against BCL-XL but showed markedly reduced activity in the presence of human serum albumin (HSA). Analysis of the crystal structures of 19 in complex with BCL-2 and HSA showed structural features that were solvent exposed in complex with BCL-2 were engaged in hydrophobic interactions in complex with HSA. To reduce affinity for HSA, these features were modified with polar groups, including the incorporation of a 2-dimethylethylamino group and a substituted piperazine. Finally, the introduction of a bis-phenyl moiety off the piperazine to probe a lipophilic pocket in the binding cleft resulted in ABT-737, which had a Ki (FP) of less than 1 nM for the highly homologous BCL-2, BCL-XL and BCL-w, with activity maintained in the presence of HSA. ABT-737 showed selectivity over the less homologous family members BCL-B, MCL-1, and A1 (Ki (FP) = 0.46 μM, >1 μM, >1 μM respectively).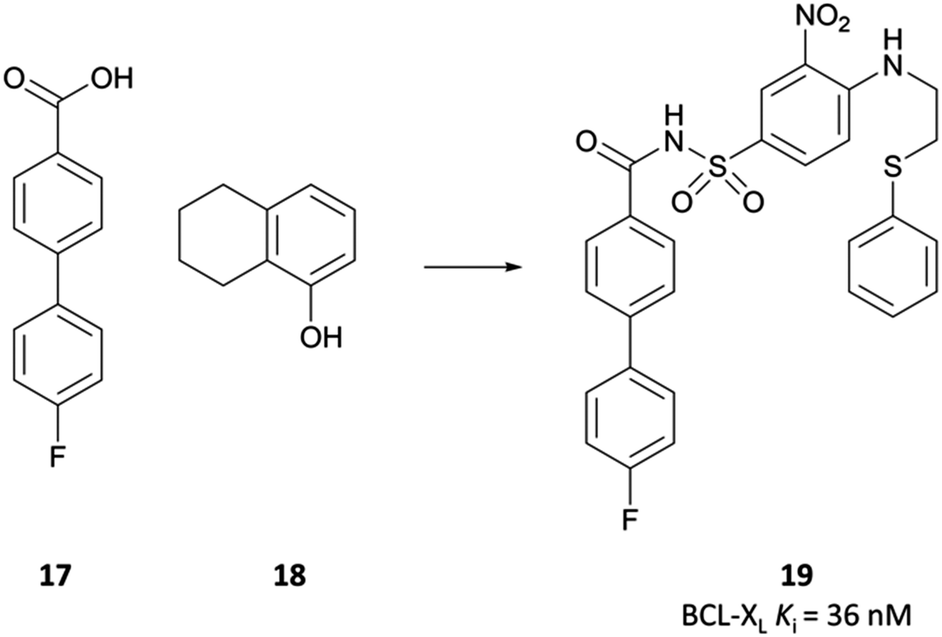 | ||
| Fig. 33 NMR screening identified compounds 17 and 18 that bound to distinct sites on BCL-XL. Linking the two, with some scaffold modulation, led to 19. | ||
 | ||
| Fig. 34 Chemical probe for the BCL-2 family of proteins ABT-737. Its derivative Navitoclax displayed DLTs in the clinic due to BCL-XL inhibition. The next iteration, Venetoclax, Is selective for BCL-2 over BCL-XL, which led to far fewer adverse events and, coupled with high efficacy, its eventual approval for use. | ||
ABT-737 was shown to increase the sensitivity of various cancer cell lines to other chemotherapeutic agents, as well as to be cytotoxic itself, inducing cell death in patient-derived lymphoma cells at sub-micromolar concentrations.315 This cytotoxicity was replicated in solid tumours, with complete tumour regression observed in a mouse xenograft model of SCLC following intraperitoneal (IP) dosing at 100 mg kg−1. This was a breakthrough moment for investigating the BCL-2 family of proteins, an incredibly potent and selective probe that demonstrated excellent efficacy in cellular and animal models of disease.
P less than five, and fewer than five hydrogen bond donors and ten hydrogen bond acceptors.79 A quick glance at ABT-737 shows it lies far outside this space, with both a high molecular weight and clogP. Nevertheless, the Abbott researchers decided to attempt to modify its scaffold in an effort to improve its physiochemical properties for oral exposure.
They began by exploring changes to the arylsulfonamide and amine functionalities, reasoning that modulation of their pKas would alter the ionisation state of the molecule at physiological pH and improve oral absorption.326,327 They also investigated modifications to potential sites of metabolism, focusing on N-demethylation of the dimethylamino functionality and oxidative metabolism of the biphenyl moiety. Several initial changes that resulted in improved oral exposure also led to reduced cellular efficacy, however provided encouraging evidence that changes at these sites could result in the desired improvements.
Metabolism of the dimethylamino group was eventually mitigated with the introduction of a morpholine group, whose lower pKa led to a modest improvement in oral exposure (4-fold) whilst maintaining cellular potency.327 Introduction of a triflone moiety to replace the aryl nitro substituent modestly increased the potency but crucially lowered the polar surface area, leading to a 7-fold increase in exposure. Finally, replacement of one of the phenyl groups on the lower half of the molecule with a gem-dimethylcyclohexene resulted in a sub-nanomolar cellular potency. Combining this with the above changes resulted in navitoclax (Fig. 34), which maintained its potency against BCL-XL, BCL-2 and BCL-w, but displayed a 22-fold higher oral exposure than ABT-727 in mice, mirrored in an improvement in drug-likeness.
As a result, navitoclax was suitable for oral dosing in mouse xenograft models of SCLC and acute lymphocytic leukaemia (ALL).325 Once-daily, 100 mg kg−1 dosing led to complete tumour regression after 21 days, demonstrating equivalent effects to IP dosing of ABT-737. Navitoclax was subsequently advanced to the clinic for the treatment of chronic lymphocytic leukaemia (CLL), SCLC and other solid tumours.328–330 Some clinical activity was observed, however it induced thrombocytopenia in a dose-limiting fashion,331 and as a result it became difficult to safely reach effective concentrations in patients.
They approached this by systematically removing key structural features to examine the effect on binding.334 It was discovered that removal of the thiophenyl motif imparted some selectivity for BCL-2, however with lower potency than navitoclax. A crystal structure of this analogue revealed that a second BCL-2 molecule had bound to form a dimer, with a tryptophan residue creating a π-stacking interaction with the trifloylaryl and a hydrogen bond with a binding site aspartate residue. This proved to be the key: this residue was one of the few not shared between the binding domains of BCL-2 and BCL-XL.
Introduction of an azaindole moiety tethered to the central core of the compound recapitulated the interaction, engaging the aspartate along with an arginine residue (Fig. 35b).334,335 Other subtle changes, for example substitution of the morpholine with a pyran, reintroduction of the nitroaryl, and a gem-dimethycyclohexene regioisomer resulted in venetoclax, which had a Ki (FP) of <0.01 nM for BCL-2 compared to 48 nM for BCL-XL and 245 nM for BCL-w, greater than 300-fold selectivity. This selectivity was also observed when the compound was tested against cell lines engineered to be dependent on either BCL-2 or BCL-XL, with substantially lower activity against the BCL-XL dependent cells.
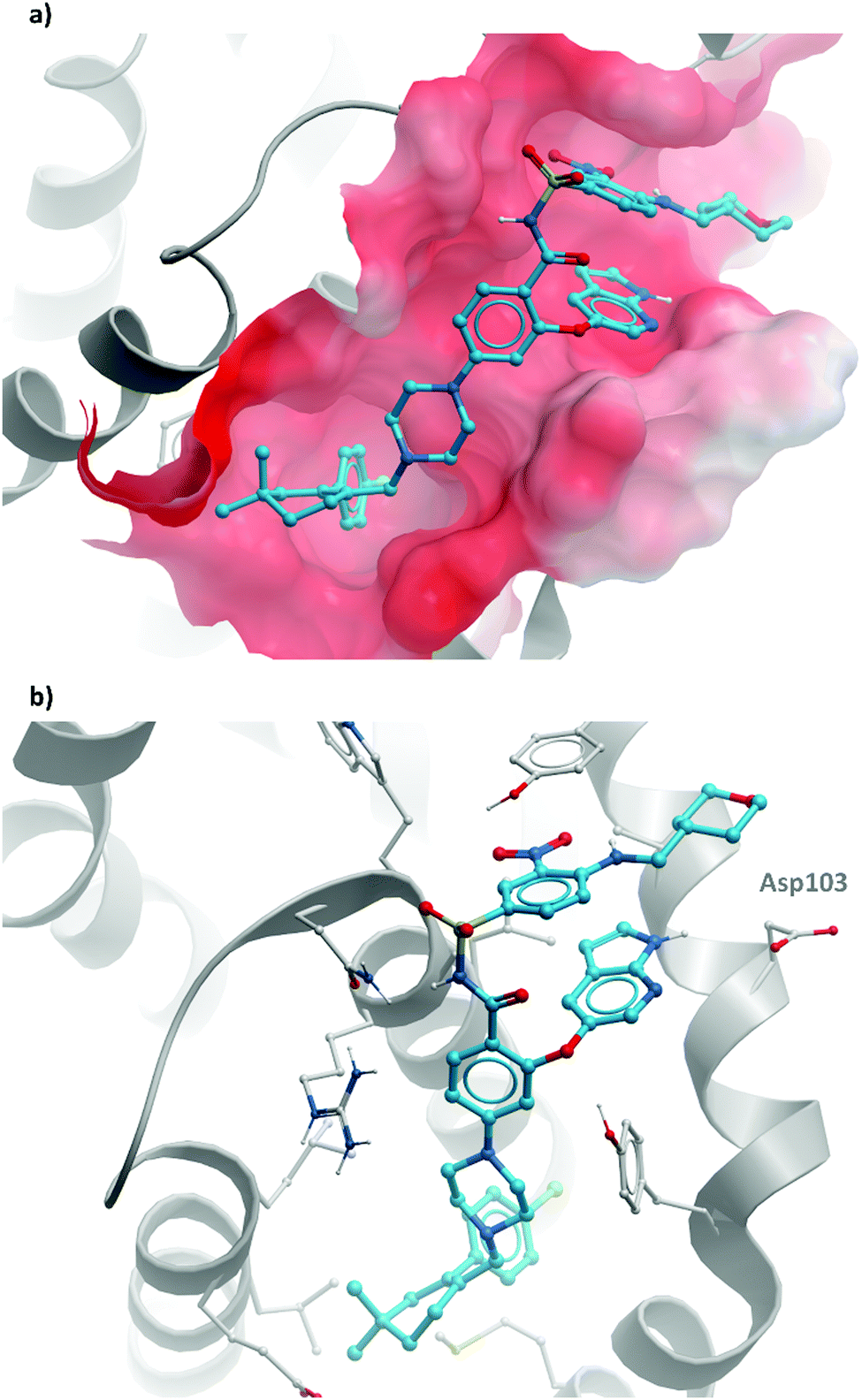 | ||
| Fig. 35 (a) Crystal structure of Venetoclax bound to the extended hydrophobic BCL-2 BH3 binding cleft; (b) the root of selectivity: Interaction of the azaindole moiety of Venetoclax with Asp103 in the BCL-2 binding site, which isn’t present in BCL-XL. | ||
Evaluation in mouse xenograft models of ALL and B cell lymphoma led to significant tumour growth inhibition following oral dosing at 100 mg kg−1 over 21 days, with no weight loss observed.334 It was also platelet sparing, with a far lower EC50 against human platelets than navitoclax (5.5 μM compared to 83 nM), and with minimal effects at high doses (100 mg kg−1) in dogs. Whilst venetoclax shows a drop in drug-likeness, this improved selectivity was the key to its clinical progression.
Venetoclax was enrolled in a phase I trial for patients with refractory CLL.334–336 The first patients receiving doses exhibited such a rapid reduction in circulating tumour levels that they developed laboratory tumour lysis syndrome (TLS), indicating such extensive tumour cell death that the cellular debris could not be cleared effectively. This was a significant safety concern, and so the dosing regimen was designed to slowly increase over several weeks to the desired dose. At the end of the phase I trial, venetoclax had an acceptable safety profile and 79% of patients had displayed objective responses, with 20% achieving complete remission. A phase II study in a larger cohort reported 79.4% of patients achieving objective responses, with 8% in complete remission.337 As a result, it was granted breakthrough designation by the FDA and subsequently approved for the treatment of CLL patients with the 17p deletion in September 2016,338 which was extended to all patients with CLL or small lymphocytic lymphoma in May 2019. It was also approved in November 2018 as part of a combination treatment for AML.
Conclusion
There is a rich history of chemical probes informing the development of clinical candidates.339 Selective probes for the two cannabinoid receptors, CB1 and CB2, enabled precise investigation of the contribution of each to analgesic and psychotropic effects, eventually informing a decision on targeting CB2 for the treatment of chronic pain.340 Probes for members of the dipeptidyl-peptidase (DPP) family of proteins identified toxicity associated with inhibition of some members, but showed that selective inhibition of DPP4 was an effective therapy for the treatment of type-2 diabetes.341 This eventually led to the discovery of sitagliptin. A probe for the dual orexin receptor helped guide understanding of the relationship between pharmacokinetics and target-engagement, informing relevant clinical exposure targets.342 These studies directly led to the discovery of suvorexant as a treatment for insomnia.Investigations of target viability, safety, and translation have all been enabled by chemical probes. In addition, obvious structural inheritance highlights the importance of chemical probes in producing drug leads. The probes described (summarised, Table 1) were often first-in-class compounds to interrogate the biology of disease-relevant targets. In the case of (+)-JQ1 (BET bromodomain probe, Fig. 1), it went on to directly inspire several structurally near-identical clinical candidates, with some showing incredible promise. Both PFI-1 (BET bromodomain probe, Fig. 7) and CBP-30 (CBP/p300 probe, Fig. 8) identified pharmacophores that robustly mimicked endogenous substrates and were recapitulated in potential drugs, with the structure of CBP-30 also being imitated to great effect. EPZ00477 and SGC0946 (DOT1L probes, Fig. 11) identified a novel binding mode to impart exquisite selectivity for a target, which was duly employed for clinical benefit. UNC0638 (G9a/GLP probe, Fig. 14), EPZ005687 (EZH2 probe, Fig. 16), EPZ020411 and MS-023 (type I PRMT probes, Fig. 21) identified key SAR around diverse scaffolds that informed further development, with EPZ005687 in particular inspiring a molecule that went on to be approved. EPZ015666 and LLY-283 (PRMT5 probes, Fig. 24 and 25, respectively) probed the same protein with different binding modes, providing multiple approaches to inhibiting disease-relevant targets. LLY-283 is also an example of how academic probe development can lead to the discovery of structures remarkably similar to clinical candidates, independent of any collaboration. GSK′481 (Fig. 27) opened the door to translating pharmacological investigation of necroptosis in vivo, whilst GW4064 (Fig. 30) was a pioneering molecule that identified a key pharmacophore for targeting the FXR. Finally, the development of ABT-737 (BCL-2 probe, Fig. 34) was a medicinal chemistry tour de force that resulted in a devastatingly effective drug, an example of the benefit of initial research into notionally “undruggable” targets with chemical probes. In general, probes have good drug-likeness scores14 but these improve following drug development, with most showing a modest increase in score compared to the probes. Overall, the drugs have much better pharmacokinetics compared to the probes, as we have shown for the examples.
| Probe | Target | Drugs | Indication |
|---|---|---|---|
| JQ1 | BET bromodomains | I-BET762 (phase I/II) | AML/breast cancer |
| OTX015 (phase I) | Glioblastoma | ||
| CPI-0610 (phase III) | Myelofibrosis | ||
| PFI-1 | BET bromodomains | ABBV-075/mivebresib (phase I) | Solid tumours |
| CBP-30 | CBP/p300 | CCS1477 (phase I) | Prostate cancer |
| EPZ004777/SGC0946 | DOT1L | Pinometostat (phase I/II) | MLL-rearranged leukaemias |
| UNC0638 | G9a/GLP | EPZ035544 (preclinical) | Sickle cell anaemia |
| EPZ005687 | EZH2 | GSK126 (phase I) | Haematologic/solid tumours |
| Tazemetostat (EPZ-6438) – approved | Advanced epithelioid sarcoma | ||
| CPI-1205 (phase Ib/II | Lymphoma/prostate cancer | ||
| EPZ020411/MS-023 | Type I PRMTs | GSK3367815 – phase I | Solid tumours/DLBCL |
| EPZ015666/EPZ015866 | PRMT5 | GSK3326595 – phase I/II | Solid tumours/lymphoma |
| LLY-283 | PRMT5 | JNJ64619178 – phase I | Solid tumours/lymphoma |
| GSK′481 | RIPK1 | GSK2982772 – phase I | Psoriasis |
| GW4064 | FXR | CIlofexor (GS-9674) – phase II | Primary sclerosing cholangitis |
| LY25621755 (TERN-101) – phase II | NASH | ||
| Tropifexor (LJN452) – phase II | PBC/NASH | ||
| ABT-737 | BCL2 | Navitoclax – phase II | Solid tumours/CLL/SCLC |
| Venetoclax – approved | CLL |
Beyond their structures, these compounds also represent a potentially transformative approach to drug discovery. The open access model that led to (+)-JQ1 being widely shared across diverse sections of the research community drove innovation,343 leading to a large number of patents being filed in relation to the scaffold. This is a model the SGC has consistently adopted with its probe collection,7 which will hopefully continue to inspire clinical candidates and go some way to attenuating the high attrition rate of new drugs.344,345 Target 203512 is thus not only a means of probing the entire proteome, but also open access to thousands of molecules that may provide the blueprints for a raft of life-changing therapeutics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Gian-Filippo Ruda, Dylan McGagh, George Tresidder, and Tom Humberstone for many helpful discussions on the subject. RQ is grateful to the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1) for a studentship, generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex. PEB was supported by Alzheimer's Research UK (ARUK2018DDI-OX).Notes and references
- J. Rauch, N. Volinsky, D. Romano and W. Kolch, Cell Commun. Signaling, 2011, 9, 23 CrossRef CAS PubMed.
- H. Aksnes, R. Ree and T. Arnesen, Mol. Cell, 2019, 73, 1097–1114 CrossRef CAS PubMed.
- C. J. Gerry and S. L. Schreiber, Nat. Chem. Biol., 2020, 16, 369–378 CrossRef CAS PubMed.
- A. Cipriano, G. Sbardella and A. Ciulli, Curr. Opin. Chem. Biol., 2020, 57, 82–94 CrossRef CAS PubMed.
- D. Whitley, S. P. Goldberg and W. D. Jordan, J. Vasc. Surg., 1999, 29, 748–751 CrossRef CAS PubMed.
- S. V. Frye, Nat. Chem. Biol., 2010, 6, 159–161 CrossRef CAS PubMed.
- C. H. Arrowsmith, J. E. Audia, C. Austin, J. Baell, J. Bennett, J. Blagg, C. Bountra, P. E. Brennan, P. J. Brown, M. E. Bunnage, C. Buser-Doepner, R. M. Campbell, A. J. Carter, P. Cohen, R. A. Copeland, B. Cravatt, J. L. Dahlin, D. Dhanak, A. M. Edwards, M. Frederiksen, S. V. Frye, N. Gray, C. E. Grimshaw, D. Hepworth, T. Howe, K. V. M. Huber, J. Jin, S. Knapp, J. D. Kotz, R. G. Kruger, D. Lowe, M. M. Mader, B. Marsden, A. Mueller-Fahrnow, S. Müller, R. C. O’Hagan, J. P. Overington, D. R. Owen, S. H. Rosenberg, B. Roth, R. Ross, M. Schapira, S. L. Schreiber, B. Shoichet, M. Sundström, G. Superti-Furga, J. Taunton, L. Toledo-Sherman, C. Walpole, M. A. Walters, T. M. Willson, P. Workman, R. N. Young and W. J. Zuercher, Nat. Chem. Biol., 2015, 11, 536–541 CrossRef CAS PubMed.
- W. A. Weiss, S. S. Taylor and K. M. Shokat, Nat. Chem. Biol., 2007, 3, 739–744 CrossRef CAS PubMed.
- D. M. C. Schwarz and J. E. Gestwicki, ACS Chem. Biol., 2018, 13, 1109–1110 CrossRef CAS PubMed.
- P. Workman and I. Collins, Chem. Biol., 2010, 17, 561–577 CrossRef CAS PubMed.
- R. F. Sweis, ACS Med. Chem. Lett., 2015, 6, 618–621 CrossRef CAS PubMed.
- A. J. Carter, O. Kraemer, M. Zwick, A. Mueller-Fahrnow, C. H. Arrowsmith and A. M. Edwards, Drug Discovery Today, 2019, 24, 2111–2115 CrossRef CAS PubMed.
- M. R. Nelson, H. Tipney, J. L. Painter, J. Shen, P. Nicoletti, Y. Shen, A. Floratos, P. C. Sham, M. J. Li, J. Wang, L. R. Cardon, J. C. Whittaker and P. Sanseau, Nat. Genet., 2015, 47, 856–860 CrossRef CAS PubMed.
- Molsoft L.L.C.: Drug-Likeness and molecular property prediction, https://molsoft.com/mprop/, (accessed March 01, 2021).
- P. Filippakopoulos, S. Picaud, M. Mangos, T. Keates, J. P. Lambert, D. Barsyte-Lovejoy, I. Felletar, R. Volkmer, S. Müller, T. Pawson, A. C. Gingras, C. H. Arrowsmith and S. Knapp, Cell, 2012, 149, 214–231 CrossRef CAS PubMed.
- C. Choudhary, C. Kumar, F. Gnad, M. L. Nielsen, M. Rehman, T. C. Walther, J. V. Olsen and M. Mann, Science, 2009, 325, 834–840 CrossRef CAS PubMed.
- E. M. Flynn, O. W. Huang, F. Poy, M. Oppikofer, S. F. Bellon, Y. Tang and A. G. Cochran, Structure, 2015, 23, 1801–1814 CrossRef CAS PubMed.
- F. Vollmuth and M. Geyer, Angew. Chem., Int. Ed., 2010, 49, 6768–6772 CrossRef CAS PubMed.
- S. Muller, P. Filippakopoulos and S. Knapp, Expert Rev. Mol. Med., 2011, 13, e29 CrossRef PubMed.
- A. G. Cochran, A. R. Conery and R. J. Sims, Nat. Rev. Drug Discovery, 2019, 18, 609–628 CrossRef CAS PubMed.
- C. Galdeano and A. Ciulli, Future Med. Chem., 2016, 8, 1655–1680 CrossRef CAS PubMed.
- R. K. Prinjha, J. Witherington and K. Lee, Trends Pharmacol. Sci., 2012, 33, 146–153 CrossRef CAS PubMed.
- I. Barbieri, E. Cannizzaro and M. A. Dawson, Briefings Funct. Genomics, 2013, 12, 219–230 CrossRef CAS PubMed.
- C. Y. Wang and P. Filippakopoulos, Trends Biochem. Sci., 2015, 40, 468–479 CrossRef CAS PubMed.
- D. Boehm, R. Conrad and M. Ott, Viruses, 2013, 5, 1571–1586 CrossRef CAS PubMed.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. la Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed.
- E. Nicodeme, K. L. Jeffrey, U. Schaefer, S. Beinke, S. Dewell, C. W. Chung, R. Chandwani, I. Marazzi, P. Wilson, H. Coste, J. White, J. Kirilovsky, C. M. Rice, J. M. Lora, R. K. Prinjha, K. Lee and A. Tarakhovsky, Nature, 2010, 468, 1119–1123 CrossRef CAS PubMed.
- S. Miyoshi, S. Ooike, K. Iwata, H. Hikawa and K. Sugahara, WO2009084693, 2009.
- G. Borbely, L. A. Haldosen, K. Dahlman-Wright and C. Zhao, Oncotarget., 2015, 6, 33623–33635 CrossRef PubMed.
- M. Pérez-Salvia, L. Simó-Riudalbas, P. Llinàs-Arias, L. Roa, F. Setien, M. Soler, M. C. de Moura, J. E. Bradner, E. Gonzalez-Suarez, C. Moutinho and M. Esteller, Oncotarget., 2017, 8, 51621–51629 CrossRef PubMed.
- Y. Zhang, S. Tian, J. Xiong, Y. Zhou, H. Song and C. Liu, Chem. Res. Toxicol., 2018, 31, 302–307 Search PubMed.
- Z. Cheng, Y. Gong, Y. Ma, K. Lu, X. Lu, L. A. Pierce, R. C. Thompson, S. Muller, S. Knapp and J. Wang, Clin. Cancer Res., 2013, 19, 1748–1759 CrossRef CAS PubMed.
- P. Bandopadhayay, G. Bergthold, B. Nguyen, S. Schubert, S. Gholamin, Y. Tang, S. Bolin, S. E. Schumacher, R. Zeid, S. Masoud, F. Yu, N. Vue, W. J. Gibson, B. R. Paolella, S. S. Mitra, S. H. Cheshier, J. Qi, K. W. Liu, R. Wechsler-Reya, W. A. Weiss, F. J. Swartling, M. W. Kieran, J. E. Bradner, R. Beroukhim and Y. J. Cho, Clin. Cancer Res., 2014, 20, 912–925 CrossRef CAS PubMed.
- J. E. Delmore, G. C. Issa, M. E. Lemieux, P. B. Rahl, J. Shi, H. M. Jacobs, E. Kastritis, T. Gilpatrick, R. M. Paranal, J. Qi, M. Chesi, A. C. Schinzel, M. R. McKeown, T. P. Heffernan, C. R. Vakoc, P. L. Bergsagel, I. M. Ghobrial, P. G. Richardson, R. A. Young, W. C. Hahn, K. C. Anderson, A. L. Kung, J. E. Bradner and C. S. Mitsiades, Cell, 2011, 146, 904–917 CrossRef CAS PubMed.
- J. A. Mertz, A. R. Conery, B. M. Bryant, P. Sandy, S. Balasubramanian, D. A. Mele, L. Bergeron and R. J. Sims, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16669–16674 CrossRef CAS PubMed.
- W. Fiskus, S. Sharma, J. Qi, B. Shah, S. G. T. Devaraj, C. Leveque, B. P. Portier, S. Iyer, J. E. Bradner and K. N. Bhalla, Mol. Cancer Ther., 2014, 13, 2315–2327 CrossRef CAS PubMed.
- M. Loosveld, R. Castellano, S. Gon, A. Goubard, T. Crouzet, L. Pouyet, T. Prebet, N. Vey, B. Nadel, Y. Collette and D. Payet-Bornet, Oncotarget., 2014, 5, 3168–3172 CrossRef PubMed.
- S. Liu, S. R. Walker, E. A. Nelson, R. Cerulli, M. Xiang, P. A. Toniolo, J. Qi, R. M. Stone, M. Wadleigh, J. E. Bradner and D. A. Frank, Mol. Cancer Ther., 2014, 13, 1194–1205 CrossRef CAS PubMed.
- S. J. Hogg, S. J. Vervoort, S. Deswal, C. J. Ott, J. Li, L. A. Cluse, P. A. Beavis, P. K. Darcy, B. P. Martin, A. Spencer, A. K. Traunbauer, I. Sadovnik, K. Bauer, P. Valent, J. E. Bradner, J. Zuber, J. Shortt and R. W. Johnstone, Cell Rep., 2017, 18, 2162–2174 CrossRef CAS PubMed.
- G. Andrieu, A. C. Belkina and G. V. Denis, Drug Discovery Today: Technol., 2016, 19, 45–50 CrossRef PubMed.
- O. Mirguet, R. Gosmini, J. Toum, C. A. Clément, M. Barnathan, J. M. Brusq, J. E. Mordaunt, R. M. Grimes, M. Crowe, O. Pineau, M. Ajakane, A. Daugan, P. Jeffrey, L. Cutler, A. C. Haynes, N. N. Smithers, C. W. Chung, P. Bamborough, I. J. Uings, A. Lewis, J. Witherington, N. Parr, R. K. Prinjha and E. Nicodème, J. Med. Chem., 2013, 56, 7501–7515 CrossRef CAS PubMed.
- C. W. Chung, H. Coste, J. H. White, O. Mirguet, J. Wilde, R. L. Gosmini, C. Delves, S. M. Magny, R. Woodward, S. A. Hughes, E. V. Boursier, H. Flynn, A. M. Bouillot, P. Bamborough, J. M. G. Brusq, F. O. J. Gellibert, E. J. Jones, A. M. Riou, P. Homes, S. L. Martin, I. J. Uings, J. Toum, C. A. Clément, A. B. Boullay, R. L. Grimley, F. M. Blandel, R. K. Prinjha, K. Lee, J. Kirilovsky and E. Nicodeme, J. Med. Chem., 2011, 54, 3827–3838 CrossRef CAS PubMed.
- M. J. Cho, T. A. Scahill and J. B. Hester, J. Pharm. Sci., 1983, 72, 356–362 CrossRef CAS PubMed.
- S. A. Piha-Paul, C. L. Hann, C. A. French, S. Cousin, I. Braña, P. A. Cassier, V. Moreno, J. S. de Bono, S. D. Harward, G. Ferron-Brady, O. Barbash, A. Wyce, Y. Wu, T. Horner, M. Annan, N. J. Parr, R. K. Prinjha, C. L. Carpenter, J. Hilton, D. S. Hong, N. B. Haas, M. C. Markowski, A. Dhar, P. J. O’Dwyer and G. I. Shapiro, JNCI Cancer Spectr., 2020, 4, pkz093 CrossRef PubMed.
- A. Chaidos, V. Caputo, K. Gouvedenou, B. Liu, I. Marigo, M. S. Chaudhry, A. Rotolo, D. F. Tough, N. N. Smithers, A. K. Bassil, T. D. Chapman, N. R. Harker, O. Barbash, P. Tummino, N. Al-Mahdi, A. C. Haynes, L. Cutler, B. C. Le, A. Rahemtulla, I. Roberts, M. Kleijnen, J. J. Witherington, N. J. Parr, R. K. Prinjha and A. Karadimitris, Blood, 2014, 123, 697–705 CrossRef CAS PubMed.
- A. Wyce, Y. Degenhardt, Y. Bai, B. C. Le, S. Korenchuk, M. C. Crouthamel, C. F. McHugh, R. Vessella, C. L. Creasy, P. J. Tummino and O. Barbash, Oncotarget., 2013, 4, 2419–2429 CrossRef PubMed.
- M. Dawson, E. M. Stein, B. J. P. Huntly, A. Karadimitris, M. Kamdar, C. Fernandez de Larrea, M. J. Dickinson, P. S.-H. Yeh, N. Daver, A. Chaidos, M. S. Tallman, R. Jiménez, T. Horner, J. Baron, J. Brennan, G. Ferron-Brady, Y. Wu, N. Karpinich, B. Kremer, A. Dhar and G. Borthakur, Blood, 2017, 130, 1377 CrossRef PubMed.
- J. A. Sparano, D. W. Cescon, M. Oliveira, D. G. Stover, E. Tan-Chiu, B. E. Kremer, O. Barbash, Y. Wu, L. Mathew, E. A. Cunningham, C. L. Carpenter, M. S. Ballas and A. Dhar, J. Clin. Oncol., 2017, 35, TPS1114 Search PubMed.
- U. N. Vaishampayan, V. Narayan, D. Wise, J. M. Lang, B. H. Lowentritt, B. Mellado, J. Carles, M. I. Sáez, W. Abida, M.-E. Taplin, A. Azad, K. Wang, O. Barbash, G. Ferron-Brady, D. Fecteau, A. H. Khaled, A. Dhar and J. S. de Bono, J. Clin. Oncol., 2018, 36, TPS391 Search PubMed.
- J. K. Noel, K. Iwata, S. Ooike, K. Sugahara, H. Nakamura and M. Daibata, Mol. Cancer Ther., 2013, 12, C244 Search PubMed.
- M. M. Coudé, T. Braun, J. Berrou, M. Dupont, S. Bertrand, A. Masse, E. Raffoux, R. Itzykson, M. Delord, M. E. Riveiro, P. Herait, A. Baruchel, H. Dombret and C. Gardin, Oncotarget., 2015, 6, 17698–17712 CrossRef PubMed.
- R. Vázquez, M. E. Riveiro, L. Astorgues-Xerri, E. Odore, K. Rezai, E. Erba, N. Panini, A. Rinaldi, I. Kwee, L. Beltrame, M. Bekradda, E. Cvitkovic, F. Bertoni, R. Frapolli and M. D’Incalci, Oncotarget., 2017, 8, 7598–7613 CrossRef PubMed.
- M. E. Riveiro, L. Astorgues-Xerri, R. Vazquez, R. Frapolli, I. Kwee, A. Rinaldi, E. Odore, K. Rezai, M. Bekradda, G. Inghirami, M. D’Incalci, K. Noel, E. Cvitkovic, E. Raymond and F. Bertoni, Oncotarget., 2016, 7, 84675–84687 CrossRef PubMed.
- C. Berenguer-Daizé, L. Astorgues-Xerri, E. Odore, M. Cayol, E. Cvitkovic, K. Noel, M. Bekradda, S. MacKenzie, K. Rezai, F. Lokiec, M. E. Riveiro and L. Ouafik, Int. J. Cancer, 2016, 139, 2047–2055 CrossRef PubMed.
- T. Braun, M. M. Coude, J. Berrou, S. Bertrand, E. Riveiro, P. Herait, A. Baruchel, H. Dombret and C. Gardin, Blood, 2013, 122, 4218 CrossRef.
- E. Odore, F. Lokiec, E. Cvitkovic, M. Bekradda, P. Herait, F. Bourdel, C. Kahatt, E. Raffoux, A. Stathis, C. Thieblemont, B. Quesnel, D. Cunningham, M. E. Riveiro and K. Rezaï, Clin. Pharmacokinet., 2016, 55, 397–405 CrossRef CAS PubMed.
- S. Amorim, A. Stathis, M. Gleeson, S. Iyengar, V. Magarotto, X. Leleu, F. Morschhauser, L. Karlin, F. Broussais, K. Rezai, P. Herait, C. Kahatt, F. Lokiec, G. Salles, T. Facon, A. Palumbo, D. Cunningham, E. Zucca and C. Thieblemont, Lancet Haematol., 2016, 3, e196–e204 CrossRef PubMed.
- C. Berthon, E. Raffoux, X. Thomas, N. Vey, C. Gomez-Roca, K. Yee, D. C. Taussig, K. Rezai, C. Roumier, P. Herait, C. Kahatt, B. Quesnel, M. Michallet, C. Recher, F. Lokiec, C. Preudhomme and H. Dombret, Lancet Haematol., 2016, 3, e186–e195 CrossRef PubMed.
- J. Lewin, J. C. Soria, A. Stathis, J. P. Delord, S. Peters, A. Awada, P. G. Aftimos, M. Bekradda, K. Rezai, Z. Zeng, A. Hussain, S. Perez, L. L. Siu and C. Massard, J. Clin. Oncol., 2018, 36, 3007–3014 CrossRef CAS PubMed.
- B. K. Albrecht, V. S. Gehling, M. C. Hewitt, R. G. Vaswani, A. Côté, Y. Leblanc, C. G. Nasveschuk, S. Bellon, L. Bergeron, R. Campbell, N. Cantone, M. R. Cooper, R. T. Cummings, H. Jayaram, S. Joshi, J. A. Mertz, A. Neiss, E. Normant, M. O’Meara, E. Pardo, F. Poy, P. Sandy, J. Supko, R. J. Sims, J. C. Harmange, A. M. Taylor and J. E. Audia, J. Med. Chem., 2016, 59, 1330–1339 CrossRef CAS PubMed.
- K. T. Siu, J. Ramachandran, A. J. Yee, H. Eda, L. Santo, C. Panaroni, J. A. Mertz, R. J. Sims, M. R. Cooper and N. Raje, Leukemia, 2017, 31, 1760–1769 CrossRef CAS PubMed.
- K. A. Blum, J. Abramson, M. Maris, I. Flinn, A. Goy, J. Mertz, R. Sims, F. Garner, A. Senderowicz and A. Younes, Ann. Oncol., 2018, 29, III7–III9 CrossRef.
- J. A. Mertz, K. A. Blum, A. Younes, J. S. Abramson, M. B. Maris, I. W. Flinn, A. Goy, D. R. Borger, M. R. Cooper and R. J. Sims, Cancer Res., 2018, 78, 3674 Search PubMed.
- BET Inhibitor CPI-0610 Is Well Tolerated and Induces Responses in Diffuse Large B-Cell Lymphoma and Follicular Lymphoma: Preliminary Analysis of an Ongoing Phase 1 Study, https://ash.confex.com/ash/2015/webprogramscheduler/Paper78125.html, (accessed 30 November 2020).
- M. Kremyanskaya, R. Hoffman, J. Mascarenhas, S. Verstovsek, J. Mertz, F. Garner and A. Senderowicz, Blood, 2018, 132, 5481 CrossRef.
- C. N. Harrison, A. Patriarca, J. Mascarenhas, M. Kremyanskaya, R. Hoffman, G. J. Schiller, B. Leber, T. Devos, S. Kabir, A. Senderowicz, J. Mertz, P. Trojer, J. Shao and V. Gupta, Blood, 2019, 134, 4164 CrossRef.
- R. A. Mesa, U. Yasothan and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2012, 11, 103–104 CrossRef CAS PubMed.
- M. Kleppe, R. Koche, L. Zou, P. van Galen, C. E. Hill, L. Dong, S. de Groote, E. Papalexi, A. V. Hanasoge Somasundara, K. Cordner, M. Keller, N. Farnoud, J. Medina, E. McGovern, J. Reyes, J. Roberts, M. Witkin, F. Rapaport, J. Teruya-Feldstein, J. Qi, R. Rampal, B. E. Bernstein, J. E. Bradner and R. L. Levine, Cancer Cell, 2018, 33, 29–43 CrossRef CAS PubMed .e7.
- M. A. Dawson, R. K. Prinjha, A. Dittmann, G. Giotopoulos, M. Bantscheff, W. I. Chan, S. C. Robson, C. W. Chung, C. Hopf, M. M. Savitski, C. Huthmacher, E. Gudgin, D. Lugo, S. Beinke, T. D. Chapman, E. J. Roberts, P. E. Soden, K. R. Auger, O. Mirguet, K. Doehner, R. Delwel, A. K. Burnett, P. Jeffrey, G. Drewes, K. Lee, B. J. P. Huntly and T. Kouzarides, Nature, 2011, 478, 529–533 CrossRef CAS PubMed.
- P. Bamborough, H. Diallo, J. D. Goodacre, L. Gordon, A. Lewis, J. T. Seal, D. M. Wilson, M. D. Woodrow and C. W. Chung, J. Med. Chem., 2012, 55, 587–596 CrossRef CAS PubMed.
- C. W. Chung, A. W. Dean, J. M. Woolven and P. Bamborough, J. Med. Chem., 2012, 55, 576–586 CrossRef CAS PubMed.
- D. S. Hewings, M. Wang, M. Philpott, O. Fedorov, S. Uttarkar, P. Filippakopoulos, S. Picaud, C. Vuppusetty, B. Marsden, S. Knapp, S. J. Conway and T. D. Heightman, J. Med. Chem., 2011, 54, 6761–6770 CrossRef CAS PubMed.
- P. v. Fish, P. Filippakopoulos, G. Bish, P. E. Brennan, M. E. Bunnage, A. S. Cook, O. Federov, B. S. Gerstenberger, H. Jones, S. Knapp, B. Marsden, K. Nocka, D. R. Owen, M. Philpott, S. Picaud, M. J. Primiano, M. J. Ralph, N. Sciammetta and J. D. Trzupek, J. Med. Chem., 2012, 55, 9831–9837 CrossRef CAS PubMed.
- S. Picaud, D. da Costa, A. Thanasopoulou, P. Filippakopoulos, P. V. Fish, M. Philpott, O. Fedorov, P. Brennan, M. E. Bunnage, D. R. Owen, J. E. Bradner, P. Taniere, B. O’Sullivan, S. Müller, J. Schwaller, T. Stankovic and S. Knapp, Cancer Res., 2013, 73, 3336–3346 CrossRef CAS PubMed.
- F. de, P. Careta, S. Gobessi, R. A. Panepucci, E. Bojnik, F. M. de Oliveira, D. M. Matos, R. P. Falcão, L. Laurenti, M. A. Zago and D. G. Efremov, Haematologica, 2012, 97, 1246–1254 CrossRef PubMed.
- L. Wang, J. K. Pratt, T. Soltwedel, G. S. Sheppard, S. D. Fidanze, D. Liu, L. A. Hasvold, R. A. Mantei, J. H. Holms, W. J. McClellan, M. D. Wendt, C. Wada, R. Frey, T. M. Hansen, R. Hubbard, C. H. Park, L. Li, T. J. Magoc, D. H. Albert, X. Lin, S. E. Warder, P. Kovar, X. Huang, D. Wilcox, R. Wang, G. Rajaraman, A. M. Petros, C. W. Hutchins, S. C. Panchal, C. Sun, S. W. Elmore, Y. Shen, W. M. Kati and K. F. McDaniel, J. Med. Chem., 2017, 60, 3828–3850 CrossRef CAS PubMed.
- K. F. McDaniel, L. Wang, T. Soltwedel, S. D. Fidanze, L. A. Hasvold, D. Liu, R. A. Mantei, J. K. Pratt, G. S. Sheppard, M. H. Bui, E. J. Faivre, X. Huang, L. Li, X. Lin, R. Wang, S. E. Warder, D. Wilcox, D. H. Albert, T. J. Magoc, G. Rajaraman, C. H. Park, C. W. Hutchins, J. J. Shen, R. P. Edalji, C. C. Sun, R. Martin, W. Gao, S. Wong, G. Fang, S. W. Elmore, Y. Shen and W. M. Kati, J. Med. Chem., 2017, 60, 8369–8384 CrossRef CAS PubMed.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- S. A. Piha-Paul, J. C. Sachdev, M. Barve, P. LoRusso, R. Szmulewitz, S. P. Patel, P. N. Lara, X. Chen, B. Hu, K. J. Freise, D. Modi, A. Sood, J. E. Hutti, J. Wolff and B. H. O’Neil, Clin. Cancer Res., 2019, 25, 6309–6319 CrossRef CAS PubMed.
- M. H. Bui, X. Lin, D. H. Albert, L. Li, L. T. Lam, E. J. Faivre, S. E. Warder, X. Huang, D. Wilcox, C. K. Donawho, G. S. Sheppard, L. Wang, S. Fidanze, J. K. Pratt, D. Liu, L. Hasvold, T. Uziel, X. Lu, F. Kohlhapp, G. Fang, S. W. Elmore, S. H. Rosenberg, K. F. McDaniel, W. M. Kati and Y. Shen, Cancer Res., 2017, 77, 2976–2989 CrossRef CAS PubMed.
- R. H. Goodman and S. Smolik, Genes Dev., 2000, 14, 1553–1577 CAS.
- N. G. Iyer, H. Özdag and C. Caldas, Oncogene, 2004, 23, 4225–4231 CrossRef CAS PubMed.
- A. J. Bannister and T. Kouzarides, Nature, 1996, 384, 641–643 CrossRef CAS PubMed.
- M. Delvecchio, J. Gaucher, C. Aguilar-Gurrieri, E. Ortega and D. Panne, Nat. Struct. Mol. Biol., 2013, 20, 1040–1046 CrossRef CAS PubMed.
- V. v. Ogryzko, R. L. Schiltz, V. Russanova, B. H. Howard and Y. Nakatani, Cell, 1996, 87, 953–959 CrossRef CAS PubMed.
- B. L. Ebert and H. F. Bunn, Mol. Cell. Biol., 1998, 18, 4089–4096 CrossRef CAS PubMed.
- P. J. Kallio, EMBO J., 1998, 17, 6573–6586 CrossRef CAS PubMed.
- M. L. Avantaggiati, V. Ogryzko, K. Gardner, A. Giordano, A. S. Levine and K. Kelly, Cell, 1997, 89, 1175–1184 CrossRef CAS PubMed.
- S. R. Grossman, Eur. J. Biochem., 2001, 268, 2773–2778 CrossRef CAS PubMed.
- L. Zeng, Q. Zhang, G. Gerona-Navarro, N. Moshkina and M. M. Zhou, Structure, 2008, 16, 643–652 CrossRef CAS PubMed.
- S. Mujtaba, Y. He, L. Zeng, S. Yan, O. Plotnikova, Sachchidanand, R. Sanchez, N. J. Zeleznik-Le, Z. Ronai and M. M. Zhou, Mol. Cell, 2004, 13, 251–263 CrossRef CAS PubMed.
- D. A. Hay, O. Fedorov, S. Martin, D. C. Singleton, C. Tallant, C. Wells, S. Picaud, M. Philpott, O. P. Monteiro, C. M. Rogers, S. J. Conway, T. P. C. Rooney, A. Tumber, C. Yapp, P. Filippakopoulos, M. E. Bunnage, S. Müller, S. Knapp, C. J. Schofield and P. E. Brennan, J. Am. Chem. Soc., 2014, 136, 9308–9319 CrossRef CAS PubMed.
- A. v. Gudkov and E. A. Komarova, Biochem. Biophys. Res. Commun., 2005, 331, 726–736 CrossRef CAS PubMed.
- S. Nayak, P. Panesar and H. Kumar, Curr. Med. Chem., 2009, 16, 2627–2640 CrossRef CAS PubMed.
- J. Li, C. A. Ghiani, Y. K. Jin, A. Liu, J. Sandoval, J. DeVellis and P. Casaccia-Bonnefil, J. Neurosci., 2008, 28, 6118–6127 CrossRef CAS PubMed.
- W. Duan, X. Zhu, B. Ladenheim, Q. S. Yu, Z. Guo, J. Oyler, R. G. Cutler, J. L. Cadet, N. H. Greig and M. P. Mattson, Ann. Neurol., 2002, 52, 597–606 CrossRef CAS PubMed.
- L. Z. Hong, X. Y. Zhao and H. L. Zhang, Neurosci. Bull., 2010, 26, 232–240 CrossRef CAS PubMed.
- J. Tashiro, S. Kikuchi, K. Shinpo, R. Kishimoto, S. Tsuji and H. Sasaki, J. Neurosci. Res., 2007, 85, 395–401 CrossRef CAS PubMed.
- N. Pietrancosta, C. Garino, Y. Laras, G. Quéléver, P. Pierre, G. Clavarino and J.-L. Kraus, Drug Dev. Res., 2005, 65, 43–49 CrossRef CAS.
- J. C. Borah, S. Mujtaba, I. Karakikes, L. Zeng, M. Muller, J. Patel, N. Moshkina, K. Morohashi, W. Zhang, G. Gerona-Navarro, R. J. Hajjar and M. M. Zhou, Chem. Biol., 2011, 18, 531–541 CrossRef CAS PubMed.
- G. Gerona-Navarro, Y. Rodríguez, S. Mujtaba, A. Frasca, J. Patel, L. Zeng, A. N. Plotnikov, R. Osman and M. M. Zhou, J. Am. Chem. Soc., 2011, 133, 2040–2043 CrossRef CAS PubMed.
- D. Hay, O. Fedorov, P. Filippakopoulos, S. Martin, M. Philpott, S. Picaud, D. S. Hewings, S. Uttakar, T. D. Heightman, S. J. Conway, S. Knapp and P. E. Brennan, MedChemComm, 2013, 4, 140–144 RSC.
- A. Hammitzsch, C. Tallant, O. Fedorov, A. O’Mahony, P. E. Brennan, D. A. Hay, F. O. Martinez, M. H. Al-Mossawi, J. de Wit, M. Vecellio, C. Wells, P. Wordsworth, S. Müller, S. Knapp and P. Bowness, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10768–10773 CrossRef CAS PubMed.
- N. A. Pegg, D. M. A. Taddei and R. Brown, WO2016170323, 2016.
- N. Brooks, M. Raja, B. W. Young, G. J. Spencer, T. C. Somervaille and N. A. Pegg, Blood, 2019, 134, 2560 CrossRef.
- N. Brooks, A. Prosser, B. Young, L. Gaughan, P. Elvin and N. Pegg, Cancer Res., 2019, 79, 3826 Search PubMed.
- Therapeutic targeting of the p300/CBP bromodomain for the treatment of castration resistant prostate cancer, https://www.cellcentric.com/media/uploads/files/AACR_Prostate_Poster_2017_Final.pdf (accessed 2 December 2020).
- J. Welti, A. Sharp, N. Brooks, W. Yuan, C. McNair, S. N. Chand, A. Pal, I. Figueiredo, R. Riisnaes, B. Gurel, J. Rekowski, D. Bogdan, W. West, B. Young, M. Raja, A. Prosser, J. Lane, S. Thomson, J. Worthington, S. Onions, J. Shannon, S. Paoletta, R. Brown, D. Smyth, G. W. Harbottle, V. S. Gil, S. Miranda, M. Crespo, A. Ferreira, R. Pereira, N. Tunariu, S. Carreira, A. J. Neeb, J. Ning, A. Swain, D. Taddei, S. U. to C.-P. International Dream Team, M. J. Schiewer, K. E. Knudsen, N. Pegg and J. S. de Bono, Cancer Discovery, 2021, CD-20-0751 Search PubMed.
- The discovery of CCS1477, first of its kind, p300/CBP inhibitor, https://www.cellcentric.com/updates/blog-new/discovery-ccs1477-first-its-kind-p300cbp-inhibitor/#:˜:text=CCS1477%20is%20first%20of%20its,twin%20proteins%2C%20p300%20and%20CBP.&text=CellCentric%20developed%20the%20compound%20all,and%20efficacy%20evaluation%20in%20patients. (accessed 2 December 2020).
- A novel small molecule inhibitor of p300/CBP for the treatment of castration resistant prostate cancer-preclinical evaluation, https://www.cellcentric.com/media/uploads/files/ASCO-GU_Poster_Final.pdf (accessed 2 December 2020).
- Targeting the bromodomain of p300/CBP for the treatment of castrate resistant prostate cancer, https://www.cellcentric.com/media/uploads/files/Brooks_PCF_Poster_Final_3_3Grcjn6.pdf (accessed 2 December 2020).
- Characterisation of CCS1477: A novel small molecule inhibitor of p300/CBP for the treatment of castration resistant prostate cancer, https://www.cellcentric.com/media/uploads/files/ASCO_Poster_2017_FINAL.pdf (accessed 2 December 2020).
- A first-in-class p300/CBP bromodomain inhibitor for the treatment of prostate cancer and hematologic malignancies, https://www.cellcentric.com/media/uploads/files/AACR_Pegg_Presentation_2019.pdf (accessed 2 December 2020).
- Novel small molecule inhibitor of p300/CBP down-regulates androgen receptor (AR) and c-Myc for the treatment of prostate cancer and beyond, https://www.cellcentric.com/media/uploads/files/CellCentric_AACR_Poster_2018.pdf (accessed 2 December 2020).
- J. S. de Bono, E. Cojocaru, E. R. Plummer, T. Knurowski, K. Clegg, F. Ashby, N. Pegg, W. West and A. N. Brooks, J. Clin. Oncol., 2019, 37, TPS5089 Search PubMed.
- T. Knurowski, K. Clegg, N. Brooks, F. Ashby, N. A. Pegg, W. West, H. S. Walter, T. C. Somervaille, S. Knapper and A. Davies, Blood, 2019, 134, 1266 CrossRef.
- D. Han, M. Huang, T. Wang, Z. Li, Y. Chen, C. Liu, Z. Lei and X. Chu, Cell Death Dis., 2019, 10, 290 CrossRef PubMed.
- C. Martin and Y. Zhang, Nat. Rev. Mol. Cell Biol., 2005, 6, 838–849 CrossRef CAS PubMed.
- J. C. Black, C. van Rechem and J. R. Whetstine, Mol. Cell, 2012, 48, 491–507 CrossRef CAS PubMed.
- Q. Feng, H. Wang, H. H. Ng, H. Erdjument-Bromage, P. Tempst, K. Struhl and Y. Zhang, Curr. Biol., 2002, 12, 1052–1058 CrossRef CAS PubMed.
- L. Godfrey, N. T. Crump, R. Thorne, I. J. Lau, E. Repapi, D. Dimou, A. L. Smith, J. R. Harman, J. M. Telenius, A. M. Oudelaar, D. J. Downes, P. Vyas, J. R. Hughes and T. A. Milne, Nat. Commun., 2019, 10, 2803 CrossRef PubMed.
- D. J. Steger, M. I. Lefterova, L. Ying, A. J. Stonestrom, M. Schupp, D. Zhuo, A. L. Vakoc, J.-E. Kim, J. Chen, M. A. Lazar, G. A. Blobel and C. R. Vakoc, Mol. Cell. Biol., 2008, 28, 2825–2839 CrossRef CAS PubMed.
- C. W. Chen, R. P. Koche, A. U. Sinha, A. J. Deshpande, N. Zhu, R. Eng, J. G. Doench, H. Xu, S. H. Chu, J. Qi, X. Wang, C. Delaney, K. M. Bernt, D. E. Root, W. C. Hahn, J. E. Bradner and S. A. Armstrong, Nat. Med., 2015, 21, 335–343 CrossRef CAS PubMed.
- J. L. Hess, Trends Mol. Med., 2004, 10, 500–507 CrossRef CAS PubMed.
- A. v. Krivtsov and S. A. Armstrong, Nat. Rev. Cancer, 2007, 7, 823–833 CrossRef CAS PubMed.
- T. A. Milne, S. D. Briggs, H. W. Brock, M. E. Martin, D. Gibbs, C. D. Allis and J. L. Hess, Mol. Cell, 2002, 10, 1107–1117 CrossRef CAS.
- T. Nakamura, T. Mori, S. Tada, W. Krajewski, T. Rozovskaia, R. Wassell, G. Dubois, A. Mazo, C. M. Croce and E. Canaani, Mol. Cell, 2002, 10, 1119–1128 CrossRef CAS PubMed.
- S. R. Daigle, E. J. Olhava, C. A. Therkelsen, C. R. Majer, C. J. Sneeringer, J. Song, L. D. Johnston, M. P. Scott, J. J. Smith, Y. Xiao, L. Jin, K. W. Kuntz, R. Chesworth, M. P. Moyer, K. M. Bernt, J. C. Tseng, A. L. Kung, S. A. Armstrong, R. A. Copeland, V. M. Richon and R. M. Pollock, Cancer Cell, 2011, 20, 53–65 CrossRef CAS PubMed.
- L. Morera, M. Lübbert and M. Jung, Clin. Epigenet., 2016, 8, 16 CrossRef PubMed.
- A. T. Nguyen, O. Taranova, J. He and Y. Zhang, Blood, 2011, 117, 6912–6922 CrossRef CAS PubMed.
- A. v. Krivtsov, Z. Feng, M. E. Lemieux, J. Faber, S. Vempati, A. U. Sinha, X. Xia, J. Jesneck, A. P. Bracken, L. B. Silverman, J. L. Kutok, A. L. Kung and S. A. Armstrong, Cancer Cell, 2008, 14, 355–368 CrossRef CAS PubMed.
- Y. Okada, Q. Feng, Y. Lin, Q. Jiang, Y. Li, V. M. Coffield, L. Su, G. Xu and Y. Zhang, Cell, 2005, 121, 167–178 CrossRef CAS PubMed.
- M. J. Chang, H. Wu, N. J. Achille, M. R. Reisenauer, C. W. Chou, N. J. Zeleznik-Le, C. S. Hemenway and W. Zhang, Cancer Res., 2010, 70, 10234–10242 CrossRef CAS PubMed.
- S. Y. Jo, E. M. Granowicz, I. Maillard, D. Thomas and J. L. Hess, Blood, 2011, 117, 4759–4768 CrossRef CAS PubMed.
- A. Basavapathruni, L. Jin, S. R. Daigle, C. R. A. Majer, C. A. Therkelsen, T. J. Wigle, K. W. Kuntz, R. Chesworth, R. M. Pollock, M. P. Scott, M. P. Moyer, V. M. Richon, R. A. Copeland and E. J. Olhava, Chem. Biol. Drug Des., 2012, 80, 971–980 CrossRef CAS PubMed.
- W. Yu, E. J. Chory, A. K. Wernimont, W. Tempel, A. Scopton, A. Federation, J. J. Marineau, J. Qi, D. Barsyte-Lovejoy, J. Yi, R. Marcellus, R. E. Iacob, J. R. Engen, C. Griffin, A. Aman, E. Wienholds, F. Li, J. Pineda, G. Estiu, T. Shatseva, T. Hajian, R. Al-Awar, J. E. Dick, M. Vedadi, P. J. Brown, C. H. Arrowsmith, J. E. Bradner and M. Schapira, Nat. Commun., 2012, 3, 1–12 Search PubMed.
- A. E. Conway, J. M. Haldeman, D. S. Wechsler and C. P. Lavau, Leukemia, 2015, 29, 423–432 CrossRef CAS PubMed.
- M. Wong, A. E. L. Tee, G. Milazzo, J. L. Bell, R. C. Poulos, B. Atmadibrata, Y. Sun, D. Jing, N. Ho, D. Ling, P. Y. Liu, X. D. Zhang, S. Hüttelmaier, J. W. H. Wong, J. Wang, P. Polly, G. Perini, C. J. Scarlett and T. Liu, Cancer Res., 2017, 77, 2522–2533 CrossRef CAS PubMed.
- K. Ishiguro, H. Kitajima, T. Niinuma, T. Ishida, R. Maruyama, H. Ikeda, T. Hayashi, H. Sasaki, H. Wakasugi, K. Nishiyama, T. Shindo, E. Yamamoto, M. Kai, Y. Sasaki, T. Tokino, H. Nakase and H. Suzuki, Haematologica, 2019, 104, 155–165 CrossRef CAS PubMed.
- T. D. D. Somerville and C. R. Vakoc, Cancer Cell, 2016, 30, 9–10 CrossRef CAS PubMed.
- S. R. Daigle, E. J. Olhava, C. A. Therkelsen, A. Basavapathruni, L. Jin, P. A. Boriack-Sjodin, C. J. Allain, C. R. Klaus, A. Raimondi, M. P. Scott, N. J. Waters, R. Chesworth, M. P. Moyer, R. A. Copeland, V. M. Richon and R. M. Pollock, Blood, 2013, 122, 1017–1025 CrossRef CAS PubMed.
- N. J. Waters, Eur. J. Drug Metab. Pharmacokinet., 2017, 42, 891–901 CrossRef CAS PubMed.
- E. M. Stein, G. Garcia-Manero, D. A. Rizzieri, R. Tibes, J. G. Berdeja, M. Jongen-Lavrencic, J. K. Altman, H. Dohner, B. Thomson, S. J. Blakemore, S. Daigle, G. Fine, N. J. Waters, A. V. Krivstov, R. Koche, S. A. Armstrong, P. T. Ho, B. Lowenberg and M. S. Tallman, Blood, 2015, 126, 2547 CrossRef.
- N. Shukla, M. M. O’Brien, L. B. Silverman, M. Pauly, C. Wetmore, M. L. Loh, J. A. Whitlock, P. Brown, B. Thomson, S. J. Blakemore, S. Daigle, P. Pimentel, N. J. Waters, A. V. Krivstov, R. Koche, S. A. Armstrong, P. T. Ho and L. Gore, Blood, 2015, 126, 3792 CrossRef.
- I. Mahmood and H. Boxenbaum, Regul. Toxicol. Pharmacol., 2014, 68, 468–474 CrossRef PubMed.
- N. Shukla, C. Wetmore, M. M. O’Brien, L. B. Silverman, P. Brown, T. M. Cooper, B. Thomson, S. J. Blakemore, S. Daigle, B. Suttle, N. J. Waters, A. V. Krivstov, S. A. Armstrong, P. T. Ho and L. Gore, Blood, 2016, 128, 2780 CrossRef.
- C. R. Klaus, D. Iwanowicz, D. Johnston, C. A. Campbell, J. J. Smith, M. P. Moyer, R. A. Copeland, E. J. Olhava, M. P. Scott, R. M. Pollock, S. R. Daigle and A. Raimondi, J. Pharmacol. Exp. Ther., 2014, 350, 646–656 CrossRef PubMed.
- K. Menghrajani, S. F. Cai, S. M. Devlin, S. A. Armstrong, R. Piekarz, M. Rudek, M. S. Tallman and E. M. Stein, Blood, 2019, 134, 2655 CrossRef.
- M. Tachibana, K. Sugimoto, M. Nozaki, J. Ueda, T. Ohta, M. Ohki, M. Fukuda, N. Takeda, H. Niida, H. Kato and Y. Shinkai, Genes Dev., 2002, 16, 1779–1791 CrossRef CAS PubMed.
- S. R. Shankar, A. G. Bahirvani, V. K. Rao, N. Bharathy, J. R. Ow and R. Taneja, Epigenetics, 2013, 8, 16–22 CrossRef CAS PubMed.
- M. Tachibana, J. Ueda, M. Fukuda, N. Takeda, T. Ohta, H. Iwanari, T. Sakihama, T. Kodama, T. Hamakubo and Y. Shinkai, Genes Dev., 2005, 19, 815–826 CrossRef CAS PubMed.
- Y. Kondo, L. Shen, S. Ahmed, Y. Boumber, Y. Sekido, B. R. Haddad and J. P. J. Issa, PLoS One, 2008, 3, e2037 CrossRef PubMed.
- J. Huang, J. Dorsey, S. Chuikov, X. Zhang, T. Jenuwein, D. Reinberg and S. L. Berger, J. Biol. Chem., 2010, 285, 9636–9641 CrossRef CAS PubMed.
- H. Watanabe, K. Soejima, H. Yasuda, I. Kawada, I. Nakachi, S. Yoda, K. Naoki and A. Ishizaka, Cancer Cell Int., 2008, 8, 15 CrossRef PubMed.
- I. Maze, H. E. Covingtoni, D. M. Dietz, Q. Laplant, W. Renthal, S. J. Russo, M. Mechanic, E. Mouzon, R. L. Neve, S. J. Haggarty, Y. Ren, S. C. Sampath, Y. L. Hurd, P. Greengard, A. Tarakhovsky, A. Schaefer and E. J. Nestler, Science, 2010, 327, 213–216 CrossRef CAS PubMed.
- A. Schaefer, S. C. Sampath, A. Intrator, A. Min, T. S. Gertler, D. J. Surmeier, A. Tarakhovsky and P. Greengard, Neuron, 2009, 64, 678–691 CrossRef CAS PubMed.
- K. Imai, H. Togami and T. Okamoto, J. Biol. Chem., 2010, 285, 16538–16545 CrossRef CAS PubMed.
- M. Vedadi, D. Barsyte-Lovejoy, F. Liu, S. Rival-Gervier, A. Allali-Hassani, V. Labrie, T. J. Wigle, P. A. Dimaggio, G. A. Wasney, A. Siarheyeva, A. Dong, W. Tempel, S. C. Wang, X. Chen, I. Chau, T. J. Mangano, X. P. Huang, C. D. Simpson, S. G. Pattenden, J. L. Norris, D. B. Kireev, A. Tripathy, A. Edwards, B. L. Roth, W. P. Janzen, B. A. Garcia, A. Petronis, J. Ellis, P. J. Brown, S. V. Frye, C. H. Arrowsmith and J. Jin, Nat. Chem. Biol., 2011, 7, 566–574 CrossRef CAS PubMed.
- S. Kubicek, R. J. O’Sullivan, E. M. August, E. R. Hickey, Q. Zhang, M. L. L. Teodoro, S. Rea, K. Mechtler, J. A. Kowalski, C. A. Homon, T. A. Kelly and T. Jenuwein, Mol. Cell, 2007, 25, 473–481 CrossRef CAS PubMed.
- Y. Chang, X. Zhang, J. R. Horton, A. K. Upadhyay, A. Spannhoff, J. Liu, J. P. Snyder, M. T. Bedford and X. Cheng, Nat. Struct. Mol. Biol., 2009, 16, 312–317 CrossRef CAS PubMed.
- Y. Shi, J. T. Do, C. Desponts, H. S. Hahm, H. R. Schöler and S. Ding, Cell Stem Cell, 2008, 2, 525–528 CrossRef CAS PubMed.
- Y. Shi, C. Desponts, J. T. Do, H. S. Hahm, H. R. Schöler and S. Ding, Cell Stem Cell, 2008, 3, 568–574 CrossRef CAS PubMed.
- K. Imai, H. Togami and T. Okamoto, J. Biol. Chem., 2010, 285, 16538–16545 CrossRef CAS PubMed.
- A. J. Carter, R. Seupel, P. E. Brennan, M. Sundström, A. Introini and A. Mueller-Fahrnow, Chemical Strategies for Evaluating New Drug Targets, in: A. T. Plowright, ed., Target Discovery and Validation: Methods and Strategies for Drug Discovery, Wiley, 2019 Search PubMed.
- F. Liu, X. Chen, A. Allali-Hassani, A. M. Quinn, G. A. Wasney, A. Dong, D. Barsyte, I. Kozieradzki, G. Senisterra, I. Chau, A. Siarheyeva, D. B. Kireev, A. Jadhav, J. M. Herold, S. V. Frye, C. H. Arrowsmith, P. J. Brown, A. Simeonov, M. Vedadi and J. Jin, J. Med. Chem., 2009, 52, 7950–7953 CrossRef CAS PubMed.
- F. Liu, X. Chen, A. Allali-Hassani, A. M. Quinn, T. J. Wigle, G. A. Wasney, A. Dong, G. Senisterra, I. Chau, A. Siarheyeva, J. L. Norris, D. B. Kireev, A. Jadhav, J. M. Herold, W. P. Janzen, C. H. Arrowsmith, S. V. Frye, P. J. Brown, A. Simeonov, M. Vedadi and J. Jin, J. Med. Chem., 2010, 53, 5844–5857 CrossRef CAS PubMed.
- F. Liu, D. Barsyte-Lovejoy, A. Allali-Hassani, Y. He, J. M. Herold, X. Chen, C. M. Yates, S. V. Frye, P. J. Brown, J. Huang, M. Vedadi, C. H. Arrowsmith and J. Jin, J. Med. Chem., 2011, 54, 6139–6150 CrossRef CAS PubMed.
- R. F. Sweis, M. Pliushchev, P. J. Brown, J. Guo, F. Li, D. Maag, A. M. Petros, N. B. Soni, C. Tse, M. Vedadi, M. R. Michaelides, G. G. Chiang and W. N. Pappano, ACS Med. Chem. Lett., 2014, 5, 205–209 CrossRef CAS PubMed.
- W. N. Pappano, J. Guo, Y. He, D. Ferguson, S. Jagadeeswaran, D. J. Osterling, W. Gao, J. K. Spence, M. Pliushchev, R. F. Sweis, F. G. Buchanan, M. R. Michaelides, A. R. Shoemaker, C. Tse and G. G. Chiang, PLoS One, 2015, 10, e0131716 CrossRef PubMed.
- M. R. Pan, M. C. Hsu, C. W. Luo, L. T. Chen, Y. S. Shan and W. C. Hung, Oncotarget., 2016, 7, 61136–61151 CrossRef PubMed.
- K. Zhang, J. Wang, L. Yang, Y. C. Yuan, T. R. Tong, J. Wu, X. Yun, M. Bonner, R. Pangeni, Z. Liu, T. Yuchi, J. Y. Kim and D. J. Raz, Mol. Cancer, 2018, 17, 153 CrossRef CAS PubMed.
- I. Krivega, C. Byrnes, J. F. de Vasconcellos, Y. T. Lee, M. Kaushal, A. Dean and J. L. Miller, Blood, 2015, 126, 665–672 CrossRef CAS PubMed.
- A. Renneville, P. van Galen, M. C. Canver, M. McConkey, J. M. Krill-Burger, D. M. Dorfman, E. B. Holson, B. E. Bernstein, S. H. Orkin, D. E. Bauer and B. L. Ebert, Blood, 2015, 126, 1930–1939 CrossRef CAS PubMed.
- J. Lampe, J. Campbell, K. Duncan, M. A. C. Foley, D. M. Harvey, M. J. Munchhof, M. Thomenius and L. A. Reiter, WO2020037079, 2020.
- E. Chan-Penebre, V. Gibaja, J. Campbell, O. Olu, E. Admirand, D. Brach, C. Tang, A. Raimondi, T. Hood, A. Cocozaki, S. Karaman, K. Stickland, C. Plescia, M. Munchhof, K. Armstrong, T. Riera, S. Orkin, A. Boriack-Sjodin, W. Janzen, S. Blakemore, S. A. Ribich, R. Chesworth, J. J. Smith and K. W. Duncan, Blood, 2017, 130, 537 CrossRef PubMed.
- E. Chan-Penebre, V. Gibaja, J. Campbell, O. Ogunbodede, E. Admirand, D. Brach, C. Tang, A. Raimondi, T. Hood, A. Cocozaki, S. Karaman, K. Stickland, C. Plescia, M. Munchhof, K. Armstrong, T. Riera, S. Orkin, A. Boriack-Sjodin, W. Janzen, S. Blakemore, S. A. Ribich, R. Chesworth, J. J. Smith and K. W. Duncan, Blood, 2017, 130, 537 CrossRef PubMed.
- Epizyme, Inc. Annual Report on Form 10-K for the Fiscal Year Ended December 31, 2017, https://www.sec.gov/Archives/edgar/data/1571498/000156459018005397/epzm-10k_20171231.htm (accessed 3 December 2020).
- Epizyme Announces Positive Pre-NDA Meeting for Tazemetostat for Follicular Lymphoma, Pipeline Updates and Third Quarter 2019 Results, https://epizyme.gcs-web.com/news-releases/news-release-details/epizyme-announces-positive-pre-nda-meeting-tazemetostat, (accessed December 3, 2020).
- G. van Mierlo, G. J. C. Veenstra, M. Vermeulen and H. Marks, Trends Cell Biol., 2019, 29, 660–671 CrossRef CAS PubMed.
- R. Margueron and D. Reinberg, Nature, 2011, 469, 343–349 CrossRef CAS PubMed.
- B. Schuettengruber, D. Chourrout, M. Vervoort, B. Leblanc and G. Cavalli, Cell, 2007, 128, 735–745 CrossRef CAS PubMed.
- C. H. Lee, M. Holder, D. Grau, R. Saldaña-Meyer, J. R. Yu, R. A. Ganai, J. Zhang, M. Wang, G. LeRoy, M. W. Dobenecker, D. Reinberg and K. J. Armache, Mol. Cell, 2018, 70, 435–448.e5 CrossRef CAS PubMed.
- R. Margueron, G. Li, K. Sarma, A. Blais, J. Zavadil, C. L. Woodcock, B. D. Dynlacht and D. Reinberg, Mol. Cell, 2008, 32, 503–518 CrossRef CAS PubMed.
- A. Kuzmichev, K. Nishioka, H. Erdjument-Bromage, P. Tempst and D. Reinberg, Genes Dev., 2002, 16, 2893–2905 CrossRef CAS PubMed.
- J. A. Simon and C. A. Lange, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2008, 647, 21–29 CrossRef CAS PubMed.
- P. A. Croonquist and B. van Ness, Oncogene, 2005, 24, 6269–6280 CrossRef CAS PubMed.
- A. P. Bracken, D. Pasini, M. Capra, E. Prosperini, E. Colli and K. Helin, EMBO J., 2003, 22, 5323–5335 CrossRef CAS PubMed.
- F. J. van Kemenade, F. M. Raaphorst, T. Blokzijl, E. Fieret, K. M. Hamer, D. P. E. Satijn, A. P. Otte and C. J. L. M. Meijer, Blood, 2001, 97, 3896–3901 CrossRef CAS PubMed.
- B. D. W. Karanikolas, M. L. Figueiredo and L. Wu, Mol. Cancer Res., 2009, 7, 1456–1465 CrossRef CAS PubMed.
- S. Varambally, S. M. Dhanasekaran, M. Zhou, T. R. Barrette, C. Kumar-Sinha, M. G. Sanda, D. Ghosh, K. J. Pienta, R. G. A. B. Sewalt, M. A. Rubin and A. M. Chinnaiyan, Nature, 2002, 419, 624–629 CrossRef CAS PubMed.
- X. Li, M. E. Gonzalez, K. Toy, T. Filzen, S. D. Merajver and C. G. Kleer, Am. J. Pathol., 2009, 175, 1246–1254 CrossRef CAS PubMed.
- C. G. Kleer, Q. Cao, S. Varambally, R. Shen, I. Ota, S. A. Tomlins, D. Ghosh, R. G. A. B. Sewalt, A. P. Otte, D. F. Hayes, M. S. Sabel, D. Livant, S. J. Weiss, M. A. Rubin and A. M. Chinnaiyan, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11606–11611 CrossRef CAS PubMed.
- G. van Haaften, G. L. Dalgliesh, H. Davies, L. Chen, G. Bignell, C. Greenman, S. Edkins, C. Hardy, S. O’Meara, J. Teague, A. Butler, J. Hinton, C. Latimer, J. Andrews, S. Barthorpe, D. Beare, G. Buck, P. J. Campbell, J. Cole, S. Forbes, M. Jia, D. Jones, C. Y. Kok, C. Leroy, M. L. Lin, D. J. McBride, M. Maddison, S. Maquire, K. McLay, A. Menzies, T. Mironenko, L. Mulderrig, L. Mudie, E. Pleasance, R. Shepherd, R. Smith, L. Stebbings, P. Stephens, G. Tang, P. S. Tarpey, R. Turner, K. Turrell, J. Varian, S. West, S. Widaa, P. Wray, V. P. Collins, K. Ichimura, S. Law, J. Wong, S. T. Yuen, S. Y. Leung, G. Tonon, R. A. Depinho, Y. T. Tai, K. C. Anderson, R. J. Kahnoski, A. Massie, S. K. Khoo, B. T. Teh, M. R. Stratton and P. A. Futreal, Nat. Genet., 2009, 41, 521–523 CrossRef CAS PubMed.
- Y. H. Chen, M. C. Hung and L. Y. Li, Am. J. Transl. Res., 2012, 4, 364–375 CAS.
- S. K. Knutson, T. J. Wigle, N. M. Warholic, C. J. Sneeringer, C. J. Allain, C. R. Klaus, J. D. Sacks, A. Raimondi, C. R. Majer, J. Song, M. P. Scott, L. Jin, J. J. Smith, E. J. Olhava, R. Chesworth, M. P. Moyer, V. M. Richon, R. A. Copeland, H. Keilhack, R. M. Pollock and K. W. Kuntz, Nat. Chem. Biol., 2012, 8, 890–896 CrossRef CAS PubMed.
- M. T. McCabe, H. M. Ott, G. Ganji, S. Korenchuk, C. Thompson, G. S. van Aller, Y. Liu, A. della Pietra, L. V. LaFrance, M. Mellinger, C. Duquenne, X. Tian, R. G. Kruger, C. F. McHugh, W. H. Miller, D. Dhanak, S. K. Verma, P. J. Tummino, C. L. Creasy, A. P. Graves and E. Diaz, Nature, 2012, 492, 108–112 CrossRef CAS PubMed.
- S. K. Verma, X. Tian, L. V. Lafrance, C. Duquenne, D. P. Suarez, K. A. Newlander, S. P. Romeril, J. L. Burgess, S. W. Grant, J. A. Brackley, A. P. Graves, D. A. Scherzer, A. Shu, C. Thompson, H. M. Ott, G. S. van Aller, C. A. MacHutta, E. Diaz, Y. Jiang, N. W. Johnson, S. D. Knight, R. G. Kruger, M. T. McCabe, D. Dhanak, P. J. Tummino, C. L. Creasy and W. H. Miller, ACS Med. Chem. Lett., 2012, 3, 1091–1096 CrossRef CAS PubMed.
- E. Diaz, C. A. MacHutta, S. Chen, Y. Jiang, C. Nixon, G. Hofmann, D. Key, S. Sweitzer, M. Patel, Z. Wu, C. L. Creasy, R. G. Kruger, L. Lafrance, S. K. Verma, M. B. Pappalardi, B. Le, G. S. van Aller, M. T. McCabe, P. J. Tummino, A. J. Pope, S. H. Thrall, B. Schwartz and M. Bandt, J. Biomol. Screening, 2012, 17, 1279–1292 CrossRef CAS PubMed.
- R. D. Morin, N. A. Johnson, T. M. Severson, A. J. Mungall, J. An, R. Goya, J. E. Paul, M. Boyle, B. W. Woolcock, F. Kuchenbauer, D. Yap, R. K. Humphries, O. L. Griffith, S. Shah, H. Zhu, M. Kimbara, P. Shashkin, J. F. Charlot, M. Tcherpakov, R. Corbett, A. Tam, R. Varhol, D. Smailus, M. Moksa, Y. Zhao, A. Delaney, H. Qian, I. Birol, J. Schein, R. Moore, R. Holt, D. E. Horsman, J. M. Connors, S. Jones, S. Aparicio, M. Hirst, R. D. Gascoyne and M. A. Marra, Nat. Genet., 2010, 42, 181–185 CrossRef CAS PubMed.
- R. D. Morin, M. Mendez-Lago, A. J. Mungall, R. Goya, K. L. Mungall, R. D. Corbett, N. A. Johnson, T. M. Severson, R. Chiu, M. Field, S. Jackman, M. Krzywinski, D. W. Scott, D. L. Trinh, J. Tamura-Wells, S. Li, M. R. Firme, S. Rogic, M. Griffith, S. Chan, O. Yakovenko, I. M. Meyer, E. Y. Zhao, D. Smailus, M. Moksa, S. Chittaranjan, L. Rimsza, A. Brooks-Wilson, J. J. Spinelli, S. Ben-Neriah, B. Meissner, B. Woolcock, M. Boyle, H. McDonald, A. Tam, Y. Zhao, A. Delaney, T. Zeng, K. Tse, Y. Butterfield, I. Birol, R. Holt, J. Schein, D. E. Horsman, R. Moore, S. J. M. Jones, J. M. Connors, M. Hirst, R. D. Gascoyne and M. A. Marra, Nature, 2011, 476, 298–303 CrossRef CAS PubMed.
- J. G. Lohr, P. Stojanov, M. S. Lawrence, D. Auclair, B. Chapuy, C. Sougnez, P. Cruz-Gordillo, B. Knoechel, Y. W. Asmann, S. L. Slager, A. J. Novak, A. Dogan, S. M. Ansell, B. K. Link, L. Zou, J. Gould, G. Saksena, N. Stransky, C. Rangel-Escareño, J. C. Fernandez-Lopez, A. Hidalgo-Miranda, J. Melendez-Zajgla, E. Hernández-Lemus, A. Schwarz-Cruzy Celis, I. Imaz-Rosshandler, A. I. Ojesina, J. Jung, C. S. Pedamallu, E. S. Lander, T. M. Habermann, J. R. Cerhan, M. A. Shipp, G. Getz and T. R. Golub, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3879–3884 CrossRef CAS PubMed.
- T. A. Yap, J. N. Winter, L. Giulino-Roth, J. Longley, J. Lopez, J. M. Michot, J. P. Leonard, V. Ribrag, M. T. McCabe, C. L. Creasy, M. Stern, T. P. Dumitrescu, X. Wang, S. Frey, J. Carver, T. Horner, C. Oh, A. Khaled, A. Dhar and P. W. M. Johnson, Clin. Cancer Res., 2019, 25, 7331–7339 CrossRef CAS PubMed.
- K. W. Kuntz, J. E. Campbell, H. Keilhack, R. M. Pollock, S. K. Knutson, M. Porter-Scott, V. M. Richon, C. J. Sneeringer, T. J. Wigle, C. J. Allain, C. R. Majer, M. P. Moyer, R. A. Copeland and R. Chesworth, J. Med. Chem., 2016, 59, 1556–1564 CrossRef CAS PubMed.
- S. K. Knutson, N. M. Warholic, T. J. Wigle, C. R. Klaus, C. J. Allain, A. Raimondi, M. P. Scott, R. Chesworth, M. P. Moyer, R. A. Copeland, V. M. Richon, R. M. Pollock, K. W. Kuntz and H. Keilhack, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7922–7927 CrossRef CAS PubMed.
- S. K. Knutson, S. Kawano, Y. Minoshima, N. M. Warholic, K. C. Huang, Y. Xiao, T. Kadowaki, M. Uesugi, G. Kuznetsov, N. Kumar, T. J. Wigle, C. R. Klaus, C. J. Allain, A. Raimondi, N. J. Waters, J. J. Smith, M. Porter-Scott, R. Chesworth, M. P. Moyer, R. A. Copeland, V. M. Richon, T. Uenaka, R. M. Pollock, K. W. Kuntz, A. Yokoi and H. Keilhack, Mol. Cancer Ther., 2014, 13, 842–854 CrossRef CAS PubMed.
- K. F. Ginn and A. Gajjar, Front. Oncol., 2012, 2, 114 Search PubMed.
- V. Ribrag, J.-C. Soria, J.-M. Michot, A. Schmitt, S. Postel-Vinay, F. Bijou, B. Thomson, H. Keilhack, S. J. Blakemore, L. Reyderman, P. Kumar, G. Fine, A. McDonald, P. T. Ho and A. Italiano, Blood, 2015, 126, 473 CrossRef.
- C. Sarkozy, F. Morschhauser, S. Dubois, T. Molina, J. M. Michot, P. Cullières-Dartigues, B. Suttle, L. Karlin, S. le Gouill, J. M. Picquenot, R. Dubois, H. Tilly, C. Herbaux, F. Jardin, G. Salles and V. Ribrag, Clin. Cancer Res., 2020, 26, 3145–3153 CrossRef CAS PubMed.
- A. Italiano, J. C. Soria, M. Toulmonde, J. M. Michot, C. Lucchesi, A. Varga, J. M. Coindre, S. J. Blakemore, A. Clawson, B. Suttle, A. A. McDonald, M. Woodruff, S. Ribich, E. Hedrick, H. Keilhack, B. Thomson, T. Owa, R. A. Copeland, P. T. C. Ho and V. Ribrag, Lancet Oncol., 2018, 19, 649–659 CrossRef CAS PubMed.
- F. Morschhauser, H. Tilly, A. Chaidos, P. McKay, T. Phillips, S. Assouline, C. L. Batlevi, P. Campbell, V. Ribrag, G. L. Damaj, M. Dickinson, W. Jurczak, M. Kazmierczak, S. Opat, J. Radford, A. Schmitt, J. Yang, J. Whalen, S. Agarwal, D. Adib and G. Salles, Lancet Oncol., 2020, 21, 1433–1442 CrossRef CAS PubMed.
- M. Gounder, P. Schöffski, R. L. Jones, M. Agulnik, G. M. Cote, V. M. Villalobos, S. Attia, R. Chugh, T. W. W. Chen, T. Jahan, E. T. Loggers, A. Gupta, A. Italiano, G. D. Demetri, R. Ratan, L. E. Davis, O. Mir, P. Dileo, B. A. van Tine, J. G. Pressey, T. Lingaraj, A. Rajarethinam, L. Sierra, S. Agarwal and S. Stacchiotti, Lancet Oncol., 2020, 21, 1423–1432 CrossRef CAS PubMed.
- S. M. Hoy, Drugs, 2020, 80, 513–521 CrossRef PubMed.
- FDA granted accelerated approval to tazemetostat for follicular lymphoma, https://www.fda.gov/drugs/fda-granted-accelerated-approval-tazemetostat-follicular-lymphoma, (accessed 3 December 2020).
- S. Garapaty-Rao, C. Nasveschuk, A. Gagnon, E. Y. Chan, P. Sandy, J. Busby, S. Balasubramanian, R. Campbell, F. Zhao, L. Bergeron, J. E. Audia, B. K. Albrecht, J. C. Harmange, R. Cummings and P. Trojer, Chem. Biol., 2013, 20, 1329–1339 CrossRef CAS PubMed.
- C. G. Nasveschuk, A. Gagnon, S. Garapaty-Rao, S. Balasubramanian, R. Campbell, C. Lee, F. Zhao, L. Bergeron, R. Cummings, P. Trojer, J. E. Audia, B. K. Albrecht and J. C. P. Harmange, ACS Med. Chem. Lett., 2014, 5, 378–383 CrossRef CAS PubMed.
- V. S. Gehling, R. G. Vaswani, C. G. Nasveschuk, M. Duplessis, P. Iyer, S. Balasubramanian, F. Zhao, A. C. Good, R. Campbell, C. Lee, L. A. Dakin, A. S. Cook, A. Gagnon, J. C. Harmange, J. E. Audia, R. T. Cummings, E. Normant, P. Trojer and B. K. Albrecht, Bioorg. Med. Chem. Lett., 2015, 25, 3644–3649 CrossRef CAS PubMed.
- R. G. Vaswani, V. S. Gehling, L. A. Dakin, A. S. Cook, C. G. Nasveschuk, M. Duplessis, P. Iyer, S. Balasubramanian, F. Zhao, A. C. Good, R. Campbell, C. Lee, N. Cantone, R. T. Cummings, E. Normant, S. F. Bellon, B. K. Albrecht, J. C. Harmange, P. Trojer, J. E. Audia, Y. Zhang, N. Justin, S. Chen, J. R. Wilson and S. J. Gamblin, J. Med. Chem., 2016, 59, 9928–9941 CrossRef CAS PubMed.
- W. Harb, J. Abramson, M. Lunning, A. Goy, K. Maddocks, C. Lebedinsky, A. Senderowicz, P. Trojer, W. D. Bradley and I. Flinn, Ann. Oncol., 2018, 29, iii7 CrossRef.
- M.-E. Taplin, A. Hussain, N. D. Shore, B. Bradley, P. Trojer, C. Lebedinsky, A. M. Senderowicz and E. S. Antonarakis, J. Clin. Oncol., 2018, 36, TPS398 Search PubMed.
- M.-E. Taplin, A. Hussain, S. Shah, N. D. Shore, W. J. Edenfield, O. A. Sartor, L. T. Nordquist, M. Agrawal, W. Clark, D. R. Wise, W. K. Oh, M. T. Fleming, J. E. Butrynski, G. S. Chatta, M. Bupathi, C. Lebedinsky, A. Senderowicz, J. Li, G. Colak, D. Nash, P. Trojer, W. D. Bradley, J. Piel and E. S. Antonarakis, Cancer Res., 2019, CT094 Search PubMed.
- R. S. Blanc and S. Richard, Mol. Cell, 2017, 65, 8–24 CrossRef CAS PubMed.
- M. T. Bedford and S. Richard, Mol. Cell, 2005, 18, 263–272 CrossRef CAS PubMed.
- M. T. Bedford and S. G. Clarke, Mol. Cell, 2009, 33, 1–13 CrossRef CAS PubMed.
- K. Tripsianes, T. Madl, M. MacHyna, D. Fessas, C. Englbrecht, U. Fischer, K. M. Neugebauer and M. Sattler, Nat. Struct. Mol. Biol., 2011, 18, 1414–1420 CrossRef CAS PubMed.
- Y. Yang and M. T. Bedford, Nat. Rev. Cancer, 2013, 13, 37–50 CrossRef CAS PubMed.
- M. Yoshimatsu, G. Toyokawa, S. Hayami, M. Unoki, T. Tsunoda, H. I. Field, J. D. Kelly, D. E. Neal, Y. Maehara, B. A. J. Ponder, Y. Nakamura and R. Hamamoto, Int. J. Cancer, 2011, 128, 562–573 CrossRef CAS PubMed.
- S. el Messaoudi, E. Fabbrizio, C. Rodriguez, P. Chuchana, L. Fauquier, D. Cheng, C. Theillet, L. Vandel, M. T. Bedford and C. Sardet, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13351–13356 CrossRef CAS PubMed.
- Y. R. Kim, B. K. Lee, R. Y. Park, N. T. X. Nguyen, J. A. Bae, D. D. Kwon and C. Jung, BMC Cancer, 2010, 10, 197 CrossRef PubMed.
- H. Hong, C. Kao, M. H. Jeng, J. N. Eble, M. O. Koch, T. A. Gardner, S. Zhang, L. Li, C. X. Pan, Z. Hu, G. T. MacLennan and L. Cheng, Cancer, 2004, 101, 83–89 CrossRef CAS PubMed.
- S. Majumder, Y. Liu, O. H. Ford, J. L. Mohler and Y. E. Whang, Prostate, 2006, 66, 1292–1301 CrossRef CAS PubMed.
- L. H. Mitchell, A. E. Drew, S. A. Ribich, N. Rioux, K. K. Swinger, S. L. Jacques, T. Lingaraj, P. A. Boriack-Sjodin, N. J. Waters, T. J. Wigle, O. Moradei, L. Jin, T. Riera, M. Porter-Scott, M. P. Moyer, J. J. Smith, R. Chesworth and R. A. Copeland, ACS Med. Chem. Lett., 2015, 6, 655–659 CrossRef CAS PubMed.
- M. S. Eram, Y. Shen, M. M. Szewczyk, H. Wu, G. Senisterra, F. Li, K. V. Butler, H. Ü. Kaniskan, B. A. Speed, C. dela Seña, A. Dong, H. Zeng, M. Schapira, P. J. Brown, C. H. Arrowsmith, D. Barsyte-Lovejoy, J. Liu, M. Vedadi and J. Jin, ACS Chem. Biol., 2016, 11, 772–781 CrossRef CAS PubMed.
- S. Dhar, V. Vemulapalli, A. N. Patananan, G. L. Huang, A. di Lorenzo, S. Richard, M. J. Comb, A. Guo, S. G. Clarke and M. T. Bedford, Sci. Rep., 2013, 3, 1311 CrossRef PubMed.
- A. Fedoriw, S. R. Rajapurkar, S. O’Brien, S. V. Gerhart, L. H. Mitchell, N. D. Adams, N. Rioux, T. Lingaraj, S. A. Ribich, M. B. Pappalardi, N. Shah, J. Laraio, Y. Liu, M. Butticello, C. L. Carpenter, C. Creasy, S. Korenchuk, M. T. McCabe, C. F. McHugh, R. Nagarajan, C. Wagner, F. Zappacosta, R. Annan, N. O. Concha, R. A. Thomas, T. K. Hart, J. J. Smith, R. A. Copeland, M. P. Moyer, J. Campbell, K. Stickland, J. Mills, S. Jacques-O’Hagan, C. Allain, D. Johnston, A. Raimondi, M. Porter Scott, N. Waters, K. Swinger, A. Boriack-Sjodin, T. Riera, G. Shapiro, R. Chesworth, R. K. Prinjha, R. G. Kruger, O. Barbash and H. P. Mohammad, Cancer Cell, 2019, 36(100-114), e25 Search PubMed.
- N. Stopa, J. E. Krebs and D. Shechter, Cell. Mol. Life Sci., 2015, 72, 2041–2059 CrossRef CAS PubMed.
- S. Pal, S. N. Vishwanath, H. Erdjument-Bromage, P. Tempst and S. Sif, Mol. Cell. Biol., 2004, 24, 9630–9645 CrossRef CAS PubMed.
- L. Alinari, K. v. Mahasenan, F. Yan, V. Karkhanis, J. H. Chung, E. M. Smith, C. Quinion, P. L. Smith, L. Kim, J. T. Patton, R. Lapalombella, B. Yu, Y. Wu, S. Roy, A. de Leo, S. Pileri, C. Agostinelli, L. Ayers, J. E. Bradner, S. Chen-Kiang, O. Elemento, T. Motiwala, S. Majumder, J. C. Byrd, S. Jacob, S. Sif, C. Li and R. A. Baiocchi, Blood, 2015, 125, 2530–2543 CrossRef CAS PubMed.
- C. Paul, C. Sardet and E. Fabbrizio, Biol. Open, 2015, 4, 312–316 CrossRef.
- S. E. LeBlanc, S. Konda, Q. Wu, Y. J. Hu, C. M. Oslowski, S. Sif and A. N. Imbalzano, Mol. Endocrinol., 2012, 26, 583–597 CrossRef CAS PubMed.
- H. Shailesh, Z. Z. Zakaria, R. Baiocchi and S. Sif, Oncotarget., 2018, 9, 36705–36718 CrossRef PubMed.
- S. Pal, R. A. Baiocchi, J. C. Byrd, M. R. Grever, S. T. Jacob and S. Sif, EMBO J., 2007, 26, 3558–3569 CrossRef CAS PubMed.
- J. Chung, V. Karkhanis, S. Tae, F. Yan, P. Smith, L. W. Ayers, C. Agostinelli, S. Pileri, G. V. Denis, R. A. Baiocchi and S. Sif, J. Biol. Chem., 2013, 288, 35534–35547 CrossRef CAS PubMed.
- T. Y. W. Wei, C. C. Juan, J. Y. Hisa, L. J. Su, Y. C. G. Lee, H. Y. Chou, J. M. M. Chen, Y. C. Wu, S. C. Chiu, C. P. Hsu, K. L. Liu and C. T. R. Yu, Cancer Sci., 2012, 103, 1640–1650 CrossRef CAS PubMed.
- X. Han, R. Li, W. Zhang, X. Yang, C. G. Wheeler, G. K. Friedman, P. Province, Q. Ding, Z. You, H. M. Fathallah-Shaykh, G. Y. Gillespie, X. Zhao, P. H. King and L. B. Nabors, J. Neuro-Oncol., 2014, 118, 61–72 CrossRef CAS PubMed.
- F. Yan, L. Alinari, M. E. Lustberg, L. K. Martin, H. M. Cordero-Nieves, Y. Banasavadi-Siddegowda, S. Virk, J. Barnholtz-Sloan, E. H. Bell, J. Wojton, N. K. Jacob, A. Chakravarti, M. O. Nowicki, X. Wu, R. Lapalombella, J. Datta, B. Yu, K. Gordon, A. Haseley, J. T. Patton, P. L. Smith, J. Ryu, X. Zhang, X. Mo, G. Marcucci, G. Nuovo, C. H. Kwon, J. C. Byrd, E. A. Chiocca, C. Li, S. Sif, S. Jacob, S. Lawler, B. Kaur and R. A. Baiocchi, Cancer Res., 2014, 74, 1752–1765 CrossRef CAS PubMed.
- M. A. Powers, M. M. Fay, R. E. Factor, A. L. Welm and K. S. Ullman, Cancer Res., 2011, 71, 5579–5587 CrossRef CAS PubMed.
- E.-C. Cho, S. Zheng, S. Munro, G. Liu, S. M. Carr, J. Moehlenbrink, Y.-C. Lu, L. Stimson, O. Khan, R. Konietzny, J. McGouran, A. S. Coutts, B. Kessler, D. J. Kerr and N. B. la Thangue, EMBO J., 2012, 31, 1785–1797 CrossRef CAS PubMed.
- J. C. Fisk and L. K. Read, Eukaryotic Cell, 2011, 10, 1013–1022 CrossRef CAS PubMed.
- C. der Liu, C. P. Cheng, J. S. Fang, L. C. Chen, B. Zhao, E. Kieff and C. W. Peng, Biochem. Biophys. Res. Commun., 2013, 430, 1097–1102 CrossRef PubMed.
- R. Doueiri, R. Anupam, M. Kvaratskhelia, K. B. Green, M. D. Lairmore and P. L. Green, Retrovirology, 2012, 9, 64 CrossRef CAS.
- E. Chan-Penebre, K. G. Kuplast, C. R. Majer, P. A. Boriack-Sjodin, T. J. Wigle, L. D. Johnston, N. Rioux, M. J. Munchhof, L. Jin, S. L. Jacques, K. A. West, T. Lingaraj, K. Stickland, S. A. Ribich, A. Raimondi, M. P. Scott, N. J. Waters, R. M. Pollock, J. J. Smith, O. Barbash, M. Pappalardi, T. F. Ho, K. Nurse, K. P. Oza, K. T. Gallagher, R. Kruger, M. P. Moyer, R. A. Copeland, R. Chesworth and K. W. Duncan, Nat. Chem. Biol., 2015, 11, 432–437 CrossRef CAS PubMed.
- K. W. Duncan, N. Rioux, P. A. Boriack-Sjodin, M. J. Munchhof, L. A. Reiter, C. R. Majer, L. Jin, L. D. Johnston, E. Chan-Penebre, K. G. Kuplast, M. Porter Scott, R. M. Pollock, N. J. Waters, J. J. Smith, M. P. Moyer, R. A. Copeland and R. Chesworth, ACS Med. Chem. Lett., 2016, 7, 162–166 CrossRef CAS PubMed.
- S. V. Gerhart, W. A. Kellner, C. Thompson, M. B. Pappalardi, X. P. Zhang, R. Montes De Oca, E. Penebre, K. Duncan, A. Boriack-Sjodin, B. Le, C. Majer, M. T. McCabe, C. Carpenter, N. Johnson, R. G. Kruger and O. Barbash, Sci. Rep., 2018, 8, 9711 CrossRef PubMed.
- L. L. Siu, D. W. Rasco, S. P. Vinay, P. M. Romano, J. Menis, F. L. Opdam, K. M. Heinhuis, J. L. Egger, S. A. Gorman, R. Parasrampuria, K. Wang, B. E. Kremer and M. M. Gounder, Ann. Oncol., 2019, 30, v159 CrossRef.
- D. Rasco, A. Tolcher, L. L. Siu, K. Heinhuis, S. Postel-Vinay, O. Barbash, J. L. Egger, S. Gorman, T. Horner, A. Dhar and B. E. Kremer, Cancer Res., 2017, 77, CT038 Search PubMed.
- J. M. Watts, T. J. Bradley, A. Thomassen, A. M. Brunner, M. D. Minden, N. Papadantonakis, S. Abedin, A. J. Baines, O. Barbash, S. Gorman, B. E. Kremer and G. M. Borthakur, Blood, 2019, 134, 2656 CrossRef.
- Z. Q. Bonday, G. S. Cortez, M. J. Grogan, S. Antonysamy, K. Weichert, W. P. Bocchinfuso, F. Li, S. Kennedy, B. Li, M. M. Mader, C. H. Arrowsmith, P. J. Brown, M. S. Eram, M. M. Szewczyk, D. Barsyte-Lovejoy, M. Vedadi, E. Guccione and R. M. Campbell, ACS Med. Chem. Lett., 2018, 9, 612–617 CrossRef CAS PubMed.
- S. Antonysamy, Z. Bonday, R. M. Campbell, B. Doyle, Z. Druzina, T. Gheyi, B. Han, L. N. Jungheim, Y. Qian, C. Rauch, M. Russell, J. M. Sauder, S. R. Wasserman, K. Weichert, F. S. Willard, A. Zhang and S. Emtage, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17960–17965 CrossRef CAS PubMed.
- T. Wu, D. Brehmer, L. Beke, A. Boeckx, G. S. M. Diels, R. A. H. J. Gilissen, E. C. Lawson, V. Pande, M. C. B. C. Parade, W. B. G. Schepens, J. W. J. F. Thuring, M. Viellevoye, W. Sun and L. Meerpoel, WO2017032840, 2017.
- D. Brehmer, T. Wu, G. Mannens, L. Beke, P. Vinken, D. Gaffney, W. Sun, V. Pande, J.-W. Thuring, H. Millar, I. Poggesi, I. Somers, A. Boeckx, M. Parade, E. van Heerde, T. Nys, C. Yanovich, B. Herkert, T. Verhulst, M. du Jardin, L. Meerpoel, C. Moy, G. Diels, M. Viellevoye, W. Schepens, A. Poncelet, J. T. Linders, E. C. Lawson, J. P. Edwards, D. Chetty, S. Laquerre and M. V. Lorenzi, Cancer Res., 2017, 77, DDT02-04 Search PubMed.
- H. Lin and J. I. Luengo, Bioorg. Med. Chem. Lett., 2019, 29, 1264–1269 CrossRef CAS PubMed.
- H. Tao, X. Yan, K. Zhu and H. Zhang, Chem. Pharm. Bull., 2019, 67, 382–388 CrossRef CAS PubMed.
- X. Li, C. Wang, H. Jiang and C. Luo, Expert Opin. Ther. Pat., 2019, 29, 97–114 CrossRef CAS PubMed.
- R. Ferreira de Freitas, D. Ivanochko and M. Schapira, Molecules, 2019, 24, 4492 CrossRef CAS PubMed.
- T. Wu, H. Millar, D. Gaffney, L. Beke, G. Mannens, P. Vinken, I. Sommers, J.-W. Thuring, W. Sun, C. Moy, V. Pande, J. Zhou, N. Haddish-Berhane, M. Salvati, S. Laquerre, M. V. Lorenzi and D. Brehmer, Cancer Res., 2018, 78, 4859 Search PubMed.
- H. J. Millar, D. Brehmer, T. Verhulst, N. Haddish-Berhane, T. Greway, D. Gaffney, A. Boeckx, E. van Heerde, T. Nys, J. Portale, U. Philippar, T. Wu, S. Laquerre and K. Packman, Cancer Res., 2019, 79, 950 Search PubMed.
- M. V. Villar, A. Spreafico, V. Moreno, I. Braña, T. Hernandez, A. A. Razak, J. Wang, N. Haddish-Berhane, J. Mehta, A. Johnson, A. Maes, J. Haslam, P. Mistry, A. Kalota, L. Lenox, J. Infante, M. Lorenzi, H. Xie, J. Lauring and M. R. Patel, Ann. Oncol., 2020, 31, S470 CrossRef.
- D. Ofengeim and J. Yuan, Nat. Rev. Mol. Cell Biol., 2013, 14, 727–736 CrossRef CAS PubMed.
- D. Ofengeim, Y. Ito, A. Najafov, Y. Zhang, B. Shan, J. P. DeWitt, J. Ye, X. Zhang, A. Chang, H. Vakifahmetoglu-Norberg, J. Geng, B. Py, W. Zhou, P. Amin, J. B. Lima, C. Qi, Q. Yu, B. Trapp and J. Yuan, Cell Rep., 2015, 10, 1836–1849 CrossRef CAS PubMed.
- Y. Ito, D. Ofengeim, A. Najafov, S. Das, S. Saberi, Y. Li, J. Hitomi, H. Zhu, H. Chen, L. Mayo, J. Geng, P. Amin, J. P. DeWitt, A. K. Mookhtiar, M. Florez, A. T. Ouchida, J. B. Fan, M. Pasparakis, M. A. Kelliher, J. Ravits and J. Yuan, Science, 2016, 353, 603–608 CrossRef CAS PubMed.
- A. Caccamo, C. Branca, I. S. Piras, E. Ferreira, M. J. Huentelman, W. S. Liang, B. Readhead, J. T. Dudley, E. E. Spangenberg, K. N. Green, R. Belfiore, W. Winslow and S. Oddo, Nat. Neurosci., 2017, 20, 1236–1246 CrossRef CAS PubMed.
- B. Shan, H. Pan, A. Najafov and J. Yuan, Genes Dev., 2018, 32, 327–340 CrossRef CAS PubMed.
- A. Kaczmarek, P. Vandenabeele and D. V. Krysko, Immunity, 2013, 38, 209–223 CrossRef CAS PubMed.
- J. Yuan, P. Amin and D. Ofengeim, Nat. Rev. Neurosci., 2019, 20, 19–33 CrossRef CAS PubMed.
- A. Degterev, J. Hitomi, M. Germscheid, I. L. Ch’en, O. Korkina, X. Teng, D. Abbott, G. D. Cuny, C. Yuan, G. Wagner, S. M. Hedrick, S. A. Gerber, A. Lugovskoy and J. Yuan, Nat. Chem. Biol., 2008, 4, 313–321 CrossRef CAS PubMed.
- A. Degterev, J. L. Maki and J. Yuan, Cell Death Differ., 2013, 20, 366 CrossRef CAS PubMed.
- X. Teng, A. Degterev, P. Jagtap, X. Xing, S. Choi, R. Denu, J. Yuan and G. D. Cuny, Bioorg. Med. Chem. Lett., 2005, 15, 5039–5044 CrossRef CAS PubMed.
- S. Berger, P. Harris, R. Nagilla, V. Kasparcova, S. Hoffman, B. Swift, L. Dare, M. Schaeffer, C. Capriotti, M. Ouellette, B. King, D. Wisnoski, J. Cox, M. Reilly, R. Marquis, J. Bertin and P. Gough, Cell Death Discovery, 2015, 1, 1 CrossRef PubMed.
- P. A. Harris, B. W. King, D. Bandyopadhyay, S. B. Berger, N. Campobasso, C. A. Capriotti, J. A. Cox, L. Dare, X. Dong, J. N. Finger, L. S. C. Grady, S. J. Hoffman, J. U. Jeong, J. Kang, V. Kasparcova, A. S. Lakdawala, R. Lehr, D. E. McNulty, R. Nagilla, M. T. Ouellette, C. S. Pao, A. R. Rendina, M. C. Schaeffer, J. D. Summerfield, B. A. Swift, R. D. Totoritis, P. Ward, A. Zhang, D. Zhang, R. W. Marquis, J. Bertin and P. J. Gough, J. Med. Chem., 2016, 59, 2163–2178 CrossRef CAS PubMed.
- P. A. Harris, S. B. Berger, J. U. Jeong, R. Nagilla, D. Bandyopadhyay, N. Campobasso, C. A. Capriotti, J. A. Cox, L. Dare, X. Dong, P. M. Eidam, J. N. Finger, S. J. Hoffman, J. Kang, V. Kasparcova, B. W. King, R. Lehr, Y. Lan, L. K. Leister, J. D. Lich, T. T. MacDonald, N. A. Miller, M. T. Ouellette, C. S. Pao, A. Rahman, M. A. Reilly, A. R. Rendina, E. J. Rivera, M. C. Schaeffer, C. A. Sehon, R. R. Singhaus, H. H. Sun, B. A. Swift, R. D. Totoritis, A. Vossenkämper, P. Ward, D. D. Wisnoski, D. Zhang, R. W. Marquis, P. J. Gough and J. Bertin, J. Med. Chem., 2017, 60, 1247–1261 CrossRef CAS PubMed.
- A. Degterev, D. Ofengeim and J. Yuan, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9714–9722 CrossRef CAS PubMed.
- K. Weisel, N. E. Scott, D. J. Tompson, B. J. Votta, S. Madhavan, K. Povey, A. Wolstenholme, M. Simeoni, T. Rudo, L. Richards-Peterson, T. Sahota, J. G. Wang, J. Lich, J. Finger, A. Verticelli, M. Reilly, P. J. Gough, P. A. Harris, J. Bertin and M. L. Wang, Pharmacol. Res. Perspect., 2017, 5, e00365 CrossRef PubMed.
- L. Mifflin, D. Ofengeim and J. Yuan, Nat. Rev. Drug Discovery, 2020, 19, 553–571 CrossRef CAS PubMed.
- K. Weisel, S. Berger, K. Papp, C. Maari, J. G. Krueger, N. Scott, D. Tompson, S. Wang, M. Simeoni, J. Bertin and P. Peter Tak, Clin. Pharmacol. Ther., 2020, 108, 808–816 CrossRef CAS PubMed.
- J. Y. L. Chiang, J. Lipid Res., 2009, 50, 1955–1966 CrossRef CAS PubMed.
- D. J. Parks, S. G. Blanchard, R. K. Bledsoe, G. Chandra, T. G. Consler, S. A. Kliewer, J. B. Stimmel, T. M. Willson, A. M. Zavacki, D. D. Moore and J. M. Lehmann, Science, 1999, 284, 1365–1368 CrossRef CAS PubMed.
- M. Makishima, A. Y. Okamoto, J. J. Repa, H. Tu, R. M. Learned, A. Luk, M. V. Hull, K. D. Lustig, D. J. Mangelsdorf and B. Shan, Science, 1999, 284, 1362–1365 CrossRef CAS PubMed.
- A. C. Calkin and P. Tontonoz, Nat. Rev. Mol. Cell Biol., 2012, 13, 213–224 CrossRef CAS PubMed.
- L. Ding, S. Pang, Y. Sun, Y. Tian, L. Yu and N. Dang, Int. J. Endocrinol., 2014, 751859 Search PubMed.
- D. C. Tully, P. v. Rucker, D. Chianelli, J. Williams, A. Vidal, P. B. Alper, D. Mutnick, B. Bursulaya, J. Schmeits, X. Wu, D. Bao, J. Zoll, Y. Kim, T. Groessl, P. McNamara, H. M. Seidel, V. Molteni, B. Liu, A. Phimister, S. B. Joseph and B. Laffitte, J. Med. Chem., 2017, 60, 9960–9973 CrossRef CAS PubMed.
- J. K. Dyson, G. M. Hirschfield, D. H. Adams, U. Beuers, D. A. Mann, K. D. Lindor and D. E. J. Jones, Nat. Rev. Gastroenterol. Hepatol., 2015, 12, 147–158 CrossRef CAS PubMed.
- A. M. Diehl and C. Day, N. Engl. J. Med., 2017, 377, 2063–2072 CrossRef CAS PubMed.
- P. R. Maloney, D. J. Parks, C. D. Haffner, A. M. Fivush, G. Chandra, K. D. Plunket, K. L. Creech, L. B. Moore, J. G. Wilson, M. C. Lewis, S. A. Jones and T. M. Willson, J. Med. Chem., 2000, 43, 2971–2974 CrossRef CAS PubMed.
- C. Gege, E. Hambruch, N. Hambruch, O. Kinzel and C. Kremoser, Handb. Exp. Pharmacol., 2019, 256, 167–205 CAS.
- C. H. Kuo, J. B. Hook and J. Bernstein, Toxicology, 1981, 22, 149–160 CrossRef CAS.
- K. Sugihara, S. Kitamura, S. Sanoh, S. Ohta, N. Fujimoto, S. Maruyama and A. Ito, Toxicol. Appl. Pharmacol., 2000, 167, 46–54 CrossRef CAS PubMed.
- A. Akwabi-Ameyaw, J. Y. Bass, R. D. Caldwell, J. A. Caravella, L. Chen, K. L. Creech, D. N. Deaton, S. A. Jones, I. Kaldor, Y. Liu, K. P. Madauss, H. B. Marr, R. B. McFadyen, A. B. Miller, F. Navas III, D. J. Parks, P. K. Spearing, D. Todd, S. P. Williams and G. B. Wisely, Bioorg. Med. Chem. Lett., 2008, 18, 4339–4343 CrossRef CAS PubMed.
- J. Y. Bass, J. A. Caravella, L. Chen, K. L. Creech, D. N. Deaton, K. P. Madauss, H. B. Marr, R. B. McFadyen, A. B. Miller, W. Y. Mills, F. Navas, D. J. Parks, T. L. Smalley, P. K. Spearing, D. Todd, S. P. Williams and G. B. Wisely, Bioorg. Med. Chem. Lett., 2011, 21, 1206–1213 CrossRef CAS PubMed.
- O. Kinzel, C. Steeneck, T. Schlüter, A. Schulz, C. Gege, U. Hahn, E. Hambruch, M. Hornberger, A. Spalwisz, K. Frick, S. Perović-Ottstadt, U. Deuschle, M. Burnet and C. Kremoser, Bioorg. Med. Chem. Lett., 2016, 26, 3746–3753 CrossRef CAS PubMed.
- E. Hambruch, S. Miyazaki-Anzai, U. Hahn, S. Matysik, A. Boettcher, S. Perović-Ottstadt, T. Schlüter, O. Kinzel, H. D. Krol, U. Deuschle, M. Burnet, M. Levi, G. Schmitz, M. Miyazaki and C. Kremoser, J. Pharmacol. Exp. Ther., 2012, 343, 556–567 CrossRef CAS PubMed.
- E. Hambruch, O. Kinzel and C. Kremoser, Nucl. Recept. Res., 2016, 3, 12 Search PubMed.
- P. A. Blomgren, K. S. Currie, J. Farand, C. Gege, J. E. Kropf and J. Xu, WO2017218337A1, 2017.
- P. Schwabl, G. Budas, E. Hambruch, P. Supper, M. Burnet, J. Liles, T. Sullivan, E. Huntzicker, M. Birkel, D. French, D. Tumas, M. Peck-Radosavljevic, W. Watkins, M. Trauner, D. Breckenridge, C. Kremoser and T. Reiberger, J. Hepatol., 2018, 68, S471–S472 CrossRef.
- M. Trauner, A. Gulamhusein, B. Hameed, S. Caldwell, M. L. Shiffman, C. Landis, B. Eksteen, K. Agarwal, A. Muir, S. Rushbrook, X. Lu, J. Xu, J. Chuang, A. N. Billin, G. Li, C. Chung, G. M. Subramanian, R. P. Myers, C. L. Bowlus and K. V. Kowdley, Hepatology, 2019, 70, 788–801 CrossRef CAS PubMed.
- K. Patel, S. A. Harrison, M. Elkhashab, J. F. Trotter, R. Herring, S. E. Rojter, Z. Kayali, V. W. Wong, S. Greenbloom, S. Jayakumar, M. L. Shiffman, B. Freilich, E. J. Lawitz, E. J. Gane, E. Harting, J. Xu, A. N. Billin, C. Chung, C. S. Djedjos, G. M. Subramanian, R. P. Myers, M. S. Middleton, M. Rinella and M. Noureddin, Hepatology, 2020, 72, 58–71 CrossRef CAS PubMed.
- B. Goodwin, S. A. Jones, R. R. Price, M. A. Watson, D. D. McKee, L. B. Moore, C. Galardi, J. G. Wilson, M. C. Lewis, M. E. Roth, P. R. Maloney, T. M. Willson and S. A. Kliewer, Mol. Cell, 2000, 6, 517–526 CrossRef CAS PubMed.
- M. J. Genin, A. B. Bueno, J. Agejas Francisco, P. R. Manninen, W. P. Bocchinfuso, C. Montrose-Rafizadeh, E. A. Cannady, T. M. Jones, J. R. Stille, E. Raddad, C. Reidy, A. Cox, M. D. Michael and L. F. Michael, J. Med. Chem., 2015, 58, 9768–9772 CrossRef CAS PubMed.
- C. Gege, E. Hambruch, N. Hambruch, O. Kinzel and C. Kremoser, Handb. Exp. Pharmacol., 2019, 256, 167–205 CAS.
- A novel farnesoid X receptor agonist, TERN-101, reduces liver steatosis, inflammation, ballooning and fibrosis in a murine model of non-alcoholic steatohepatitis, https://static1.squarespace.com/static/5ca5527392441ba90c555077/t/5ced485124a694fe23372d28/1559054421299/2618+TERNS-101+Poster+to+EASL.pdf, (accessed 01 March 2021).
- Pharmacokinetics of two oral formulations of liver-directed, nonsteroidal farnesoid X-receptor agonist TERN-101 in healthy volunteers, https://static1.squarespace.com/static/5ca5527392441ba90c555077/t/5fbea14f7acac6192a9d1ae8/1606328655634/TERN-101_ParisNASH_2020_POSTER_FINAL.pdf, (accessed 01 March 2021).
- M. K. Badman, J. Chen, S. Desai, S. Vaidya, S. Neelakantham, J. Zhang, L. Gan, K. Danis, B. Laffitte and L. B. Klickstein, Clin. Pharmacol. Drug Dev., 2020, 9, 395–410 CrossRef CAS PubMed.
- Clinical trial results summary, https://www.novctrd.com/ctrdweb/patientsummary/patientsummaries?patientSummaryId=322, (accessed March 1, 2021).
- K. J. Lucas, P. Lopez, E. Lawitz, A. Sheikh, D. Aizenberg, S. Hsia, G. G. B. Bee, J. Vierling, J. Frias, J. White, Y. Eguchi, D. Lazas, G. Neff, M. Yoneda, S. Augustin, W. Kim, Y. Loeffler, F. Schaefer, S. Lamle, M. Martic, C. Brass and A. Sanyal, Dig. Liver Dis., 2020, 52, e38 CrossRef.
- T. Oltersdorf, S. W. Elmore, A. R. Shoemaker, R. C. Armstrong, D. J. Augeri, B. A. Belli, M. Bruncko, T. L. Deckwerth, J. Dinges, P. J. Hajduk, M. K. Joseph, S. Kitada, S. J. Korsmeyer, A. R. Kunzer, A. Letai, C. Li, M. J. Mitten, D. G. Nettesheim, S. C. Ng, P. M. Nimmer, J. M. O’Connor, A. Oleksijew, A. M. Petros, J. C. Reed, W. Shen, S. K. Tahir, C. B. Thompson, K. J. Tomaselli, B. Wang, M. D. Wendt, H. Zhang, S. W. Fesik and S. H. Rosenberg, Nature, 2005, 435, 677–681 CrossRef CAS PubMed.
- J. D. Leverson, D. Sampath, A. J. Souers, S. H. Rosenberg, W. J. Fairbrother, M. Amiot, M. Konopleva and A. Letai, Cancer Discovery, 2017, 7, 1376–1393 CrossRef CAS PubMed.
- N. N. Danial and S. J. Korsmeyer, Cell, 2004, 116, 205–219 CrossRef CAS PubMed.
- T. J. McDonnell, N. Deane, F. M. Platt, G. Nunez, U. Jaeger, J. P. McKearn and S. J. Korsmeyer, Cell, 1989, 57, 79–88 CrossRef CAS PubMed.
- D. L. Vaux, S. Cory and J. M. Adams, Nature, 1988, 335, 440–442 CrossRef CAS PubMed.
- L. H. Boise, M. González-García, C. E. Postema, L. Ding, T. Lindsten, L. A. Turka, X. Mao, G. Nuñez and C. B. Thompson, Cell, 1993, 74, 597–608 CrossRef CAS PubMed.
- K. M. Kozopas, T. Yang, H. L. Buchan, P. Zhou and R. W. Craig, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3516–3520 CrossRef CAS PubMed.
- V. Kirkin, S. Joos and M. Zörnig, Biochim. Biophys. Acta, Mol. Cell Res., 2004, 1644, 229–249 CrossRef CAS PubMed.
- J. C. Reed, J. Natl. Cancer Inst., 1997, 89, 988–990 CrossRef CAS PubMed.
- M. Sattler, H. Liang, D. Nettesheim, R. P. Meadows, J. E. Harlan, M. Eberstadt, H. S. Yoon, S. B. Shuker, B. S. Chang, A. J. Minn, C. B. Thompson and S. W. Fesik, Science, 1997, 275, 983–986 CrossRef CAS PubMed.
- C. Tse, A. R. Shoemaker, J. Adickes, M. G. Anderson, J. Chen, S. Jin, E. F. Johnson, K. C. Marsh, M. J. Mitten, P. Nimmer, L. Roberts, S. K. Tahir, Y. Xiao, X. Yang, H. Zhang, S. Fesik, S. H. Rosenberg and S. W. Elmore, Cancer Res., 2008, 68, 3421–3428 CrossRef CAS PubMed.
- M. D. Wendt, Curr. Top. Med. Chem., 2012, 8, 231–258 CAS.
- C. M. Park, M. Bruncko, J. Adickes, J. Bauch, H. Ding, A. Kunzer, K. C. Marsh, P. Nimmer, A. R. Shoemaker, X. Song, S. K. Tahir, C. Tse, X. Wang, M. D. Wendt, X. Yang, H. Zhang, S. W. Fesik, S. H. Rosenberg and S. W. Elmore, J. Med. Chem., 2008, 51, 6902–6915 CrossRef CAS.
- W. H. Wilson, O. A. O’Connor, M. S. Czuczman, A. S. LaCasce, J. F. Gerecitano, J. P. Leonard, A. Tulpule, K. Dunleavy, H. Xiong, Y. L. Chiu, Y. Cui, T. Busman, S. W. Elmore, S. H. Rosenberg, A. P. Krivoshik, S. H. Enschede and R. A. Humerickhouse, Lancet Oncol., 2010, 11, 1149–1159 CrossRef CAS.
- L. Gandhi, D. R. Camidge, M. R. de Oliveira, P. Bonomi, D. Gandara, D. Khaira, C. L. Hann, E. M. McKeegan, E. Litvinovich, P. M. Hemken, C. Dive, S. H. Enschede, C. Nolan, Y. L. Chiu, T. Busman, H. Xiong, A. P. Krivoshik, R. Humerickhouse, G. I. Shapiro and C. M. Rudin, J. Clin. Oncol., 2011, 29, 909–916 CrossRef CAS.
- A. W. Roberts, J. F. Seymour, J. R. Brown, W. G. Wierda, T. J. Kipps, S. L. Khaw, D. A. Carney, S. Z. He, D. C. S. Huang, H. Xiong, Y. Cui, T. A. Busman, E. M. McKeegan, A. P. Krivoshik, S. H. Enschede and R. Humerickhouse, J. Clin. Oncol., 2012, 30, 488–496 CrossRef CAS.
- T. J. Kipps, H. Eradat, S. Grosicki, J. Catalano, W. Cosolo, I. S. Dyagil, S. Yalamanchili, A. Chai, S. Sahasranaman, E. Punnoose, D. Hurst and H. Pylypenko, Leuk. Lymphoma, 2015, 56, 2826–2833 CrossRef CAS PubMed.
- H. Zhang, P. M. Nimmer, S. K. Tahir, J. Chen, R. M. Fryer, K. R. Hahn, L. A. Iciek, S. J. Morgan, M. C. Nasarre, R. Nelson, L. C. Preusser, G. A. Reinhart, M. L. Smith, S. H. Rosenberg, S. W. Elmore and C. Tse, Cell Death Differ., 2007, 14, 943–951 CrossRef CAS.
- K. D. Mason, M. R. Carpinelli, J. I. Fletcher, J. E. Collinge, A. A. Hilton, S. Ellis, P. N. Kelly, P. G. Ekert, D. Metcalf, A. W. Roberts, D. C. S. Huang and B. T. Kile, Cell, 2007, 128, 1173–1186 CrossRef CAS PubMed.
- A. J. Souers, J. D. Leverson, E. R. Boghaert, S. L. Ackler, N. D. Catron, J. Chen, B. D. Dayton, H. Ding, S. H. Enschede, W. J. Fairbrother, D. C. S. Huang, S. G. Hymowitz, S. Jin, S. L. Khaw, P. J. Kovar, L. T. Lam, J. Lee, H. L. Maecker, K. C. Marsh, K. D. Mason, M. J. Mitten, P. M. Nimmer, A. Oleksijew, C. H. Park, C. M. Park, D. C. Phillips, A. W. Roberts, D. Sampath, J. F. Seymour, M. L. Smith, G. M. Sullivan, S. K. Tahir, C. Tse, M. D. Wendt, Y. Xiao, J. C. Xue, H. Zhang, R. A. Humerickhouse, S. H. Rosenberg and S. W. Elmore, Nat. Med., 2013, 19, 202–208 CrossRef CAS PubMed.
- W. J. Fairbrother, J. D. Leverson, D. Sampath and A. J. Souers, Discovery and Development of Venetoclax, a Selective Antagonist of BCL-2 in: J. Fischer, C. Klein and W. E. Childers, ed., Successful Drug Discovery, Wiley, 2019. vol. 4 Search PubMed.
- A. W. Roberts, M. S. Davids, J. M. Pagel, B. S. Kahl, S. D. Puvvada, J. F. Gerecitano, T. J. Kipps, M. A. Anderson, J. R. Brown, L. Gressick, S. Wong, M. Dunbar, M. Zhu, M. B. Desai, E. Cerri, S. Heitner Enschede, R. A. Humerickhouse, W. G. Wierda and J. F. Seymour, N. Engl. J. Med., 2016, 374, 311–322 CrossRef CAS PubMed.
- S. Stilgenbauer, B. Eichhorst, J. Schetelig, S. Coutre, J. F. Seymour, T. Munir, S. D. Puvvada, C. M. Wendtner, A. W. Roberts, W. Jurczak, S. P. Mulligan, S. Böttcher, M. Mobasher, M. Zhu, M. Desai, B. Chyla, M. Verdugo, S. H. Enschede, E. Cerri, R. Humerickhouse, G. Gordon, M. Hallek and W. G. Wierda, Lancet Oncol., 2016, 17, 768–778 CrossRef CAS PubMed.
- L. M. Juárez-Salcedo, V. Desai and S. Dalia, Drugs Cont., 2019, 8, 212574 Search PubMed.
- R. M. Garbaccio and E. R. Parmee, Cell Chem. Biol., 2016, 23, 10–17 CrossRef CAS PubMed.
- M. Beltramo, Mini-Rev. Med. Chem., 2009, 9, 11–25 CrossRef CAS PubMed.
- N. Thornberry and A. Weber, Curr. Top. Med. Chem., 2007, 7, 557–568 CrossRef CAS PubMed.
- A. J. Roecker, C. D. Cox and P. J. Coleman, J. Med. Chem., 2016, 59, 504–530 CrossRef CAS PubMed.
- Z. Arshad, J. Smith, M. Roberts, W. H. Lee, B. Davies, K. Bure, G. A. Hollander, S. Dopson, C. Bountra and D. Brindley, Expert Opin. Drug Discovery, 2016, 11, 321–332 CrossRef CAS PubMed.
- J. M. Fernandez, R. M. Stein and A. W. Lo, Nat. Biotechnol., 2012, 30, 964–975 CrossRef CAS PubMed.
- M. J. Waring, J. Arrowsmith, A. R. Leach, P. D. Leeson, S. Mandrell, R. M. Owen, G. Pairaudeau, W. D. Pennie, S. D. Pickett, J. Wang, O. Wallace and A. Weir, Nat. Rev. Drug Discovery, 2015, 14, 475–486 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |