
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Strategies to expand peptide functionality through hybridisation with a small molecule component
Yuteng
Wu†
 ab,
Jack
Williams†
ab,
Ewen D. D.
Calder†
ab and
Louise J.
Walport
*ab
ab,
Jack
Williams†
ab,
Ewen D. D.
Calder†
ab and
Louise J.
Walport
*ab
aProtein-Protein Interaction Laboratory, The Francis Crick Institute, London, UK. E-mail: Louise.walport@crick.ac.uk
bDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College London, London, UK
First published on 8th December 2020
Abstract
Combining different compound classes gives molecular hybrids that can offer access to novel chemical space and unique properties. Peptides provide ideal starting points for such molecular hybrids, which can be easily modified with a variety of molecular entities. The addition of small molecules can improve the potency, stability and cell permeability of therapeutically relevant peptides. Furthermore, they are often applied to create peptide-based tools in chemical biology. In this review, we discuss general methods that allow the discovery of this compound class and highlight key examples of peptide–small molecule hybrids categorised by the application and function of the small molecule entity.
 Yuteng Wu | Yuteng Wu obtained his PhD (2016) in chemical biology from the University of Cambridge with Prof. David Spring. He then carried out postdoctoral research at École Polytechnique Fédérale de Lausanne with Prof. Christian Heinis. In 2019, he joined the group of Dr Louise Walport at The Francis Crick Institute and Imperial College London, focusing on developing new techniques to expand the functionality of RaPID encoded peptides. |
 Jack Williams | Jack Williams obtained his MSci in Chemistry (2019) from Imperial College London where he completed his MSci project abroad at École Polytechnique Fédérale de Lausanne under the supervision of Prof. Christian Heinis. He then started his PhD at the Francis Crick Institute on a joint project between the groups of Dr Louise Walport and Prof. Ed Tate, focusing on using chemical and structural biology to elucidate the function of protein arginine deiminases. |
 Ewen D. D. Calder | Ewen Calder received his MChem in Chemistry (2011) from the University of St Andrews before obtaining his PhD (2015) from the University of Glasgow with Prof. Andrew Sutherland where he worked on methodology for the synthesis of heterocycles and natural products. He subsequently carried out postdoctoral research at Oxford University with Prof. Stuart Conway on hypoxia-activated pro-drugs before joining the group of Dr Louise Walport in 2019 at The Francis Crick Institute and Imperial College London. His projects focus on developing new methods to expand the use of the RaPID system. |
 Louise J. Walport | Louise Walport obtained her doctorate under the supervision of Prof. Chris Schofield and Prof. Christina Redfield focussing on mechanistic studies of histone demethylases. Following further postdoctoral work in Oxford, she was awarded a Marie Skłodowska-Curie Global Fellowship to work in the group of Hiroaki Suga at the University of Tokyo, where her interest in cyclic peptides arose. Since 2018, she is a lecturer in the Chemistry Department at Imperial College London and a group leader at the Francis Crick Institute. She continues to be interested in enzyme-catalysed post-translational modifications and developing new approaches to probe these with cyclic peptide-based tools. |
Introduction
Peptides are a promising class of drug molecule that are also particularly useful as tools in chemical biology. With a size range between small molecules and biologics, they occupy a Goldilocks region, that when optimised, have the potential to adopt some of the favourable properties from each: the high potency and selectivity of biologics, and the higher bioavailability and cellular permeability associated with smaller molecules.1,2 This unique place in chemical space, makes them particularly applicable to modulating targets previously thought of as undruggable, such as protein–protein interactions.3–5Despite this great potential, achieving the optimal balance of favourable properties is challenging, and as yet their development and use in the clinic or in vivo systems has been limited.2,6 Smaller peptides, which may have favourable pharmacokinetic properties, often have inferior binding affinity to biologics. By contrast, larger peptides with enhanced potency, tend to have reduced stability and cell permeability as a result of their size, limiting them to extracellular targets.
Strategies to optimise the drug-like qualities of peptides are continually being developed.7–10 Their inherent modularity means their structure and sequence can be easily tweaked to enhance their efficacy. Conjugation to natural biomolecules such as fatty acids, steroids or sugars is a well-established technique to modulate peptide properties.11–16 Small molecule components have also been incorporated into peptides to generate ‘peptide–small molecule hybrids’. Depending on the intended function, conjugation of the small molecule motif can occur directly on an amino acid side chain, through the amide backbone or, for cyclic peptides, through the cyclisation linkage itself.17–19 These additions modulate the properties of the parent peptide, producing hybrids that show significant potential in addressing the limitations of peptides as therapeutic agents or tools in chemical biology.
The aim of this review is to illustrate the potential of peptide–small molecule hybrids in both the development of pharmaceuticals and as tools for basic research. Following an overview of screening strategies by which peptide–small molecule hybrids can be identified, we detail prominent hybrid examples classified by the function of the small molecule component. First, we describe examples where the hybrid exhibits enhanced binding affinity attributed to the incorporation of the small molecule component. In the second section, we discuss the use of small molecule tags to improve the pharmacokinetics of peptides through prolonging their in vivo half-life or improving their cell permeability. Finally, we highlight examples where the hybrids have applications as tools in chemical biology, primarily as switchable molecules and photoaffinity probes. There are various standard peptide–small molecule hybrids that are not covered in this review. In particular, the well-established use of peptides to improve the efficacy of a known small molecule or drug, in the form of a peptide–drug conjugate is not covered and the interested reader is directed to one of the many excellent reviews on this topic.20–24
1. Discovery strategies
When developing hybrid molecules, the conceptually simplest approach is to begin with a small molecule or peptide which is known to bind to the target protein. These validated ligands can provide the starting point for the rational design of peptide hybrid molecules with enhanced properties, often with assistance from structural data. At present this approach is the most widely used, and examples of this approach are discussed below. However, validated binders and high-resolution crystal structures are not always available. A more generalisable approach requiring no prior knowledge of the target is therefore to screen vast small molecule–peptide hybrid libraries. This allows the de novo discovery of lead compounds for almost any target of choice.An established method for generating diverse hybrid libraries is the split-and-pool technique, where libraries of up to 107 molecules can be produced through combinatorial chemistry.25 As these libraries are constructed synthetically, highly diverse building blocks can be incorporated into library members. In a notable example of this approach, Liu and co-workers reported a ring-closing metathesis-based strategy to generate a library of 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rapamycin-like hybrid peptide macrocycles.26 Rapamycin and FK506 are macrocyclic natural products which share an FK506-binding protein (FKBP) binding motif as well as an effector domain which binds mTOR (Rapamycin) or calcineurin (FK506). By generating ternary complexes between FKBP12 and their target protein, Rapamycin and FK506 allosterically block substrates from reaching the active site of mTOR and calcineurin respectively. In a successful attempt to hijack this interesting mode of action, Liu and co-workers individually fused two optimised FKBP-binding domains (FKBD10, FKBD11, Fig. 1a) with a combinatorial library of solid-phase synthesised tetrapeptides intended to replace the effector domain and so alter their selectivity. Screening of the library in human cells identified a potent inhibitor (A15-34-8, IC50 = 426 nM) of human equilibrative nucleoside transporter 1 (hENT1). Further optimisation of the tetrapeptide sequence in the hybrid improved the potency by 85.2-fold.
000 rapamycin-like hybrid peptide macrocycles.26 Rapamycin and FK506 are macrocyclic natural products which share an FK506-binding protein (FKBP) binding motif as well as an effector domain which binds mTOR (Rapamycin) or calcineurin (FK506). By generating ternary complexes between FKBP12 and their target protein, Rapamycin and FK506 allosterically block substrates from reaching the active site of mTOR and calcineurin respectively. In a successful attempt to hijack this interesting mode of action, Liu and co-workers individually fused two optimised FKBP-binding domains (FKBD10, FKBD11, Fig. 1a) with a combinatorial library of solid-phase synthesised tetrapeptides intended to replace the effector domain and so alter their selectivity. Screening of the library in human cells identified a potent inhibitor (A15-34-8, IC50 = 426 nM) of human equilibrative nucleoside transporter 1 (hENT1). Further optimisation of the tetrapeptide sequence in the hybrid improved the potency by 85.2-fold.
 | ||
| Fig. 1 Strategies for constructing vast small molecule–peptide hybrid libraries. (a) Split-pool library of Rapamycin-like hybrid macrocycles comprised of an optimised FKBP-binding domain fused with a tetrapeptide effector domain. For A15-34-8: X = NHCOCH2, R1 = Phenylalanine, R2 = N-methyl-D-Phenylalanine, R3 = Proline, R4 = N-methyl-Leucine. (b) DNA encoded library bearing a structurally-defined cyclic peptide scaffold with three encoded small molecule diversification sites. (c) Phage-displayed peptide library functionalised with mannose, biotin or sulphonamide each encoded by a silent barcode. [X]4 = randomised (NNK)4 library. | ||
DNA-encoded libraries are an extension of the split-and-pool approach which can generate and screen libraries of up to 1012 compounds.27 In a DNA-encoded library, each combinatorial element is assigned a unique nucleotide barcode which is attached in tandem with each synthetic step. After affinity panning of the library against an immobilised protein target, molecules that bind can be identified by sequencing of the attached unique DNA barcode. This allows for pooled screening of much larger libraries than can be achieved with traditional tag-free combinatorial libraries, where library members must be screened individually. In an impressive application of this technique Scheuermann, Neri and co-workers reported a strategy to display multiple diverse chemical elements, each encoded by a DNA barcode, on a structurally-defined macrocyclic scaffold which adopts an antiparallel beta-sheet conformation (Fig. 1b).28 Various chemical elements were incorporated through three diversity sites resulting in the construction of an impressive 35393112 compound library. Subsequent selections with this library yielded specific binders against eight proteins, including carbonic anhydrase IX, calmodulin and prostate-specific antigen. By exchanging the DNA-barcode for fluorescein and modifying the scaffold to add a fourth functionalised diversity site, the identified hybrid peptide binders were converted to chemical probes. For example, a phenyl azide moiety was fused to a calmodulin binder identified from the library to yield a compound that preferentially photo-crosslinked to calmodulin in the presence of human serum albumin.
By contrast to the above methods, phage display and mRNA display use DNA to directly produce peptide libraries by translation, removing the need for numerous synthetic steps. In each case, the translated peptides remain associated with their encoding DNA, allowing the generation of libraries that are orders of magnitude larger than those produced by DNA-encoded synthesis.1,29,30
In phage display libraries, small molecule components can easily be incorporated as the cyclising linker or as a post-translational modification. This allows for the construction and screening of enormous hybrid libraries.31
One of the first applications producing peptide hybrids using phage display was the pioneering work of Christian Heinis and co-workers in developing bicyclic peptide hybrids. By incorporating three cysteine residues into the phage displayed peptides, linear peptides were bi-cyclised with a set of tri-electrophilic linkers for screening against targets of interest.31 Varying ring size,32 linker,33 and number of cyclising cysteines34 can enhance diversity in the resulting peptide macrocycles. Such libraries have been used successfully to identify binders of a wide range of proteins including plasma kallikrein,31,35 urokinase,36 β-catenin,37 and coagulation factor XII.38 Additionally, these libraries can be used to develop chemical probes with the incorporation of a photoswitching motif, as discussed in Section 2.3. Recently, the approach has been modified to introduce protease pressure during selection, with the aim of identifying therapeutic peptides that are resistant to gastrointestinal proteases after oral administration.39 The strategy was successful in identifying a peptide inhibitor of coagulation Factor XIa and an antagonist for the interleukin-23 receptor, which were proteolytically stable in the gastrointestinal tract and more suitable for oral administration.
Subsequently, Derda and co-workers expanded the phage display toolbox by using phage libraries incorporating “silent barcodes” (unique combinations of redundant codons) to identify peptides that had been post-translationally modified with small molecule fragments.40 The barcodes enable mixing of libraries with different fragment additions which can be deconvoluted following DNA sequencing. In a proof-of-concept study, phage-displayed peptide libraries were individually functionalised with one of three small molecule modifications (mannose, biotin, or sulphonamide, Fig. 1c) and the libraries combined. Hits were identified for concanavalin A, streptavidin, or carbonic anhydrase (known binders of these functionalities) through affinity panning with the pooled libraries. This approach has since been used with particular effect to target carbohydrate-binding proteins.41,42 In a recent extension of this approach, the post-translational incorporation of a 1,3-diketone greatly expanded the functionality that could be incorporated in the library.43 Having installed the diketone on their “silent barcode” phage-displayed libraries, Derda and co-workers then diversified by using a Knorr pyrazole synthesis with a wide range of hydrazine containing reagents. Using this methodology, they were able to incorporate fluorophores, lipids, PEG chains, metal chelators and groups known to bind certain proteins. Specifically, by installing a sulphonamide, known to bind carbonic anhydrase IX, they use their method to find a nanomolar potency peptide–small molecule hybrid. This “silent barcode” approach has the potential to be applied more widely in both phage and mRNA display technologies and could allow the screening of much larger libraries with a wide variety of post-translational modifications.
Another prominent screening method for the development of peptide–small molecule hybrids is mRNA display and in particular the random non-standard peptide integrated discovery (RaPID) system. The RaPID system can incorporate a wide range of non-canonical amino acids at multiple sites in the final peptide library with relative ease. Successful examples include N-alkyl, β- or D-amino acids, as well as more exotic hybrid-like amino acids such as carboranylalanine.44–46
Such an approach also allows rational incorporation of designed small molecule warheads, to customise libraries for a specific target. In an early example, Suga and co-workers produced RaPID libraries containing an ε-N-trifluoroacetyl group, a weak mechanism-based inhibitor of SIRT2, to search for improved inhibitors.47 The study resulted in the identification of two potent inhibitors of SIRT2 (KD and IC50 in the single digit nanomolar region), which importantly displayed impressive isoform selectivity for SIRT2 over other isoforms SIRT1 and SIRT3. In a more recent example, Payne and co-workers employed a similar strategy to screen for cyclic peptide binders for chemokine CCL11, with peptides possessing key sulfo-Tyrosine (sTyr) residues.48 Four high affinity binders were identified (Kd < 30 nM) that inhibited CCL11 activation of its cognate receptor CCR3, with the sulfated residue found to be vital for achieving inhibition.
The use of foldamers to mimic peptide secondary structures is a well-established field,49–51 but only recently have the two classes of molecule been combined to form hybrid species. In a prominent example, Gellman and co-workers designed peptides that contain β-amino acid residues in addition to standard α-residues. When optimised, the replacement of α with β-amino acids can lead to improvements in proteolytic stability as well as target selectivity.52–56 In another example, Huc, Suga and co-workers demonstrated the ribosomal synthesis of helical aromatic foldamer–peptide hybrid molecules, with the large foldamer component, based on quinoline and pyridine monomers, incorporated at the peptide N-terminus.57 In follow up work, the methodology was extended to incorporate foldamers within and at the C-terminus of the peptide sequence.58 Though not yet validated in a protein selection, incorporation of such large non-peptidic entities may give rise to desirable properties such as greater water solubility, protease resistance and cell permeability.
As the field of peptide–small molecule hybrids grows, the ability to develop these molecules without relying on known peptide binding sequences will only increase in importance. The expansion of techniques that allow the construction and screening of vast (106 to 1015 members) libraries of diverse hybrid molecules is already beginning to show great promise in the delivery of this class of compound. Further expansion of these approaches will be key to the development of this field.
2. Functionalities imparted by small molecule component
The addition of small molecules can impart a variety of different functions to the resulting hybrid. In the context of enhancing the drug-like properties of peptides and their functionality as chemical tools, we have chosen to focus on three main areas: (1) enhanced target binding, (2) improved pharmacokinetics and (3) switchable activity. In the following section, we discuss key examples of peptide–small molecule hybrids categorised by the application and function of the small molecule entity.2.1 Binding
A major application of peptide–small molecule hybrids is to improve upon the binding affinity achieved with peptides alone. Though peptides often display greater binding affinities for their target than smaller molecules (due in part to their larger size), it is rare they can reach the potency and specificity of biologics. Conjugation of a small molecule to a validated binding peptide (linear or cyclic) can enhance affinity through additional interactions provided by the small molecule component. Alternatively, the incorporation of reactive moieties can lead to improved potency through covalent bonding with the target. Finally, pre-organising the peptide via a small molecule-mediated macrocyclisation can enhance ligand affinity by reducing the entropic penalty upon target binding. In these cases, the cyclisation linkage can also participate in binding by forming direct interactions with the target.In a notable example, where improved affinity was achieved by expanding the binding interface of a validated peptide, Ottmann and co-workers rationally developed a hybrid inhibitor (3b, Fig. 2a) of the 14-3-3/Tau interaction.59 Comparison of crystal structures of 14-3-3 bound to an inhibitory Tau epitope or a small molecule natural product, Fusicoccin A (FC), led to the identification of a region of overlap between the two binding interfaces. Based on this information, peptide–small molecule derivatives were designed. By starting from the Tau pS213 phosphopeptide epitope and attempting to also probe the proximal FC binding pocket, the interaction interface was extended. In particular, addition of a C-terminal benzhydryl pyrrolidine group significantly improved 14-3-3 binding affinity (∼225-fold) by addressing the hydrophobic pocket targeted by the A-ring of FC. The work was further developed by varying the amino acid adjacent to the small molecule and further exploring substituents on the aromatic rings, which resulted in the discovery of a library of low micromolar hybrid inhibitors of the 14-3-3/Tau interaction.60
 | ||
| Fig. 2 Small molecule fragments for generating high affinity hybrids. (a) Benzhydryl pyrrolidine group for targeting the fusicoccin A binding pocket in 14-3-3. pS = phosphoserine (b) N-aryl pyrrole moiety for interacting with the hydrophobic pocket of HIV-1 gp41. P26 = NNYTSLIHSLIEESQNQQEKNEQELL. βA = β-Alanine (c) Nanomolar thrombin inhibitor with an exocyclic hydroxymethyl-benzyl group which induced a new hydrophobic cavity in the protein binding pocket. βHPro = L-β-Homoproline. (d) Selective MC5R binding hybrid (K1 major) generated by stereoselective 1,3-dipolar cycloaddition. The diastereoisomer, K1 minor, was unable to bind to the target. NMePhe = N-methyl-L-phenylalanine. f = D-phenylalanine. In each case the small molecule component is highlighted in red. | ||
Apart from engaging in additional interactions with the target, small molecules are often explored to replace segments of a binding peptide. Ideally, the resulting hybrid would retain the high potency of the parent peptide whilst simultaneously imparting other favourable properties, such as improved stability. For example, Liu and co-workers investigated N-aryl pyrroles for developing potent HIV-1 gp41 binding hybrids with improved stability.61 Hybrid development started from a peptide sequence known as C34, a nanomolar potency binder, containing a short region which interacts with a deep, hydrophobic pocket on HIV-1 gp41. A truncated version of C34 (P26, 26-AAs) lacking this region was designed and coupled with a range of N-aryl pyrrole moieties. The N-substituted pyrroles were chosen based on a series of known low micromolar HIV-1 gp41 fusion inhibitors. Whilst the best hybrid (Aoc-βAla-P26, IC50 = 14.9 nM, Fig. 2b) showed around a 10-fold loss in potency when compared to the full length C34 peptide, it had a 170-fold gain when compared to the truncated peptide. Importantly, Aoc-βAla-P26 also displayed better stability in a proteinase K digestion assay compared to C34. This highlights the ability of hybrids to combine the advantages and minimise the respective shortcomings of the two classes of compounds: loss of potency from peptide truncation was restored by conjugation of small molecules, simultaneously improving the stability of the resulting compounds.
Covalent cross-linking to the target protein represents a distinctive approach for enhancing the binding of peptides. Current strategies involve the rational incorporation of reactive motifs into validated binding sequences. For example, Hoppmann and Wang developed covalent inhibitors of MDM4 by addition of an exocyclic aryl sulfonyl fluoride group.62 The reactive group was attached to the side chain of an unnatural amino acid and inserted within a known MDM4 binding sequence. The peptide–small molecule hybrid was able to bind MDM4 covalently through proximity-enabled bioreactivity, resulting in a 10-fold improvement in p53/MDM4 inhibition over the unmodified peptide sequence. Similarly, Spring and co-workers utilised an alternative electrophilic warhead, a sulfotetrafluorophenyl (STP) ester group, for cross-linking to MDM2.63 Successful cross-linking relies on an appropriately positioned, suitably reactive amino acid. Fortunately, there are a wide range of options beyond electrophilic warheads including photoreactive groups (see Section 2.3), making this technique widely applicable. Screening methods to incorporate the reactive motifs in the starting libraries would extend this even further.
Finally, macrocyclisation is an established methodology for improving the binding affinity of peptides. Whilst this can be achieved directly through forming a head-to-tail peptide bond or a side-chain mediated disulfide bond, small molecule moieties are often used to achieve this. A notable example of such a methodology was reported by Fasan and co-workers who developed macrocyclic organo-peptide hybrids (MOrPHs).64 The first step involves the copper-catalyzed azide–alkyne cycloaddition (CuAAC) of an azido bearing synthetic precursor with an alkyne on the backbone of an intein-fused polypeptide. In the second step, the thioester bond at the intein junction is then intercepted by a hydrazide moiety on the synthetic precursor to simultaneously form the cyclic peptide and expel the intein protein. Variations of the technique include replacing the synthetic precursor with an oxyamino/amino-thiol trifunctional alternative,65 allowing catalyst-free cyclisation, and the incorporation of two cysteines in the polypeptide backbone to form bicyclic macrocycles through a disulfide bridge.66 In an application of the MOrPH strategy, HDM2/X binding peptides were constrained into their alpha-helical conformation by the small molecule synthetic precursor.67 The resulting macrocycles displayed inhibitory activity of the p53/HDM2 interaction, however the small molecule motif did not directly participate in binding. This cyclisation strategy has recently been integrated with phage display with the displayed macrocyclic libraries successfully applied to identify a high-affinity binder for streptavidin (KD = 20 nM), and potent inhibitors of Keap 1 (KD = 40 nM) and Sonic Hedgehog (KD = 550 nM).68
Of the large variety of macrocyclisation chemistries currently available, the majority focus only on cross-links that constrain the peptide fragment of the molecule.17–19,69 For example, constraining the peptide through stapling can promote secondary structure formation in the form of alpha helices.18,70 However, in some cases, the cyclisation linkage can also contribute directly to target engagement, either by serendipity or design. In a notable example reported by Sawyer and co-workers,71 the hydrocarbon linkage in an alpha helical MDM2/MDMX dual inhibitor was itself found to form hydrophobic interactions on the surface of the target protein. The optimised peptide ATSP-7041 bound both MDM2 (Ki = 0.9 nM) and MDMX (Ki = 6.8 nM) with nanomolar affinities. Similar linkages have since been designed to participate in binding for other alpha helical peptides,72,73 as well as irregularly structured peptides.74 For example, apart from the structurally minimal hydrocarbon motif, Spring and co-workers demonstrated a relatively large and complex bis(triazolyl) linkage can also engage in favourable hydrophobic binding interactions with MDM2, analogous to that of the hydrocarbon cross-link.75
Incorporation of structurally diverse or complex linkages can generate peptide libraries with unique backbone structures, that may contribute to enhanced binding. Heinis and co-workers recently reported an efficient macrocyclisation reaction based on thiol-to-amine ligations using a set of diverse bis-electrophiles.76 A library of 8988 macrocycles was constructed by reacting 1284 unique tetrapeptides with seven different linkers. Subsequent screening against thrombin led to a selective nanomolar inhibitor (Ki = 42 nM). Interestingly, the most potent compound (P2, Fig. 2c) had undergone a second addition of the linker, connecting an exocyclic N-(2-(hydroxymethyl)benzyl) substituent to the macrocycle via the secondary amine in the backbone. A structure–activity relationship analysis showed the unforeseen hydroxymethyl-benzyl group was essential for binding, such that a shift of the hydroxymethyl group from the two to the four-position resulted in a ∼100-fold drop in potency. X-ray crystallography studies revealed a new hydrophobic cavity was created in the protein binding pocket to accommodate the hydroxymethyl-benzyl group. In a follow up study, it was found that the simultaneous substitution of L-β-homoproline with D-β-homoproline, and hydroxymethyl-benzyl with furfuryl resulted in a 5.5-fold improvement in potency over P2.77
Many cyclisation chemistries are currently available to add functionalisation in a diversity orientated fashion.78–82 In a recent example, Waldmann and co-workers developed a strategy to add natural product inspired small molecule structures via the cyclisation linkage.83 The structurally complex fragments were incorporated into macrocyclic hot loop-derived peptides by imine formation and subsequent stereoselective 1,3-dipolar cycloaddition using nine unique dipolarophiles. Screening against two protein–protein interactions demonstrated that the absolute configuration of the small molecule addition was essential in achieving optimal potency. In the first example, natural product like structures were cyclised with peptides derived from the iNOS hot loop to yield a series of nanomolar binders of the hSPSB2 adaptor protein. The second example utilised ARGP RFF hot spot sequences to develop selective ligands and agonists for the melanocortin 5 receptor (MC5R). Importantly, a major diastereomer (K1 major, Fig. 2d) was found to bind to MC5R (IC50 = 4.1 μM) with 7-, 5-, and 13-fold selectivity over MC1R, MC3R and MC4R respectively, whereas the minor diastereomer (K1 minor, Fig. 2d) did not display binding to any of the receptors. Whilst the current work focused solely on using the small molecule scaffolds to conformationally constrain the peptide part of the macrocycle, such diverse natural product-like linkages show great potential in also contributing favourable binding interactions themselves.
Many of the described peptide–small molecule hybrids successfully improve target binding affinity. In the few cases where binding affinity is only retained when comparing the hybrid to the peptide starting point, the hybrid holds other advantages such as superior stability by reducing the susceptibility to proteases. Overall, they demonstrate the potential of such hybrids in tailoring the binding affinity of therapeutic peptides. Optimal hybrid structure can be determined through rational design when a crystal structure of a peptide starting point is readily available. However, we have also highlighted a number of examples where serendipitous features of hybrid molecules have been identified, emphasising that inclusion of such hybrids in the initial screening strategy is likely to be an effective way of identifying high affinity binders.
2.2 Pharmacokinetics
The earliest reported small molecule-based strategies involved conjugation of naphthalene acylsulfonamide or phosphate ester-based tags to improve in vivo half-lives of peptide anticoagulants.95,96 More recent strategies have mostly focussed on improving the in vivo pharmacokinetic properties of Glucagon like peptide-1 (GLP-1) analogues, such as extendin-4, which are used in the treatment of type 2 diabetes.
Towards this goal, Chen and co-workers have developed two tag variants that are based on a maleimide-modified version of Evans blue dye (MEB), a small molecule with high affinity for serum albumin.97 MEB was conjugated to Cys40 of extendin-4 to give Abextide, which displayed superior bio-distribution over extendin-4 with both intravenous and subcutaneous injection. The in vivo half-life of Abextide (t1/2 = 36.28 ± 7.01 h) was 7-fold longer than that of extendin-4 (t1/2 = 5.16 ± 5.23 h), with concomitant increase in hypoglycaemic duration in Abextide-treated type 2 diabetic mice compared with extendin-4-treated mice (36 h and 10 h respectively). Despite the significantly improved distribution, pharmacokinetic and anti-diabetic properties of Abextide, the thiol–maleimide tag is susceptible to hydrolysis, prompting Liu et al. to develop a more stable MEB tag (MEB-C3, Fig. 3a).98 Conjugation of MEB-C3 to extendin-4 gave abextide II, which displayed superior stability to hydrolysis over Abextide I. Not only was Abextide II an improvement on extendin-4, but also Albiglutide, a commercial extendin-4-albumin fusion, over which it maintains a longer hypoglycaemic state and better blood glucose control.
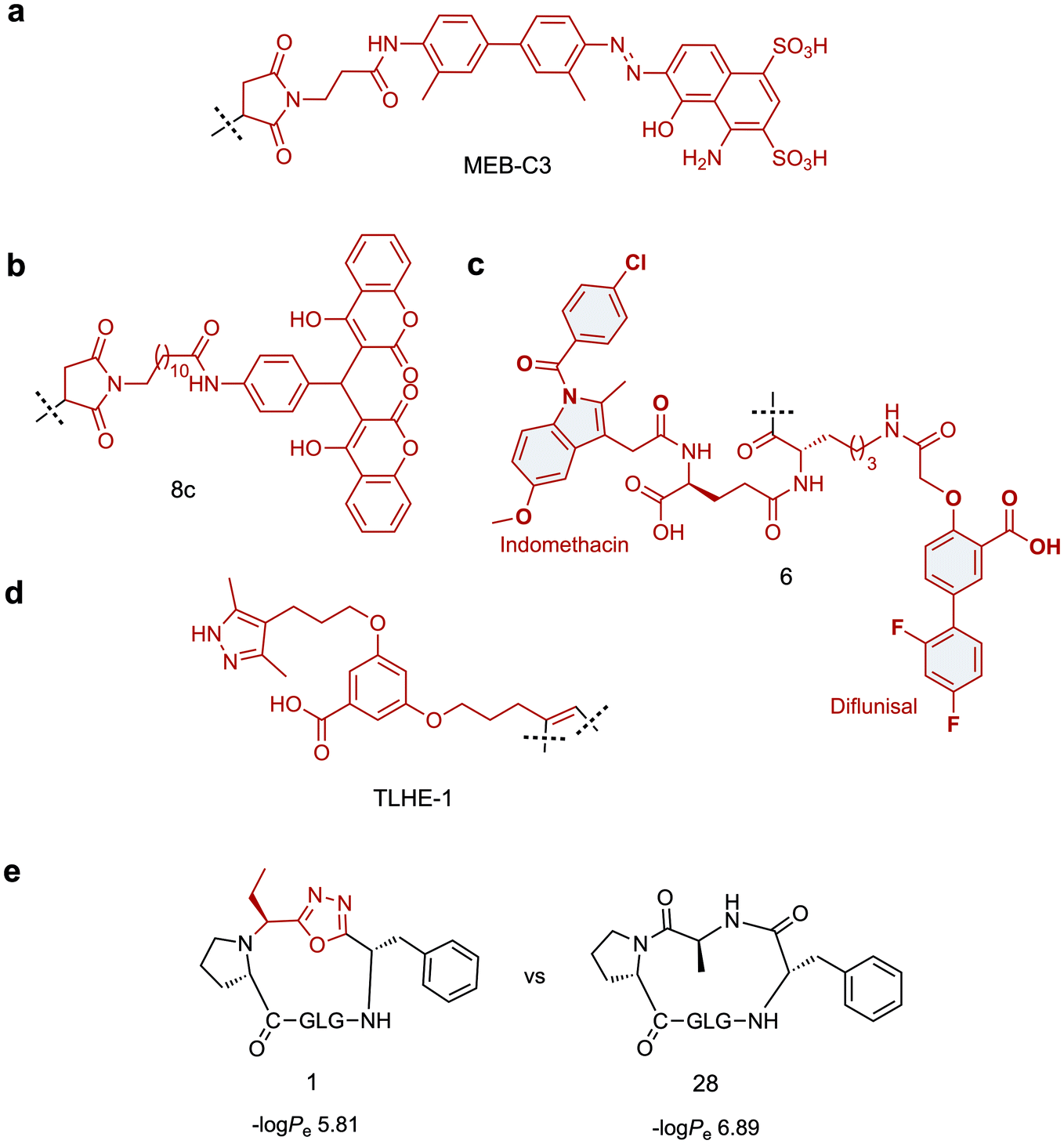 | ||
| Fig. 3 Small molecule scaffolds for prolonging in vivo half-life or cell permeability of peptides. (a) Stable maleimide modified version of Evans blue dye with high affinity for serum albumin. (b) Dicoumarol maleimide-based tag for enhancing the in vivo half-life of Glucagon like peptide-1 analogues. (c) Divalent albumin binding tag comprised of Diflunisal and Indomethacin. (d) Transthyretin-binding small molecule ligand for improving the in vivo half-life of Gonadotropin-releasing hormone. (e) Oxadiazole-containing cyclic peptide displaying higher passive membrane permeability compared to its homodetic counterpart. Small molecule components are highlighted in red. | ||
An alternative strategy for improving the in vivo half-life and efficacy of GLP-1 analogues was devised by Han et al. who exploited the potent binding of 4-hydroxycoumarin to human serum albumin.99 The approach involved conjugating a dicoumarol maleimide-based tag (8c, Fig. 3b) to cysteine residues in the peptide backbone of potent GLP-1 analogues. Two compounds were identified that displayed significantly improved in vivo half lives in rats (t1/2 = 22.07 ± 1.1 h, t1/2 = 18.78 ± 2.79 h) compared with that of extendin-4 (t1/2 = 2.82 ± 0.21 h), whilst maintaining comparable antidiabetic properties.
A novel approach involving divalent albumin binding was employed by Bech et al. who developed a tag comprised of two small molecule albumin binding drugs, Diflunisal and Indomethacin (6, Fig. 3c).100 When conjugated to GLP-1 analogues, the avidity of the divalent peptide–small molecule hybrid for albumin resulted in increased in vivo half-life over a monovalent analogue where only the Indomethacin tag was attached (t1/2 = 299 min vs. 55 min) as well as displaying protracted absorption into the blood after sub-cutaneous injection.
The strategies discussed so far have all employed albumin as the serum protein target for delaying renal clearance. In contrast, Penchala et al. developed a strategy targeting the 55 kDa homo-tetramer Transthyretin (TTR) utilising a potent, selective and reversible small molecule TTR ligand (AG10).84 Model peptides were conjugated to an AG10 based tag (TLHE1, Fig. 3d) using click chemistry. Conjugation of TLHE1 to Gonadotropin-releasing hormone (GnRH, t1/2 = 2–6 min) dramatically improved its in vivo half-life giving an initial distribution phase of 12 min followed by an extended terminal phase (46 ± 3 min, 13-fold longer than GnRH t1/2). Furthermore, tag addition to a proteolytically stable analogue of GnRH (GnRH-A, t1/2 = 55 ± 11 min) increased its in vivo half-life more than 3-fold, with an associated improvement in in vivo efficacy over the un-tagged peptide. Importantly, it was demonstrated that the peptide–TLHE1/TTR interaction was reversible and did not interfere with the peptide binding to its target, GnRH-R.
An innovative strategy for promoting cellular uptake has been developed by Matile and co-workers, utilising the ability of constrained cyclic disulphides to pass through a lipid bilayer by dynamic covalent thiol–disulphide exchange.111,112 The methodology was then extended to diselenolanes, which showed improvement over dithiolanes in delivering fluorophores to the cytosol of HeLa Kyoto cells.113 Recently, it has been demonstrated that these diselenolanes can be used to transport a range of cargos into the cytosol, including peptides, proteins and quantum dots, suggesting that this approach may be widely applicable.114
Whilst the introduction of conformational constraint in the form of macrocyclisation can in itself improve membrane permeability,115,116 enhancement is not guaranteed. Strategic design of the cyclising moiety can promote enhanced uptake, however. In an impressive example reported by Yudin and co-workers, the addition of a 1,3,4-oxadiazole scaffold within the peptide backbone was found to improve the passive membrane permeability of cyclic peptides.117 The approach utilises a multicomponent cyclisation reaction between a linear peptide, an aldehyde and a (N-isocyanimino)triphenylphosphorane. The reaction has a broad substrate scope, tolerating various sequences and ring sizes (15-, 18-, 21- and 24-membered rings), although amino acid side chain protection is required for the ring closure step and a proline is preferred as the N-terminal residue. In a parallel artificial membrane permeability assay, the oxadiazole-containing macrocycles showed higher permeability compared with their homodetic counterparts (e.g. compound 1vs.28, Fig. 3e). The noncanonical backbone region was shown to serve as an endocyclic control element that promotes a unique intramolecular hydrogen bond network, stabilising beta-turn motifs and facilitating passive membrane permeability. Applying this strategy to biologically relevant peptides and evaluating their uptake properties in a biological context will permit wider application of this approach.
In an example where peptide–small molecule hybrids were investigated in a biological context, Spring and co-workers found a small molecule carrier (SMoC) group incorporated through the cyclising linker can enhance the cellular uptake of a series MDM2-binding peptides.118 SMoCs are molecules that possess guanidinium cations linked to a lipophilic biphenyl system; they were designed by Selwood and co-workers to be a smaller and potentially more proteolytically stable mimic of guanidine-rich cell-penetrating peptides.119 Higher uptake for the SMoC-bearing MDM2-binding peptide was visualised by confocal microscopy, which correlated with an improvement in p53 activation in cells. However, the SMoC group was found to induce toxicity in some sequence variants in a lactate dehydrogenase leakage assay. This highlights that care should be taken when appending small molecule moieties to peptides, and that sequence optimisation to avoid unwanted toxicity may be required.
The above strategies highlight the potential of peptide–small molecule hybrids for improving the pharmacokinetic properties of therapeutic peptides. Strategies to improve in vivo half-life are well validated and whilst many have been exclusively applied to GLP-1 analogues, they have the potential to be applicable to other peptide therapeutics. Furthermore, it is likely that there are many more serum-binding small molecule ligands that could be employed in a similar fashion and this is a topic that warrants further investigation. Importantly, these ligands must bind reversibly to the serum protein, without compromising the function of either the carrier protein or the therapeutic peptide. Small-molecules are less likely to negatively impact peptide solubility than fusion proteins, fatty-acid chains and PEGylation, and their small size means they rarely interfere with in vivo peptide function. Linker length is an important factor to be considered and likely needs optimisation on a case by case basis. Small molecule strategies to improve cellular uptake are more limited and the majority of existing approaches highlighted here are not yet generalisable. With better understanding of permeability mechanisms, new methods and small molecules can be developed to overcome this challenge.
2.3 Chemical tools
Chemical biology probes are powerful reagents for studying the mechanistic and phenotypic roles of protein targets. The majority of these molecules comprise a section which binds the target of choice, combined with a group or molecule that imparts the additional functionality of the probe. We have chosen here to focus on switchable molecules, photoaffinity probes and chemically-triggered tools. Conjugation of small molecule dyes, such as rhodamine, is a standard method for peptide visualisation and thus will not be discussed.120Incorporation of a switch into a molecule makes it possible to “turn-on”—and in some cases “turn-off”—a probe's activity in a highly spatio-temporally controlled manner. Switchable small molecules allow the study of topics such as rapid kinetic responses121–124 and the activation or inhibition of a protein in a specific region of a cell.125–127 The investigation of switchable probe molecules also opens the way for switchable drug molecules,128–130 and the development of pro-drugs.131 There are numerous switchable small molecule probe motifs and the expansion of these into peptides has yielded some fascinating results.
Photo-switchable probes represent one of the largest classes of switchable molecules. In general, the switchable part of these probes is centred around a double bond which can be isomerised by irradiation with particular wavelengths of light. Irradiation with one wavelength sets the geometry to cis or trans, while irradiation with a different wavelength switches the geometry to the other. cis- and trans-isomerisation of a moiety embedded in a macrocycle will have a profound effect on the structure of the system as a whole and the affinity with which it can bind a target protein. The most commonly utilised examples of this system are azobenzenes where the trans-isomer is sterically preferred. In these systems, irradiation with UV light switches the molecule to its cis-isomer and irradiation with visible light or heating reverts the molecule to the trans-state.
In a prominent example, Nevola et al. created photo-switchable peptides to regulate clathrin-mediated endocytosis (CME) by targeting the beta-adaptin subunit of the AP2 complex, the most well characterised hub of the CME interactome.132 An azobenzene-based linkage (Fig. 4a) was used to alter the secondary structure of the macrocyclic peptides, and photo-switching of this moiety in vitro led to between 3- and 12-fold changes in affinity. The strategy was employed in a cell-based model where cell permeable analogues were found to photoregulate endocytosis of transferrin receptor protein in living cells.
 | ||
| Fig. 4 Switchable small molecule motifs for controlling peptide conformation. (a) Azobenzene-based linker for peptide cyclisation, UV light induces formation of the cis-isomer, which reverts to the trans-state when subjected to visible light. (b) Diarylethene-based cyclisation linker which adopts the “closed” form upon UV irradiation, switching back to the “open” form facilitated by visible light. (c) Iminoboronate-mediated peptide cyclisation, linkage readily cleaved by acidification (pH 6), oxidation (peroxynitrite) or treatment with α-nucleophiles (hydrazine). Small molecule components are highlighted in red. | ||
Following this work, Wegner, Heinis and co-workers devised an in vitro evolution methodology to identify light-activatable macrocyclic peptide using the same azobenzene-based linker (Fig. 4a).133 Peptide binders of streptavidin were selected using phage-display. Crucially, the azobenzene motif was photo-switched to the cis conformation on the phage surface, prior to panning against the target protein. This facilitated the isolation of activatable peptides with an up to 3-fold binding selectivity over the trans form. More recently, Derda and coworkers extended this methodology by developing an azobenzene-based linchpin that could be used to create photoswitchable bicyclic peptides, though further work will be required to combine this methodology with phage display libraries.134
Whilst azobenzenes are by far the most heavily studied photo-switchable motifs, there are other groups which exhibit similar properties. Inspired by earlier studies,135–137 the Spring lab recently reported a photo-switchable hybrid molecule using a diarylethene (DAE, Fig. 4b) as the photo-switch.138 Incorporation of the DAE photo-switch into a known MDM2 binding sequence (pDI) maintained the low nanomolar binding affinity of the parent peptide in the “open” form, whilst reducing it up to 8-fold in the “closed” form.
Other methods for the development of switchable molecules rely on the use of chemical or environmental triggers139 such as pH,140,141 oxygen tension142–144 or the presence of an oxidising agent.145,146 These methods are now also being applied to peptides through the formation of peptide–small molecule hybrids. Gao and co-workers reported a reversible iminoboronate-mediated peptide cyclisation for tuning peptide activity.147 The imine product is stabilised by forming a dative bond with an adjacent boronic acid, resulting in robust stability at neutral pH against commonly seen biomolecules. Crucially, the iminoboronate linkage can be readily cleaved, with acidification to below pH 6.8, oxidation with peroxynitrite or addition of exogenous small α-nucleophilic molecules (Fig. 4c). The potential of this technique for biological applications was demonstrated by creating an iminoboronate-cyclised peptide which contained a potent and selective αvβ3 integrin binding motif (RGDf, f = D-Phe). The binding interaction was successfully ‘switched-off’ in cells by lowering the cellular pH to induce peptide linearization, as the RGDf motif is only capable of binding integrin when placed in a suitable cyclic scaffold.
A well-established methodology related to photo-switchable molecules is photoaffinity crosslinking (or labelling). This approach has many advantages over other covalent methodologies. It is highly spatio-temporally resolved, crosslinking occurs rapidly and irreversibly, and unlike many other covalent approaches, it does not require a specific nucleophile for crosslinking. In the context of small molecules, it has been used for the identification of unknown binding partners, target validation,148–150 and fragment-based screening151 in both cell-based and purified enzyme settings. Once crosslinking has occurred the resulting species is usually characterised by proteomic mass spectrometry. This method allows investigation of both the target protein and the site of the protein which has been labelled. We have chosen to highlight a few articles from the most recent literature to demonstrate the different types of small molecule modification which are used with peptide hybrid photoaffinity probes. Other more general reviews which include photoaffinity probes are available.152
A recent example of a photo-crosslinking peptide probe was reported by Tate and co-workers during their investigation into the mode of action of the peptide Huwentoxin-IV and other cysteine knot peptides on voltage gated sodium (Nav) channels.153 To investigate the molecular mechanism of the potent modulation of Nav1.7, a library of aziridine (L-photomethionine) containing Huwentoxin-IV peptides was screened. Using proteomic mass spectrometry, a new model for binding of Huwentoxin-IV to the Nav1.7 channel was proposed.
A benzophenone functionality is a common alternative for photo-crosslinking. A recent example of this functionality being used to identify transient PPIs was demonstrated by An et al. in their investigation of phosphotyrosine binding modules such as the Src homology 2 (SH2) domain.154 By appending a benzophenone containing amino acid at the N-terminus and biotin at the C-terminus, a known SH2 domain was successfully captured and pulled-down. Screening for an optimised peptide sequence, using a combinatorial peptide ligand library subsequently identified a probe with which 24 SH2 domain proteins were captured.155,156 In addition, the technique identified six phosphotyrosine binding domains and five C2 domains which bind to phosphotyrosine. This study has laid the groundwork for a new mining strategy to identify putative binders of this PTM.
While the previous example incorporated the photo-crosslinking group on an amino acid side chain, inclusion of these motifs through the cyclising linker has also been described. Using their previously developed multi-functional dialkynyl cyclisation platform, Spring and co-workers were able to incorporate a benzophenone photoaffinity labelling motif into the linker. This allowed the development of peptides which preferentially cross-linked to MDM2 in the presence of BSA.157
Switchable and triggerable small molecule probes are an important class of compounds for chemical biology studies. The methods utilised to make these compounds are now being applied to peptide-based compounds through conjugation of small molecule fragments into peptide structures. This emerging class of peptide–small molecule hybrids is already showing potential in addressing complex questions and allowing the development of more potent peptide binders. Further developments in this field will undoubtedly continue to yield interesting and important molecules.
Outlook
Peptides show immense promise as novel therapeutics or tools to investigate complex biological questions.6,7,158 A plethora of approaches are being developed to address the final hurdles to applying them more widely in the clinic and in vivo.8,10 In this review we highlight the potential peptide–small molecule hybrids provide both to tackle these limitations and to further extend peptide functionalities, including through enhancing binding potency, cellular stability or creating switchable tools.The potential of hybrid approaches to extend peptide functionality is only just beginning to be realised and there is much scope for their expansion and general implementation. For example, conjugation of serum-binding small molecule tags to peptides is now a well-established approach to improve in vivo half-life.89,93,159 So far, however, the biological targets explored have all been extracellular, and it will be interesting to see the effect of these tags on cell permeability. A widely applicable small molecule tag that lengthens a peptide's in vivo t1/2 whilst retaining or even improving cell permeability would be highly coveted.
Similarly, switchable and triggerable small molecule probes are widely applied and immensely useful both for probing functionality and also as pro-drugs.128,131 Some of these modalities are already being included in peptide probes. Further incorporation of such small molecules into already validated bioactive peptides poses as a profitable opportunity to expand this class of compound and permit the investigation of more complex biological questions.
What is notable however, is that the majority of hybrid molecules are still tailored on a case by case basis. This often requires laborious rational design and multiple iterations of synthesis and testing to identify desirable compounds. Whilst effective, though slow, when applied to well characterised targets (where a crystal structure and small molecule or peptide binders are known) application to novel targets is more challenging. The lack of validated ‘one size fits all’ strategies remains a key hinderance in their wider use. With the growing ability to include these hybrid molecules in library screening strategies, however, the scope of targets available will increase exponentially, unleashing the full potential of this powerful class of molecules.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Francis Crick Institute which receives its core funding from Cancer Research UK (FC001748), the UK Medical Research Council (FC001748), and the Wellcome Trust (FC001748).References
- Y. Huang, M. M. Wiedmann and H. Suga, Chem. Rev., 2019, 119, 10360–10391 CrossRef CAS.
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS.
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef CAS.
- H. Yin and A. D. Hamilton, Angew. Chem., Int. Ed., 2005, 44, 4130–4163 CrossRef CAS.
- G. L. Verdine and L. D. Walensky, Clin. Cancer Res., 2007, 13, 7264–7270 CrossRef CAS.
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS.
- L. Di, AAPS J., 2015, 17, 134–143 CrossRef CAS.
- M. Goldflam and C. G. Ullman, Front. Chem., 2015, 3, 69 Search PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS.
- M. C. Morejón, A. Laub, B. Westermann, D. G. Rivera and L. A. Wessjohann, Org. Lett., 2016, 18, 4096–4099 CrossRef.
- M. C. Morejón, A. Laub, G. N. Kaluderović, A. R. Puentes, A. N. Hmedat, A. J. Otero-González, D. G. Rivera and L. A. Wessjohann, Org. Biomol. Chem., 2017, 15, 3628–3637 RSC.
- J. J. Koh, S. Lin, R. W. Beuerman and S. Liu, Amino Acids, 2017, 49, 1653–1677 CrossRef CAS.
- P. Singla and D. B. Salunke, Eur. J. Med. Chem., 2020, 187, 111909 CrossRef CAS.
- S. V. Moradi, W. M. Hussein, P. Varamini, P. Simerska and I. Toth, Chem. Sci., 2016, 7, 2492–2500 RSC.
- M. R. Pratt and C. R. Bertozzi, Chem. Soc. Rev., 2005, 34, 58–68 RSC.
- E. Valeur, S. M. Guéret, H. Adihou, R. Gopalakrishnan, M. Lemurell, H. Waldmann, T. N. Grossmann and A. T. Plowright, Angew. Chem., Int. Ed., 2017, 56, 10294–10323 CrossRef CAS.
- Y. H. Lau, P. De Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS.
- Y. Wang, A. G. Cheetham, G. Angacian, H. Su, L. Xie and H. Cui, Adv. Drug Delivery Rev., 2017, 110–111, 112–126 CrossRef CAS.
- I. Vhora, S. Patil, P. Bhatt and A. Misra, Protein- and Peptide-Drug Conjugates: An Emerging Drug Delivery Technology, Elsevier Inc., 1st edn, 2015, vol. 98 Search PubMed.
- L. Ma, C. Wang, Z. He, B. Cheng, L. Zheng and K. Huang, Curr. Med. Chem., 2017, 24, 3373–3396 CrossRef CAS.
- P. Hoppenz, S. Els-Heindl and A. G. Beck-Sickinger, Front. Chem., 2020, 8, 571 CrossRef CAS.
- E. I. Vrettos, G. Mező and A. G. Tzakos, Beilstein J. Org. Chem., 2018, 14, 930–954 CrossRef CAS.
- Z. Qian, P. Upadhyaya and D. Pei, Peptide Libraries: Methods and Protocols, 2015, vol. 1248, pp. 39–53 Search PubMed.
- Z. Guo, S. Y. Hong, J. Wang, S. Rehan, W. Liu, H. Peng, M. Das, W. Li, S. Bhat, B. Peiffer, B. R. Ullman, C. M. Tse, Z. Tarmakova, C. Schiene-Fischer, G. Fischer, I. Coe, V. O. Paavilainen, Z. Sun and J. O. Liu, Nat. Chem., 2019, 11, 254–263 CrossRef CAS.
- Z. Zhu, A. Shaginian, L. C. Grady, T. O’Keeffe, X. E. Shi, C. P. Davie, G. L. Simpson, J. A. Messer, G. Evindar, R. N. Bream, P. P. Thansandote, N. R. Prentice, A. M. Mason and S. Pal, ACS Chem. Biol., 2018, 13, 53–59 CrossRef CAS.
- Y. Li, R. de Luca, S. Cazzamalli, F. Pretto, D. Bajic, J. Scheuermann and D. Neri, Nat. Chem., 2018, 10, 441–448 CrossRef CAS.
- S. Mimmi, D. Maisano, I. Quinto and E. Iaccino, Trends Pharmacol. Sci., 2019, 40, 87–91 CrossRef CAS.
- C. H. Wu, I. J. Liu, R. M. Lu and H. C. Wu, J. Biomed. Sci., 2016, 23 DOI:10.1186/s12929-016-0223-x.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS.
- I. R. Rebollo, A. Angelini and C. Heinis, MedChemComm, 2013, 4, 145–150 RSC.
- S. Chen, J. Morales-Sanfrutos, A. Angelini, B. Cutting and C. Heinis, ChemBioChem, 2012, 13, 1032–1038 CrossRef CAS.
- S. S. Kale, C. Villequey, X. D. Kong, A. Zorzi, K. Deyle and C. Heinis, Nat. Chem., 2018, 715–723 CrossRef CAS.
- V. Baeriswyl and C. Heinis, Protein Eng., Des. Sel., 2013, 26, 81–89 CrossRef CAS.
- A. Angelini, L. Cendron, S. Chen, J. Touati, G. Winter, G. Zanotti and C. Heinis, ACS Chem. Biol., 2012, 7, 817–821 CrossRef CAS.
- D. Bertoldo, M. M. G. Khan, P. Dessen, W. Held, J. Huelsken and C. Heinis, ChemMedChem, 2016, 11, 834–839 CrossRef CAS.
- J. Wilbs, X. D. Kong, S. J. Middendorp, R. Prince, A. Cooke, C. T. Demarest, M. M. Abdelhafez, K. Roberts, N. Umei, P. Gonschorek, C. Lamers, K. Deyle, R. Rieben, K. E. Cook, A. Angelillo-Scherrer and C. Heinis, Nat. Commun., 2020, 11 DOI:10.1038/s41467-020-17648-w.
- X. D. Kong, J. Moriya, V. Carle, F. Pojer, L. A. Abriata, K. Deyle and C. Heinis, Nat. Biomed. Eng., 2020, 4, 560–571 CrossRef CAS.
- K. F. Tjhung, P. I. Kitov, S. Ng, E. N. Kitova, L. Deng, J. S. Klassen and R. Derda, J. Am. Chem. Soc., 2016, 138, 32–35 CrossRef CAS.
- Y. Chou, E. N. Kitova, M. Joe, R. Brunton, T. L. Lowary, J. S. Klassen and R. Derda, Org. Biomol. Chem., 2018, 16, 223–227 RSC.
- D. F. Vinals, P. I. Kitov, Z. Tu, C. Zou, C. W. Cairo, H. C. H. Lin and R. Derda, Pept. Sci., 2019, 111, e24097 CrossRef.
- A. Ekanayake, L. Sobze, J. Youk, N. J. Bennett, R. Mukherjee, A. Bhardwaj, F. Wuest and R. Derda, ChemRxiv DOI:10.26434/chemrxiv.12644225.v1.
- T. Passioura and H. Suga, Chem. Commun., 2017, 53, 1931–1940 RSC.
- C. Tsiamantas, J. M. Rogers and H. Suga, Chem. Commun., 2020, 56, 4265–4272 RSC.
- Y. Yin, N. Ochi, T. W. Craven, D. Baker, N. Takigawa and H. Suga, J. Am. Chem. Soc., 2019, 141, 19193–19197 CrossRef CAS.
- J. Morimoto, Y. Hayashi and H. Suga, Angew. Chem., Int. Ed., 2012, 51, 3423–3427 CrossRef CAS.
- J. Johansen-Leete, T. Passioura, T. Passioura, T. Passioura, T. Passioura, S. R. Foster, R. P. Bhusal, D. J. Ford, M. Liu, S. A. K. Jongkees, H. Suga, M. J. Stone, R. J. Payne and R. J. Payne, J. Am. Chem. Soc., 2020, 142, 9141–9146 CrossRef CAS.
- T. A. Martinek and F. Fülöp, Chem. Soc. Rev., 2012, 41, 687–702 RSC.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252–262 CrossRef CAS.
- R. Gopalakrishnan, A. I. Frolov, L. Knerr, W. J. Drury and E. Valeur, J. Med. Chem., 2016, 59, 9599–9621 CrossRef CAS.
- J. W. Checco, D. F. Kreitler, N. C. Thomas, D. G. Belair, N. J. Rettko, W. L. Murphy, K. T. Forest and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4552–4557 CrossRef CAS.
- J. W. Checco, E. F. Lee, M. Evangelista, N. J. Sleebs, K. Rogers, A. Pettikiriarachchi, N. J. Kershaw, G. A. Eddinger, D. G. Belair, J. L. Wilson, C. H. Eller, R. T. Raines, W. L. Murphy, B. J. Smith, S. H. Gellman and W. D. Fairlie, J. Am. Chem. Soc., 2015, 137, 11365–11375 CrossRef CAS.
- M. V. Hager, L. M. Johnson, D. Wootten, P. M. Sexton and S. H. Gellman, J. Am. Chem. Soc., 2016, 138, 14970–14979 CrossRef CAS.
- R. W. Cheloha, B. Chen, N. N. Kumar, T. Watanabe, R. G. Thorne, L. Li, T. J. Gardella and S. H. Gellman, J. Med. Chem., 2017, 60, 8816–8833 CrossRef CAS.
- S. Liu, R. W. Cheloha, T. Watanabe, T. J. Gardella and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12383–12388 CrossRef CAS.
- J. M. Rogers, S. Kwon, S. J. Dawson, P. K. Mandal, H. Suga and I. Huc, Nat. Chem., 2018, 10, 405–412 CrossRef CAS.
- C. Tsiamantas, S. Kwon, J. M. Rogers, C. Douat, I. Huc and H. Suga, Angew. Chem., Int. Ed., 2020, 59, 4860–4864 CrossRef CAS.
- L. G. Milroy, M. Bartel, M. A. Henen, S. Leysen, J. M. C. Adriaans, L. Brunsveld, I. Landrieu and C. Ottmann, Angew. Chem., Int. Ed., 2015, 54, 15720–15724 CrossRef CAS.
- S. A. Andrei, F. A. Meijer, J. F. Neves, L. Brunsveld, I. Landrieu, C. Ottmann and L. G. Milroy, ACS Chem. Neurosci., 2018, 9, 2639–2654 CrossRef CAS.
- C. Wang, W. Shi, L. Cai, L. Lu, Q. Wang, T. Zhang, J. Li, Z. Zhang, K. Wang, L. Xu, X. Jiang, S. Jiang and K. Liu, J. Med. Chem., 2013, 56, 2527–2539 CrossRef CAS.
- C. Hoppmann and L. Wang, Chem. Commun., 2016, 52, 5140–5143 RSC.
- J. Charoenpattarapreeda, Y. S. Tan, J. Iegre, S. J. Walsh, E. Fowler, R. S. Eapen, Y. Wu, H. F. Sore, C. S. Verma, L. Itzhaki and D. R. Spring, Chem. Commun., 2019, 55, 7914–7917 RSC.
- J. M. Smith, F. Vitali, S. A. Archer and R. Fasan, Angew. Chem., Int. Ed., 2011, 50, 5075–5080 CrossRef CAS.
- M. Satyanarayana, F. Vitali, J. R. Frost and R. Fasan, Chem. Commun., 2012, 48, 1461–1463 RSC.
- J. M. Smith, N. C. Hill, P. J. Krasniak and R. Fasan, Org. Biomol. Chem., 2014, 12, 1135–1142 RSC.
- J. M. Smith, J. R. Frost and R. Fasan, Chem. Commun., 2014, 50, 5027–5030 RSC.
- A. E. Owens, J. A. Iannuzzelli, Y. Gu and R. Fasan, ACS Cent. Sci., 2020, 6, 368–381 CrossRef CAS.
- L. Reguera and D. G. Rivera, Chem. Rev., 2019, 119, 9836–9860 CrossRef CAS.
- X. Li, S. Chen, W. D. Zhang and H. G. Hu, Chem. Rev., 2020, 120, 10079–10144 CrossRef CAS.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K. H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, e3445–e3454 CrossRef CAS.
- T. E. Speltz, S. W. Fanning, C. G. Mayne, C. Fowler, E. Tajkhorshid, G. L. Greene and T. W. Moore, Angew. Chem., Int. Ed., 2016, 55, 4252–4255 CrossRef CAS.
- C. Phillips, L. R. Roberts, M. Schade, R. Bazin, A. Bent, N. L. Davies, R. Moore, A. D. Pannifer, A. R. Pickford, S. H. Prior, C. M. Read, A. Scott, D. G. Brown, B. Xu and S. L. Irving, J. Am. Chem. Soc., 2011, 133, 9696–9699 CrossRef CAS.
- A. Glas, D. Bier, G. Hahne, C. Rademacher, C. Ottmann and T. N. Grossmann, Angew. Chem., Int. Ed., 2014, 53, 2489–2493 CrossRef CAS.
- Y. H. Lau, Y. Wu, M. Rossmann, B. X. Tan, P. De Andrade, Y. S. Tan, C. Verma, G. J. McKenzie, A. R. Venkitaraman, M. Hyvönen and D. R. Spring, Angew. Chem., Int. Ed., 2015, 54, 15410–15413 CrossRef CAS.
- S. S. Kale, M. Bergeron-Brlek, Y. Wu, M. G. Kumar, M. V. Pham, J. Bortoli, J. Vesin, X. D. Kong, J. Franco Machado, K. Deyle, P. Gonschorek, G. Turcatti, L. Cendron, A. Angelini and C. Heinis, Sci. Adv., 2019, 5, eaaw2851 CrossRef CAS.
- G. K. Mothukuri, S. S. Kale, C. L. Stenbratt, A. Zorzi, J. Vesin, J. Bortoli Chapalay, K. Deyle, G. Turcatti, L. Cendron, A. Angelini and C. Heinis, Chem. Sci., 2020, 11, 7858–7863 RSC.
- Y. H. Lau, Y. Wu, P. De Andrade, W. R. J. D. Galloway and D. R. Spring, Nat. Protoc., 2015, 10, 585–594 CrossRef CAS.
- A. V. Vasco, Y. Méndez, A. Porzel, J. Balbach, L. A. Wessjohann and D. G. Rivera, Bioconjugate Chem., 2019, 30, 253–259 CrossRef CAS.
- N. Assem, D. J. Ferreira, D. W. Wolan and P. E. Dawson, Angew. Chem., Int. Ed., 2015, 54, 8665–8668 CrossRef CAS.
- S. J. Walsh, J. Iegre, H. Seki, J. D. Bargh, H. F. Sore, J. S. Parker, J. S. Carroll and D. R. Spring, Org. Biomol. Chem., 2020, 18, 4224–4230 RSC.
- S. S. Kale, C. Villequey, X. D. Kong, A. Zorzi, K. Deyle and C. Heinis, Nat. Chem., 2018, 10, 715–723 CrossRef CAS.
- S. M. Guéret, S. Thavam, R. J. Carbajo, M. Potowski, N. Larsson, G. Dahl, A. Dellsén, T. N. Grossmann, A. T. Plowright, E. Valeur, M. Lemurell and H. Waldmann, J. Am. Chem. Soc., 2020, 142, 4904–4915 CrossRef.
- S. C. Penchala, M. R. Miller, A. Pal, J. Dong, N. R. Madadi, J. Xie, H. Joo, J. Tsai, P. Batoon, V. Samoshin, A. Franz, T. Cox, J. Miles, W. K. Chan, M. S. Park and M. M. Alhamadsheh, Nat. Chem. Biol., 2015, 11, 793–798 CrossRef CAS.
- L. J. Walport, R. Obexer and H. Suga, Curr. Opin. Biotechnol., 2017, 48, 242–250 CrossRef CAS.
- A. Zorzi, S. J. Middendorp, J. Wilbs, K. Deyle and C. Heinis, Nat. Commun., 2017, 8, 16092 CrossRef CAS.
- B. Meibohm and H. Zhou, J. Clin. Pharmacol., 2012, 52, 54–62 CrossRef.
- A. N. Zaykov, J. P. Mayer and R. D. Dimarchi, Nat. Rev. Drug Discovery, 2016, 15, 425–439 CrossRef CAS.
- S. B. van Witteloostuijn, S. L. Pedersen and K. J. Jensen, ChemMedChem, 2016, 11, 2474–2495 CrossRef CAS.
- M. S. Dennis, M. Zhang, Y. Gloria Meng, M. Kadkhodayan, D. Kirchhofer, D. Combs and L. A. Damico, J. Biol. Chem., 2002, 277, 35035–35043 CrossRef CAS.
- J. Milton Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS.
- L. Pollaro and C. Heinis, MedChemComm, 2010, 1, 319–324 RSC.
- S. Mitragotri, P. A. Burke and R. Langer, Nat. Rev. Drug Discovery, 2014, 13, 655–672 CrossRef CAS.
- M. F. T. Koehler, K. Zobel, M. H. Beresini, L. D. Caris, D. Combs, B. D. Paasch and R. A. Lazarus, Bioorg. Med. Chem. Lett., 2002, 12, 2883–2886 CrossRef CAS.
- K. Zobel, M. F. T. Koehler, M. H. Beresini, L. D. Caris and D. Combs, Bioorg. Med. Chem. Lett., 2003, 13, 1513–1515 CrossRef CAS.
- H. Chen, G. Wang, L. Lang, O. Jacobson, D. O. Kiesewetter, Y. Liu, Y. Ma, X. Zhang, H. Wu, L. Zhu, G. Niu and X. Chen, Theranostics, 2016, 6, 243–253 CrossRef CAS.
- Y. Liu, G. Wang, H. Zhang, Y. Ma, L. Lang, O. Jacobson, D. O. Kiesewetter, L. Zhu, S. Gao, Q. Ma and X. Chen, Bioconjugate Chem., 2016, 27, 54–58 CrossRef CAS.
- J. Han, L. Sun, Y. Chu, Z. Li, D. Huang, X. Zhu, H. Qian and W. Huang, J. Med. Chem., 2013, 56, 9955–9968 CrossRef CAS.
- E. M. Bech, M. C. Martos-Maldonado, P. Wismann, K. K. Sørensen, S. B. Van Witteloostuijn, M. B. Thygesen, N. Vrang, J. Jelsing, S. L. Pedersen and K. J. Jensen, J. Med. Chem., 2017, 60, 7434–7446 CrossRef CAS.
- L. Peraro and J. A. Kritzer, Angew. Chem., Int. Ed., 2018, 57, 11868–11881 CrossRef CAS.
- G. H. Bird, E. Mazzola, K. Opoku-Nsiah, M. A. Lammert, M. Godes, D. S. Neuberg and L. D. Walensky, Nat. Chem. Biol., 2016, 12, 845–852 CrossRef CAS.
- A. Gräslund, F. Madani, S. Lindberg, Ü. Langel and S. Futaki, J. Biophys., 2011, 414729 Search PubMed.
- S. Silva, A. J. Almeida and N. Vale, Biomolecules, 2019, 9, 22, DOI:10.3390/biom9010022.
- G. Guidotti, L. Brambilla and D. Rossi, Trends Pharmacol. Sci., 2017, 38, 406–424 CrossRef CAS.
- P. G. Dougherty, J. Wen, X. Pan, A. Koley, J. G. Ren, A. Sahni, R. Basu, H. Salim, G. Appiah Kubi, Z. Qian and D. Pei, J. Med. Chem., 2019, 62, 10098–10107 CrossRef CAS.
- T. L. Sun, Y. Sun, C. C. Lee and H. W. Huang, Biophys. J., 2013, 104, 1923–1932 CrossRef CAS.
- Q. Chu, R. E. Moellering, G. J. Hilinski, Y. W. Kim, T. N. Grossmann, J. T. H. Yeh and G. L. Verdine, MedChemComm, 2015, 6, 111–119 RSC.
- K. Welser, F. Campbell, L. Kudsiova, A. Mohammadi, N. Dawson, S. L. Hart, D. J. Barlow, H. C. Hailes, M. J. Lawrence and A. B. Tabor, Mol. Pharmaceutics, 2013, 10, 127–141 CrossRef CAS.
- S. Kannan, P. G. A. Aronica, S. Ng, D. T. Gek Lian, Y. Frosi, S. Chee, J. Shimin, T. Y. Yuen, A. Sadruddin, H. Y. K. Kaan, A. Chandramohan, J. H. Wong, Y. S. Tan, Z. W. Chang, F. J. Ferrer-Gago, P. Arumugam, Y. Han, S. Chen, L. Rénia, C. J. Brown, C. W. Johannes, B. Henry, D. P. Lane, T. K. Sawyer, C. S. Verma and A. W. Partridge, Chem. Sci., 2020, 11, 5577–5591 RSC.
- G. Gasparini, G. Sargsyan, E. K. Bang, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2015, 54, 7328–7331 CrossRef CAS.
- L. Zong, E. Bartolami, D. Abegg, A. Adibekian, N. Sakai and S. Matile, ACS Cent. Sci., 2017, 3, 449–453 CrossRef CAS.
- N. Chuard, A. I. Poblador-Bahamonde, L. Zong, E. Bartolami, J. Hildebrandt, W. Weigand, N. Sakai and S. Matile, Chem. Sci., 2018, 9, 1860–1866 RSC.
- E. Bartolami, D. Basagiannis, L. Zong, R. Martinent, Y. Okamoto, Q. Laurent, T. R. Ward, M. Gonzalez-Gaitan, N. Sakai and S. Matile, Chem. – Eur. J., 2019, 25, 4047–4051 CrossRef CAS.
- T. Rezai, B. Yu, G. L. Millhauser, M. P. Jacobson and R. S. Lokey, J. Am. Chem. Soc., 2006, 128, 2510–2511 CrossRef CAS.
- P. S. Burton, R. A. Conradi, N. F. H. Ho, A. R. Hilgers and R. T. Borchardt, J. Pharm. Sci., 1996, 85, 1336–1340 CrossRef CAS.
- J. R. Frost, C. C. G. Scully and A. K. Yudin, Nat. Chem., 2016, 8, 1105–1111 CrossRef CAS.
- Y. Wu, A. Kaur, E. Fowler, M. M. Wiedmann, R. Young, W. R. J. D. Galloway, L. Olsen, H. F. Sore, A. Chattopadhyay, T. T. L. Kwan, W. Xu, S. J. Walsh, P. De Andrade, M. Janecek, S. Arumugam, L. S. Itzhaki, Y. H. Lau and D. R. Spring, ACS Chem. Biol., 2019, 14, 526–533 CrossRef CAS.
- A. S. Rebstock, C. Visintin, E. Leo, C. G. Posada, S. R. Kingsbury, G. H. Williams, K. Stoeber and D. L. Selwood, ChemBioChem, 2008, 9, 1787–1796 CrossRef CAS.
- S. Lee, J. Xie and X. Chen, Biochemistry, 2010, 49, 1364–1376 CrossRef CAS.
- A. Reiner and E. Y. Isacoff, Nat. Chem. Biol., 2014, 10, 273–280 CrossRef CAS.
- P. Gorostiza, M. Volgraf, R. Numano, S. Szobota, D. Trauner and E. Y. Isacoff, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10865–10870 CrossRef CAS.
- H. Higuchi, E. Muto, Y. Inoue and T. Yanagida, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4395–4400 CrossRef CAS.
- G. C. Faas and I. Mody, Biochim. Biophys. Acta, Gen. Subj., 1820, 2012, 1195–1204 Search PubMed.
- N. Wagner, M. Stephan, D. Höglinger and A. Nadler, Angew. Chem., Int. Ed., 2018, 57, 13339–13343 CrossRef CAS.
- H. Ando, T. Furuta, R. Y. Tsien and H. Okamoto, Nat. Genet., 2001, 28, 317–325 CrossRef CAS.
- A. Rodríguez-Moreno, M. M. Kohl, J. E. Reeve, T. R. Eaton, H. A. Collins, H. L. Anderson and O. Paulsen, J. Neurosci., 2011, 31, 8564–8569 CrossRef.
- M. J. Fuchter, J. Med. Chem., 2020, 63, 11436–11447 CrossRef CAS.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS.
- O. Babii, S. Afonin, T. Schober, L. V. Garmanchuk, L. I. Ostapchenko, V. Yurchenko, S. Zozulya, O. Tarasov, I. Pishel, A. S. Ulrich and I. V. Komarov, Future Drug Discovery, 2020, 2 DOI:10.4155/fdd-2019-0033.
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS.
- L. Nevola, A. Martín-Quirõs, K. Eckelt, N. Camarero, S. Tosi, A. Llobet, E. Giralt and P. Gorostiza, Angew. Chem., Int. Ed., 2013, 52, 7704–7708 CrossRef CAS.
- S. Bellotto, S. Chen, I. Rentero Rebollo, H. A. Wegner and C. Heinis, J. Am. Chem. Soc., 2014, 136, 5880–5883 CrossRef CAS.
- M. R. Jafari, H. Yu, J. M. Wickware, Y. S. Lin and R. Derda, Org. Biomol. Chem., 2018, 16, 7588–7594 RSC.
- K. Fujimoto, M. Kajino, I. Sakaguchi and M. Inouye, Chem. – Eur. J., 2012, 18, 9834–9840 CrossRef CAS.
- K. Fujimoto, T. Maruyama, Y. Okada, T. Itou and M. Inouye, Tetrahedron, 2013, 69, 6170–6175 CrossRef CAS.
- O. Babii, S. Afonin, M. Berditsch, S. Reier, P. K. Mykhailiuk, V. S. Kubyshkin, T. Steinbrecher, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2014, 53, 3392–3395 CrossRef CAS.
- A. V. Strizhak, O. Babii, S. Afonin, I. Bakanovich, T. Pantelejevs, W. Xu, E. Fowler, R. Eapen, K. Sharma, M. O. Platonov, V. V. Hurmach, L. Itzhaki, M. Hyvönen, A. S. Ulrich, D. R. Spring and I. V. Komarov, Org. Biomol. Chem., 2020, 18, 5359–5369 RSC.
- F. Kratz, I. A. Müller, C. Ryppa and A. Warnecke, ChemMedChem, 2008, 3, 20–53 CrossRef CAS.
- P. Czupiel, V. Delplace and M. Shoichet, Sci. Rep., 2020, 10, 8726 CrossRef CAS.
- Z. Wang, X. Deng, J. Ding, W. Zhou, X. Zheng and G. Tang, Int. J. Pharm., 2018, 535, 253–260 CrossRef CAS.
- W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393–410 CrossRef CAS.
- I. N. Mistry, M. Thomas, E. D. D. Calder, S. J. Conway and E. M. Hammond, Int. J. Radiat. Oncol., Biol., Phys., 2017, 98, 1183–1196 CrossRef CAS.
- E. D. D. Calder, A. Skwarska, D. Sneddon, L. K. Folkes, I. N. Mistry, S. J. Conway and E. M. Hammond, Tetrahedron, 2020, 76, 131170 CrossRef CAS.
- J. Peiró Cadahiá, V. Previtali, N. S. Troelsen and M. H. Clausen, MedChemComm, 2019, 10, 1531–1549 RSC.
- G. Saravanakumar, J. Kim and W. J. Kim, Adv. Sci., 2017, 4, 1600124 CrossRef.
- A. Bandyopadhyay and J. Gao, J. Am. Chem. Soc., 2016, 138, 2098–2101 CrossRef CAS.
- J. Sumranjit and S. J. Chung, Molecules, 2013, 18, 10425–10451 CrossRef CAS.
- E. Smith and I. Collins, Future Med. Chem., 2015, 7, 159–183 CrossRef CAS.
- S. Y. Seo and T. W. Corson, Small molecule target identification using photo-affinity chromatography, Elsevier Inc., 1st edn, 2019, vol. 622 Search PubMed.
- C. G. Parker, A. Galmozzi, Y. Wang, B. E. Correia, K. Sasaki, C. M. Joslyn, A. S. Kim, C. L. Cavallaro, R. M. Lawrence, S. R. Johnson, I. Narvaiza, E. Saez and B. F. Cravatt, Cell, 2017, 168(527–541), e29 Search PubMed.
- A. Tuley and W. Fast, Biochemistry, 2018, 57, 3326–3337 CrossRef CAS.
- F. Tzakoniati, H. Xu, T. Li, N. Garcia, C. Kugel, J. Payandeh, C. M. Koth and E. W. Tate, Cell Chem. Biol., 2020, 27, 306–313 CrossRef CAS . e4.
- J. An, G. Zhai, Z. Guo, X. Bai, P. Chen, H. Dong, S. Tian, D. Ai, Y. Zhang and K. Zhang, Anal. Chem., 2019, 91, 3221–3226 CrossRef CAS.
- P. G. Righetti, G. Candiano, A. Citterio and E. Boschetti, Anal. Chem., 2015, 87, 293–305 CrossRef CAS.
- P. G. Righetti, E. Boschetti, A. Zanella, E. Fasoli and A. Citterio, J. Chromatogr. A, 2010, 1217, 893–900 CrossRef CAS.
- Y. Wu, L. B. Olsen, Y. H. Lau, C. H. Jensen, M. Rossmann, Y. R. Baker, H. F. Sore, S. Collins and D. R. Spring, ChemBioChem, 2016, 17, 689–692 CrossRef CAS.
- X. Jing and K. Jin, Med. Res. Rev., 2020, 40, 753–810 CrossRef CAS.
- A. Zorzi, S. Linciano and A. Angelini, MedChemComm, 2019, 10, 1068–1081 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |