
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vivo modulation of ubiquitin chains by N-methylated non-proteinogenic cyclic peptides†
Joseph M.
Rogers‡
 ab,
Mickal
Nawatha‡
c,
Betsegaw
Lemma
d,
Ganga B.
Vamisetti
c,
Ido
Livneh
e,
Uri
Barash
e,
Israel
Vlodavsky
e,
Aaron
Ciechanover
e,
David
Fushman
d,
Hiroaki
Suga
*a and
Ashraf
Brik
*c
ab,
Mickal
Nawatha‡
c,
Betsegaw
Lemma
d,
Ganga B.
Vamisetti
c,
Ido
Livneh
e,
Uri
Barash
e,
Israel
Vlodavsky
e,
Aaron
Ciechanover
e,
David
Fushman
d,
Hiroaki
Suga
*a and
Ashraf
Brik
*c
aDepartment of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: hsuga@chem.s.u-tokyo.ac.jp
bDepartment of Drug Design and Pharmacology, University of Copenhagen, Copenhagen 2100, Denmark
cSchulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa 3200008, Israel. E-mail: abrik@technion.ac.il
dDepartment of Chemistry and Biochemistry, Center for Biomolecular Structure and Organization, University of Maryland, College Park, MD 20742, USA
eThe Rappaport Faculty of Medicine and Research Institute, Technion-Israel Institute of Technology, Haifa 31096, Israel
First published on 16th December 2020
Abstract
Cancer and other disease states can change the landscape of proteins post-translationally tagged with ubiquitin (Ub) chains. Molecules capable of modulating the functions of Ub chains are potential therapeutic agents, but their discovery represents a significant challenge. Recently, it was shown that de novo cyclic peptides, selected from trillion-member random libraries, are capable of binding particular Ub chains. However, these peptides were overwhelmingly proteinogenic, so the prospect of in vivo activity was uncertain. Here, we report the discovery of small, non-proteinogenic cyclic peptides, rich in non-canonical features like N-methylation, which can tightly bind Lys48-linked Ub chains. These peptides engage three Lys48-linked Ub units simultaneously, block the action of deubiquitinases and the proteasome, induce apoptosis in vitro, and attenuate tumor growth in vivo. This highlights the potential of non-proteinogenic cyclic peptide screening to rapidly find in vivo-active leads, and the targeting of ubiquitin chains as a promising anti-cancer mechanism of action.
Introduction
Ubiquitination is an abundant post-translational modification.1 Indeed, hundreds of enzymes exist to attach the small, 76 amino acid protein ubiquitin (Ub) mainly to lysine side-chains of particular cellular proteins.2 Ub itself has multiple lysines, onto which further Ub can be attached. As a result, there are many possible Ub chains of various lengths and linkage types. These Ub chains can differently perturb the tagged protein3,4 or be recognized by different cellular receptors, allowing them to perform different functions.5,6 Lys48-linked Ub chains are particularly important, as these direct their tagged proteins for degradation by the 26S proteasome.7Cancer cells use the ubiquitin–proteasome system to remove critical proteins and evade programmed cell death.8 Consequently, inhibitors of the 26S proteasome have proved powerful research tools and successful anticancer therapeutics,9 although resistance can emerge.10 An attractive alternative is to target the signal for degradation: find molecules to bind to and interfere with the recognition of Lys48-linked Ub chains.11–15 However, given the breadth of cellular processes that rely on differently linked Ub chains, specificity is crucial5 and also a challenge due to the following reasons: Ub chains are assembled from the same monomer, and the differences in structure and dynamics of differently linked-Ub chains can be subtle.16,17 Moreover, chain length is also important. For example, tetra Ub (Lys48-linked, K48Ub4) is postulated to be the minimal chain length to label a protein for degradation,7 although there is some evidence suggesting shorter chains can also perform this function.18,19 Molecules specific for long (e.g. ≥3), Lys48-linked Ub chains are required to specifically modulate protein degradation.
Ub chains have proved challenging targets for traditional drug discovery using small molecules. Those discovered to date suffer from weak binding and poor specificity for linkage and length.12–15 Typically, these are symmetric molecules that bind monomeric Ub weakly, and then achieve what affinity they have for Ub chains through multimeric interactions. However, true recognition of Ub chains with a specific linkage was achieved recently using an alternative modality – de novo cyclic peptides.11
Nawatha et al. discovered de novo cyclic peptides capable of specifically binding Lys48-linked Ub chains. To achieve this, they first used solid-phase peptide synthesis (SPPS) coupled to Lys analogue native chemical ligation20,21 to generate Lys48-linked Ub chains.11 These Ub chains were of sufficient purity for use as targets for the RaPID system, a method that uses reprogrammed ribosome peptide synthesis to synthesize, and then screen, extremely large (>1012) cyclic peptide libraries for binding molecules.22 Tight-binding, having low nM KD, and linkage-specific cyclic peptides were discovered, and their novel Ub recognition abilities were explained by their binding modes. The cyclic peptide bound in one orientation on the symmetric Ub chain target, simultaneously interacting with two or more Ub in the chains. Importantly, these cyclic peptides inhibited Ub chain recognition in vitro and had activity when added to cells, causing Ub-conjugate accumulation and apoptosis.11
Unfortunately, these first-generation cyclic peptides did not necessarily exhibit physicochemical properties comparable to known drug-like cyclic peptides. Orally-available, membrane-permeable, macrocyclic and cyclic peptides, such as the natural product cyclosporin, tend to be small (≤1.2 kDa)23,24 and have few H-bond acceptors, fewer H-bond donors, and no formal charges at pH 7.25–27 Key to accessing this property space using peptides is the use of non-canonical amino acids, such as amine N-methylation,28 which have the added benefit of conferring protease-resistance. By contrast, the first-generation Ub chain binding cyclic peptides were large (∼1.7 kDa) and made almost entirely of H-bond-rich, protease-recognizable, canonical amino acids.11
A better subclass of cyclic peptides with improved bioactivity targeting the ubiquitin–proteasome system is needed to provide research tools and drug leads. A potential route to such molecules is to screen cyclic peptide libraries which include from the beginning non-canonical amino acids known to confer desirable physical properties.22,29 However, it is not yet known if cyclic peptides from such a library can succeed at a challenging molecular recognition task like specific Ub chain binding. Here, we constructed and screened a highly non-proteinogenic peptide library to discover de novo cyclic peptides capable of modulating specific Lys48-linked ubiquitin chains.
Results
Target Ub chain synthesized by total chemical synthesis
To discover binders specific for the Lys48-linked Ub tetramer, while also avoiding binders that use di- or mono-Ub as the recognition element, high purity K48Ub4 and K48Ub2 chains were required. Ub chains were assembled by total chemical synthesis, as described previously,11,20,21 for use as the target and anti-target respectively. Each was biotinylated at the N-terminus for attachment to a streptavidin solid support, enabling screening of pooled libraries of peptides.Construction of a highly non-proteinogenic cyclic peptide library
We sought to create a large cyclic peptide library containing multiple non-canonical amino acids, to emulate the properties of drug-like, highly non-proteinogenic cyclic peptides. To form cyclic peptides, the genetic code was reprogrammed so that ribosomal peptide synthesis was initiated using a chloroacetyled amino acid.30 We withdrew the natural initiator amino acid Met and its cognate aaRS during the construction of a custom reconstituted in vitro translation system,31 and supplemented this with initiator tRNA loaded with ClAc-D-Phe,22 using an artificial ribozyme flexizyme.32 We then further reprogrammed the genetic code to remove the charged amino acids Glu, Asp, and Arg, and add the following non-canonical amino acids:22,29,33 three N-methylated: MeGly, MeAla and MePhe; two D-stereochemistry: D-Phe and D-Ala, and the linear hydrophobic side-chain Aoc (Fig. 1A). Each was flexizyme-loaded onto tRNA and supplied to the translation system. The two D-amino acids were loaded onto an optimized tRNAPro and the translation system supplemented with the E. coli translation factor EF-P to enhance translation efficiency.34,35 To test the fidelity of this extensively reprogrammed 22 amino-acid genetic code (Fig. S1A, ESI†), we confirmed the ribosomal synthesis of peptides containing these non-canonical amino acids, with the major products detected by MALDI-TOF MS having the expected masses (Fig. S1B, ESI†).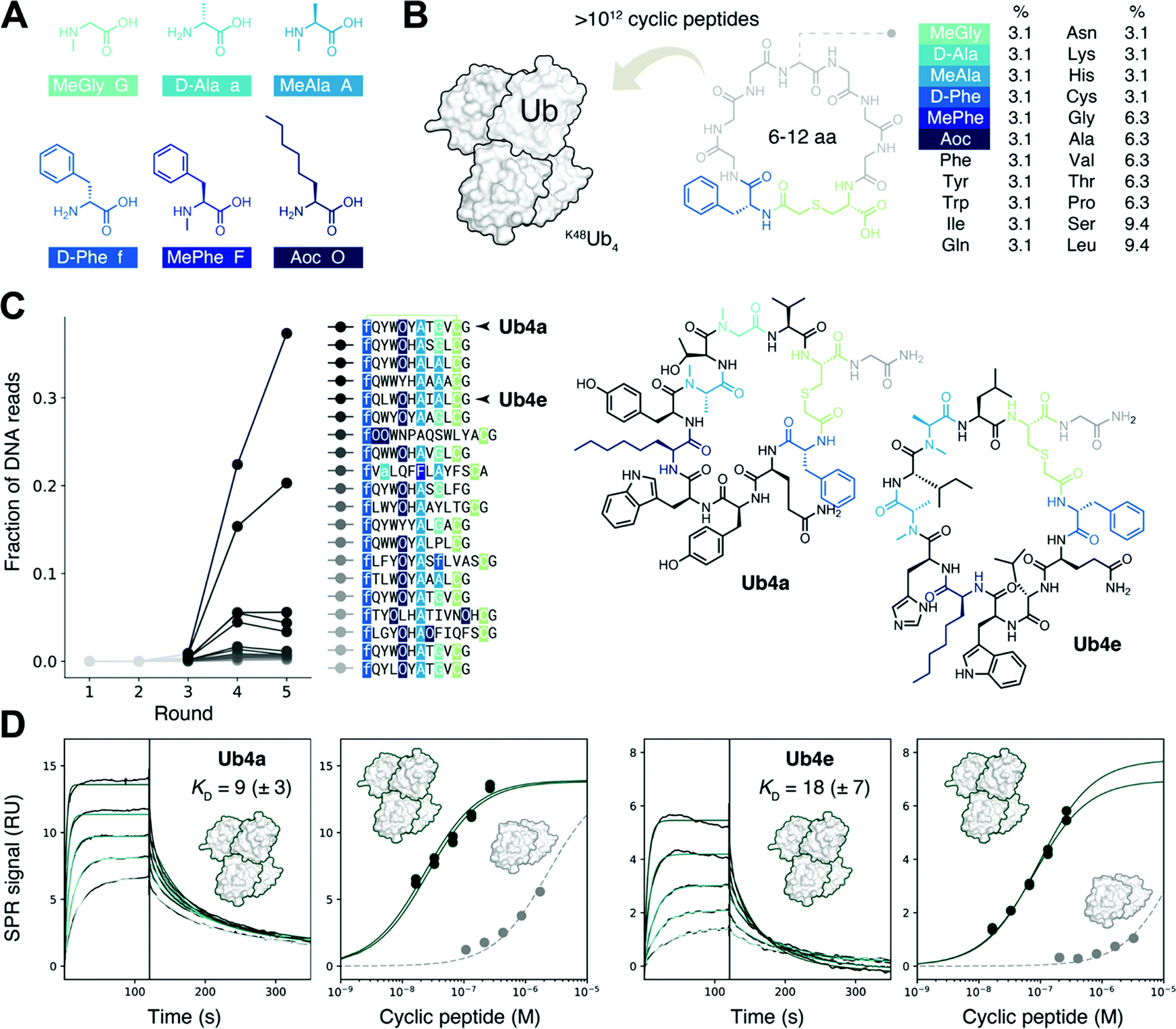 | ||
| Fig. 1 Discovery of tight-binding non-proteinogenic cyclic peptides for the K48-linked ubiquitin tetramer. (A) The six non-canonical amino acids, replacing Met, Glu, Asp and Arg, used in the extensively reprogrammed genetic code used for cyclic peptide discovery. In addition, the initiation codon was reprogrammed to chloroacetyl-D-Phe which, after reaction with Cys, allows for the formation of cyclic peptides. (B) Amino acid distribution in the randomized sequence of the trillion-member cyclic peptide library screened against K48Ub4. (C) Cyclic peptide library composition, inferred from next generation sequencing of the DNA library, after five rounds of RaPID selection with K48Ub4 as the target and K48Ub2 as the anti-target. The library became enriched in certain cyclic peptide sequences, such as the highlighted Ub4a and Ub4e. (D) Surface plasmon resonance (SPR) kinetics (left) reveals tight (nM KD) binding of cyclic peptide Ub4a to K48Ub4. Binding traces shown for 17, 33, 66, 130 and 260 nM cyclic peptide. SPR amplitudes upon titration (center left) suggests binding to K48Ub2 (grey) has a KD in the μM range, significantly weaker binding than to K48Ub4 (data from two titrations shown, dark green). Similar selective binding observed for cyclic peptide Ub4e (centre right and right). | ||
Non-proteinogenic cyclic peptide library construction and selection
Following the RaPID protocol,22 we assembled a greater than trillion-member cyclic peptide library using the ribosome and our reprogrammed genetic code, using degenerate DNA codons to encode cyclic peptides with 6–12 randomized amino acids each (Fig. 1B). mRNA display36 was used to covalently tag each unique cyclic peptide with its encoding mRNA/DNA, for later resynthesis of the library, sequencing, or both. In the first round of screening, the cyclic peptide library was incubated with the target K48Ub4, the bound mRNA/DNA amplified and the enriched library resynthesized. Subsequent rounds included a ‘negative selection’ step where K48Ub2 and streptavidin binders were discarded before incubation with K48Ub4. This was to avoid a problem encountered in the first cyclic peptide screenings effort,11 where the majority of the cyclic peptides recovered used K48Ub2 as the main recognition element, then using multimeric interaction to bind the longer chain target K48Ub4.After each round, the fraction of mRNA/DNA recovered increased, suggesting an enrichment for desired K48Ub4-binding peptides (Fig. S2, ESI†). Next-generation sequencing (NGS) was then used to assess the library composition after each round of selection, and this showed the progressive enrichment of particular, putative K48Ub4-binding, peptide sequences (Fig. 1C). The top DNA sequence, and that of an analogue, were resubmitted to the in vitro translation system to confirm the integrity of the reprogrammed genetic code. The expected cyclic peptides, named Ub4a and Ub4e respectively (Fig. 1C), were detected by MALDI-TOF MS (Fig. S3, ESI†). Ub4a and Ub4e each contain an 11 amino acid macrocycle, with two N-methyl amino acids, a non-canonical side-chain as well as a D-amino acid. Ub4e contains a His, which may be partially protonated at pH 7. Otherwise, as hoped, Ub4a and Ub4e are uncharged peptides with no formally charged amino acids at neutral pH.
Cyclic peptides preferentially bind longer K48-linked Ub chains
Cyclic peptides Ub4a and Ub4e were both synthesized by SPPS and tested for binding to the target K48Ub4 by SPR, and both displayed high affinity, nM KD, for the Ub chain (Fig. 1D and Table S1, ESI†). As expected, binding to the anti-target K48Ub2 was considerably weaker, with >100 fold higher KD, (Fig. 1D and Fig. S4 and Table S2, ESI†). To further confirm binding, we synthesized Ub4a labelled with a fluorescein dye (Fig. S5, ESI†) and used this as a probe for Ub-chains transferred onto nitrocellulose membranes (Fig. S6A and B, ESI†). Under these denaturing conditions, K48Ub2 and K48Ub4 bound Ub4a-fluorescein, with greater intensity then alternatively linked dimers K63Ub2 and K11Ub2.NMR reveals unit- and residue-specific interactions between Ub chains and cyclic peptide Ub4a
We used NMR to confirm the physical interaction between cyclic peptide Ub4a and Ub chains as well as to map the Ub units and residues involved in binding. The unlabeled peptide was titrated into a solution of selectively 15N-labeled Ub chains, and the binding was monitored by NMR. For this purpose, K48Ub4, K48Ub3, and K48Ub2 with specific Ub units uniformly 15N-enriched were assembled from respective recombinant Ub monomers using controlled enzymatic chain assembly methodology.11,37,38 The addition of Ub4a to K48Ub4 caused strong residue-specific perturbations in the 1H–15N NMR spectra of the proximal Ub unit (the one bearing free C terminus) (Fig. 2A). We observed a gradual decrease in intensity of the ‘unbound’ signals accompanied by the appearance and increase of new (‘bound’) signals at a different position in the spectrum, a textbook example of slow exchange, consistent with the slow dissociation rates (∼1 per minute) detected by SPR (Table S1, ESI†). The perturbed residues are located primarily in and around the L8-I44-V70 hydrophobic patch on Ub surface, as well as the α-helix, and for most residues, the spectral changes saturated at about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 peptide:K48Ub4 molar ratio. However, substantially weaker changes in the NMR spectra were observed for the distal Ub (the opposite end of the chain) of K48Ub4, where the signal shifts were substantially smaller. Furthermore, most residues exhibited gradual signal shifts characteristic for fast exchange on the NMR time scale. The noncovalent Ub–Ub interactions in K48-linked chains are sufficiently weak to allow fast interconversion between closed (possessing Ub–Ub interface) and open conformations.38–40 Thus, it is possible that Ub4a binding disrupts the interactions between distal Ub and the rest of the Ub chain (Fig. 2B). These results indicate that interactions with the distal Ub in the tetramer do not significantly contribute to Ub4a binding and suggests that K48Ub3 is the actual peptide-binding element.
:1 peptide:K48Ub4 molar ratio. However, substantially weaker changes in the NMR spectra were observed for the distal Ub (the opposite end of the chain) of K48Ub4, where the signal shifts were substantially smaller. Furthermore, most residues exhibited gradual signal shifts characteristic for fast exchange on the NMR time scale. The noncovalent Ub–Ub interactions in K48-linked chains are sufficiently weak to allow fast interconversion between closed (possessing Ub–Ub interface) and open conformations.38–40 Thus, it is possible that Ub4a binding disrupts the interactions between distal Ub and the rest of the Ub chain (Fig. 2B). These results indicate that interactions with the distal Ub in the tetramer do not significantly contribute to Ub4a binding and suggests that K48Ub3 is the actual peptide-binding element.
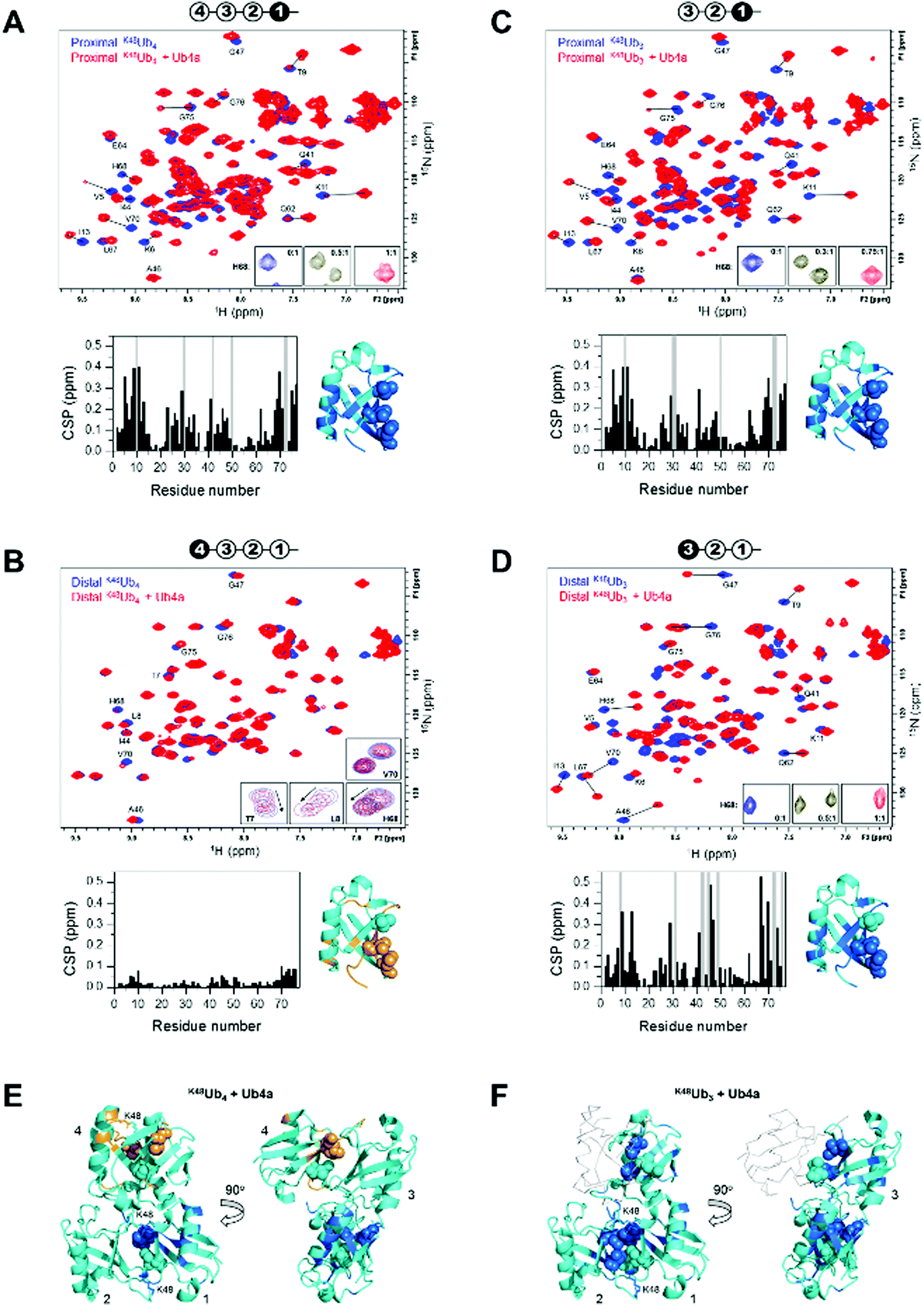 | ||
| Fig. 2 Cyclic peptide Ub4a binds to hydrophobic patch residues in K48-linked tri- and tetra-Ub and engages first three Ub units in the chain. Shown are overlays of 1H–15N NMR spectra of free (blue) and Ub4a-bound (red) states of the proximal (A) and distal (B) Ub units of K48Ub4 and proximal (C) and distal (D) Ub units of K48Ub3. Select residues are indicated and their signals in the free and peptide-bound states are connected by lines. Insets in A, C, and D zoom on the signals of H68 to illustrate the slow-exchange behavior (the numbers indicate peptide/polyUb molar ratio). Insets in B are examples of signal behavior in the course of titration (up to 2:1 molar ratio) for the indicated residues: fast exchange for T7, L8, H68 and slow exchange for V70. Cartoon drawings on the top of each spectrum indicate which Ub unit in each chain is 15N-labeled (colored black) and acts as the reporter; the numbering of Ub units starts from the proximal Ub. The plots below each spectrum show residue-specific chemical shift perturbations (CSPs) for each Ub unit; the grey bars correspond to residues for which unbound signals disappeared but the bound signals could not be unambiguously identified. Shown to the right of each plot is the structure of Ub monomer (PDB ID: 1UBQ) with residues exhibiting significant CSPs mapped, colored marine (light orange for the distal Ub of K48Ub4); the side-chains of the hydrophobic patch residues L8, I44, V70 are shown as spheres. The threshold for CSP-based mapping was set to 0.15 ppm for all Ub units studied here except for the distal Ub (unit 4) of K48Ub4 where it was 0.03 ppm. The distal Ub of K48Ub4 is colored differently to emphasize the markedly different level of NMR spectral perturbations. (E and F) Map of the residues that showed significant CSPs upon Ub4a binding to K48Ub4 (E) and K48Ub3 (F) on the cartoon representation of the structure of the compact state of K48Ub4 (PDB ID: 2O6V39). Only Ub units analyzed in this work are mapped; the CSP threshold and coloring are the same as in A–D. The Ub units as well as K48 side chains involved in the isopeptide linkages are indicated. In panel F, Ub unit 4 which is absent in K48Ub3 is shown as a light grey backbone trace, for comparison. | ||
To test this hypothesis, we analyzed Ub4a binding to the proximal, middle (endo), and distal Ub units of K48Ub3. All three Ub units exhibited slow-exchange behavior upon Ub4a titration indicative of tight binding. The observed spectral changes in the proximal Ub of K48Ub3 were almost indistinguishable from those in the proximal Ub of K48Ub4 (Fig. 2C and Fig. S7, ESI†), pointing to strong similarity in the peptide's contacts with the proximal Ub in both chains. Also, the distal Ub of K48Ub3 exhibited strong NMR signal shifts (Fig. 2D), in stark contrast with the distal Ub of K48Ub4 (see also Fig. S8, ESI†). Similar strong NMR signal shifts were observed for the middle Ub of K48Ub3 (Fig. S9, ESI†). While the structure of K48Ub3:Ub4a complex awaits determination, these results indicate that all three Ub units in K48Ub3 are engaged in tight interactions with Ub4a (Fig. 2E and F).
To further verify Ub4a selectivity for Ub chains longer than the dimer, Ub4a peptide was titrated into K48Ub2 with 15N-labeled proximal Ub. In stark contrast with the proximal Ub in K48Ub4 and K48Ub3, here we observed little or no signal shifts in the NMR spectra as well as strong signal broadening/disappearance for several residues located on the hydrophobic patch surface of Ub (Fig. S10, ESI†). This behavior is indicative of intermediate exchange on the NMR time scale, consistent with faster dissociation rates hence weaker binding to K48Ub2 detected by SPR (Fig. S4 and Table S2, ESI†).
To summarize, the NMR results corroborate tight Ub4a binding to K48-linked Ub chains of length ≥3 and revealed differential and residue-specific physical interactions between the peptide and various Ub units in these chains. The observed spectral perturbations generally map Ub4a binding sites to the hydrophobic patch surfaces of the Ub units involved. However, there are clear differences in the strength, direction, and exchange regime of these perturbations depending on the chain length and the position of the Ub unit within each chain. In particular, the strikingly weaker changes in NMR spectra observed for the distal Ub of K48Ub4 suggest that it is the trimer (or the first three Ub units in the tetramer) that is the Ub4a recognition element in K48-linked chains.
Cyclic peptides prevent recognition of Ub chains by specific proteases and the proteasome
We chose to investigate whether binding of the discovered cyclic peptides can prevent their recognition of Ub-chains by interacting proteins. For example, deubiquitinating enzymes (DUBs) recognize Ub-chains and cleave the connecting isopeptide bond. The DUB OTUB1 is specific for K48-linked Ub-chains41 and can digest K48Ub4 to shorter chains and monomer in vitro (Fig. 3A). However, in the presence of equimolar Ub4a, K48Ub4 is protected from the activity of OTUB1 (Fig. 3A). This suggests that binding to the Ub chain by the cyclic peptide can interfere with recognition by the DUB, protecting against DUB cleavage. Ub4a also offers some protection to K48Ub2 from cleavage by OTUB1 (Fig. S11A, ESI†). However, the cyclic peptide was not able to protect K11Ub2 or K63Ub2 from cleavage by the K11 linkage-specific DUB Cezanne42 and the linkage non-specific USP2, respectively (Fig. S11B and C, ESI†). Likely this is due to the specificity of these cyclic peptides binding to Ub-chains with the K48-linkage.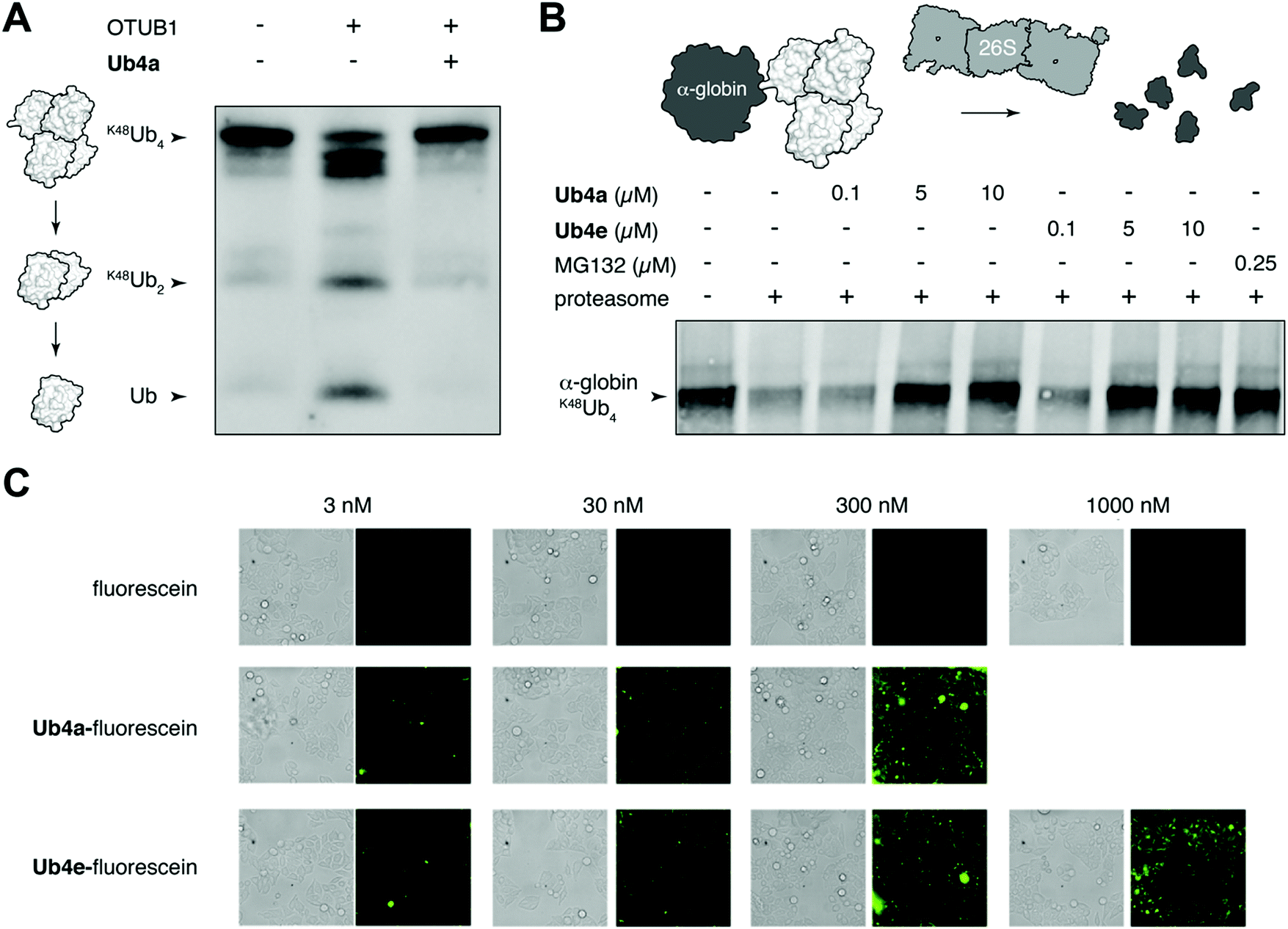 | ||
| Fig. 3 Cyclic peptides inhibit Ub-dependent proteolysis and can enter cells. (A) Cyclic peptide Ub4a (2 μM) inhibits the cleavage of K48Ub4 (2 μM) by the K48-specific DUB OTUB1 (0.25 μM) in vitro. Western blot with anti-Ub antibody. (B) Cyclic peptides Ub4a and Ub4e inhibit degradation of K48Ub4-tagged HA-tagged α-globin (5 μM) by the 26S proteasome (150 nM) in vitro. Similar inhibition of degradation is observed in the presence of direct proteasome inhibitor MG132. Western blot with anti-HA antibody. (C) Cyclic peptides Ub4a and Ub4e, fluorescein labelled, are internalized by live HeLa cells. | ||
A critical Ub-chain interacting protein is the 26S proteasome, which recognizes K48-linked Ub-chains in order to degrade the tagged protein. It is possible that the cyclic peptides, by binding the Ub-chain, can inhibit this recognition and degradation. To test this, we added synthetic α-globin-K48Ub443 to purified 26S proteasome in vitro, with and without Ub4a or Ub4e. Similar to the addition of the direct proteasome inhibitor MG132,44 the cyclic peptides were able to protect α-globin-K48Ub443 from degradation by the 26S proteasome (Fig. 3B). Presumably, these cyclic peptides bind K48-linked Ub chains in a manner that prevents recognition by the 26S proteasome (Fig. 3B).
The NMR finding that only first three Ub units (counting from the proximal end) in K48Ub4 bind Ub4a tightly, was independently verified by comparing the ability of the peptide to protect K48Ub4 and K48Ub3 against disassembly by OTUB1 that preferentially cleaves from the distal end of a poly-Ub chain. The results (Fig. S12A, ESI†) show that Ub4a effectively shields K48Ub3 from disassembly by OTUB1, consistent with the NMR data showing tight peptide binding to all Ub units in the trimer. Interestingly, in the presence of Ub4a, OTUB1 cleaves only one Ub molecule from K48Ub4 resulting in visible and sustained accumulation of a trimer, thus supporting the conclusion that the distal Ub unit in the tetramer binds Ub4a loosely. A slightly different trend was observed for Ub4a protection against proteasome-associated DUB Ubp6, where some amount of K48Ub4 was reduced to a trimer but a significant portion of it remained unaffected (Fig. S12B, ESI†), likely reflecting Ubp6's preference for cleaving the endo isopeptide bond (between two middle Ubs) in K48Ub4.45 By contrast, Ub4a effectively blocked K48Ub4 disassembly by a general linkage-nonspecific DUB USP2 (Fig. S12C, ESI†).
Cyclic peptides can enter cells
Having shown that cyclic peptides can modulate the function of Ub-chains in vitro, we next sought to test their effects in live cells. However, as the ubiquitin system is largely intracellular, it was important to check their ability to enter these cells. We used fluorescein-labelled cyclic peptides (Fig. S5, ESI†) and live-cell imaging to monitor their entrance into HeLa cells. Uptake of Ub4a-fluorescein and Ub4e-fluorescein peptides could be observed after incubation for 2 hours with 3 nM peptide, whereas fluorescein alone showed negligible uptake even at 1000 nM (Fig. 3C). To quantify the uptake, we incubated HeLa cells with Ub4a-fluorescein for 16 h, the cells were washed and the residual fluorescence indicates significant retention of peptide-fluorescein relative to fluorescein alone (Fig. S13, ESI†).Cyclic peptides lead to Ub-conjugates accumulation
We have shown using purified proteins that cyclic peptide binding can inhibit Ub-chain cleavage by DUB activity, and therefore might be expected to cause an accumulation of Ub-chains and proteins tagged with Ub-chains in the cell. We incubated HeLa cells with cyclic peptides Ub4a or Ub4e, as well as MG132, lysed the cells, separated the proteins and stained using anti-Ub western blot to detect Ub-conjugates. In a dose-dependent manner, the cyclic peptides Ub4a and Ub4e caused accumulation of intracellular Ub-conjugates, producing a similar effect to the proteasome inhibitor MG132 (Fig. 4A). Incidentally, cyclic peptide Ub4a could be used to detect these Ub-conjugates on the western blot membrane (Fig. S6C and D, ESI†).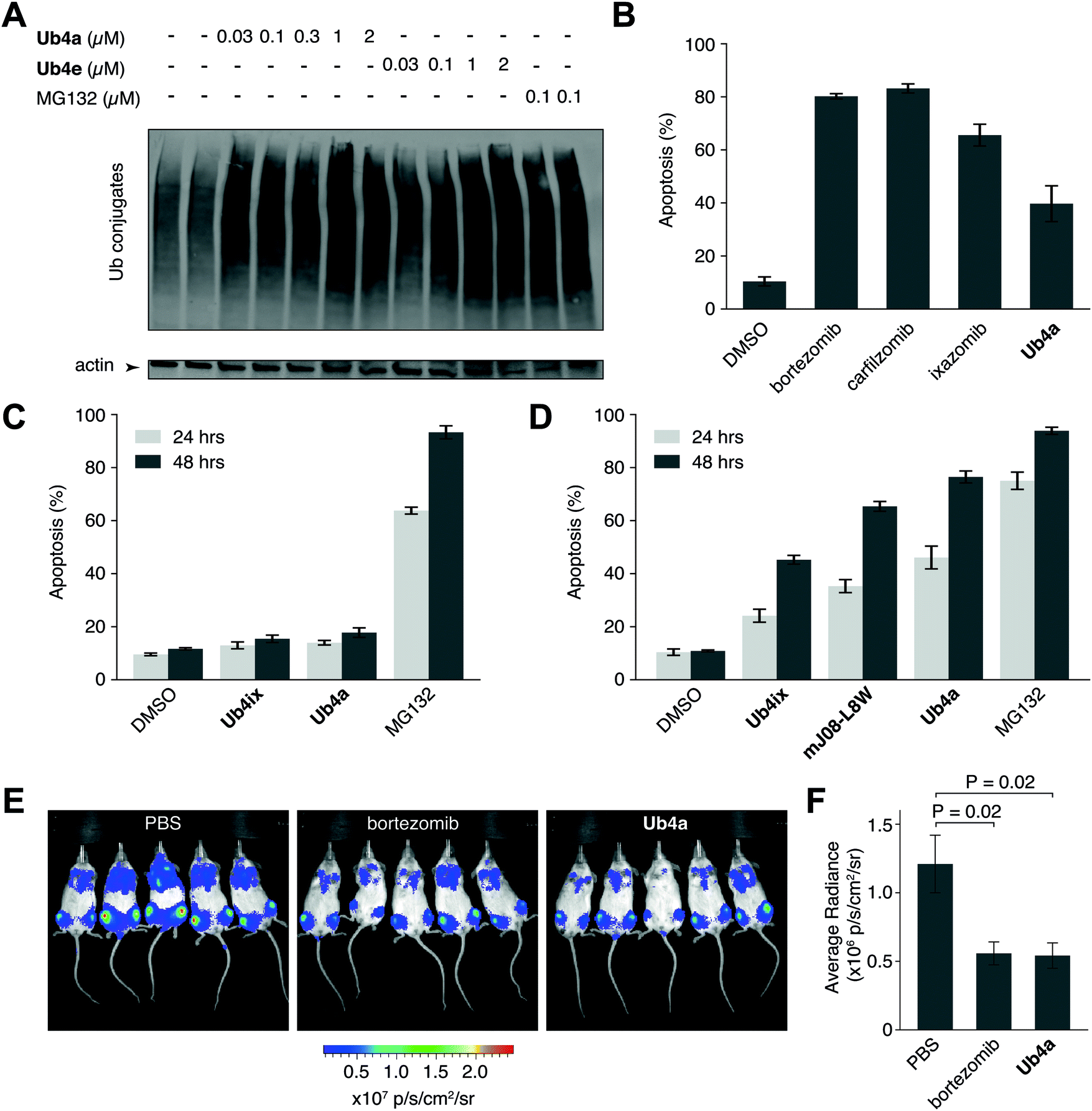 | ||
| Fig. 4 Cyclic peptides cause intracellular accumulation of Ub-conjugates, induce apoptosis in vitro and show anti-cancer activity in vivo. (A) Addition of cyclic peptides Ub4a or Ub4e induced the accumulation of Ub-conjugates in live cells, in a dose-dependent manner. A similar effect was seen upon addition of the direct proteasome inhibitor MG132. (B) Ub4a (100 nM), induced apoptosis in the U87 cell line (brain cancer cell line, primary glioblastoma) after 24 h incubation. Apoptotic cells were detected using fluorescently labelled annexin-V and FACS. A similar induction of apoptosis was seen upon addition of (100 nM) direct proteasome inhibitors bortezomib, carfilzomib and ixazomib. (C) HEK-293 non-cancer derived (embryonic kidney) cells were exposed to the cyclic peptides Ub4a, Ub4ix or MG132 (10 μM), only MG132 showed significant induction of apoptosis. (D) Cyclic peptide Ub4a induces more apoptosis in U87 cells than previous generation Ub-binding peptides Ub4ix or mJ08-L8W, approaching the potency of direct proteasome inhibitor MG132 (all compounds at 100 nM). (E) Imaging of luciferase-expressing human CAG myeloma cells in mice shows that treatment with either the approved anti-cancer drug bortezomib, or the cyclic peptide Ub4a, inhibit the growth of tumors in vivo. (F) Quantification of luminescence from human tumor cells in mice shows that 15 days of treatment with Ub4a causes a reduction in tumor growth similar to treatment with bortezomib. | ||
Ub4a can induce apoptosis in cancer cell lines
Inhibition of the ubiquitin system using direct proteasome inhibitors can induce apoptosis, enabling these inhibitors to be used as cancer therapeutics.46 We wanted to examine whether our cyclic peptides, through their Ub-chain binding, could also induce apoptosis.47,48 We incubated Ub4a with the cancer-derived U87 cells, which are a human primary glioblastoma cell line (brain cancer), and cells sorted for their ability to bind fluorescently labelled annexin-V using Fluorescence Activated Cell Sorting (FACS), detecting both early and late apoptotic cells. Ub4a at 100 nM exhibited a clear increase in apoptosis after 24 h treatment, similar to the direct proteasome inhibitors: bortezomib, ixazomib, and carfilzomib46 (Fig. 4B). Furthermore, we tested the effects of the cyclic peptides on three other cancer cell lines, SH-SY5Y cells (neuroblastoma), MDA-MB-231 cells (epithelial human breast cancer) and HeLa cells (cervical cancer) which all showed similar induction of apoptosis upon treatment with cyclic peptides (Fig. S14, ESI†). Interestingly, under these conditions the Ub4a cyclic peptide did not induce apoptosis in the HEK-293 cell line (embryonic kidney derived), whereas treatment with the direct proteasome inhibitor MG132 did induce apoptosis (Fig. 4C).Finally, we compared the apoptotic induction abilities of the non-proteinogenic cyclic peptide Ub4a with two Ub-binding largely proteinogenic cyclic peptides from previous studies Ub4ix11 and mJ08-L8W.49Ub4a was able to induce apoptosis in a greater proportion of U87 (Fig. 4D), SH-SY5Y, MDA-MB-231 and HeLa cells (Fig. S14, ESI†). One possible reason for the efficacy of Ub4a is that it is resistant to proteases. We tested the stability of Ub4a in human plasma and found that the non-proteinogenic cyclic peptide is very stable in human plasma, with a half-life of multiple days (∼60 h) (Fig. S15, ESI†).
Ub4a shows anti-cancer activity in vivo
The plasma stability of Ub4a combined with its ability to induce apoptosis in vitro in cancer cells, led us to investigate if this de novo cyclic peptide might show anti-cancer activity in vivo. We sought to investigate whether Ub4a could inhibit the growth of human tumors in mice.50 Briefly, SCID/NOD mice were injected intravenously with luciferase-expressing human CAG myeloma cells,51 and after a week of growth, Ub4a (1 mg kg−1 twice a week) or the approved anti-cancer medication bortezomib52 (0.5 mg kg−1 twice a week) were injected intraperitoneally. Both Ub4a and bortezomib treated mice showed a marked reduction in tumor progression relative to untreated mice (Fig. 4E, F and Fig. S16, ESI†). Even in this first test of in vivo activity, 15 days of Ub4a treatment resulted in a reduction in tumor growth similar to the bortezomib positive control (Fig. 4E and F).Discussion
Ubiquitin chains are attractive targets for therapeutic intervention, given their central role in cellular activities and their frequent dysregulation in pathology.53 Here, we report the discovery of non-proteinogenic cyclic peptides able to specifically bind and modulate long (>3) K48-linked Ub chains: the tag for protein degradation by the proteasome. In contrast to previous efforts,11 the cyclic peptide library was built to include amino acids known to confer drug-like properties onto peptides, with the aim of generating Ub chain modulating molecules which are proteolytically stable and able to engage their intracellular target in vivo.Using the RaPID system methodology, a cyclic peptide library was constructed to include D-stereocenteres, N-methylated backbones, non-canonical side chains, while being depleted in canonical charged amino acids, This library was screened against K48Ub4, and two hit non-proteinogenic de novo cyclic peptides were chosen for characterization in detail: Ub4a and Ub4e. Both tightly bound K48Ub4, and showed significantly weaker binding for shorter chain K48Ub2. NMR studies revealed that Ub4a uses K48Ub3 as the main recognition element. Interestingly, while K48Ub4 contains two overlapping K48Ub3 units, 1–3 and 2–4, it is specifically the proximal trimer, 1–3, that engages the cyclic peptide. Chemical shift perturbations upon binding map the binding interactions to residues in and around the surface hydrophobic patch on the Ub units involved (Fig. 2E and F). Overlaying these CSPs on the “closed” K48Ub4 structure, which is the predominant conformation in the apo form, suggests that cyclic peptide binding involves intercalation into the Ub–Ub interface. The binding mode of Ub4a is broadly similar to that of Ub4ix, a cyclic peptide from the previous study, which also used the proximal K48Ub3.11 However, given the dissimilar chemical structures of Ub4a and Ub4ix (Fig. S17, ESI†), their interfaces with K48Ub3 are likely very different and await further characterization.
Natural product bioactive cyclic peptides are often highly non-proteinogenic: they are rich in non-canonical amino acids which confer protease resistance and passive membrane permeability. Moreover, these peptides tend to be small (<1000 Da), and lack amino acids that are charged at neutral pH. An archetype for this class is the orally available immunosuppressant cyclosporin.33 Previously characterized Ub chain binding cyclic peptides, like Ub4ix, were quite unlike cyclosporin: they were highly proteinogenic, larger and rich in charged amino acids at neutral pH (Fig. S17, ESI†). Promisingly, the cyclic peptides characterized here, Ub4a and Ub4e, are highly non-proteinogenic, containing two N-methyl, a D-stereochemistry and a non-canonical aliphatic side-chain amino acid. They are small, with just 11-amino acids in the macrocycle, making them undecamer cyclic peptides similar to cyclosporin. Lastly, similar to cyclosporin, these peptides lack formal charges, having zero, or close to zero net charge in the case of Ub4e, at neutral pH (Fig. S17, ESI†).
Importantly, the physicochemical make-up of the peptide library, the non-canonical amino acids added and the charged canonical amino acids removed, did not preclude the discovery of cyclic peptides with impressive binding strength and specificity for the challenging K48Ub4 target. Indeed, the non-canonical rich cyclic peptides described here have similar binding affinities to previously discovered, largely canonical, cyclic peptides.11 Yet the former have smaller macrocycles and had appreciably lower molecular mass, suggesting that the non-canonical amino acid collection is in fact superior at producing high-affinity, selective cyclic peptides.
We showed that Ub4a binding to Lys48-chain Ub chains prevents their recognition by deubiquitinates and the proteasome. Moreover, Ub4a can induce apoptosis in cancer cell lines in vitro, and attenuate the growth of tumor cells in vivo. These anti-cancer effects are likely due to disruption of Ub chain recognition processes which are part of the ubiquitin–proteasome system. Cancer cells are particularly dependent on the proteasome activity to remove certain proteins and evade programmed cell death, and this is the rationalization behind the anti-cancer effects observed for direct proteasome inhibitors such as bortezomib, carfilzomib, and ixazomib, which have been approved for the treatment of multiple myeloma.54 Indeed, throughout our in vitro and in vivo trials, Ub4a and direct proteasome inhibitors show broadly similar effects and comparable efficacy, the latter is striking considering Ub4a is a hit from a screen and the proteasome inhibitors are the product of extensive optimization.
While Ub4a showed similar effects to the direct proteasome inhibitors, it is important to emphasize that these cyclic peptides act through a different mechanism, targeting a different component of the ubiquitin–proteasome system. Ub-chain binders might therefore overcome the shortcomings of current proteasome inhibitors, the acquired resistance and adverse side-effects which decrease patients’ quality of life.55,56 Non-proteinogenic cyclic peptide targeting of Ub chains, therefore, have great potential for the development of new anti-cancer therapeutics.
Author contributions
J. M. R. designed and performed the cyclic peptide discovery, carried out SPR assays, analysed the data, and co-wrote the manuscript and the ESI.† M. N. assisted in the chemical synthesis of cyclic peptides, carried out in vitro and cellular assays and co-wrote the manuscript and the ESI.† B. L. synthesized isotope-labelled Ub chains, conducted the NMR studies and assisted with writing the paper. G. B. V. assisted in the synthesis of cyclic peptides. I. L. carried out the confocal microscopy assay and assisted with cellular studies. U. B. carried out the in vivo assays and I. V. assisted in the design of the in vivo assays. A. C. assisted in the design of the confocal microscopy assay and in vitro and cellular studies. D. F. designed and supervised the NMR studies, carried out data analysis and assisted with writing the manuscript and the ESI.† Hi. S. supervised the RaPID study and assisted in the writing of the paper. A. B. designed and supervised the entire project and the writing of the paper.Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. B. holds the Jordan and Irene Tark Academic Chair. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. [831783]). This work was also supported by Japan Agency for Medical Research and Development (AMED), Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research) under JP20am0101090 to H. S. J. M. R. was supported by Grants-in-aid for JSPS Fellows (P13766), a joint ANR-JST grant (ANR-14-JITC-2014-003 and JST-SICORP) and the Novo Nordisk Foundation (0054441).References
- A. Hershko and A. Ciechanover, Annu. Rev. Biochem., 1998, 67, 425–479 CrossRef CAS PubMed.
- C. E. Berndsen and C. Wolberger, Nat. Struct. Mol. Biol., 2014, 21, 301–307 CrossRef CAS PubMed.
- D. Morimoto, E. Walinda, H. Fukada, K. Sugase and M. Shirakawa, Sci. Rep., 2016, 6, 39453 CrossRef CAS PubMed.
- T. Hagai and Y. Levy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2001–2006 CrossRef CAS PubMed.
- D. Komander and M. Rape, Annu. Rev. Biochem., 2012, 81, 203–229 CrossRef CAS PubMed.
- R. Yau and M. Rape, Nat. Cell Biol., 2016, 18, 579–586 CrossRef CAS PubMed.
- J. S. Thrower, L. Hoffman, M. Rechsteiner and C. M. Pickart, EMBO J., 2000, 19, 94–102 CrossRef CAS PubMed.
- J. Adams, Cancer Cell, 2004, 5, 417–421 CrossRef CAS PubMed.
- X. Huang and V. M. Dixit, Cell Res., 2016, 26, 484–498 CrossRef CAS PubMed.
- S. Lu and J. Wang, Biomark Res., 2013, 1, 13 CrossRef PubMed.
- M. Nawatha, J. M. Rogers, S. M. Bonn, I. Livneh, B. Lemma, S. M. Mali, G. B. Vamisetti, H. Sun, B. Bercovich, Y. Huang, A. Ciechanover, D. Fushman, H. Suga and A. Brik, Nat. Chem., 2019, 11, 644–652 CrossRef CAS PubMed.
- T. Nguyen, M. Ho, K. Kim, S. I. Yun, P. Mizar, J. W. Easton, S. S. Lee and K. K. Kim, Molecules, 2019, 24, 1073 CrossRef CAS PubMed.
- T. Nguyen, M. Ho, A. Ghosh, T. Kim, S. I. Yun, S. S. Lee and K. K. Kim, Biochem. Biophys. Res. Commun., 2016, 479, 33–39 CrossRef CAS.
- M. A. Nakasone, T. A. Lewis, O. Walker, A. Thakur, W. Mansour, C. A. Castaneda, J. L. Goeckeler-Fried, F. Parlati, T. F. Chou, O. Hayat, D. Zhang, C. M. Camara, S. M. Bonn, U. K. Nowicka, S. Krueger, M. H. Glickman, J. L. Brodsky, R. J. Deshaies and D. Fushman, Structure, 2017, 25(1839–1855), e1811 Search PubMed.
- R. Verma, N. R. Peters, M. D'Onofrio, G. P. Tochtrop, K. M. Sakamoto, R. Varadan, M. Zhang, P. Coffino, D. Fushman, R. J. Deshaies and R. W. King, Science, 2004, 306, 117–120 CrossRef CAS PubMed.
- Y. Ye, G. Blaser, M. H. Horrocks, M. J. Ruedas-Rama, S. Ibrahim, A. A. Zhukov, A. Orte, D. Klenerman, S. E. Jackson and D. Komander, Nature, 2012, 492, 266–270 CrossRef CAS PubMed.
- C. A. Castaneda, A. Chaturvedi, C. M. Camara, J. E. Curtis, S. Krueger and D. Fushman, Phys. Chem. Chem. Phys., 2016, 18, 5771–5788 RSC.
- Y. Lu, B. H. Lee, R. W. King, D. Finley and M. W. Kirschner, Science, 2015, 348, 1250834 CrossRef PubMed.
- I. Livneh, Y. Kravtsova-Ivantsiv, O. Braten, Y. T. Kwon and A. Ciechanover, BioEssays, 2017, 39, 1700027 CrossRef PubMed.
- K. S. Ajish Kumar, M. Haj-Yahya, D. Olschewski, H. A. Lashuel and A. Brik, Angew. Chem., Int. Ed., 2009, 48, 8090–8094 CrossRef CAS PubMed.
- S. N. Bavikar, L. Spasser, M. Haj-Yahya, S. V. Karthikeyan, T. Moyal, K. S. Kumar and A. Brik, Angew. Chem., Int. Ed., 2012, 51, 758–763 CrossRef CAS PubMed.
- Y. Yamagishi, I. Shoji, S. Miyagawa, T. Kawakami, T. Katoh, Y. Goto and H. Suga, Chem. Biol., 2011, 18, 1562–1570 CrossRef CAS PubMed.
- C. R. Pye, W. M. Hewitt, J. Schwochert, T. D. Haddad, C. E. Townsend, L. Etienne, Y. Lao, C. Limberakis, A. Furukawa, A. M. Mathiowetz, D. A. Price, S. Liras and R. S. Lokey, J. Med. Chem., 2017, 60, 1665–1672 CrossRef CAS PubMed.
- P. Matsson and J. Kihlberg, J. Med. Chem., 2017, 60, 1662–1664 CrossRef CAS PubMed.
- B. C. Doak, B. Over, F. Giordanetto and J. Kihlberg, Chem. Biol., 2014, 21, 1115–1142 CrossRef CAS PubMed.
- B. Over, P. Matsson, C. Tyrchan, P. Artursson, B. C. Doak, M. A. Foley, C. Hilgendorf, S. E. Johnston, M. D. T. Lee, R. J. Lewis, P. McCarren, G. Muncipinto, U. Norinder, M. W. Perry, J. R. Duvall and J. Kihlberg, Nat. Chem. Biol., 2016, 12, 1065–1074 CrossRef CAS PubMed.
- M. R. Naylor, A. T. Bockus, M. J. Blanco and R. S. Lokey, Curr. Opin. Chem. Biol., 2017, 38, 141–147 CrossRef CAS PubMed.
- J. Chatterjee, F. Rechenmacher and H. Kessler, Angew. Chem., Int. Ed., 2013, 52, 254–269 CrossRef CAS PubMed.
- T. Passioura, W. Liu, D. Dunkelmann, T. Higuchi and H. Suga, J. Am. Chem. Soc., 2018, 140, 11551–11555 CrossRef CAS PubMed.
- Y. Goto, A. Ohta, Y. Sako, Y. Yamagishi, H. Murakami and H. Suga, ACS Chem. Biol., 2008, 3, 120–129 CrossRef CAS.
- Y. Shimizu, A. Inoue, Y. Tomari, T. Suzuki, T. Yokogawa, K. Nishikawa and T. Ueda, Nat. Biotechnol., 2001, 19, 751–755 CrossRef CAS PubMed.
- Y. Goto, T. Katoh and H. Suga, Nat. Protoc., 2011, 6, 779–790 CrossRef CAS PubMed.
- C. L. Ahlbach, K. W. Lexa, A. T. Bockus, V. Chen, P. Crews, M. P. Jacobson and R. S. Lokey, Future Med. Chem., 2015, 7, 2121–2130 CrossRef CAS PubMed.
- T. Katoh, Y. Iwane and H. Suga, Nucleic Acids Res., 2017, 45, 12601–12610 CrossRef CAS PubMed.
- T. Katoh, I. Wohlgemuth, M. Nagano, M. V. Rodnina and H. Suga, Nat. Commun., 2016, 7, 11657 CrossRef CAS PubMed.
- R. W. Roberts and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12297–12302 CrossRef CAS PubMed.
- V. Ranjani, M. Assfalg and D. Fushman, Methods Enzymol., 2005, 399, 177–192 Search PubMed.
- R. Varadan, O. Walker, C. Pickart and D. Fushman, J. Mol. Biol., 2002, 324, 637–647 CrossRef CAS.
- M. J. Eddins, R. Varadan, D. Fushman, C. M. Pickart and C. Wolberger, J. Mol. Biol., 2007, 367, 204–211 CrossRef CAS PubMed.
- Y. E. Ryabov and D. Fushman, J. Am. Chem. Soc., 2007, 129, 3315–3327 CrossRef CAS PubMed.
- T. E. Mevissen, M. K. Hospenthal, P. P. Geurink, P. R. Elliott, M. Akutsu, N. Arnaudo, R. Ekkebus, Y. Kulathu, T. Wauer, F. El Oualid, S. M. Freund, H. Ovaa and D. Komander, Cell, 2013, 154, 169–184 CrossRef CAS PubMed.
- A. Bremm, S. M. Freund and D. Komander, Nat. Struct. Mol. Biol., 2010, 17, 939–947 CrossRef CAS PubMed.
- H. Sun, S. M. Mali, S. K. Singh, R. Meledin, A. Brik, Y. T. Kwon, Y. Kravtsova-Ivantsiv, B. Bercovich and A. Ciechanover, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7805–7812 CrossRef CAS PubMed.
- A. L. Goldberg, J. Cell Biol., 2012, 199, 583–588 CrossRef CAS PubMed.
- W. Mansour, M. A. Nakasone, M. von Delbruck, Z. Yu, D. Krutauz, N. Reis, O. Kleifeld, T. Sommer, D. Fushman and M. H. Glickman, J. Biol. Chem., 2015, 290, 4688–4704 CrossRef CAS PubMed.
- R. J. Deshaies, BMC Biol., 2014, 12, 94 CrossRef.
- L. J. Crawford, B. Walker and A. E. Irvine, J. Cell Commun. Signaling, 2011, 5, 101–110 CrossRef PubMed.
- E. E. Manasanch and R. Z. Orlowski, Nat. Rev. Clin. Oncol., 2017, 14, 417–433 CrossRef CAS PubMed.
- Y. Huang, M. Nawatha, I. Livneh, J. M. Rogers, H. Sun, S. K. Singh, A. Ciechanover, A. Brik and H. Suga, Chemistry, 2020, 26, 8022–8027 CrossRef CAS.
- U. Barash, M. Lapidot, Y. Zohar, C. Loomis, A. Moreira, S. Feld, C. Goparaju, H. Yang, E. Hammond, G. Zhang, J. P. Li, N. Ilan, A. Nagler, H. I. Pass and I. Vlodavsky, J. Natl. Cancer Inst., 2018, 110, 1102–1114 CrossRef PubMed.
- M. Weissmann, G. Arvatz, N. Horowitz, S. Feld, I. Naroditsky, Y. Zhang, M. Ng, E. Hammond, E. Nevo, I. Vlodavsky and N. Ilan, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 704–709 CrossRef CAS PubMed.
- J. Adams, V. J. Palombella, E. A. Sausville, J. Johnson, A. Destree, D. D. Lazarus, J. Maas, C. S. Pien, S. Prakash and P. J. Elliott, Cancer Res., 1999, 59, 2615–2622 CAS.
- D. Vilchez, I. Saez and A. Dillin, Nat. Commun., 2014, 5, 5659 CrossRef CAS PubMed.
- L. D. Fricker, Annu. Rev. Pharmacol. Toxicol., 2020, 60, 457–476 CrossRef CAS PubMed.
- G. S. Kaplan, C. C. Torcun, T. Grune, N. K. Ozer and B. Karademir, Free Radical Biol. Med., 2017, 103, 1–13 CrossRef CAS PubMed.
- F. S. Liu, Taiwan. J. Obstet. Gynecol., 2009, 48, 239–244 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The data that support the findings of this study are available from the corresponding authors upon reasonable request. See DOI: 10.1039/d0cb00179a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |