
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe chemical biology of coronavirus host–cell interactions
Suprama
Datta
a,
Erik C.
Hett
b,
Kalpit A.
Vora
c,
Daria J.
Hazuda
bc,
Rob C.
Oslund
 *b,
Olugbeminiyi O.
Fadeyi
*b and
Andrew
Emili
*a
*b,
Olugbeminiyi O.
Fadeyi
*b and
Andrew
Emili
*a
aCenter for Network Systems Biology, Department of Biochemistry, Boston University School of Medicine, Boston, MA, USA. E-mail: aemili@bu.edu
bExploratory Science Center, Merck & Co., Inc., Cambridge, Massachusetts, USA. E-mail: rob.oslund@merck.com; olugbeminiyi.fadeyi@merck.com
cInfectious Diseases and Vaccine Research, Merck & Co., Inc., West Point, Pennsylvania, USA
First published on 23rd December 2020
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for the current coronavirus disease 2019 (COVID-19) pandemic that has led to a global economic disruption and collapse. With several ongoing efforts to develop vaccines and treatments for COVID-19, understanding the molecular interaction between the coronavirus, host cells, and the immune system is critical for effective therapeutic interventions. Greater insight into these mechanisms will require the contribution and combination of multiple scientific disciplines including the techniques and strategies that have been successfully deployed by chemical biology to tease apart complex biological pathways. We highlight in this review well-established strategies and methods to study coronavirus–host biophysical interactions and discuss the impact chemical biology will have on understanding these interactions at the molecular level.
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) strain that causes coronavirus disease 2019 (COVID-19) has infected over 60 million people with nearly 1.5 million deaths worldwide as of late November 2020 leading to not only severe impact on human health, but widespread global economic and social disruption.1 While no vaccines are currently approved to prevent SARS-CoV-2 infection or spread of COVID-19, more than 100 coronavirus vaccine candidates based on various platform technologies are currently in preclinical and clinical development.2 Furthermore, several clinical trials are currently under investigation to evaluate potential therapeutics to alleviate COVID-19 symptoms and disease progression.3 Thus far, remdesivir4 (a nucleoside-analogue RNA-dependent RNA polymerase (RdRP) inhibitor), is the only approved antiviral shown to be beneficial to patients who don’t require oxygen ventilation. With the surging number of cases this Fall, normalcy will likely not return until safe and efficacious vaccines and therapeutics become widely available.SARS-CoV-2 originated in the Wuhan province in China after the summer of 2019, rapidly spread across the globe, and was eventually declared a pandemic by the World Health Organization (WHO) in early 2020. The virus has subsequently shown some genetic changes, particularly the D614G mutation in the spike protein has been associated with higher infectivity but probably lower mortality.5 Mutations at position 614 are unlikely to affect antibodies that bind to the neutralizing epitopes. The immunological sequalae during and after infection remain incompletely understood.6 The ensuing immune response can cause a variety of outcomes. These range from severe disease initially in some patients, exemplified by acute respiratory distress syndrome (ARDS), especially those at older age or with comorbidities, to recovery and resolution in most others with the potential for establishing immunity to subsequent exposure.
The portal of entry for SARS-CoV-2 is through nose, mouth, and eyes establishing an early infection in the upper respiratory tract (Fig. 1). In the majority of individuals this virus replication in the upper respiratory tract does not progress to severe disease and is cleared quickly.7 It is postulated that a robust innate immune response and/or trained immunity, and likely pre-existing cellular immunity (T-cells generated to prior exposures to common cold coronaviruses) could account for the limited infection in the upper respiratory tract.8 Subsequently if the virus is not cleared from the upper respiratory tract, the virus will travel to the lower respiratory tract and establish the infection in lung airway or bronchiole cells leading to moderate or severe COVID-19 symptoms (Fig. 1). It is also hypothesized that the ensuing immune response in the lung to clear the infection causes pathogenic inflammation and, in some instances, manifests into ARDS.9
 | ||
| Fig. 1 SARS-CoV-2 infection lifecycle. Following entry into host cells by engagement of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral envelope glycoprotein spike (S) with receptors on the surface of epithelial cells located in distal lung, e.g. angiotensin-converting enzyme 2 (ACE2) on alveolar cells, and establishment of upper respiratory tract infection via nose, eyes and mouth, the viral lifecycle proceeds with the fusion of viral envelope with cell membrane and subsequent release of viral RNA into the cell. The RNA is then translated to produce viral pre-proteins that are processed by its own proteases to release over two dozen effector proteins which eventually participate in viral replication, hijacking of host pathways, and assembly of virions followed by release. | ||
Immunity to several respiratory viral pathogens, including respiratory syncytial virus (RSV), human metapneumovirus (hMPV), parainfluenza virus type 3 (PIV3) and Rhino viruses in the upper respiratory tract is considered short-lived and partial.7 Likewise, protective immunity to coronavirus infection may also be short-lived, as has been observed with common cold corona, severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) viruses.10 Protection from severe, lower respiratory tract disease may be more robust and durable but is unlikely to be complete, especially in at-risk populations. Any form of prior specific or non-specific immune education by way of vaccination or exposure may slow down the march of the virus to the lower respiratory tract. Therefore, vaccination against SARS-CoV-2 is highly desirable to avoid serious disease.11 Most prophylactic vaccines block viral entry by eliciting antibodies to the viral surface glycoproteins responsible for viral entry into the host cells and hence the spike protein of SARS-CoV-2 has become an attractive vaccine candidate antigen.12 In addition to eliciting neutralizing antibodies, the spike protein has several epitopes that can elicit CD4-T cell responses and some CD8-T cells.13 Additional viral proteins like nucleocapsid and proteases are more conserved than the spike protein and therefore are candidate antigens to elicit additional T-cell responses or boost pre-existing T cell responses.14
The goal of the SARS-CoV-2 vaccine is to raise robust spike specific humoral responses that prevent viral infection. Addition of CD4 T-cell responses can also help the process, but on their own may not afford protection. Requirement of CD8 T-cell responses is controversial as the potential for collateral damage to the lung by inducing cytotoxicity, re-modeling and fibrosis while clearing infected cells is a concern. Additionally, any vaccine developed will have to deal with the perceived risk of enhanced disease by vaccinations that raise suboptimal antibody and/or Th2 T-cell responses. Therefore, understanding the molecular interaction between the virus, host cells and the immune system is of paramount importance for developing prophylactic and therapeutic interventions.
With a history deeply rooted in understanding how biomolecules engage within the complexity of biological systems, chemical biology is well-suited for interrogating the dynamic repertoire of SARS-CoV-2–host molecular interactions. The successful integration of chemical biology within the drug discovery pipeline through the development and application of chemical probe technologies enables the elucidation of ligand–target pairs and enhances exploration of biological pathway interactions through targeted protein modulation.15,16 The potential utility of chemical biology-based approaches for understanding viral pathobiology have been further enhanced through the development of a portfolio of protein activity-based probes and labeling methods that allow for high-resolution investigation of cellular enzyme function and proximal protein communities in cellular environments, respectively.17 Mass spectrometry-based proteomic endeavors to identify viral–host protein engagement within cells have also been improved through chemical biology-based affinity tags, enrichment strategies, and selective labeling methods.18
The successful demonstration of these chemical biology-based strategies and technologies have propelled expansion into other functional characterization initiatives such as the elucidation of metabolite–protein interactions,19 understanding protein community environments within different cellular regions,20 and detailing mechanisms behind cell–cell engagement.21–24 The field of chemical biology is ideally suited to address these and other fundamental questions that are central to unlocking a more detailed understanding of coronavirus–host interactions at all phases of a viral replication life cycle. In this review, we highlight previous efforts to study coronavirus host molecular interactions and discuss the impact chemical biology will have on understanding these interactions at the molecular level.
Rationale behind studying virus–host small molecule interactions
Viruses are obligatory intracellular parasites that hijack key biochemical processes and regulatory pathways of infected host cells to favor their replication. Since the number of genes encoded by the host genome is exponentially larger than that of a viral genome, upon infection, a relatively few different types of viral components interact with a complex pool of host factors to take advantage of the host–cell machinery. Perturbation of host pathways results in rewired intracellular signaling, transcription, translation, metabolism, and dysregulated immunity and stress responses.25–27Central metabolic pathways, including glycolysis, tricarboxylic acid (TCA) cycle and lipid metabolism are important targets for energy resources for viral replication (Fig. 2).28 Mapping the host–virus interaction interface impacting metabolic systems is crucial to understanding the: (1) cellular pathways co-opted to support viral replication and infection, (2) function of uncharacterized viral components (guilt-by-association), and (3) reveal potentially druggable targets to counter infection. Chemical probes targeting key host pathways co-opted during viral replication, or other potential ligand binders involved in antiviral immune response, could leverage metabolic responses and pathways that are comprised of actionable proteins (e.g. enzymes and transcription factors) and small molecules metabolites (glutamine, citrate, palmitate, etc.) that natively engage with bioactive compounds. The dynamic interplay between host proteins and metabolites is therefore key to the metabolic reprogramming that occurs upon infection. For example, pattern recognition receptors such as Toll-like receptors (TLRs), retinoic-acid-inducible protein I (RIG-I), RIG-1-like receptors (RLRs) and cytosolic sensors (cGAS) sense pathogen-associated molecular patterns, such as small molecules, and dimerize with their cognate adaptor molecules to activate IKK and TBK-1/IKK.29 These kinases in turn activate the transcription factors IRF3 and NF-kB to promote the production of cytokines.
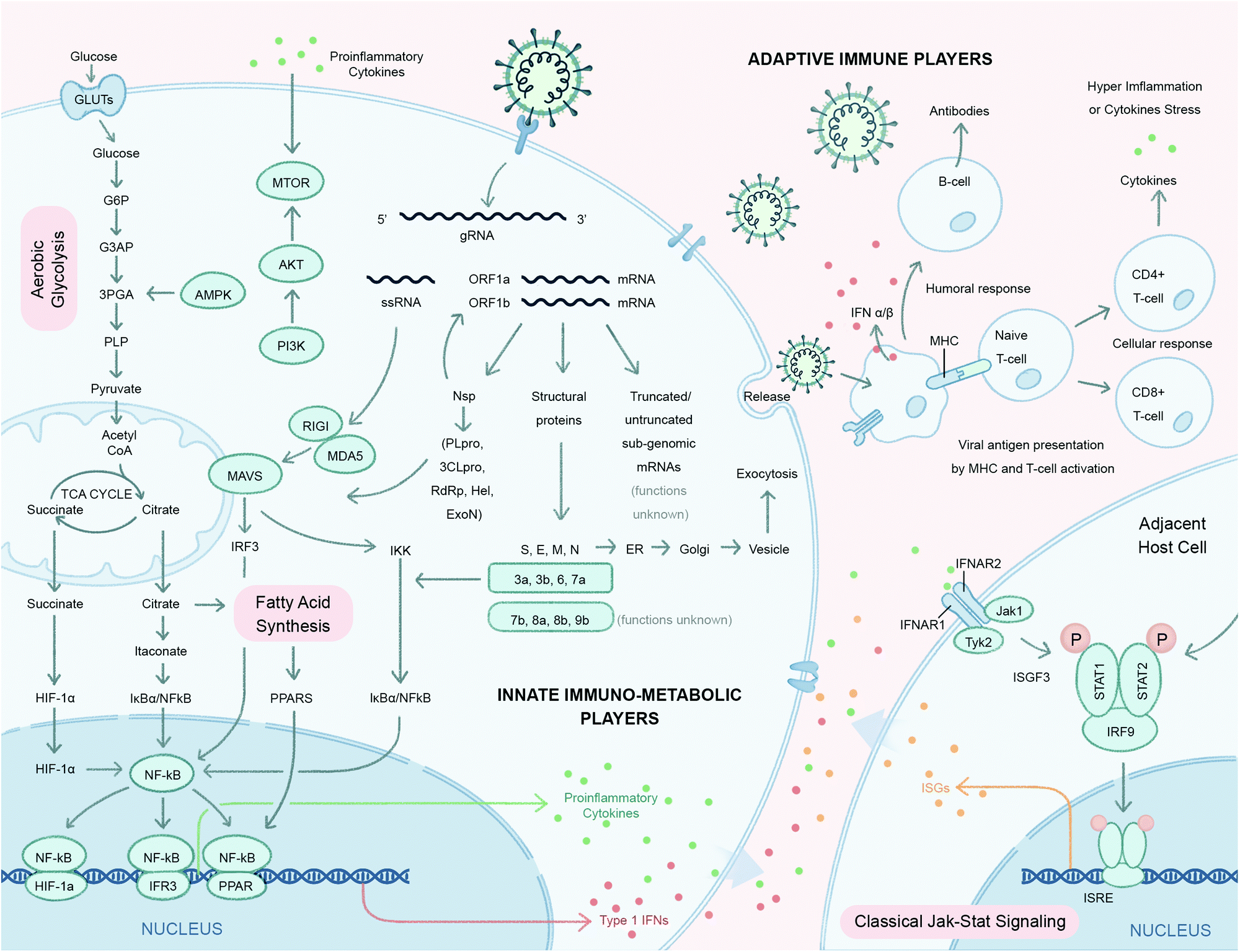 | ||
| Fig. 2 Host signaling and metabolic responses to SARS-CoV-2 infection predicted based on strong similarities with two previous highly pathogenic human β-coronaviruses SARS-CoV and MERS-CoV. After viral entry, the single-stranded RNA genome (gRNA) of SARS-CoV-2 is released in the host cell cytoplasm. ORF1a and ORF1ab mRNAs are translated to produce two large polyproteins, pp1a and pp1ab, which are proteolytically cleaved into 16 non-structural proteins (nsps), including papain-like protease (PLpro), 3C-like protease (3CLpro), RNA-dependent RNA polymerase (RdRp), helicase (Hel), exonuclease (ExoN), and 4 structural proteins. The envelope glycoprotein spike (S) forms a layer of glycoproteins that protrude from the envelope. Two additional transmembrane glycoproteins are incorporated in the virion: envelope (E) and membrane (M). Inside the viral envelope resides the helical nucleocapsid, which consists of the viral positive-sense RNA ((+)RNA) genome encapsidated by protein nucleocapsid (N). An additional 9–12 ORFs are encoded through the transcription of a nested set of sub-genomic RNAs. Functions of these truncated or untruncated sub-genomic RNAs are still unknown. The viral elements (ssRNA or sub-genomic RNAs) are recognized by pattern recognition receptors such as Toll-like receptors (TLRs), retinoic-acid-inducible protein I (RIG-I), RIG-I like receptor, and melanoma differentiation-associated protein 5 (MDA-5) that recruit adapter mitochondrial antiviral-signaling protein (MAVS) and leads to activation of interferon regulatory factor 3 (IRF3) and NF-κB and the subsequent production of type I Interferons (IFNs) and pro-inflammatory cytokines (e.g. IL-1β and IL-6), respectively. Itaconate, an anti-inflammatory metabolite transported by TCA cycle intermediates (e.g. citrate), results in activation of pathways involved in proinflammatory cytokines. Proinflammatory cytokines activate the mammalian target of rapamycin (mTOR) signaling in addition to the increased energy metabolism through aerobic glycolysis. The increased production of tricarboxylic acid cycle (TCA) intermediates e.g. succinate and citrate activate NF-κB downstream signaling resulting in the production of proinflammatory cytokines. The antiviral activity of type I IFNs limit viral replication and modulate the innate and adaptive immune responses. They bind to interferon-α/β receptors (IFNARs), which are expressed on a number of different cells including macrophages and activate the JAK/STAT signaling pathway. This signaling leads to the formation of the STAT1/2/IRF9 complex and the induction of a plethora of IFN-stimulated genes (ISGs). Cytokines released by infected cells modulate the adaptive immune response by recruiting and activating immune cells such as macrophages, B-cells, and T-cells to orchestrate the elimination of the virus. However, an unbalanced immune response has been reported to cause hyper-inflammation, a condition termed as ‘cytokine storm’ in cases of severe clinical symptoms of COVID-19. Many, but not all, aspects of this model have been directly verified in the rapidly emerging SARS-CoV-2 experimental literature. | ||
Energy precursors like citrates, succinates and other glycolysis and TCA cycle intermediates also modulate production of inflammatory cytokines during host immune response.30 Lipids and fatty acids play active roles in protein modifications and the formation of the viral envelope.31 Amino acids like glutamine can serve as an alternative carbon source for viral infected cells.32 Endogenous nucleotides like cGMP and cAMP act as second messengers that signal interferon gene expression.33 Specialized organelles such as mitochondria and lysosomes serve as important subcellular hubs that integrate diverse players at the interface of immune response and metabolism.34,35 Conversely, the metabolic-reprogramming activity of the cytokine response can originate from immune signaling components whereby metabolites, such as succinate and citrate, can directly or indirectly regulate the canonical NF-κB pathway (Fig. 2). Studies suggest that itaconate, a structurally similar small molecule metabolite to malate, is transported across the mitochondrial inner membrane by the TCA cycle intermediate carriers (e.g. citrate) and activate the anti-inflammatory transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) following lipopolysaccharide (LPS) stimulation.36 A recent study by Olagnier et al. reported the antiviral effects of a chemically synthesized cell permeable derivative of itaconate and fumarate, 4-octyl itaconate (4-OI) and dimethyl fumarate (DMF) respectively, in SARS-CoV-2 infection37 and will be discussed further in the strategy section of this review. The antiviral effects of metabolic sensing enzymes (e.g., mammalian target of rapamycin (mTOR)38 and adenosine monophosphate-activated protein kinase (AMPK)39) play major regulatory roles in both metabolism and immune responses with the onset of proinflammatory signals.
With the havoc created by the COVID-19 pandemic coupled with the recent history of other zoonotic coronavirus infections, MERS and SARS-CoV, it has become imperative to understand the pathophysiology of host–SARS-CoV-2 and coronavirus infections more generally with the goal of uncovering the mechanisms associated with host cell invasion, replication and persistence. Although most of the structural and non-structural coronaviral proteins have been assigned viral replication and cell/immune modulatory functions, respectively, the functions of the accessory proteins (7b, 8a, 8b and 9b) and truncated/untruncated sub-genomic mRNAs are still unknown (Fig. 2). We also have limited knowledge of the immune players involved in SARS-CoV-2 infection. In a conventional viral infection model, innate immune metabolic players like nuclear hormone receptors (e.g. PPARs) and TCA cycle metabolites (e.g. succinate, citrate) activate the NF-kB signaling cascade which release proinflammatory cytokines (IL-1, IL-6, TNF-α) and interferons (IFNs) (Fig. 2).40,41 These proinflammatory cytokines in turn activate the mTOR signaling pathway of adjacent host cells (Fig. 2).38,42 IFNs are critical to both innate and adaptive immunity, and function as the primary activator of macrophages, in addition to stimulating natural killer cells and neutrophils.43 IFNs also take part in activating the JAK-STAT signaling pathway upon binding to its receptor on the adjacent host cell membrane.43 While all these aspects are still to be figured out for SARS-CoV-2 infection, their elucidation will be possible upon employing suitable strategies to map these virus–host interactions. The following section describes some of the well-established methods and strategies to address coronavirus–host interactions.
Strategies to profile coronavirus–host interactions
Since identification and characterization of key host–viral protein interactions is crucial towards understanding how inter/intra-cellular signaling cascades and biochemical processes are initiated to discover new therapeutic targets, a number of chemical biology strategies have been developed and employed over the years to address this challenge. The advancement of experimental strategies to map these interactions using monoclonal and anti-idiotypic antibodies, solid-phase assays, and affinity purification with receptor antibodies44–48 to more recent high-throughput screening (HTS) technologies based on gene targeting methods and new generation high resolution mass spectrometry49 has brought the flexibility of choosing workflows based on the desired output for mapping these interactions. In this section, these strategies are discussed in detail with respect to the principle of the workflows and the impact each is contributing towards unravelling the mystery around the pathophysiological and chemical biology of coronavirus–host interactions.1. Imaging-based interaction analysis
Microscopy is an important tool in virology and infection biology. Different microscopic imaging strategies and workflows are employed to understand the underlying principles of receptor binding, genome release, replication, assembly, and virus budding, as well as the response of the host immune system. Virologists deploy a broad range of light microscopy techniques, with the primary choice being fluorescence microscopy. Fluorescence microscopy can be roughly subdivided into immunofluorescence analysis (IFA) and the utilization of fluorescent proteins. IFA utilizes fixed cells or tissues to stain a protein of interest with fluorescently marked antibodies, while fluorescent proteins can be expressed in vivo for utilization in live-cell imaging (Fig. 3). One of the examples in the context of virology is to image reporter viruses expressing fluorescent proteins as described in the next section.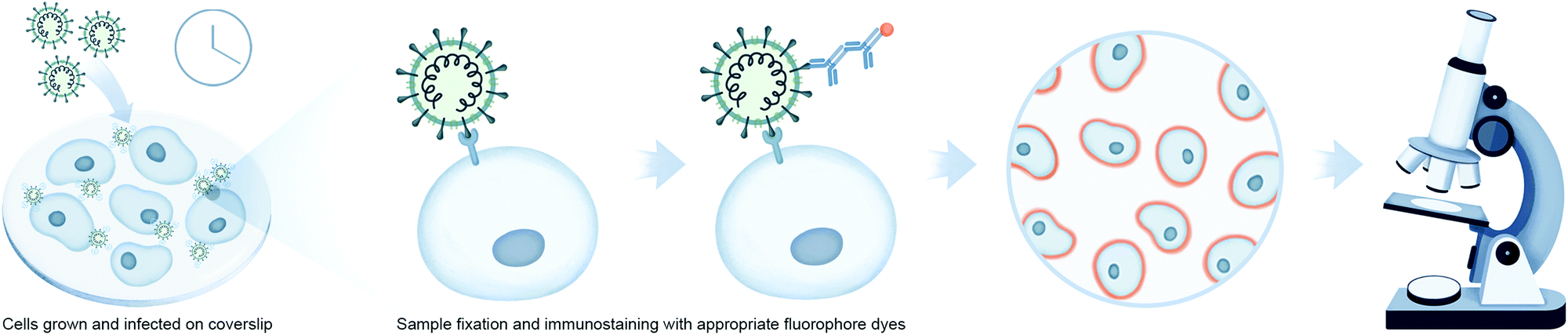 | ||
| Fig. 3 Imaging-based interaction analysis using viral reporter strains for monitoring virus–host interactions. Workflow illustrates fluorescence imaging using immunolabeling of fixed cells or tissues to stain a protein of interest, or reporter viruses stained with antibodies labeled with fluorophores. | ||
Notably, recent IFA studies report tracking membranous structures colocalizing with coronaviral replication/transcriptional complexes. Müller et al. studied the influence of cellular lipid metabolism on human coronavirus replication in culture using both confocal and transmission electron microscopy.50 Specifically, the group was interested in understanding the colocalization of viral-mediated dsRNA production and lysophospholipids generated by cytosolic phospholipase A2α (cPLA2α) activity. They infected Huh-7 cells with HCov-229E or MERS-CoV, and stained these cells with antibodies against dsRNA, the coronavirus Nucleocapsid (N) and NSP8 protein. To monitor lysophospholipid generation and localization, they employed the use of a fluorogenic cPLA2α active probe. In this cell system, viral replication/transcription complexes (RTC) were observed to co-localize with lysophospholipids. Notably, when cells were treated with the cPLA2α inhibitor pyrrolidine-2 to disrupt lysophospholipid production, a corresponding decrease in viral RNA and protein accumulation was detected in coronavirus infected cells.
Another study by Poppe et al. investigated the influence of HCoV-229E infection on cellular NF-κB signaling and the cellular transcriptome landscape.51 The authors used IFA to track coronavirus N protein expression to monitor viral infection and its spread in an A549 lung-derived epithelial cell line. Other microscopic methods have involved the detection of viral RNA or DNA by fluorescence in situ hybridization (FISH) whereby a fluorescent probe is used to target viral sequences.52
A recent imaging technique for visualizing SARS-CoV-2 infected nasopharyngeal epithelial cells was developed by Rut et al. using activity-based probes (ABPs) for the SARS-CoV-2 main protease (Mpro) that were developed from a hybrid combinatorial substrate library.53 An ABP from this library screen was able to detect SARS-CoV-2 Mpro at 5 nM enzyme concentration when incubated in the presence of 2.5 μM probe for 5 min. This study also resulted in the development of a potent SARS-CoV-2 Mpro inhibitor with half-maximal effective concentration (EC50) of 3.7 μM in a human cell line-based viral infection assay.
2. Viral reporter strains for virus–host interaction analysis
Recombinant reporter viruses are vital tools for studying the pathogenicity and transmission of viruses in animal models. They are generated to incorporate a marker, such as a fluorescent protein whose expression can be readily followed, into a specific gene/locus (or multiple loci) of the viral genome. The simplest recombinants are those generated by insertion of a reporter gene into a nonessential site on the viral genome, which can either cause a disruption of a particular gene that is not required for viral replication in tissue culture (and/or in vivo) or alternatively an insertion of an intergenic site such that the repertoire of viral genes is unaffected. Disruption of a specific gene enables the ability to characterize the functional significance of the perturbed viral gene. The presence of the reporter allows for facile tracking of viral infection to provide a better understanding of pathogenesis. However, the genetic manipulations required to achieve reporter gene expression comes with the caveats of altering the viral genome and potential downstream function as well as creating potential instability of reporter expression and detection depending on the tissue type.54 However, despite these limitations, this technology has proved to be instrumental in understanding viral infections in both living cells and animals.Classically, one of the most commonly used reporter genes has been lacZ (encoding beta-galactosidase), which can either be placed under the control of a viral promoter specific to the virus being studied or under a strong universal promoter such as HCMV immediate early enhancer element or the SV40 promoter.55 The choice of a promoter is dependent on the intended application for the recombinant virus. Although lacZ is an effective reporter, the infected cells/tissues require downstream processing/staining to enable the detection of beta-galactosidase, and this can add an additional 6–18 h to the protocol for detection of virus. An additional concern is the large size of the lacZ gene, which might be an important consideration for viruses with limited genome oversizing potential because of packaging constraints. This could potentially result in second site mutations (e.g. gene deletion) unrelated to the reporter gene insertion locus, which would also alter the phenotype of the virus.
Fluorescent proteins have become extremely popular for incorporation into recombinant viruses based on their sensitive detection in live cells in real time via fluorescent microscopy. The most common fluorescent reporter is green fluorescent protein (GFP) from the jelly fish (Aequorea victoria) and engineered variants that produce high levels of fluorescent signal and consequently easy detection and real-time tracking. For example, Sims and co-workers generated recombinant SARS-CoV constructs by deletion of open reading frame 7a/7b (ORF7a/7b) and insertion of GFP to study the infectivity of SARS-CoV virus in human angiotensin 1 converting enzyme 2 (hACE2) mediated invasion of an in vitro model of human ciliated airway epithelia (HAE) derived from nasal and tracheobronchial airway regions.56
An additional advantage of fluorescent proteins is that their small size enables viral protein tagging and construction of virus expressing fluorescently tagged structural proteins. One disadvantage of using GFP as a reporter gene in the context of an animal model is that some tissues have an endogenous autofluorescence that makes the detection of the reporter difficult relative to the background. The liver in particular can be very problematic due high autofluorescence background. Antibodies to GFP and other fluorescent proteins partly overcome this problem via the use of immunohistochemical staining of tissue.
A potential alternative for in vivo detection of viruses is the use of bioluminescence imaging (BLI). This particular technique is extremely sensitive and enables the detection of virus in real time in the same animal over days to weeks post-infection. BLI requires the introduction of the firefly (Photinus pyralis) luciferase reporter gene into the viral genome of interest. After interaction with their substrate, firefly luciferase (FLuc) enzymes emit photons; the specific wavelength (400–615 nm) is dependent upon the enzyme. A fairly recent introduction of a smaller variant of the 62 kDa FLuc called nanoLuciferase (nLuc), a 19 kDa enzyme from deep sea shrimp, now comprises a popular reporter assay system which produces a luminescent signal >100-fold brighter than FLuc. The key feature that makes nLuc an ideal reporter is that its luminescent signal exhibits a long lifetime, resulting in glow kinetics rather than a rapid flash signal typically produced by other small, bright luciferases such as Gaussia luciferase. nLuc produces blue light with maximum emission at 460 nm, is thermally stable, active over a broad pH range, and requires no post-translational modifications or maturation after translation to make an active enzyme. Agostini et al. used nLuc fusions to study the susceptibility of β-coronaviruses such as mouse heptatitis virus, MERS-CoV, and SARS-CoV to remdesivir in cell-based assays and observed RNA polymerase-mediated inhibition of viral replication.57
3. Bioinformatic-based interaction analysis
A thorough analysis of virus–host protein interactomes can identify potential drug targets while deciphering the molecular etiology underlying viral replication and pathogenesis. With the application of high-throughput methods, described below, or literature mining, researchers have collected reasonably complete and high quality virus–host protein–protein interactomes (PPIs), generating invaluable virus–host PPI databases58,59 while providing a global view of human cellular processes controlled by viruses. These assays are typically performed using individual viral proteins expressed in isolation in transformed cell lines that may be quite dissimilar phenotypically from native host target cells, such as lung epithelia.While most recent efforts have pivoted towards more targeted proteomic approaches to study viral–host protein interactions in a cellular context, as described further below, structure guided computational approaches aim to predict the conserved set of putative interactions of virus and host (Fig. 4). This has deepened understanding of virus-specific cellular targets that lead to rewiring of host pathways and are illuminating promising new aspects of disease intervention. These computational inferences involve data mining from literature-curated binary PPIs from public databases e.g. Biomolecular Interaction Network Database (BIND),60 the Database of Interaction Proteins (DIP),61 Human Protein Reference Database (HPRD),62 IntAct,63 the Molecular INTeraction database (MINT),64 Virus–Host Network (VirHost-Net),65 Biological General Repository for Interaction Datasets (BioGrid),66 InnateDB,67 and the Pathogen–Host Interaction Search Tool (PHISTO).68 These PPI datasets can be scanned for conserved Pfam-A domains present in both virus and host encoded proteins, which are mapped to form putative domain–domain interaction networks (DDIs) that can be assessed for the topological parameters using R packages like igraph or Cytoscape.
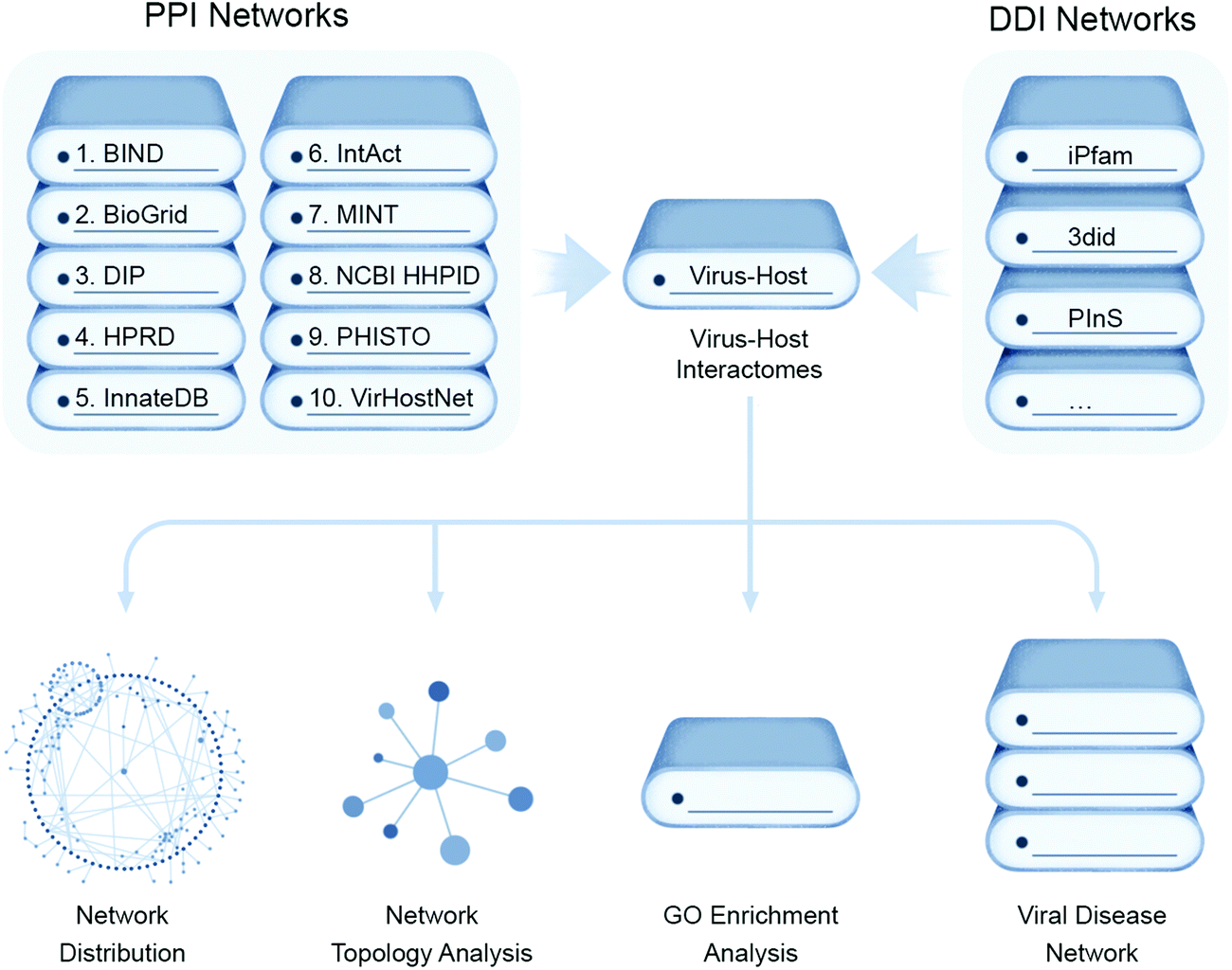 | ||
| Fig. 4 Computational approaches for virus–host interaction analysis. Bioinformatics pipeline for identifying virus–host interactions by data parsing from high quality virus–host protein–protein interactomes (PPIs) databases and mapped to form putative domain–domain interaction networks (DDIs) for conserved domains of both virus and host proteins that can be assessed for their network distribution properties, such as node topology, as well as enrichment for gene ontology (GO) functional annotations and disease associations. | ||
To analyze the functional impact of predicted targeting of host protein domains by viruses, enrichment analysis of GO functional annotation terms can be carried out using R and the Bioconductor topGO package. Finally, the DDIs, along with their assigned functionality, can be assessed for disease relevance from public databases (e.g. Online Mendelian Inheritance in Man (OMIM), Human Gene Mutation Database (HGMD Public)69 and Catalogue of Somatic Mutations in Cancer (COSMIC)70) to make a comprehensive list of disease-associated viral host targets. For example, Ostaszewski et al. built a comprehensive COVID-19 disease map as a standardized knowledge repository of all putative SARS-CoV-2 virus–host interactions.71 This project was an open collaboration between scientists across the globe curating SARS-CoV-2 infection related information in one platform to support the research community in understanding SARS-CoV-2 pathophysiology and aid the development of efficient diagnostics and therapies. This platform has enabled the visual exploration and computational analyses of molecular processes involved in SARS-CoV-2 entry, replication, and host–pathogen interactions, as well as elucidated immune response, host cell recovery, and repair mechanisms.
4. Molecular biology-based interaction analysis
Advances in functional genomics and proteomics have allowed for the unbiased identification of cellular factors involved in viral infection. Diverse screening techniques have been applied to this field, including loss-of-function and gain-of-function screens. When applied proteome-wide, these techniques allow for the interrogation of cellular requirements for viral infection, generating information on those factors that are most important for viral infection.The yeast two-hybrid assay (Y2H), devised by Fields and Song for mapping binary protein–protein interactions (PPI)72 has been used in multiple high throughput viral–host PPI screens for unbiased viral genome-wide interactome exploration or focusing on a subset of viral proteins.73 These studies are based on an initial construction of a viral ORFeome, comprising clone ORFs encoding distinct viral proteins fused to a DNA binding domain (Fig. 5A). Interactions with human proteins expressed as activation domain fusions can reconstitute a functional transcriptional factor, driving expression of a reporter gene(s). This genetic assay is prone to false positives (and negatives), and so putative interactions discovered using Y2H screens must be confirmed by a secondary, biochemical method to generate high confidence data.
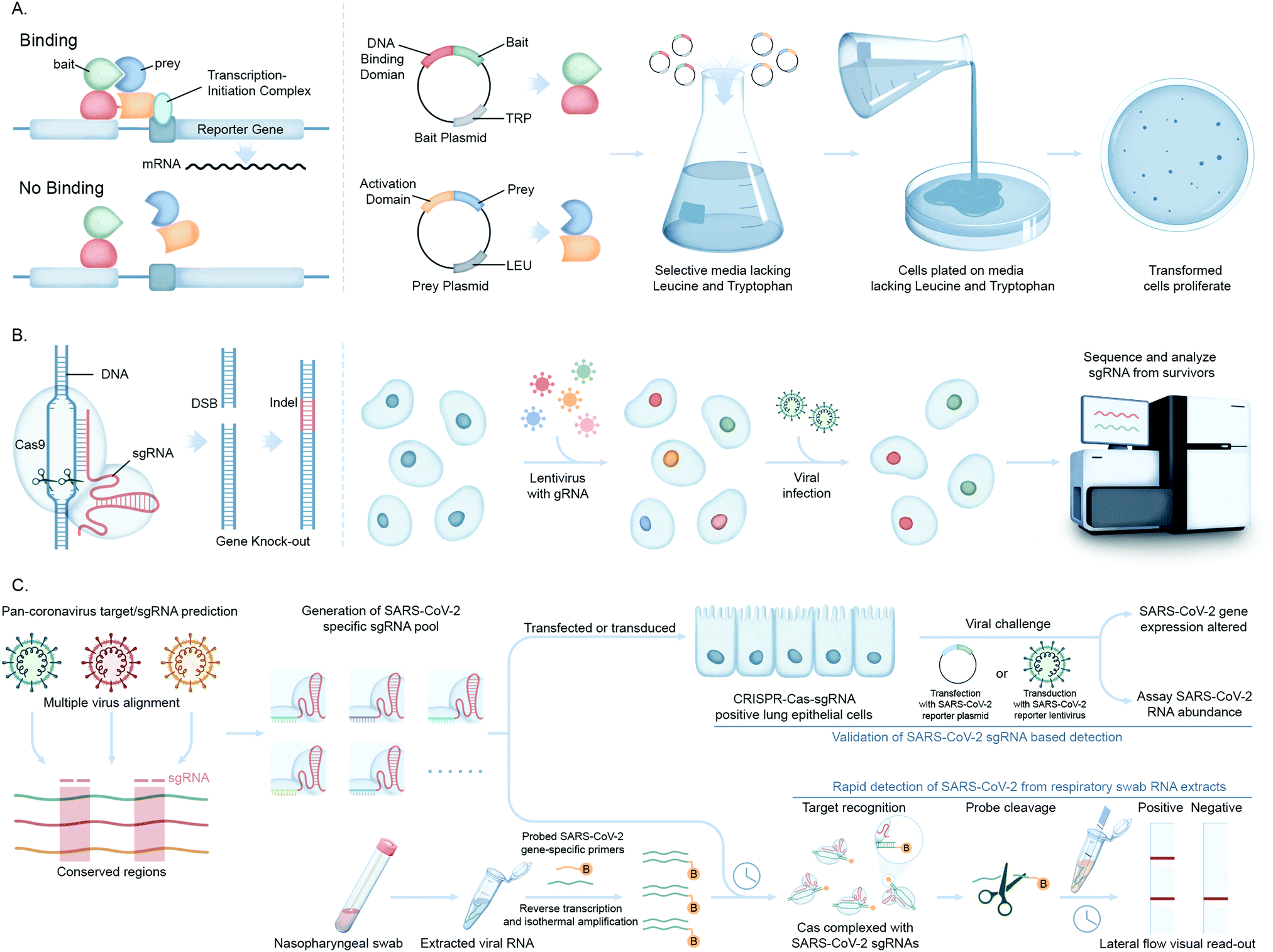 | ||
| Fig. 5 Molecular biology-based investigations of coronavirus–host interactions. (A) The yeast 2-hybrid methods of screening PPIs involve mating two haploid complementary yeast strains each expressing a distinct expression plasmid. The first strain expresses a protein bait fused to a DNA-binding domain (DBD) that binds to its cognate binding site, usually upstream of a reporter gene. The second strain expresses a protein prey fused to a transcription activation domain (AD). If there is interaction between bait and prey, the AD is brought into the proximity of the DBD which causes transcriptional activation of the reporter gene leading to selection. (B) Engineered CRISPR systems contain two components, a single guide RNA (sgRNA) and a CRISPR-associated endonuclease (Cas9 protein) to edit (insert or delete) DNA sequences by generating double stranded breaks (DSB) and introducing indels (insertions/deletions) for generation of gene knock-out libraries. In CRISPR–Cas9-pooled screen methods, single-guide RNA (sgRNA) libraries are initially synthesized as lentiviral vectors and amplified and packaged into lentiviruses. Lentivirus pools are then used to transduce Cas9 positive target cells at low multiplicity of infection (MOI). The genomic DNA is extracted from input versus selected cell pools, and the integrated sgRNA sequences are PCR-amplified. The sgRNA abundance is then determined by next-generation DNA sequencing. (C) Workflow showing the application of the CRISPR–Cas screening platform in detection of SARS-CoV-2 infection. A SARS-CoV-2 gene specific CRISPR–Cas sgRNA library is created from identifying the conserved gene sequences of pan-coronaviral genome and validated for detection of SARS-CoV-2 RNAs (PAC-MAN method). This library is then used to generate lateral flow strip-based assays to detect the viral RNAs in nasopharyngeal swabs from COVID-19 patients. | ||
Using the Y2H assay, Xiao et al. reported the interaction of the N-terminal region of SARS-CoV Spike (S) protein with eIF3f, a subunit of the eukaryotic initiation factor 3 (eIF3), which they subsequently confirmed by co-immunoprecipitation (co-IP) and IFA.74 Another systems biology study employing a proteome-wide Y2H screen to identify immunophilins as interaction partners of the SARS-CoV non-structural protein 1 (Nsp1) was performed by Pfefferle et al.75 Immunophilins act as receptors of immunosuppressive drugs such as cyclosporin A (CsA) and tacrolimus and thus this study suggests non-immunosuppressive derivatives of CsA may serve as potential broad-range inhibitors of SARS-CoV infection.
Another powerful tool to investigate the biological function of specific proteins either in vitro or in vivo is the RNA interference (RNAi) platform. The introduction of small interfering RNAs (siRNA), 20–25 nucleotide short double-stranded RNAs that are specific to target mRNA sequences, into cells allow for sequence-specific degradation of the mRNA. This method is a relatively fast, simple, and robust approach to specifically downregulate protein expression and study resulting cellular function. Expression knockdown has been successfully used in virology to study the role of specific host factors in the infectious life cycle and replication of viruses. Millet et al. used this platform to successfully validate the functional impact of ezrin, an actin binding protein, that they identified by Y2H screens as an interactor with the SARS-CoV spike (S) protein.76
Recently, clustered regularly interspaced short palindromic repeats (CRISPR/Cas) technology has greatly expanded the ability to genetically probe virus–host functional dependencies by means of initial screening and loss/gain-of-function based confirmation of targets (Fig. 5B).77 Exploiting the characteristics of virus–host interactions and the basic rules of nucleic acid cleavage or, most recently, gene regulation via the CRISPR–Cas system, it can be used to target components associated with both the virus genome and host factors to define their roles in virus infection or replication. These include CRISPR interference (CRISPRi) technology that can inhibit expression of target genes, or CRISPR activation (CRISPRa) that can increase target gene expression to test contrasting impact on viral proliferation.
In a notable recent study, Abbot et al. reported a CRISPR–Cas13-based antiviral strategy, PAC-MAN (prophylactic antiviral CRISPR in human cells), for viral inhibition that can effectively degrade SARS-CoV-2 RNA sequences in human lung epithelial cells.78 They designed and screened multiple single guide RNAs (sgRNAs) targeting conserved viral genomic regions to identify functional sgRNAs potently targeting SARS-CoV-2 (Fig. 5C). Another study by Broughton et al. developed a CRISPR–Cas12-based lateral flow assay for the rapid detection of SARS-CoV-2 from respiratory swab RNA extracts (Fig. 5C).79 Daly et al. reported the interaction of host cell receptor Neuropilin-1 (NRP-1) with CendR motif of SARS-CoV-2 spike (S), a polybasic Arg-Arg-Ala-Arg C-terminal sequence on the S1 subunit upon cleavage by host protease furin. NRP-1 is a cell-surface receptor that plays an essential role in angiogenesis, regulation of vascular permeability, and the development of the nervous system. This group probed the functional relevance of this interaction through CRISPR/Cas-mediated knockout of NRP-1.80
5. Biochemical-based interaction assays
Surface plasmon resonance (SPR) is an established, label-free optical technique used to study the strength of binary biomolecular interactions in real time by detecting changes in the reflection of light from proteins affixed to a prism–gold film interface. SPR facilitates investigation of the biophysical nature of binding interactions, and provides detailed binding kinetic information across a wide range of molecular weights, including small molecule ligands, all without the use of labels. Automated instrument platforms for SPR analysis are commercially available that can be used for a wide variety of protein interaction assays, including specificity, active concentration measurement, and profiling thermodynamic parameters. SPR is increasingly used in the field of virology, spanning from the study of biological interactions to the identification of putative antiviral drugs.The receptor recognition mechanism of SARS-CoV spike (S) protein towards hACE2, which defines host cell infectivity, pathogenesis and host range, was assessed using SPR by Wu et al. (2012).81 They measured the binding affinities between the receptor binding domain (RBD) of S and hACE2 variants by immobilizing serial dilutions of the proteins on a sensor chip through covalent coupling via amine groups. An alternate biophysical approach was likewise employed to explore the RBD–hACE2 interface by Shang et al. (2020).82 They generated a crystal structure of SARS-CoV-2 RBD complexed with hACE2 and compared it to the interface for SARS-CoV. This study found the ACE2-binding ridge in SARS-CoV-2 RBD assumes a more compact conformation compared to SARS-CoV RBD along with several residue changes that stabilize binding pockets at the RBD–hACE2 interface, thereby increasing the binding affinity for ACE2.
6. Omic-based interaction analysis methods
During the last decade, a multitude of additional omic approaches have emerged as powerful tools in basic, translational, and clinical research for the study of biological pathways involved in pathogen replication, host response, and disease progression. Advancement of proteomic technologies offering sensitive protein detection and quantification and integration of proteomics with other biochemical and molecular biology methods, as well as with other omic approaches, has expanded the repertoire of tools to study pathogen infections (Fig. 6A). Proteomics, the study of the protein component of biological systems, has emerged to the frontline for the discovery and understanding of host–pathogen interactions in SARS-CoV-2.49 Genomic, transcriptomic, and metabolomic profiling studies provide orthogonal information that complements the data generated by proteomic analyses to achieve a global, systems-level understanding of the infection process (Fig. 6B). For example, Olagnier et al. reported suppression of Nrf2 signaling events upon SARS-CoV-2 infection by transcriptomic analysis of COVID-19 patient lung biopsies. To further reinforce the therapeutic potential of modulating this pathway as an antiviral approach, they performed in vitro assays of cell lines treated with Nrf2 agonists, 4-OI and DMF, and reported significant reduction in the SARS-CoV-2 RNA abundance in the treated cell lines when compared with untreated infected ones.37 Also, given that the majority of proteins do not function in isolation and their interactions with other proteins define their cellular functions, a detailed understanding of PPIs and larger protein interaction environments are key for deciphering regulation of cellular networks and pathways.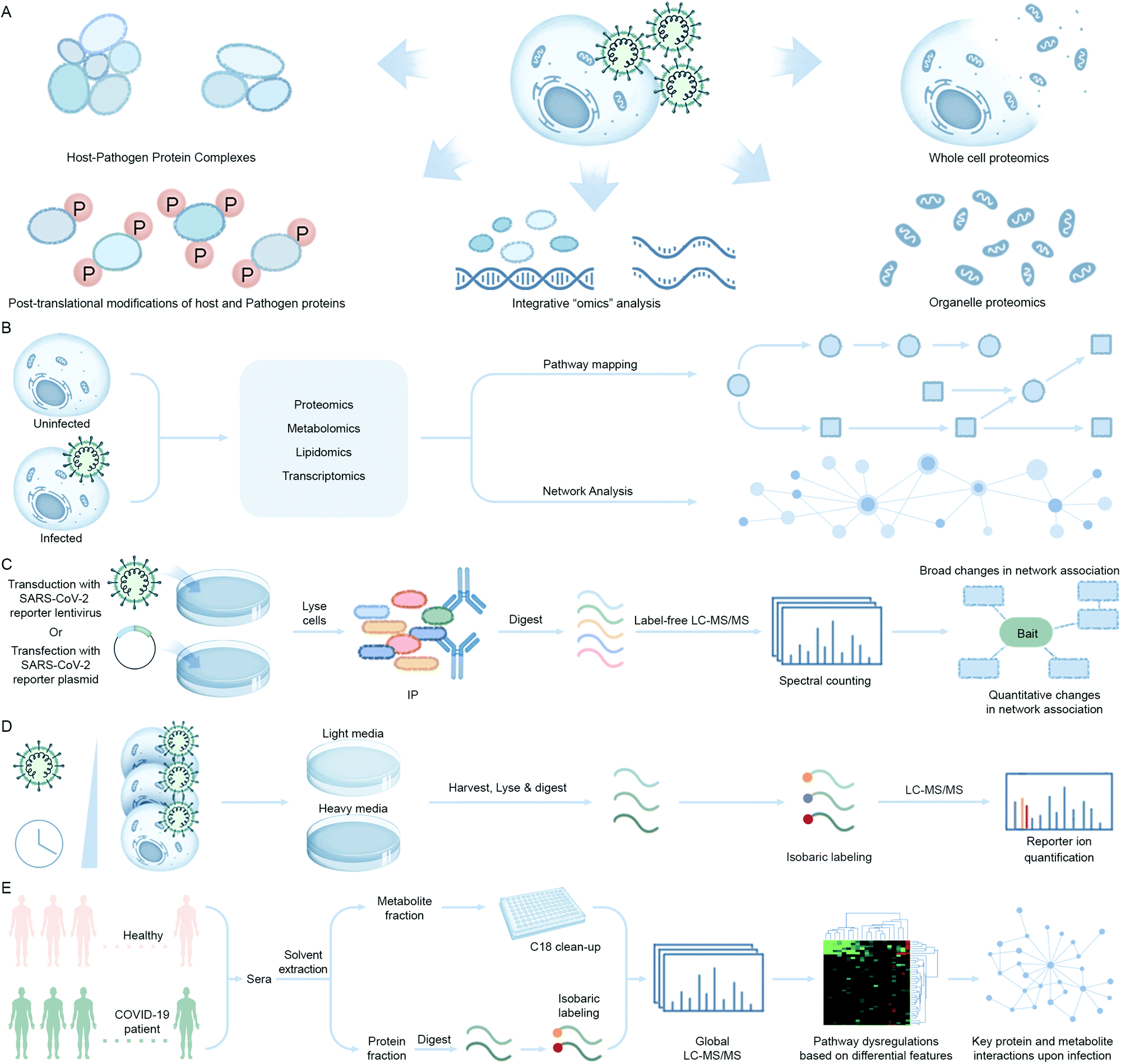 | ||
| Fig. 6 Mass spectrometry-based techniques used to identify virus–host interaction networks. (A) Overview of proteomic tools utilized in the of study host–pathogen interactions and their integration with omics approaches. (B) Quantitative multi-omic analysis workflow to define the host response to an infection. The resulting datasets are mapped to known metabolic pathways to measure the up- or downregulation upon infection and integrated with phenotype data to construct correlation networks. (C) Schematic of immunoaffinity or epitope tag-based affinity purification coupled to mass spectrometry (IP-MS/AP-MS) workflow. Components include immunoaffinity purification of protein complexes, enzymatic digestion of proteins, nano-liquid chromatography coupled to mass spectrometry (nLC-MS/MS), and bioinformatic analysis to identify proteins. Label-free protein quantification may be performed by MS/MS spectral counting or precursor ion intensity (MS1) integration. (D) Global proteomic approaches can be used to study alterations throughout infection in protein abundance. These changes can be quantified at the MS level, by pulse labeling via Stable Isotope Labeling in Cell Culture (SILAC) and/or isobaric tagging (such as tandem mass tagging, TMT) of samples after proteolysis and comparing the ion intensities to define proteome alterations at multiple time points of infection. In this workflow, cells are harvested at different infection times after pulse labeling and digested peptides from each sample are labeled with isobaric tags consisting of unique reporter masses. The samples are mixed together for MS analysis, and peptide quantification is assessed at the MS/MS level using the reporter ion intensities. Peptide quantitative values derived from sequences assigned to the same protein are used to calculate the overall relative protein abundance. (E) Workflow showing differential analysis of global proteomic and metabolomic profiles of COVID-19 patient cohorts vs. healthy individuals via an untargeted LC-MS/MS platform. (A–D) adapted from ref. 95. | ||
The method that has seen the widest implementation in host–pathogen interaction studies is immunoaffinity or epitope tag-based affinity purification coupled to mass spectrometry (Fig. 6C). In immunoprecipitation mass spectrometry (IP-MS), a target of interest is isolated using an antibody raised against the endogenous protein whereas in the case of affinity purification coupled to mass spectrometry (AP-MS) the target protein of interest is co-expressed with an epitope tag which is isolated using antibodies raised against the epitope tag. This immunoprecipitated or affinity-purified protein of interest, along with co-isolated interacting proteins, is then identified by MS. IP-MS and AP-MS studies can be performed from both the pathogen and host perspective. For example, selective enrichment of a viral protein can facilitate understanding of the host factors it interacts with to promote replication or suppress host defense pathways. Alternatively, enrichment of host cellular protein can be performed to identify interactions with surrounding protein partners during viral infection to characterize possible changes in the hijacked host protein function(s). Both IP-MS and AP-MS studies that capture differential interactions are often performed in conjunction with fluorescent tagging and microscopy. These studies provide complementary spatial–temporal information about host–pathogen interactions to follow through the temporal cascade of cellular events that occur during a pathogen infection. One advantage of IP-MS over AP-MS is that experiments can be performed in a native cellular context to enable unbiased detection of PPIs whereas AP-MS requires overexpression of the protein of interest to capture interacting proteins and can result in background binding artifacts. Furthermore, in both methods, weakly bound protein interactors or localized, but non-interacting, by-stander proteins cannot be captured.
A recent example by Gordon et al. used AP-MS to investigate the SARS-CoV-2 virus–host protein interactions. The authors devised a viral–host SARS-CoV-2 protein interaction map by cloning, tagging, and expressing 26 of the 29 proteins encoded by the SARS-CoV-2 genome in HEK293T cell lines and identified the human proteins that physically associate with each viral protein bait.83 They identified 332 putative viral–host PPIs encompassing 66 druggable human proteins targeted by 69 small molecule compounds (including 29 drugs approved by the US Food and Drug Administration, 12 in clinical trials, and 28 in preclinical studies), and established several antiviral candidates potentially suitable for repurposing as antiviral therapeutics. This group also did a comparative coronavirus – human PPI study to understand the conservation of target proteins and cellular processes between SARS-CoV-2, SARS-CoV, and MERS-CoV.84
Global proteomics approaches that quantify changes in protein abundance and post-translational modifications (PTMs) (e.g. phosphorylation) are powerful tools to elucidate mechanisms of viral pathogenesis by providing a snapshot of how cellular pathways are rewired upon infection. Importantly, the functional outcomes of many protein and PTM event changes are well annotated, especially for kinases as drug targets where phosphorylation directly regulates their activity. These approaches employ mass-spectrometry analysis coupled with bioinformatics-based tools to quantitatively assess changes to protein and/or PTM levels. Bojkova et al. performed a global quantitative proteomic analysis of SARS-CoV-2 infected Caco-2 cell lines and revealed that the virus rewires host cellular pathways such as translation, splicing, carbon metabolism, protein homeostasis (proteostasis) and nucleic acid metabolism.85 The authors used a previously developed method known as multiplexed enhanced protein dynamics (mePROD) proteomics that combined both Stable Isotope Labeling in Cell Culture (SILAC) and Tandem Mass Tags (TMT) labeling methodologies to enhance quantification of protein level changes with high temporal resolution (Fig. 6D).86 A quantitative profiling of the global phosphorylation and protein abundance landscape of SARS-CoV-2 infection was also reported by Bouhaddou et al. where they mapped phosphorylation changes to disrupted kinases and pathways and used this information to identify potential anti-viral small molecules.87 Collectively, the combination of these quantitative methods with other chemogenomic screening efforts88 have the potential to rapidly prioritize proteins, PTMs, and associated drugs and compounds for treating SARS-CoV-2 infection.
Since SARS-CoV-2 infects pneumocytes, leading to acute lung injury and impaired gas exchange, the molecular mechanisms driving infection and pathology remain unclear. To address this gap, our team performed a quantitative phosphoproteomic survey of pluripotent stem cell-derived human alveolar epithelial type 2 cells cultured as an air–liquid interface during infection with SARS-CoV-2 (Hekman et al., in press). The resulting time-course profiles revealed rapid remodeling of diverse host cell systems, including signal transduction machinery, RNA processing, translation, metabolism, nuclear integrity, protein trafficking, cytoskeletal-microtubule organization, leading to cell cycle arrest, genotoxic stress, and innate immunity. Comparison with other proteomic studies of undifferentiated transformed cell lines highlighted convergent and divergent responses, reflected by differential sensitivity to antiviral compounds, providing a rich perspective for targeting respiratory processes hijacked by SARS-CoV-2 as potential therapeutic avenues.
Global proteomic and metabolomic profiling of COVID-19 patient sera is another approach adopted to address the differential expression of proteins and metabolites upon infection when compared to healthy cohorts (Fig. 6E). Shen et al. found 105 differentially expressed proteins in the sera of COVID-19 patients.89 They correlated expression profiles with clinical disease severity that showed 93 proteins to be specifically modulated in severe patients. Activation of the complement system, macrophage function, and platelet degranulation were the major pathways correlated to 50 out of the 93 differentially expressed proteins as shown by their network enrichment analyses. 373 metabolites were significantly changed in COVID-19 patients and 204 metabolites were correlated with disease severity. Activation of the complement system, macrophage function, and platelet degranulation were the three major pathways correlated to the key dysregulated proteins and metabolites from the pathway and network enrichment analyses in this study.
7. Emerging chemical proteomic-based interaction technologies
Mapping protein–metabolite interactions (PMI) and identifying the endogenous host metabolites that complex with proteins is of utmost importance to address the gap that still lies in understanding functional association networks. Small molecule metabolites are key to allosteric regulations of enzymes and signaling molecules for receptors involved in nuclear or cellular transport. In the context of viral infection, the discovery of small molecule ligands of viral proteins upon infection or viral–host protein complexes will not only shed light on structural and functional configuration of binding sites to facilitate the design of novel inhibitors for these complexes, but will also led to identification of pathways and cellular events involved in these interactions. While these chemical proteomic approaches are challenging in terms of confident identification of small molecules due to the diverse structural variation, they can be overcome with stringent filters for false discoveries.Our group has been building a ligand discovery pipeline employing the AP-MS approach for screening host endogenous metabolite ligands of diverse protein baits, including SARS-CoV-2 proteases (Fig. 7A). We believe that this study will contribute to basic understanding of novel aspects of the chemical biology of SARS-CoV-2–host interactions and possibly connect the dots as to how the cellular and immunological responses are so varied innately to this virus.
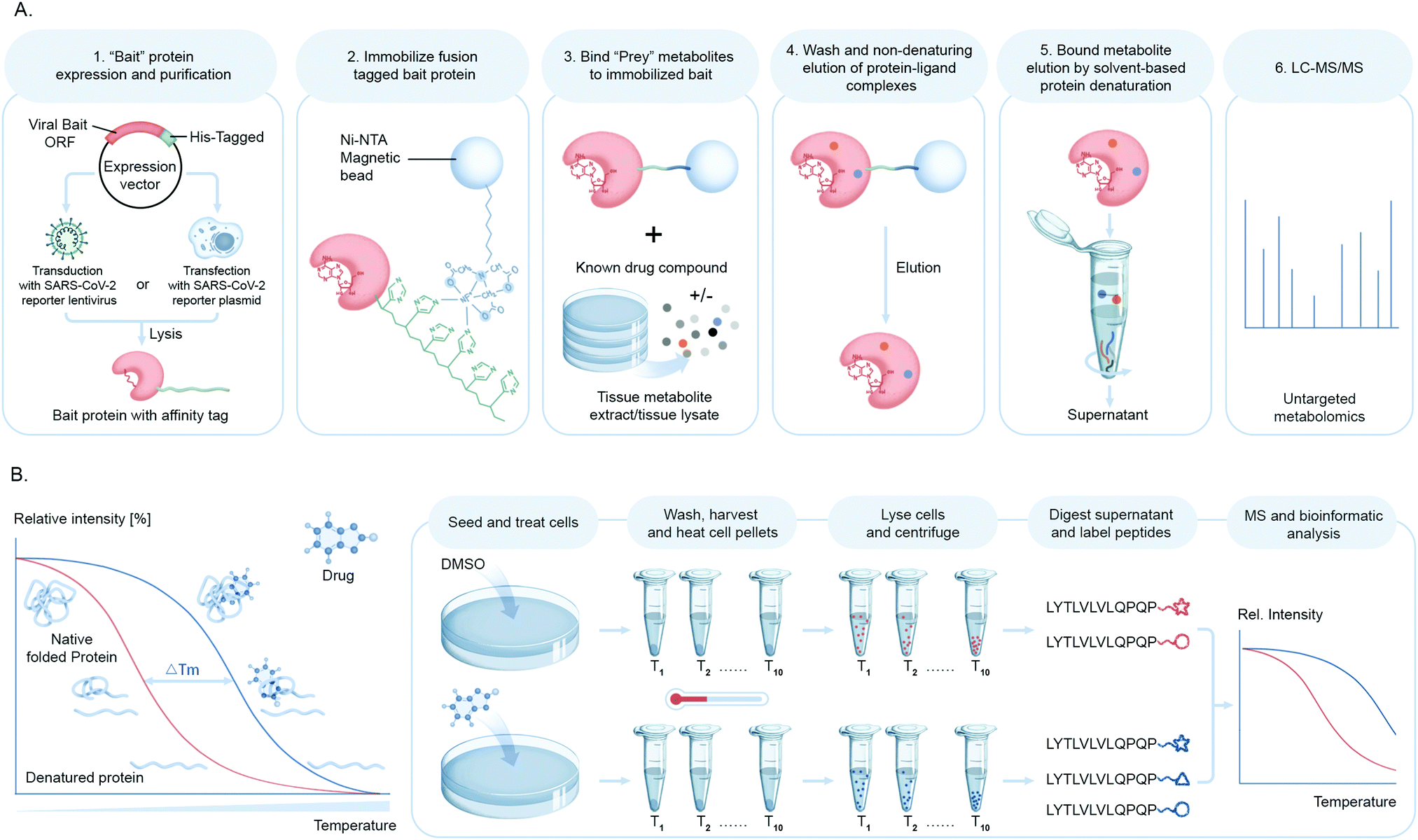 | ||
| Fig. 7 Chemo-proteomic analysis of drug or cellular metabolites in coronavirus host interactions. (A) Schematic illustrating small molecule ligand pull down workflow. A SARS-CoV-2 protein of interest or bait fused to an epitope tag is expressed in cells either by transfection of bait plasmid or transduction of SARS-CoV-2 typed lentivirus and immobilized on an affinity matrix specific for the epitope tag. The protein bound matrix is then exposed to metabolites or synthetic compounds to identify high-affinity binders which are then recovered and eluted from the bait protein and identified by untargeted metabolomics. (B) The principle of cellular thermal shift assay coupled to mass spectrometry (CETSA-MS) for the identification of the endogenous target(s) of a bioactive compound is based on measuring changes in the enthalpy of SARS-CoV-2 protein structures upon binding with its ligand, which is reflected in a shift in its melting temperature (Tm). The assay involves treatment of cells or cell free extracts with desired compound/s and subsequent sample heating followed by quantitative proteomic analysis. | ||
Another chemical biology approach to measure targeted small molecule–protein engagement is the cellular thermal shift assay (CETSA).90 This assay involves treatment of cells with one or more compounds of interest, followed by heating to denature and precipitate proteins, with the premise that ligand binding will stabilize proteins and hence preserve their solubility. After removing aggregated protein from the soluble fraction, a quantitative proteomics workflow is applied to measure ligand-induced changes in protein solubility to infer the target of a compound of interest such as a bioactive small molecule from a cell-based antiviral phenotypic screen or a drug lead in order to deduce or confirm their mechanism of action (Fig. 7B).
A recent addition to the affinity purification-based interactomics toolkit are proximity-based labeling techniques, wherein either an engineered enzyme or, in a recent advance, a photocatalyst, is used to chemically label cellular protein(s) in close proximity to a protein of interest. Established methods are centered on BioID, in which a promiscuous biotin ligase is fused to a target protein leading to the biotinylation of cellular factors bound to, or in close proximity, to the bait.91 The biotinylated proteins are then selectively recovered by binding to an affinity capture matrix (e.g. streptavidin). Related approaches based on a similar engineered enzyme principle, i.e. ascorbate peroxidase (APEX, APEX2) and horseradish peroxidase (HRP) have been described,92 as has another peroxidase-based assay known as selective proximity labeling assay using tyramide (SPPLAT)93 for mapping PPIs. When compared to AP-MS based approaches, proximity labeling methods can map cellular networks consisting of both stable and transient PPIs, whereas the former tend to be biased towards identifying stable protein complexes. Proximity labeling systems are finding increased utility for profiling entire cellular compartments and to monitor cell–cell contacts that mediate cell engagement.
Proximity labeling efforts to understand coronavirus–host protein interactions have been employed by V'kovski et al. using BioID to map the microenvironment of the replication and transcription complex of murine coronavirus (MHV).94 By fusing the biotin ligase enzyme to MHV-nsp2 followed by subsequent infection of L929 murine fibroblasts, this group identified the close association of replicase gene products nsp2–10, RNA-dependent RNA polymerase (nsp12), the NTPase/helicase (nsp13), the 5′-cap methyltransferases (nsp14, nsp16), the proof-reading exonuclease (nsp14), and the nucleocapsid protein. They proposed that nsp2–16 and the nucleocapsid protein collectively constitute a functional coronavirus replication and transcription complex in infected cells. In addition to these enriched viral gene products, >500 human host proteins were also enriched from this labeling experiment suggesting their proximity to the MHV replication/transcription complex.
Recently, we developed a microenvironment-mapping platform (μMap) to identify protein interacting partners through photocatalytic-mediated carbene generation to selectively tag and identify neighboring proteins on cell membranes.24 This technology was used to identify proximal protein environments of the immune cell surface proteins CD45, CD47, CD29, and PD-L1 in a visible light dependent manner. Furthermore, synaptic labeling between interacting cells within a co-culture environment was achieved with the μMap technology. Successful application of this technology in mammalian cell systems highlights the potential for utilizing photocatalyst-based strategies to map viral–host protein interaction environments relevant to coronaviral infection. This can involve attaching a photocatalyst to SARS-CoV-2 spike, protease, or replicase proteins to identify their respective interacting host proteins on the cell surface or within cytosolic environments (Fig. 8). A more detailed understanding of these dynamic interactions may provide alternative therapeutic targets towards inhibiting SARS-CoV-2 infection and disease spread.
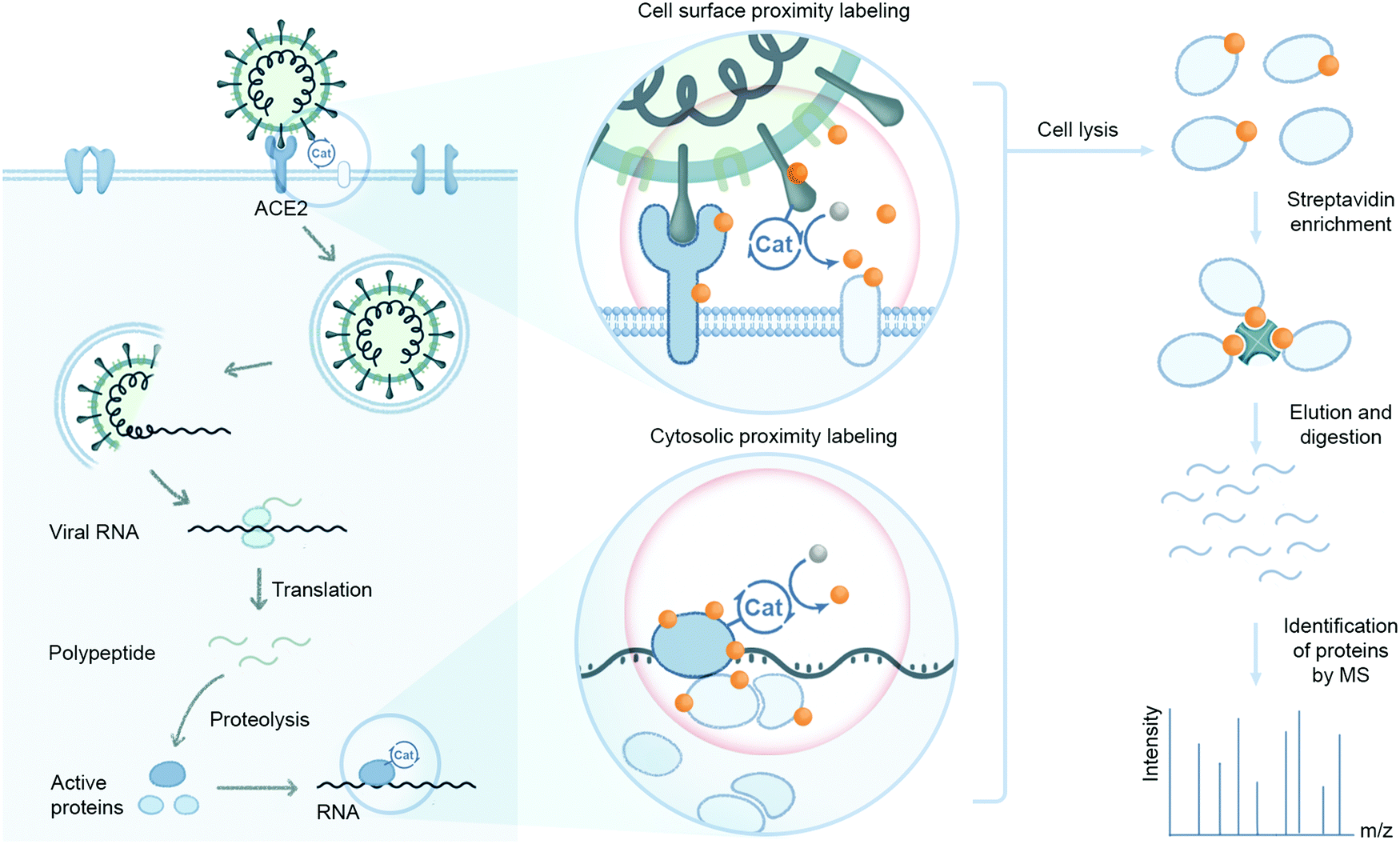 | ||
| Fig. 8 Virus–host protein microenvironment mapping. Protein environments associated with coronavirus–host cell interactions can be assessed using proximity labeling technologies that rely on proximal protein labeling via an enzyme or small molecule catalyst. The catalyst can be used to probe cell surface and cytosolic environments through attachment to viral components such as the spike protein that engages host cell surfaces or viral replication and transcription proteins responsible for production of RNA. Catalytic proximity labeling can be induced enzymatically or through visible light activation of small molecule photocatalysts to covalently capture host protein interaction environments. Labeled proteins can then be affinity enriched and identified through LC-MS based proteomics. | ||
Future outlook
The devastating impact of SARS-CoV-2 viral infections on human health coupled with previous health threats from other coronaviruses (SARS-CoV and MERS-CoV) over the last two decades have raised an important biomedical need to explore and exploit the basic molecular mechanisms by which coronaviruses interact with human hosts. This will be facilitated by the application of the chemical biology strategies described here to achieve a more detailed molecular level understanding of the many interactions that occur within the life cycle of a coronavirus and a host cell that it encounters. Reducing the complexity of information obtained from these efforts to characterize host–viral protein interaction networks and cellular signaling pathways into identifying therapeutic targets can be greatly aided by follow up validation. This includes functional testing of enriched proteins or pathways, and/or the expansion of phenomenon observed within basic model systems into more physiologically relevant settings. The information obtained from these approaches will continue to unlock our understanding of the complexity of viral–host interactions and enable the broader infectious disease research community in combatting the ongoing pandemic and potential future viral infections.Public database links
DIP – https://dip.doe-mbi.ucla.eduHPRD – https://www.hprd.org/
IntAct – https://www.ebi.ac.uk/intact/
MINT – https://mint.bio.uniroma2.it
VirHost-Net – https://virhostnet.prabi.fr
BioGrid – https://thebiogrid.org
InnateDB – https://www.innatedb.com
PHISTO – https://phisto.org
OMIM – https://www.ncbi.nlm.nih.gov/omim
HGMD – http://www.hgmd.cf.ac.uk/ac/index.php
COSMIC – https://cancer.sanger.ac.uk/cosmic
Conflicts of interest
E. C. H., K. A. V., D. J. H., R. C. O., and O. O. F. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.References
- F. Wu, S. Zhao, B. Yu, Y. M. Chen, W. Wang, Z. G. Song, Y. Hu, Z. W. Tao, J. H. Tian, Y. Y. Pei, M. L. Yuan, Y. L. Zhang, F. H. Dai, Y. Liu, Q. M. Wang, J. J. Zheng, L. Xu, E. C. Holmes and Y. Z. Zhang, Nature, 2020, 579, 265–269 CrossRef CAS.
- F. Krammer, Nature, 2020, 586, 516–527 CrossRef CAS.
- J. Corum, K. J. Wu and C. Zimmer, Coronavirus Drug and Treatment Tracker, http://nytimes.com/interactive/2020/science/coronavirus-drugs-treatments.html.
- T. P. Sheahan, A. C. Sims, S. R. Leist, A. Schafer, J. Won, A. J. Brown, S. A. Montgomery, A. Hogg, D. Babusis, M. O. Clarke, J. E. Spahn, L. Bauer, S. Sellers, D. Porter, J. Y. Feng, T. Cihlar, R. Jordan, M. R. Denison and R. S. Baric, Nat. Commun., 2020, 11, 222 CrossRef CAS.
- L. Yurkovetskiy, X. Wang, K. E. Pascal, C. Tomkins-Tinch, T. P. Nyalile, Y. Wang, A. Baum, W. E. Diehl, A. Dauphin, C. Carbone, K. Veinotte, S. B. Egri, S. F. Schaffner, J. E. Lemieux, J. B. Munro, A. Rafique, A. Barve, P. C. Sabeti, C. A. Kyratsous, N. V. Dudkina, K. Shen and J. Luban, Cell, 2020, 183, 739–751 CrossRef CAS.
- N. D. Grubaugh, W. P. Hanage and A. L. Rasmussen, Cell, 2020, 182, 794–795 CrossRef CAS.
- K. Subbarao and S. Mahanty, Immunity, 2020, 52, 905–909 CrossRef CAS.
- F. J. Ibarrondo, J. A. Fulcher, D. Goodman-Meza, J. Elliott, C. Hofmann, M. A. Hausner, K. G. Ferbas, N. H. Tobin, G. M. Aldrovandi and O. O. Yang, N. Engl. J. Med., 2020, 383, 1085–1087 CrossRef.
- D. Mathew, J. R. Giles, A. E. Baxter, D. A. Oldridge, A. R. Greenplate, J. E. Wu, C. Alanio, L. Kuri-Cervantes, M. B. Pampena, K. D’Andrea, S. Manne, Z. Chen, Y. J. Huang, J. P. Reilly, A. R. Weisman, C. A. G. Ittner, O. Kuthuru, J. Dougherty, K. Nzingha, N. Han, J. Kim, A. Pattekar, E. C. Goodwin, E. M. Anderson, M. E. Weirick, S. Gouma, C. P. Arevalo, M. J. Bolton, F. Chen, S. F. Lacey, H. Ramage, S. Cherry, S. E. Hensley, S. A. Apostolidis, A. C. Huang, L. A. Vella, U. P. C. P. Unit, M. R. Betts, N. J. Meyer and E. J. Wherry, Science, 2020, 369 DOI:10.1126/science.abc8511.
- A. W. D. Edridge, J. Kaczorowska, A. C. R. Hoste, M. Bakker, M. Klein, K. Loens, M. F. Jebbink, A. Matser, C. M. Kinsella, P. Rueda, M. Ieven, H. Goossens, M. Prins, P. Sastre, M. Deijs and L. van der Hoek, Nat. Med., 2020, 26, 1691–1693 CrossRef.
- M. Peiris and G. M. Leung, Lancet, 2020, 396, 1467–1469 CrossRef CAS.
- G. Salvatori, L. Luberto, M. Maffei, L. Aurisicchio, G. Roscilli, F. Palombo and E. Marra, J. Transl. Med., 2020, 18, 222 CrossRef CAS.
- A. Grifoni, D. Weiskopf, S. I. Ramirez, J. Mateus, J. M. Dan, C. R. Moderbacher, S. A. Rawlings, A. Sutherland, L. Premkumar, R. S. Jadi, D. Marrama, A. M. de Silva, A. Frazier, A. F. Carlin, J. A. Greenbaum, B. Peters, F. Krammer, D. M. Smith, S. Crotty and A. Sette, Cell, 2020, 181, 1489–1501 CrossRef CAS.
- J. Mateus, A. Grifoni, A. Tarke, J. Sidney, S. I. Ramirez, J. M. Dan, Z. C. Burger, S. A. Rawlings, D. M. Smith, E. Phillips, S. Mallal, M. Lammers, P. Rubiro, L. Quiambao, A. Sutherland, E. D. Yu, R. da Silva Antunes, J. Greenbaum, A. Frazier, A. J. Markmann, L. Premkumar, A. de Silva, B. Peters, S. Crotty, A. Sette and D. Weiskopf, Science, 2020, 370, 89–94 CrossRef CAS.
- M. E. Bunnage, E. L. Chekler and L. H. Jones, Nat. Chem. Biol., 2013, 9, 195–199 CrossRef CAS.
- M. J. C. Long, X. Liu and Y. Aye, Front. Chem., 2019, 7, 125 CrossRef CAS.
- K. Shiraiwa, R. Cheng, H. Nonaka, T. Tamura and I. Hamachi, Cell Chem. Biol., 2020, 27, 970–985 CrossRef CAS.
- A. Leitner, Anal. Chim. Acta, 2018, 1000, 2–19 CrossRef CAS.
- M. Grammel and H. C. Hang, Nat. Chem. Biol., 2013, 9, 475–484 CrossRef CAS.
- H. G. Budayeva and I. M. Cristea, Adv. Exp. Med. Biol., 2014, 806, 263–282 CrossRef CAS.
- G. Pasqual, A. Chudnovskiy, J. M. J. Tas, M. Agudelo, L. D. Schweitzer, A. Cui, N. Hacohen and G. D. Victora, Nature, 2018, 553, 496–500 CrossRef CAS.
- Q. Liu, J. Zheng, W. Sun, Y. Huo, L. Zhang, P. Hao, H. Wang and M. Zhuang, Nat. Methods, 2018, 15, 715–722 CrossRef CAS.
- K. F. Cho, T. C. Branon, S. Rajeev, T. Svinkina, N. D. Udeshi, T. Thoudam, C. Kwak, H. W. Rhee, I. K. Lee, S. A. Carr and A. Y. Ting, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12143–12154 CrossRef CAS.
- J. B. Geri, J. V. Oakley, T. Reyes-Robles, T. Wang, S. J. McCarver, C. H. White, F. P. Rodriguez-Rivera, D. L. Parker Jr, E. C. Hett, O. O. Fadeyi, R. C. Oslund and D. W. C. MacMillan, Science, 2020, 367, 1091–1097 CrossRef CAS.
- S. Singh, P. K. Singh, H. Suhail, V. Arumugaswami, P. E. Pellett, S. Giri and A. Kumar, J. Immunol., 2020, 204, 1810–1824 CrossRef CAS.
- J. B. Black, C. Lopez and P. E. Pellett, Virus Res., 1992, 22, 13–23 CrossRef CAS.
- M. Fu, Y. Gao, Q. Zhou, Q. Zhang, Y. Peng, K. Tian, J. Wang and X. Zheng, Gene, 2014, 536, 272–278 CrossRef CAS.
- C. M. Goodwin, S. Xu and J. Munger, Trends Microbiol., 2015, 23, 789–798 CrossRef CAS.
- S. Zhang, J. Carriere, X. Lin, N. Xie and P. Feng, Viruses, 2018, 10, 521 CrossRef.
- S. C. Cheng, L. A. Joosten and M. G. Netea, Cytokine Growth Factor Rev., 2014, 25, 707–713 CrossRef CAS.
- N. S. Heaton and G. Randall, Trends Microbiol., 2011, 19, 368–375 CrossRef CAS.
- J. W. Chambers, T. G. Maguire and J. C. Alwine, J. Virol., 2010, 84, 1867–1873 CrossRef CAS.
- J. Wu, L. Sun, X. Chen, F. Du, H. Shi, C. Chen and Z. J. Chen, Science, 2013, 339, 826–830 CrossRef CAS.
- S. E. Weinberg, L. A. Sena and N. S. Chandel, Immunity, 2015, 42, 406–417 CrossRef CAS.
- B. L. Wei, P. W. Denton, E. O'Neill, T. Luo, J. L. Foster and J. V. Garcia, J. Virol., 2005, 79, 5705–5712 CrossRef CAS.
- E. L. Mills, D. G. Ryan, H. A. Prag, D. Dikovskaya, D. Menon, Z. Zaslona, M. P. Jedrychowski, A. S. H. Costa, M. Higgins, E. Hams, J. Szpyt, M. C. Runtsch, M. S. King, J. F. McGouran, R. Fischer, B. M. Kessler, A. F. McGettrick, M. M. Hughes, R. G. Carroll, L. M. Booty, E. V. Knatko, P. J. Meakin, M. L. J. Ashford, L. K. Modis, G. Brunori, D. C. Sévin, P. G. Fallon, S. T. Caldwell, E. R. S. Kunji, E. T. Chouchani, C. Frezza, A. T. Dinkova-Kostova, R. C. Hartley, M. P. Murphy and L. A. O'Neill, Nature, 2018, 556, 113–117 CrossRef CAS.
- D. Olagnier, E. Farahani, J. Thyrsted, J. Blay-Cadanet, A. Herengt, M. Idorn, A. Hait, B. Hernaez, A. Knudsen, M. B. Iversen, M. Schilling, S. E. Jørgensen, M. Thomsen, L. S. Reinert, M. Lappe, H. D. Hoang, V. H. Gilchrist, A. L. Hansen, R. Ottosen, C. G. Nielsen, C. Møller, D. van der Horst, S. Peri, S. Balachandran, J. Huang, M. Jakobsen, E. B. Svenningsen, T. B. Poulsen, L. Bartsch, A. L. Thielke, Y. Luo, T. Alain, J. Rehwinkel, A. Alcamí, J. Hiscott, T. H. Mogensen, S. R. Paludan and C. K. Holm, Nat. Commun., 2020, 11, 4938 CrossRef CAS.
- T. Weichhart, M. Hengstschläger and M. Linke, Nat. Rev. Immunol., 2015, 15, 599–614 CrossRef CAS.
- L. A. O'Neill and D. G. Hardie, Nature, 2013, 493, 346–355 CrossRef.
- K. Kotzamanis, A. Angulo and P. Ghazal, Med. Microbiol. Immunol., 2015, 204, 395–407 CrossRef CAS.
- J. Zhao, S. He, A. Minassian, J. Li and P. Feng, Curr. Opin. Virol., 2015, 15, 103–111 CrossRef CAS.
- A. W. Thomson, H. R. Turnquist and G. Raimondi, Nat. Rev. Immunol., 2009, 9, 324–337 CrossRef CAS.
- T. Decker, S. Stockinger, M. Karaghiosoff, M. Müller and P. Kovarik, J. Clin. Invest., 2002, 109, 1271–1277 CrossRef CAS.
- D. M. Bass and H. B. Greenberg, J. Clin. Invest., 1992, 89, 3–9 CrossRef CAS.
- R. J. Colonno, P. L. Callahan and W. J. Long, J. Virol., 1986, 57, 7–12 CrossRef CAS.
- G. N. Gaulton, M. S. Co, H. D. Royer and M. I. Greene, Mol. Cell. Biochem., 1984, 65, 5–21 CrossRef CAS.
- D. L. Krah and R. L. Crowell, Virology, 1982, 118, 148–156 CrossRef CAS.
- E. V. Protopopova, A. V. Sorokin, S. N. Konovalova, A. V. Kachko, S. V. Netesov and V. B. Loktev, Zentralbl. Bakteriol., 1999, 289, 632–638 CrossRef CAS.
- K. K. Lum and I. M. Cristea, Expert Rev. Proteomics, 2016, 13, 325–340 CrossRef CAS.
- C. Muller, M. Hardt, D. Schwudke, B. W. Neuman, S. Pleschka and J. Ziebuhr, J. Virol., 2018, 92, e01463–17 CrossRef.
- M. Poppe, S. Wittig, L. Jurida, M. Bartkuhn, J. Wilhelm, H. Muller, K. Beuerlein, N. Karl, S. Bhuju, J. Ziebuhr, M. L. Schmitz and M. Kracht, PLoS Pathog., 2017, 13, e1006286 CrossRef.
- K. Vyboh, L. Ajamian and A. J. Mouland, J. Visualized Exp., 2012, e4002, DOI:10.3791/4002.
- W. Rut, K. Groborz, L. Zhang, X. Sun, M. Zmudzinski, B. Pawlik, X. Wang, D. Jochmans, J. Neyts, W. Młynarski, R. Hilgenfeld and M. Drag, Nat. Chem. Biol., 2020 DOI:10.1038/s41589-020-00689-z.
- D. Falzarano, A. Groseth and T. Hoenen, Antiviral Res., 2014, 103, 78–87 CrossRef CAS.
- A. McGregor, K. Y. Choi, S. Schachtele and J. Lokensgard, in Animal Models for the Study of Human Disease, ed. P. M. Conn, Academic Press, Boston, 2013, pp. 905–925 DOI:10.1016/B978-0-12-415894-8.00037-3.
- A. C. Sims, R. S. Baric, B. Yount, S. E. Burkett, P. L. Collins and R. J. Pickles, J. Virol., 2005, 79, 15511–15524 CrossRef CAS.
- M. L. Agostini, E. L. Andres, A. C. Sims, R. L. Graham, T. P. Sheahan, X. Lu, E. C. Smith, J. B. Case, J. Y. Feng, R. Jordan, A. S. Ray, T. Cihlar, D. Siegel, R. L. Mackman, M. O. Clarke, R. S. Baric and M. R. Denison, mBio, 2018, 9, e00221–18 CrossRef CAS.
- A. Chatr-aryamontri, A. Ceol, D. Peluso, A. Nardozza, S. Panni, F. Sacco, M. Tinti, A. Smolyar, L. Castagnoli, M. Vidal, M. E. Cusick and G. Cesareni, Nucleic Acids Res., 2009, 37, D669–D673 CrossRef CAS.
- V. Navratil, B. de Chassey, L. Meyniel, S. Delmotte, C. Gautier, P. Andre, V. Lotteau and C. Rabourdin-Combe, Nucleic Acids Res., 2009, 37, D661–D668 CrossRef CAS.
- G. D. Bader, I. Donaldson, C. Wolting, B. F. Ouellette, T. Pawson and C. W. Hogue, Nucleic Acids Res., 2001, 29, 242–245 CrossRef CAS.
- I. Xenarios, D. W. Rice, L. Salwinski, M. K. Baron, E. M. Marcotte and D. Eisenberg, Nucleic Acids Res., 2000, 28, 289–291 CrossRef CAS.
- T. S. Keshava Prasad, R. Goel, K. Kandasamy, S. Keerthikumar, S. Kumar, S. Mathivanan, D. Telikicherla, R. Raju, B. Shafreen, A. Venugopal, L. Balakrishnan, A. Marimuthu, S. Banerjee, D. S. Somanathan, A. Sebastian, S. Rani, S. Ray, C. J. Harrys Kishore, S. Kanth, M. Ahmed, M. K. Kashyap, R. Mohmood, Y. L. Ramachandra, V. Krishna, B. A. Rahiman, S. Mohan, P. Ranganathan, S. Ramabadran, R. Chaerkady and A. Pandey, Nucleic Acids Res., 2009, 37, D767–D772 CrossRef CAS.
- H. Hermjakob, L. Montecchi-Palazzi, C. Lewington, S. Mudali, S. Kerrien, S. Orchard, M. Vingron, B. Roechert, P. Roepstorff, A. Valencia, H. Margalit, J. Armstrong, A. Bairoch, G. Cesareni, D. Sherman and R. Apweiler, Nucleic Acids Res., 2004, 32, D452–D455 CrossRef CAS.
- A. Chatr-aryamontri, A. Ceol, L. M. Palazzi, G. Nardelli, M. V. Schneider, L. Castagnoli and G. Cesareni, Nucleic Acids Res., 2007, 35, D572–D574 CrossRef CAS.
- T. Guirimand, S. Delmotte and V. Navratil, Nucleic Acids Res., 2015, 43, D583–D587 CrossRef CAS.
- C. Stark, B. J. Breitkreutz, T. Reguly, L. Boucher, A. Breitkreutz and M. Tyers, Nucleic Acids Res., 2006, 34, D535–D539 CrossRef CAS.
- K. Breuer, A. K. Foroushani, M. R. Laird, C. Chen, A. Sribnaia, R. Lo, G. L. Winsor, R. E. Hancock, F. S. Brinkman and D. J. Lynn, Nucleic Acids Res., 2013, 41, D1228–D1233 CrossRef CAS.
- S. Durmuş Tekir, T. Çakır, E. Ardiç, A. S. Sayılırbaş, G. Konuk, M. Konuk, H. Sarıyer, A. Uğurlu, İ. Karadeniz, A. Özgür, F. E. Sevilgen and K. Ülgen, Bioinformatics, 2013, 29, 1357–1358 CrossRef.
- P. D. Stenson, M. Mort, E. V. Ball, K. Evans, M. Hayden, S. Heywood, M. Hussain, A. D. Phillips and D. N. Cooper, Hum. Genet., 2017, 136, 665–677 CrossRef CAS.
- J. G. Tate, S. Bamford, H. C. Jubb, Z. Sondka, D. M. Beare, N. Bindal, H. Boutselakis, C. G. Cole, C. Creatore, E. Dawson, P. Fish, B. Harsha, C. Hathaway, S. C. Jupe, C. Y. Kok, K. Noble, L. Ponting, C. C. Ramshaw, C. E. Rye, H. E. Speedy, R. Stefancsik, S. L. Thompson, S. Wang, S. Ward, P. J. Campbell and S. A. Forbes, Nucleic Acids Res., 2019, 47, D941–D947 CrossRef CAS.
- M. Ostaszewski, A. Mazein, M. E. Gillespie, I. Kuperstein, A. Niarakis, H. Hermjakob, A. R. Pico, E. L. Willighagen, C. T. Evelo, J. Hasenauer, F. Schreiber, A. Drager, E. Demir, O. Wolkenhauer, L. I. Furlong, E. Barillot, J. Dopazo, A. Orta-Resendiz, F. Messina, A. Valencia, A. Funahashi, H. Kitano, C. Auffray, R. Balling and R. Schneider, Sci. Data, 2020, 7, 136 CrossRef CAS.
- S. Fields and O. Song, Nature, 1989, 340, 245–246 CrossRef CAS.
- B. de Chassey, L. Meyniel-Schicklin, J. Vonderscher, P. Andre and V. Lotteau, Genome Med., 2014, 6, 115 CrossRef.
- H. Xiao, L. H. Xu, Y. Yamada and D. X. Liu, PLoS One, 2008, 3, e1494 CrossRef.
- S. Pfefferle, J. Schopf, M. Kogl, C. C. Friedel, M. A. Muller, J. Carbajo-Lozoya, T. Stellberger, E. von Dall’Armi, P. Herzog, S. Kallies, D. Niemeyer, V. Ditt, T. Kuri, R. Zust, K. Pumpor, R. Hilgenfeld, F. Schwarz, R. Zimmer, I. Steffen, F. Weber, V. Thiel, G. Herrler, H. J. Thiel, C. Schwegmann-Wessels, S. Pohlmann, J. Haas, C. Drosten and A. von Brunn, PLoS Pathog., 2011, 7, e1002331 CrossRef CAS.
- J. K. Millet, F. Kien, C. Y. Cheung, Y. L. Siu, W. L. Chan, H. Li, H. L. Leung, M. Jaume, R. Bruzzone, J. S. Peiris, R. M. Altmeyer and B. Nal, PLoS One, 2012, 7, e49566 CrossRef CAS.
- M. Gebre, J. L. Nomburg and B. E. Gewurz, Viruses, 2018, 10, 55 CrossRef.
- T. R. Abbott, G. Dhamdhere, Y. Liu, X. Lin, L. Goudy, L. Zeng, A. Chemparathy, S. Chmura, N. S. Heaton, R. Debs, T. Pande, D. Endy, M. F. La Russa, D. B. Lewis and L. S. Qi, Cell, 2020, 181, 865–876 CrossRef CAS.
- J. P. Broughton, X. Deng, G. Yu, C. L. Fasching, V. Servellita, J. Singh, X. Miao, J. A. Streithorst, A. Granados, A. Sotomayor-Gonzalez, K. Zorn, A. Gopez, E. Hsu, W. Gu, S. Miller, C. Y. Pan, H. Guevara, D. A. Wadford, J. S. Chen and C. Y. Chiu, Nat. Biotechnol., 2020, 38, 870–874 CrossRef CAS.
- J. L. Daly, B. Simonetti, K. Klein, K. E. Chen, M. K. Williamson, C. Antón-Plágaro, D. K. Shoemark, L. Simón-Gracia, M. Bauer, R. Hollandi, U. F. Greber, P. Horvath, R. B. Sessions, A. Helenius, J. A. Hiscox, T. Teesalu, D. A. Matthews, A. D. Davidson, B. M. Collins, P. J. Cullen and Y. Yamauchi, Science, 2020, 370, 861–865 CrossRef CAS.
- K. Wu, G. Peng, M. Wilken, R. J. Geraghty and F. Li, J. Biol. Chem., 2012, 287, 8904–8911 CrossRef CAS.
- J. Shang, G. Ye, K. Shi, Y. Wan, C. Luo, H. Aihara, Q. Geng, A. Auerbach and F. Li, Nature, 2020, 581, 221–224 CrossRef CAS.
- D. E. Gordon, G. M. Jang, M. Bouhaddou, J. Xu, K. Obernier, K. M. White, M. J. O'Meara, V. V. Rezelj, J. Z. Guo, D. L. Swaney, T. A. Tummino, R. Huttenhain, R. M. Kaake, A. L. Richards, B. Tutuncuoglu, H. Foussard, J. Batra, K. Haas, M. Modak, M. Kim, P. Haas, B. J. Polacco, H. Braberg, J. M. Fabius, M. Eckhardt, M. Soucheray, M. J. Bennett, M. Cakir, M. J. McGregor, Q. Li, B. Meyer, F. Roesch, T. Vallet, A. Mac Kain, L. Miorin, E. Moreno, Z. Z. C. Naing, Y. Zhou, S. Peng, Y. Shi, Z. Zhang, W. Shen, I. T. Kirby, J. E. Melnyk, J. S. Chorba, K. Lou, S. A. Dai, I. Barrio-Hernandez, D. Memon, C. Hernandez-Armenta, J. Lyu, C. J. P. Mathy, T. Perica, K. B. Pilla, S. J. Ganesan, D. J. Saltzberg, R. Rakesh, X. Liu, S. B. Rosenthal, L. Calviello, S. Venkataramanan, J. Liboy-Lugo, Y. Lin, X. P. Huang, Y. Liu, S. A. Wankowicz, M. Bohn, M. Safari, F. S. Ugur, C. Koh, N. S. Savar, Q. D. Tran, D. Shengjuler, S. J. Fletcher, M. C. O'Neal, Y. Cai, J. C. J. Chang, D. J. Broadhurst, S. Klippsten, P. P. Sharp, N. A. Wenzell, D. Kuzuoglu-Ozturk, H. Y. Wang, R. Trenker, J. M. Young, D. A. Cavero, J. Hiatt, T. L. Roth, U. Rathore, A. Subramanian, J. Noack, M. Hubert, R. M. Stroud, A. D. Frankel, O. S. Rosenberg, K. A. Verba, D. A. Agard, M. Ott, M. Emerman, N. Jura, M. von Zastrow, E. Verdin, A. Ashworth, O. Schwartz, C. d'Enfert, S. Mukherjee, M. Jacobson, H. S. Malik, D. G. Fujimori, T. Ideker, C. S. Craik, S. N. Floor, J. S. Fraser, J. D. Gross, A. Sali, B. L. Roth, D. Ruggero, J. Taunton, T. Kortemme, P. Beltrao, M. Vignuzzi, A. Garcia-Sastre, K. M. Shokat, B. K. Shoichet and N. J. Krogan, Nature, 2020, 583, 459–468 CrossRef CAS.
- D. E. Gordon, J. Hiatt, M. Bouhaddou, V. V. Rezelj, S. Ulferts, H. Braberg, A. S. Jureka, K. Obernier, J. Z. Guo, J. Batra, R. M. Kaake, A. R. Weckstein, T. W. Owens, M. Gupta, S. Pourmal, E. W. Titus, M. Cakir, M. Soucheray, M. McGregor, Z. Cakir, G. Jang, M. J. O'Meara, T. A. Tummino, Z. Zhang, H. Foussard, A. Rojc, Y. Zhou, D. Kuchenov, R. Hüttenhain, J. Xu, M. Eckhardt, D. L. Swaney, J. M. Fabius, M. Ummadi, B. Tutuncuoglu, U. Rathore, M. Modak, P. Haas, K. M. Haas, Z. Z. C. Naing, E. H. Pulido, Y. Shi, I. Barrio-Hernandez, D. Memon, E. Petsalaki, A. Dunham, M. C. Marrero, D. Burke, C. Koh, T. Vallet, J. A. Silvas, C. M. Azumaya, C. Billesbølle, A. F. Brilot, M. G. Campbell, A. Diallo, M. S. Dickinson, D. Diwanji, N. Herrera, N. Hoppe, H. T. Kratochvil, Y. Liu, G. E. Merz, M. Moritz, H. C. Nguyen, C. Nowotny, C. Puchades, A. N. Rizo, U. Schulze-Gahmen, A. M. Smith, M. Sun, I. D. Young, J. Zhao, D. Asarnow, J. Biel, A. Bowen, J. R. Braxton, J. Chen, C. M. Chio, U. S. Chio, I. Deshpande, L. Doan, B. Faust, S. Flores, M. Jin, K. Kim, V. L. Lam, F. Li, J. Li, Y. L. Li, Y. Li, X. Liu, M. Lo, K. E. Lopez, A. A. Melo, F. R. Moss 3rd, P. Nguyen, J. Paulino, K. I. Pawar, J. K. Peters, T. H. Pospiech Jr, M. Safari, S. Sangwan, K. Schaefer, P. V. Thomas, A. C. Thwin, R. Trenker, E. Tse, T. K. M. Tsui, F. Wang, N. Whitis, Z. Yu, K. Zhang, Y. Zhang, F. Zhou, D. Saltzberg, A. J. Hodder, A. S. Shun-Shion, D. M. Williams, K. M. White, R. Rosales, T. Kehrer, L. Miorin, E. Moreno, A. H. Patel, S. Rihn, M. M. Khalid, A. Vallejo-Gracia, P. Fozouni, C. R. Simoneau, T. L. Roth, D. Wu, M. A. Karim, M. Ghoussaini, I. Dunham, F. Berardi, S. Weigang, M. Chazal, J. Park, J. Logue, M. McGrath, S. Weston, R. Haupt, C. J. Hastie, M. Elliott, F. Brown, K. A. Burness, E. Reid, M. Dorward, C. Johnson, S. G. Wilkinson, A. Geyer, D. M. Giesel, C. Baillie, S. Raggett, H. Leech, R. Toth, N. Goodman, K. C. Keough, A. L. Lind, R. J. Klesh, K. R. Hemphill, J. Carlson-Stevermer, J. Oki, K. Holden, T. Maures, K. S. Pollard, A. Sali, D. A. Agard, Y. Cheng, J. S. Fraser, A. Frost, N. Jura, T. Kortemme, A. Manglik, D. R. Southworth, R. M. Stroud, D. R. Alessi, P. Davies, M. B. Frieman, T. Ideker, C. Abate, N. Jouvenet, G. Kochs, B. Shoichet, M. Ott, M. Palmarini, K. M. Shokat, A. García-Sastre, J. A. Rassen, R. Grosse, O. S. Rosenberg, K. A. Verba, C. F. Basler, M. Vignuzzi, A. A. Peden, P. Beltrao and N. J. Krogan, Science, 2020 DOI:10.1126/science.abe9403.
- D. Bojkova, K. Klann, B. Koch, M. Widera, D. Krause, S. Ciesek, J. Cinatl and C. Münch, Nature, 2020, 583, 469–472 CrossRef CAS.
- K. Klann, G. Tascher and C. Münch, Mol. Cell, 2020, 77, 913–925 CrossRef CAS.
- M. Bouhaddou, D. Memon, B. Meyer, K. M. White, V. V. Rezelj, M. Correa Marrero, B. J. Polacco, J. E. Melnyk, S. Ulferts, R. M. Kaake, J. Batra, A. L. Richards, E. Stevenson, D. E. Gordon, A. Rojc, K. Obernier, J. M. Fabius, M. Soucheray, L. Miorin, E. Moreno, C. Koh, Q. D. Tran, A. Hardy, R. Robinot, T. Vallet, B. E. Nilsson-Payant, C. Hernandez-Armenta, A. Dunham, S. Weigang, J. Knerr, M. Modak, D. Quintero, Y. Zhou, A. Dugourd, A. Valdeolivas, T. Patil, Q. Li, R. Huttenhain, M. Cakir, M. Muralidharan, M. Kim, G. Jang, B. Tutuncuoglu, J. Hiatt, J. Z. Guo, J. Xu, S. Bouhaddou, C. J. P. Mathy, A. Gaulton, E. J. Manners, E. Felix, Y. Shi, M. Goff, J. K. Lim, T. McBride, M. C. O'Neal, Y. Cai, J. C. J. Chang, D. J. Broadhurst, S. Klippsten, E. De Wit, A. R. Leach, T. Kortemme, B. Shoichet, M. Ott, J. Saez-Rodriguez, B. R. tenOever, R. D. Mullins, E. R. Fischer, G. Kochs, R. Grosse, A. Garcia-Sastre, M. Vignuzzi, J. R. Johnson, K. M. Shokat, D. L. Swaney, P. Beltrao and N. J. Krogan, Cell, 2020, 182, 685–712 CrossRef CAS.
- L. Riva, S. Yuan, X. Yin, L. Martin-Sancho, N. Matsunaga, L. Pache, S. Burgstaller-Muehlbacher, P. D. De Jesus, P. Teriete, M. V. Hull, M. W. Chang, J. F. Chan, J. Cao, V. K. Poon, K. M. Herbert, K. Cheng, T. H. Nguyen, A. Rubanov, Y. Pu, C. Nguyen, A. Choi, R. Rathnasinghe, M. Schotsaert, L. Miorin, M. Dejosez, T. P. Zwaka, K. Y. Sit, L. Martinez-Sobrido, W. C. Liu, K. M. White, M. E. Chapman, E. K. Lendy, R. J. Glynne, R. Albrecht, E. Ruppin, A. D. Mesecar, J. R. Johnson, C. Benner, R. Sun, P. G. Schultz, A. I. Su, A. García-Sastre, A. K. Chatterjee, K. Y. Yuen and S. K. Chanda, Nature, 2020, 586, 113–119 CrossRef CAS.
- B. Shen, X. Yi, Y. Sun, X. Bi, J. Du, C. Zhang, S. Quan, F. Zhang, R. Sun, L. Qian, W. Ge, W. Liu, S. Liang, H. Chen, Y. Zhang, J. Li, J. Xu, Z. He, B. Chen, J. Wang, H. Yan, Y. Zheng, D. Wang, J. Zhu, Z. Kong, Z. Kang, X. Liang, X. Ding, G. Ruan, N. Xiang, X. Cai, H. Gao, L. Li, S. Li, Q. Xiao, T. Lu, Y. Zhu, H. Liu, H. Chen and T. Guo, Cell, 2020, 182, 59–72 CrossRef CAS.
- R. Jafari, H. Almqvist, H. Axelsson, M. Ignatushchenko, T. Lundback, P. Nordlund and D. Martinez Molina, Nat. Protoc., 2014, 9, 2100–2122 CrossRef CAS.
- K. J. Roux, D. I. Kim, M. Raida and B. Burke, J. Cell Biol., 2012, 196, 801–810 CrossRef CAS.
- J. D. Martell, T. J. Deerinck, S. S. Lam, M. H. Ellisman and A. Y. Ting, Nat. Protoc., 2017, 12, 1792–1816 CrossRef CAS.
- X. W. Li, J. S. Rees, P. Xue, H. Zhang, S. W. Hamaia, B. Sanderson, P. E. Funk, R. W. Farndale, K. S. Lilley, S. Perrett and A. P. Jackson, J. Biol. Chem., 2014, 289, 14434–14447 CrossRef CAS.
- P. V’Kovski, M. Gerber, J. Kelly, S. Pfaender, N. Ebert, S. Braga Lagache, C. Simillion, J. Portmann, H. Stalder, V. Gaschen, R. Bruggmann, M. H. Stoffel, M. Heller, R. Dijkman and V. Thiel, eLife, 2019, 8(e42037) Search PubMed.
- P. M. Jean Beltran, J. D. Federspiel, X. Sheng and I. M. Cristea, Mol. Syst. Biol., 2017, 13, 922 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |