
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptogenetic modulation of real-time nanoscale dynamics of HCN channels using photoactivated adenylyl cyclases†
Meenakshi
Tanwar
 a,
Suneel
Kateriya
b,
Deepak
Nair
a and
Mini
Jose
*a
a,
Suneel
Kateriya
b,
Deepak
Nair
a and
Mini
Jose
*a
aCentre for Neuroscience, Indian Institute of Science, Bangalore-560012, India. E-mail: mini@iisc.ac.in; minijose2000@gmail.com
bLaboratory of Optobiology, School of Biotechnology, Jawaharlal Nehru University, New Delhi-110067, India
First published on 8th March 2021
Abstract
Adenosine 3′,5′-cyclic monophosphate (cAMP) is a key second messenger that activates several signal transduction pathways in eukaryotic cells. Alteration of basal levels of cAMP is known to activate protein kinases, regulate phosphodiesterases and modulate the activity of ion channels such as Hyper polarization-activated cyclic nucleotide gated channels (HCN). Recent advances in optogenetics have resulted in the availability of novel genetically encoded molecules with the capability to alter cytoplasmic profiles of cAMP with unprecedented spatial and temporal precision. Using single molecule based super-resolution microscopy and different optogenetic modulators of cellular cAMP in both live and fixed cells, we illustrate a novel paradigm to report alteration in nanoscale confinement of ectopically expressed HCN channels. We characterized the efficacy of cAMP generation using ensemble photoactivation of different optogenetic modulators. Then we demonstrate that local modulation of cAMP alters the exchange of membrane bound HCN channels with its nanoenvironment. Additionally, using high density single particle tracking in combination with both acute and chronic optogenetic elevation of cAMP in the cytoplasm, we show that HCN channels are confined to sub 100 nm sized functional domains on the plasma membrane. The nanoscale properties of these domains along with the exchange kinetics of HCN channels in and out of these molecular zones are altered upon temporal changes in the cytoplasmic cAMP. Using HCN2 point mutants and a truncated construct of HCN2 with altered sensitivity to cAMP, we confirmed these alterations in lateral organization of HCN2 to be specific to cAMP binding. Thus, combining these advanced non-invasive paradigms, we report a cAMP dependent ensemble and single particle behavior of HCN channels mediated by its cyclic nucleotide binding domain, opening innovative ways to dissect biochemical pathways at the nanoscale and real-time in living cells.
Introduction
Cells are continually perceiving and responding to environmental stimuli. The intracellular signal transduction pathways integrate the information from extracellular stimuli into various biochemical or physiological responses. Signalling molecules categorized as secondary messengers act as mediators to commence and coordinate the diverse intracellular signalling cascades. The cyclic nucleotide cAMP, a prototypical second messenger, modulates various physiological processes ranging from learning and memory in the brain, neuronal signalling, gene expression, cell proliferation and apoptosis, metabolism, heart contraction, and relaxation.1–5 Adenylyl cyclase catalyzes the synthesis of cAMP from ATP. In eukaryotes, alteration in the basal level of cAMP has a direct influence on the molecular regulation of several proteins involved in near membrane signalling. Changes in the intracellular cAMP levels are sensed by the effector molecules which regulate numerous cellular responses. Some of the prominent effectors are protein kinase A (PKA),6 exchange protein directly activated by cAMP (EPAC),7 cyclic nucleotide-gated ion channels (CNG)8 and hyperpolarization-activated cyclic nucleotide-gated ion channels (HCN).9 HCN channels are trans-membrane molecules comprised of four identical or non-identical subunits together forming an ion-conducting pore. These channels are mainly responsible for the rhythmic pacemaker activity in cardiac cells and for regulating integration of synaptic potentials and transmission in neurons.10 Each subunit is made up of six trans-membrane domains containing a putative voltage sensor, a pore region permeable to K+ and Na+ and a cyclic nucleotide binding domain (CNBD) in the C-terminus. HCN channels consist of four subunits, namely HCN1–4, which assemble in various combinations and conformations. Among the four different isoforms of HCN, HCN2 is known to have the highest affinity for cAMP. It is well understood that cAMP can generate a bias in the voltage dependence of activation of HCN2 to more positive membrane voltages.9 However, not much is known about the alteration of lateral organization or trafficking of HCN2 on the membrane as a result of elevated cAMP.Development of optogenetic probes that alter the cAMP concentration has improved our understanding of local cAMP dependent signalling cascades within organelles and in cytoplasmic compartments in heterologous cell lines and animal models as well as in neurons. They provide a significant advantage over the conventional pharmacological approaches which lack the spatio-temporal control to manipulate local fluctuations of cAMP at submicron resolution or with a temporal precision of a few milliseconds. Adenylyl cyclases are a family of proteins which have been evolutionarily modified to synthesize cAMP by different kinds of stimulations. Photoactivated adenylyl cyclases (PACs) are photoreceptors that combine activation of a light sensing domain (Blue Light Using FAD; BLUF) with blue light and gating of synthesis of cAMP with a C-terminus coupled adenylyl cyclase homology domain (CHD). PACs first reported from unicellular flagellate Euglena gracilis (EuPAC) were composed of PACα and PACβ subunits, responsible for the step-up photophobic response.11 Recently, smaller PACs encoding a single BLUF domain as a sensor and CHD as an effector module have been identified and characterized from Beggiatoa sp. (bPAC),12Naegleria gruberi (NgPACs),13–15Turneriella parva (TpPAC),16Leptonema illini (LiPAC)17 and Oscillatoria acuminata (OaPAC),18 bestowing a wide repertoire of PACs to modulate cAMP levels in living cells.
PACs have also been successfully deployed in diverse biological systems to modulate the intracellular cAMP levels to control signal transduction of selected molecular pathways, to program cellular responses, to modulate cellular morphology and to control animal behavior.19–25 Despite these advances, there has been limited understanding on how the local or global activation of PACs can control the lateral organization and real-time trafficking of trans-membrane molecules such as HCN channels whose activity is directly controlled by cAMP. This is in part due to the lack of information on the characterization of biophysical properties of PACs in living cells and due to the lack of development of novel paradigms to investigate real-time changes in nanoscale localization and dynamics of molecules in combination with optogenetic approaches.
Here, we have bridged this gap using a battery of paradigms to evaluate cAMP dependent temporal fluctuations in the molecular organization of HCN channels. First, we studied the photophysical characteristics of a cAMP sensor by altering the intracellular cAMP levels in a controlled manner. Then, by using targeted photoactivation, we characterized different optogenetic modulators of cAMP namely, bPAC, TpPAC and native variants of NgPACs in heterologous cell lines. We then used spatially confined local elevation of cAMP as well as global acute and chronic photoactivation of PACs to investigate the transient mobility and nano-organization of ectopically expressed trans-membrane ion channel HCN2, whose function is directly modulated by intracellular cAMP levels. We show that the stimulation patterns of different PACs influence the mobility of HCN2 channels differentially, and that the elevation of cAMP can transiently immobilize HCN2 channels on the plasma membrane in live cells by optogenetic modulation with blue light. Using a truncated construct of HCN2 lacking the nucleotide binding domain as well as two point mutants of HCN2 (R591A and R591E) displaying altered sensitivity to cAMP, we verified that the dynamic distribution of HCN2 is a result of direct cAMP binding. Thus, we show that different classes of optogenetic probes for cAMP modulate the Brownian diffusion of membrane molecules differentially, altering the nanoscale dynamics of HCN2 channel segregation on the plasma membrane. These studies not only provide novel paradigms to evaluate the nanoscale organization of molecules using a non-invasive approach, but also allows alteration of the biochemical map of protein segregation in live cells. Our study opens unique ways to dissect information of complexes at the molecular scale within their native environment, paving the way for therapeutic interventions that can alter molecular pathways with unprecedented spatial and temporal control.
Results and discussion
Targeted optogenetic modulation and measurement of relative changes in cAMP concentration in live heterologous cells
In order to understand the efficacy of cAMP modulation by optogenetics using the different modulators, we opted for co-expression strategies using a single vector. This allowed us to monitor the cAMP dynamics by optical modulation along with simultaneous sensing of the cyclic nucleotide level in cells. We chose photoactivated adenylyl cyclases (PACs) to optically manipulate the cAMP level (Fig. 1). Photoactivation of the BLUF domain of PAC activated the downstream adenylyl cyclase enzyme which produces cAMP11 (Fig. S1A, ESI†). The biosensor, Pink Flamindo, which reports an increase in fluorescence intensity upon cAMP binding was used.26 We characterized the fluorescence response of Pink Flamindo upon altering the cAMP level in Neuro-2a (N2a) cells expressing the sensor. An increase in fluorescence intensity of Pink Flamindo was observed with a cumulative increase of cAMP level in the cells (Fig. S1B, ESI†). We then cloned PAC and Flamindo into a pUltra plasmid, with PAC separated from the sensor by a self-cleaving 2A peptide, which ensured the expression of both genes in the same cells (Fig. S1A, ESI†). Photoactivation of PACs co-expressed with Flamindo (referred to as PAC-Flamindo) in N2a cells results in an increase in cAMP level, detected by the sensor which displays an increase in fluorescence intensity (Fig. 1A). Local photoactivation of cells expressing PACs-Flamindo by a 30 ms 405 nm light pulse induced increased fluorescence intensity, signifying an increase in the cAMP level that lasted for several seconds and decayed exponentially (Fig. 1B). For local stimulation, a region corresponding to the point spread function of the system was randomly selected at a distance of 2–5 μm from the periphery of the cell. The details of photoactivation and imaging have been provided in the ESI,† Section 1.1.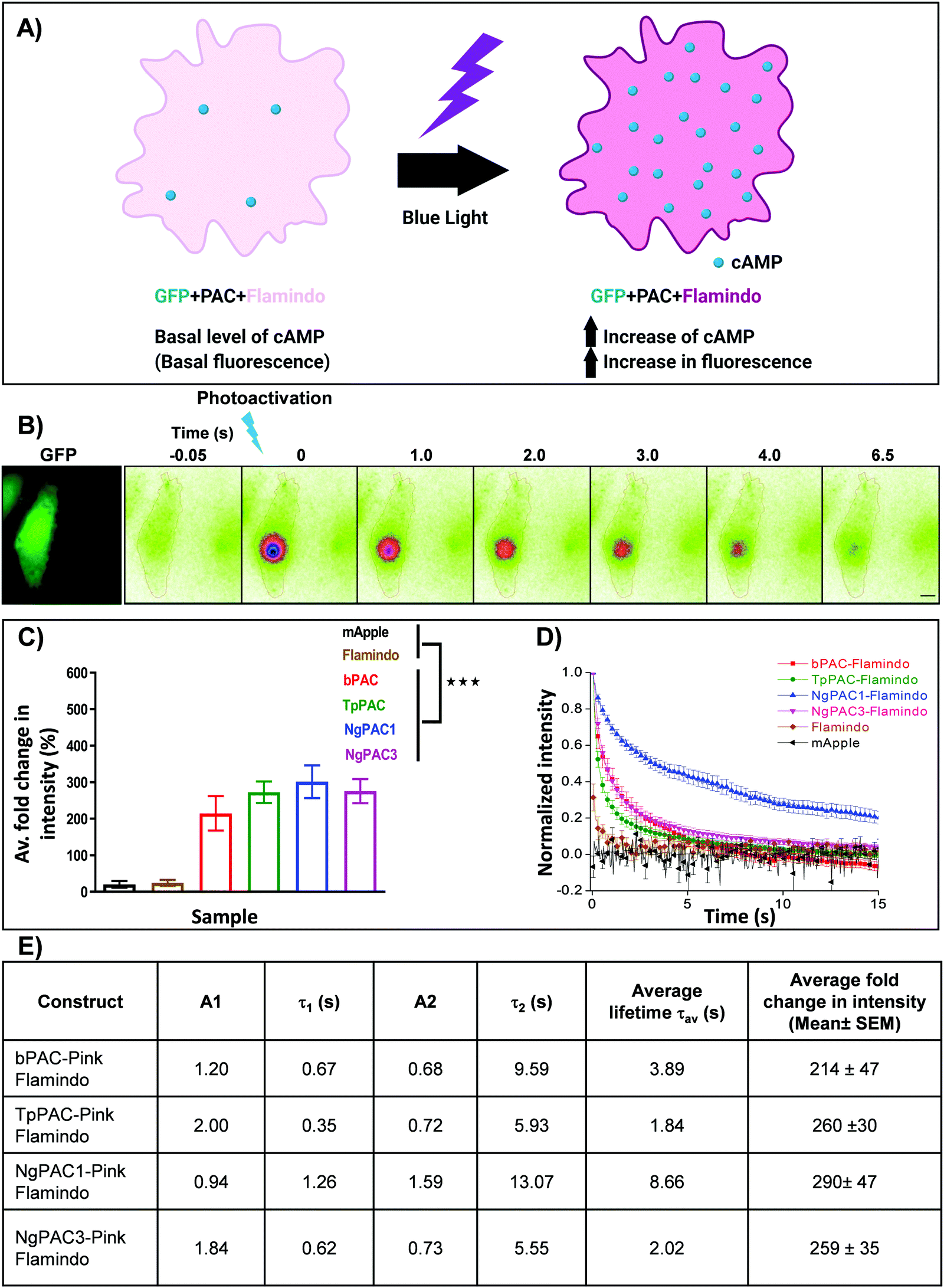 | ||
| Fig. 1 Optogenetic activation of photoactivated adenylyl cyclase (PAC) and sensing of transient changes in cAMP levels by Pink Flamindo in live Neuro-2a cells. (A) Schematic representation of a cell expressing photoactivated adenylyl cyclase (PAC) and Flamindo for sensing the cAMP level. Photoactivation of PACs increases the cAMP level in the cells, which is reported as an increase in the fluorescence intensity of Flamindo. (B) Neuro-2a cells expressing GFP-P2A-PAC-T2A-Pink Flamindo imaged in the GFP channel indicate the expression of photoactivated adenylyl cyclase (PAC), where P2A and T2A represent self-cleaving sites (ESI†). Pseudocolour images indicate transient changes in the fluorescence of Flamindo imaged at 560 nm upon targeted photoactivation of PAC with 405 nm. Fluorescence intensity in the photoactivated region decayed rapidly with time. Scale bar: 10 μm. (C) Comparison of intensity fold change of Flamindo upon photoactivation of PACs with 405 nm (n = 10, mean ± s.e.m from 2 biological replicates, ***p < 0.001, Mann–Whitney test). Average intensity of GFP::PAC in the corresponding cells is depicted in Fig. S1C, ESI.† (D) Curves representing the temporal changes in fluorescence intensity upon photoactivation of the PACs (bPAC, TpPAC, NgPAC1, and NgPAC3) with 405 nm. Fold increase in fluorescence intensity is normalized to 1. (E) Table illustrating the biophysical properties of fluorescence decay computed from (D). A1 and A2 represent the fractional components corresponding to lifetimes τ1 and τ2, respectively. All PACs-Flamindo (bPAC, TpPAC, NgPAC1 and NgPAC3) exhibited an increase in average fold intensity upon targeted photoactivation, implying a surge in cAMP level in a localized illuminated region, which decayed exponentially with varying kinetics. The controls mApple and Flamindo did not show a similar increase upon photoactivation. Consistently, the decay kinetics of PACs-Flamindo were significantly slower compared to control Flamindo, whereas mApple did not display any difference in its kinetics. | ||
Variants of PACs from Beggiatoa12 (referred to as bPAC), Naegleria gruberi (referred to as NgPAC1 and NgPAC3)13,15 and Turneriella parva (referred to as TpPAC)16 were assessed to study the dynamics of cAMP in a light regulated manner. All PACs-Flamindo (bPAC, TpPAC, NgPAC1 and NgPAC3) exhibited an increase in average fold intensity after targeted photoactivation, implying a surge in cAMP level in a localized illuminated region, which decayed exponentially with varying kinetics (Fig. 1C and D). bPAC-Flamindo showed the lowest average fold intensity change or least generation of cAMP, which increased for TpPAC and NgPAC1 (Fig. 1C and Table S1, ESI†). On the contrary, upon photoactivation no significant change was observed in the average fold intensity for control cells expressing mApple or Flamindo alone (Fig. 1C). To ensure a similar expression of the different PACs, fluorescence intensity changes in Flamindo were studied only in cells with similar GFP::PAC expression (Fig. S1C, ESI†).
The decay in fluorescence intensity at the region of photoactivation for the various PACs during photobleaching was analyzed by fitting their fluorescence decay curves using a bi-exponential decay model (Fig. S2B, ESI†), which fit better compared to a single exponential decay model (Fig. S2A, ESI†). The fluorescence decay curves indicated the temporal change in cAMP level in the cells. All PACs-Flamindo systems exhibited varying fluorescence decay kinetics with distinct short (τ1) and long (τ2) lifetime components (Fig. 1E and Table S1, ESI†). Among these, TpPAC displayed the fastest kinetics, followed by bPAC, NgPAC3, and NgPAC1. The temporal cAMP level after photoactivation depends on the recovery kinetics of the BLUF domain of the PACs. The fast kinetics of cAMP by TpPAC-Flamindo and bPAC-Flamindo resembled the fast recovery kinetics of TpPAC16 and bPAC12 photoreceptors, respectively. Compared to PAC-Flamindo expressing cells, control cells expressing Flamindo alone displayed a fast average lifetime (τav = 0.688 ± 0.286 s) with minimal increase in the residual intensity after photoactivation with 405 nm, whereas the control mApple displayed no changes in its kinetics over time (Fig. 1D). Apart from 405 nm, PACs were also photoactivated using 488 nm, resulting in an increase in the average fold fluorescence intensity and decay time (Fig. S1D, ESI†). The decay kinetics (τav) of the different PACs on photoactivation with 488 nm (Fig. S1E, ESI†) varied as compared to activation with 405 nm (Fig. 1E). However, photoactivation using either 405 nm or 488 nm revealed the slowest kinetics for NgPAC1 and fastest kinetics for TpPAC (Fig. 1E, Fig. S1E and Table S1, ESI†). All PACs showed distinct characteristics in terms of fold change, duration of recovery (recovery kinetics), and fractional components of lifetimes (A1 and A2 corresponding to τ1 and τ2, respectively). In summary, the PAC variants altered the cAMP level in a localized manner with varying amplitude and dynamics.
Using an activator-sensor strategy which utilises an equimolar concentration of PAC and the fluorescence sensor Flamindo, we were able to determine the biophysical properties of each photoactivated adenylyl cyclase variant. Expression of bi-cistronic constructs with photoactivated adenylyl cyclase (PAC) and the fluorescence sensor Flamindo ensured an equimolar concentration of PAC and Flamindo, respectively. This strategy of using an equimolar concentration of activator and sensor along with observing relative fluorescence intensity changes allowed us to rule out any changes in the ensemble diffusional behaviour of the sensor. However, the rate of synthesis of cAMP depended on the photoactivation kinetics and enzymatic activity of the different PAC variants. Therefore, the amount of cAMP generated would be proportional, but not equimolar to the PACs nor to the available Flamindo sensor.
Though the observed biophysical characteristics of PACs were comparable to their reported values, it is not well understood if the hygrodynamic radius of different PACs have a direct influence on the instantaneous generation of cAMP and persistence of enzymatic activity generating cAMP within live cells. The difference in biophysical properties of PACs which altered upon changing the photoactivation wavelength from 405 nm to 488 nm could be attributed to the differences in the absorption cross-section of PACs at different wavelengths. Since cAMP is generated as a result of the enzymatic activity of PACs, engineering PACs that are either Stokes shifted or with altered sensitivity to selected wavelengths would provide a broader spectrum of activation and flexibility for the experimenter.
Spatially confined activation of PACs results in differential mobility of hyperpolarization-activated cyclic nucleotide gated channels (HCN2)
Activity and kinetics of HCN channels are regulated by environmental stimuli/intracellular signalling cascades. HCN channels are activated by hyperpolarization of the membrane voltage and are also regulated by the cyclic nucleotide cAMP.27,28 Utilizing the property of HCN2 channel to bind with cAMP, we determined the consequences of altered cAMP level via optical stimulation on the trafficking dynamics of HCN2 channels. The mobility of HCN2 molecules on the membrane was determined after targeted photoactivation of PACs in this approach. To examine the membrane dynamics of HCN2, the protein was fused to the photoactivable fluorescent protein mEos (referred to as mEos::HCN2), which gets photoconverted from a green to red state upon photoirradiation at 405 nm. Cells expressing mEos::HCN2 imaged in total internal reflection fluorescence (TIRF) mode (Fig. 2Ai and iii) portrayed the distribution of the protein on the membrane. TIRF images processed with Super-Resolution Radial Fluctuations (SRRF) analysis enabled visualization of protein distribution on the membrane with higher lateral resolution (Fig. 2Aii and iv, ESI,† Section 1.2). Cells expressing mEos::HCN2 were exposed to a 16 ms targeted light pulse of 405 nm, which increased the fluorescence intensity when illuminated by a 561 nm laser and imaged between 575–635 nm, representing the photoconverted red state of mEos::HCN2 (Fig. 2B). To assess the mobility of mEos::HCN2 molecules, the fluorescence decay curve of the photoconverted state was fit by a single exponential decay model defined by the decay time constant τ (Fig. 2C). Cells expressing mEos::HCN2 displayed fluorescence decay curves with τ = 1.3 ± 0.489 s (Fig. 2D).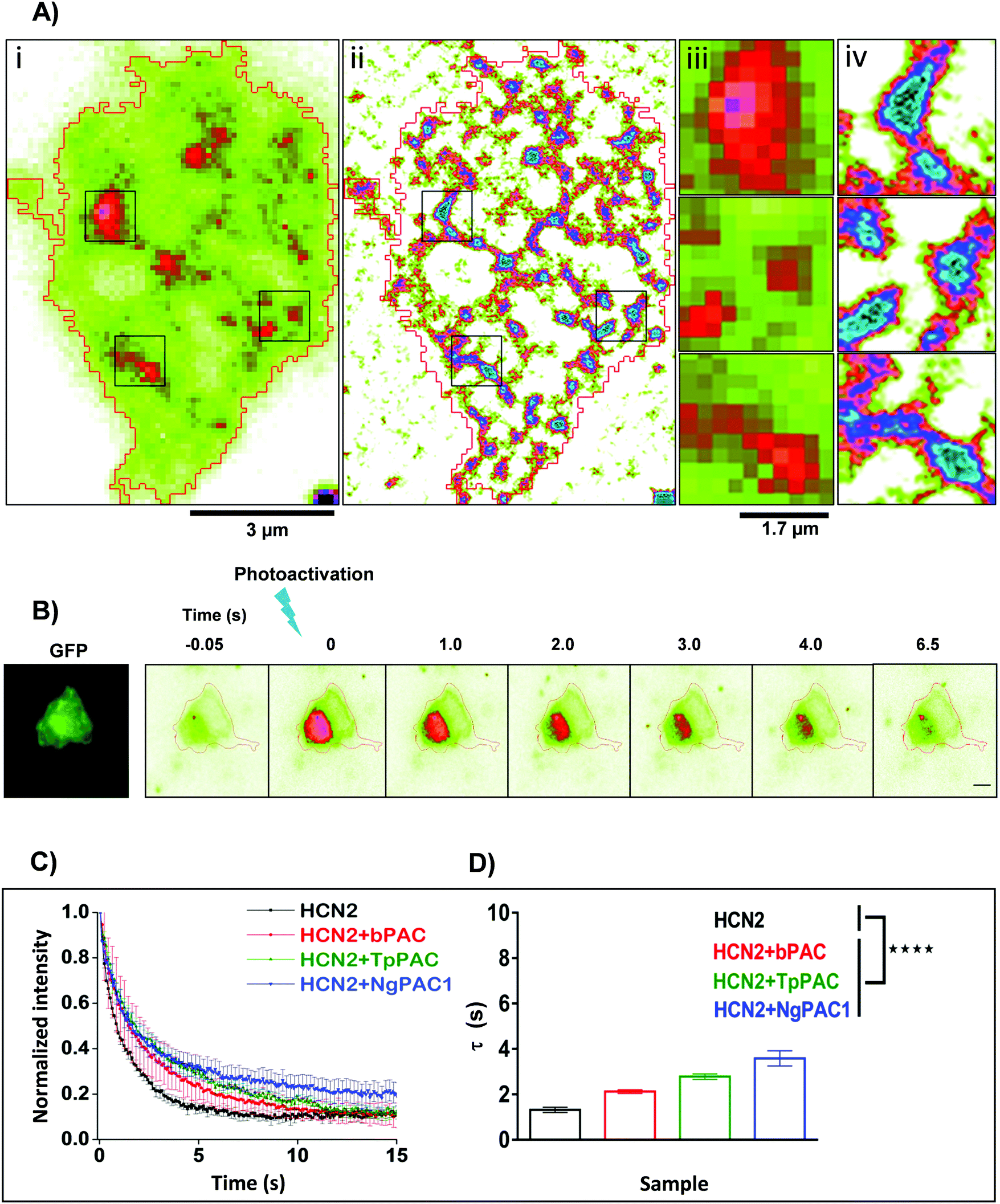 | ||
| Fig. 2 Targeted control of HCN2 dynamics upon optogenetic modulation of cAMP levels. (A) Wide field (i) and SRRF (ii) images of Neuro-2a cells expressing mEos::HCN2. To achieve high spatial resolution, wide field images were processed using SRRF (ESI†). Scale bar: 3 μm. Zoomed diffraction limited (iii) and SRRF (iv) images of selected regions outlined in black. Scale bar: 1.7 μm. (B) Neuro-2a cells expressing mEos::HCN2 + PAC imaged in the GFP channel (left panel). Representative temporal changes in fluorescence intensity within the photoactivated region upon activation of PAC and photoconversion of mEos molecules with 405 nm are depicted (right series). Scale bar: 10 μm. (C) Fluorescence decay curves representative of the temporal changes in fluorescence intensity upon photoactivation of PAC and photoconversion of mEos molecules are shown. (D) Comparison of fluorescence decay of mEos::HCN2 fit by a single exponential decay model under control condition and upon optogenetically increasing the cAMP level using PAC variants namely, bPAC, TpPAC and NgPAC1. τ represents the decay time constant (n = 11, mean ± s.e.m. from 3 biological replicates, ****p < 0.0001, Mann–Whitney test). The mobility of mEos::HCN2 decreased significantly upon elevating the intracellular cAMP by targeted photoactivation of PACs. The PAC variants modulated the mobility of HCN2, correlated with their photodynamics and amplitude of the light-gated cAMP level altered by the respective PAC. | ||
For optogenetic modulation of the cAMP level, targeted photoactivation was performed on cells co-expressing mEos::HCN2 and PACs (referred to as mEos::HCN2 + PACs), which transiently increased their fluorescence intensity that decayed exponentially (Fig. 2B and C). Targeted irradiation with 405 nm photoactivated PACs and simultaneously photoconverted mEos molecules from the dark to bright state. For targeted photoactivation, a region corresponding to the point spread function of the system was randomly selected at a distance of 2–5 μm from the periphery of the cell. It resulted in an increased concentration of cAMP in the cells, which bound to the HCN2 channels and regulated their mobility. We used PACs with the fastest kinetics namely, TpPAC and bPAC as well as the slowest PAC namely, NgPAC1 to further assess the effect of modulation of cAMP on the membrane dynamics of HCN2. Cells expressing mEos::HCN2 + TpPAC and mEos::HCN2 + bPAC displayed delayed fluorescence decay curves with τ = 2.9 ± 0.499 s and τ = 2.4 ± 0.618 s, respectively in contrast to cells expressing control mEos::HCN2, which did not induce any change in cAMP (Fig. 2C and D). On the other hand, mEos::HCN2 + NgPAC1 exhibited the slowest decay kinetics (τ = 3.4 ± 0.946 s, Fig. 2C and D). The diffusion of HCN2 depended on the amplitude and duration of increased cAMP level in the cells, defined by the photorecovery kinetics of the expressed PAC protein. The PAC variants with varying photorecovery kinetics altered the mobility of HCN2 and its corresponding fluorescence decay (τ) differentially (Fig. 2D and Table S2, ESI†). Among the different PACs that were photoactivated, HCN2 diffusion was the fastest for bPAC (Fig. 2C), which reduced for TpPAC and significantly reduced further for NgPAC1 (Table S2, ESI†), consistent with our previous observations where cAMP levels increased from bPAC to NgPAC1 (Fig. 1C and Table S1, ESI†). In summary, the PAC variants modulated the mobility of HCN2, correlated with their photodynamics and amplitude of the light-gated cAMP level altered by the respective PAC. The transient local increase of cAMP due to activation in the focal column could hinder the mobility of HCN2 molecules, which exchanged significantly slower in the presence of PACs compared to when mEos::HCN2 was expressed alone.
Acute and chronic photoactivation of PACs alters lateral diffusion of HCN2 channels
We extended the established optogenetic approaches to assess the effect of cAMP modulation on the lateral diffusion of HCN2 molecules by single particle tracking (SPT). In contrast to targeted illumination for optogenetics and photoconversion, we used a low intensity widefield excitation for Photoactivation Localization Microscopy (PALM) experiments which was performed on Neuro-2a cells expressing mEos::HCN2 and mEos::HCN2 + PAC. For single particle tracking of mEos::HCN2, molecules were photoconverted stochastically using a low intensity of 405 nm, and tracked by observing them in the photoconverted red channel as single molecules in TIRF mode. Diffraction limited TIRF images of mEos::HCN2 + PAC portrayed the membrane distribution of the protein (Fig. 3Ai). The super-resolved intensity image (Fig. 3Aii) and the single molecule trajectory map (Fig. 3Aiii and iv) obtained from individual localization and trajectories of mEos::HCN2 molecules of sequential images displayed the distribution of mEos::HCN2 with the formation of discrete HCN clusters. | ||
Fig. 3 Global spatio-temporal control of HCN2 mobility by modulating cAMP levels in live cells. (A) TIRF image (i), corresponding super-resolution intensity image (ii) and single particle trajectory map (iii) of Neuro-2a cells expressing mEos::HCN2 and bPAC. Super-resolution intensity image was procured from 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 images of single molecules from the photo-converted mEos red state. Single particle trajectory map displaying the individual trajectories of photo converted mEos::HCN2 molecules and thereby their mobility in Neuro-2a cells. Scale bar: 10 μm. Enlarged trajectory map corresponding to the boxed regions (iv). Scale bar: 2.8 μm. (B) Diffusion coefficient Dm of mEos::HCN2 under control conditions and upon elevation of cAMP level by activation of PAC variants namely, bPAC, TpPAC, and NgPAC1. Prior to PALM imaging, acute photoactivation of PACs was done using 488 nm (median, **p < 0.01, ***p < 0.001, Mann–Whitney test). (C) Effect of increase in cAMP level on the cumulative distribution of instantaneous diffusion coefficient of mEos::HCN2 compared to control conditions (n = 10, mean ± s.e.m from 2 biological replicates, ****p < 0.0001, 2-way Anova). (D) Mean square displacement (MSD) plots of mEos::HCN2 single molecule trajectories under control conditions and upon optogenetically increasing the cAMP level by photoactivation of different PACs (bPAC, TpPAC and NgPAC1). Error bars indicate cell to cell variability (n = 10, mean ± s.e.m from 2 biological replicates, ****p < 0.0001, 2-way Anova). (E) Temporal changes in the single molecule trajectories of mEos::HCN2 molecules with increase in cAMP level by chronic photoactivation of bPAC and simultaneous photoconversion of mEos with 405 nm. Representative data set from a single cell showing single particle trajectories of mEos::HCN2 between the indicated time points namely, 80, 160, 240, 320 and 400 s are displayed from left to right. With continuous increase in cAMP level, the diffusion of HCN2 molecules is decreased. (F) Temporal change in the diffusion coefficient (Dm) of mEos::HCN2 with simultaneous light regulated elevation in the cAMP level via photoactivation of bPAC (computed from E). (G) Cumulative distribution of instantaneous diffusion coefficient of mEos::HCN2 trajectories with a temporal increase in cAMP level for five subsequent time windows (computed from E, **p < 0.01, ***p < 0.001, 2-way Anova). (H) Time dependent decrease in MSD with temporal increase in cAMP level (computed from E, mean ± s.e.m of trajectories/time window frame, **p < 0.01, ***p < 0.001, 2-way Anova). Alteration in cellular cAMP levels by acute photoactivation of PACs by saturation stimulation or by chronic optical stimulation regulated the mobility of cAMP effector HCN2 channels in a spatio-temporal manner. 000 images of single molecules from the photo-converted mEos red state. Single particle trajectory map displaying the individual trajectories of photo converted mEos::HCN2 molecules and thereby their mobility in Neuro-2a cells. Scale bar: 10 μm. Enlarged trajectory map corresponding to the boxed regions (iv). Scale bar: 2.8 μm. (B) Diffusion coefficient Dm of mEos::HCN2 under control conditions and upon elevation of cAMP level by activation of PAC variants namely, bPAC, TpPAC, and NgPAC1. Prior to PALM imaging, acute photoactivation of PACs was done using 488 nm (median, **p < 0.01, ***p < 0.001, Mann–Whitney test). (C) Effect of increase in cAMP level on the cumulative distribution of instantaneous diffusion coefficient of mEos::HCN2 compared to control conditions (n = 10, mean ± s.e.m from 2 biological replicates, ****p < 0.0001, 2-way Anova). (D) Mean square displacement (MSD) plots of mEos::HCN2 single molecule trajectories under control conditions and upon optogenetically increasing the cAMP level by photoactivation of different PACs (bPAC, TpPAC and NgPAC1). Error bars indicate cell to cell variability (n = 10, mean ± s.e.m from 2 biological replicates, ****p < 0.0001, 2-way Anova). (E) Temporal changes in the single molecule trajectories of mEos::HCN2 molecules with increase in cAMP level by chronic photoactivation of bPAC and simultaneous photoconversion of mEos with 405 nm. Representative data set from a single cell showing single particle trajectories of mEos::HCN2 between the indicated time points namely, 80, 160, 240, 320 and 400 s are displayed from left to right. With continuous increase in cAMP level, the diffusion of HCN2 molecules is decreased. (F) Temporal change in the diffusion coefficient (Dm) of mEos::HCN2 with simultaneous light regulated elevation in the cAMP level via photoactivation of bPAC (computed from E). (G) Cumulative distribution of instantaneous diffusion coefficient of mEos::HCN2 trajectories with a temporal increase in cAMP level for five subsequent time windows (computed from E, **p < 0.01, ***p < 0.001, 2-way Anova). (H) Time dependent decrease in MSD with temporal increase in cAMP level (computed from E, mean ± s.e.m of trajectories/time window frame, **p < 0.01, ***p < 0.001, 2-way Anova). Alteration in cellular cAMP levels by acute photoactivation of PACs by saturation stimulation or by chronic optical stimulation regulated the mobility of cAMP effector HCN2 channels in a spatio-temporal manner. | ||
We relied on two different strategies to study the nanoscale mobility of HCN2. In the first case, we used a high power of 488 nm (acute photoactivation) which would saturate the optogenetic activity of PACs and increase the cAMP content in the cytoplasm. Then, we compared the mobility of mEos::HCN2 in unstimulated conditions versus upon saturation stimulation of cAMP. Saturation stimulation of cAMP was performed prior to PALM imaging by acute photoactivation of PACs with 100% of 488 nm laser. The mobility of mEos::HCN2 in live cells measured as the instantaneous diffusion coefficient (Dm = 0.105 ± 0.014 μm2 s−1) significantly reduced upon optical cAMP induction, compared to the basal condition (Fig. 3B). The instantaneous diffusion coefficients of mEos::HCN2 upon photostimulation of bPAC (mEos::HCN2 + bPAC), TpPAC (mEos::HCN2 + TpPAC), and NgPAC1 (mEos::HCN2 + NgPAC1) were 0.041 ± 0.004 μm2 s−1, 0.039 ± 0.007 μm2 s−1, and 0.029 ± 0.004 μm2 s−1, respectively (Fig. 3B). The cumulative distributions of the instantaneous diffusion coefficients of mEos::HCN2 + PACs demonstrated a left shift towards lower mobility upon stimulation, compared to the control mEos::HCN2 (Fig. 3C and Fig. S3A, ESI†). The plateau reached by the mean square displacement (MSD) curve of mEos::HCN2 + bPAC, mEos::HCN2 + TpPAC, and mEos::HCN2 + NgPAC1 represented the confined movement of molecules upon optically saturated cAMP stimulation via acute photoactivation of PACs (Fig. 3D; MSD plateau = 0.08 ± 0.009 μm2, 0.08 ± 0.011 μm2, 0.05 ± 0.006 μm2 for bPAC, TpPAC, and NgPAC1, respectively) in contrast to unstimulated conditions (Fig. 3D; MSD plateau = 0.14 ± 0.012 μm2). Between the PACs, there was no significant difference in the instantaneous diffusion coefficient (Dm) of mEos::HCN2 upon photoactivation (Table S2, ESI†). However, photoactivation of NgPAC1 resulted in a significant decrease in the mean square displacement and shift in cumulative frequency of the instantaneous diffusion coefficient of mEos::HCN2, compared to the other PACs (Table S2, ESI†). These data were in agreement with the ensemble diffusion, where a targeted stimulation resulted in slower mobility of HCN2 in the presence of PACs, especially NgPAC1, indicating confinement of HCN2 resulting from immobilization or trapping (Fig. 2C and D).
In the second case, we verified if a chronic increase in the cAMP levels by continuous optical stimulation using low illumination intensities could result in a progressive increase in the immobilization and trapping of HCN2 channels. We used a low intensity wide field illumination of 405 nm (10%) during PALM imaging, allowing both sparse photoconversion of Eos molecules and continuous synthesis of cAMP by mild photoactivation of PACs. This paradigm showed a progressive decrease in mobility as confirmed from the instantaneous diffusion coefficient and mean square displacement for all the trajectories, when observed in temporal blocks of 80 seconds. The trajectory map of different time windows from five continuous streams of PALM data displayed slower diffusion of mEos::HCN2 molecules (Fig. 3E), with the diffusion coefficient (Dm) of mEos::HCN2 + PACs decreasing with time (Fig. 3F). In accordance with this, the cumulative frequency distribution of the instantaneous diffusion coefficients of mEos::HCN2 + PAC trajectories demonstrated a shift towards lower mobility with time, showing an increased confinement upon exposure to 405 nm illumination (Fig. 3G). The MSD curves also displayed the confined movement of mEos::HCN2 + PACs, correlated with a continuous increase in cAMP level (Fig. 3H). Alternately, no significant change in the diffusion coefficient (Dm) and mean square displacement was observed with time for the control, namely mEos::HCN2 (Fig. S3B–D, ESI†). These studies indicated a temporal increase in the fraction of HCN2 molecules being immobilized at low illumination intensities of optogenetic activation, which tended to saturate with longer duration of activation.
The mobility changes of mEos::HCN2 on altering cAMP levels were also assessed by pharmacological application of forskolin29,30 to induce cAMP, as well as by directly adding a membrane permeable cAMP analog (8-Br-cAMP). Single particle experiments were performed in live cells expressing mEos::HCN2 before and after forskolin or 8-Br-cAMP application. The diffusion coefficient (Dm) of mEos::HCN2 decreased after elevating cAMP levels by forskolin or 8-Br-cAMP application, in accordance with the optical cAMP stimulation experiments (Fig. S4A, ESI†). The cumulative frequency distribution of instantaneous diffusion coefficient and mean square displacement of mEos::HCN2 molecules revealed decreased mobility of HCN2 after forskolin or 8-Br-cAMP application (Fig. S4B and C, ESI†). The results asserted our earlier observations that the diffusional modifications of HCN2 were indeed due to alterations in cellular cAMP levels whether by acute photoactivation of PACs by saturation stimulation or by chronic optical stimulation. Thus, the mobility of cAMP effector HCN2 channels was optogenetically modulated in a spatio-temporal manner.
To investigate further whether the dynamic distribution of HCN2 was a direct response to cAMP through cAMP-binding, we generated a deletion construct of mEos::HCN2 lacking the C-terminus cyclic nucleotide binding domain (CNBD) of HCN2 (denoted as mEos::HCN2ΔCNBD). The mobility of mEos::HCN2ΔCNBD was studied by optical stimulation as well as by pharmacological application of membrane permeable cAMP (Fig. S5, ESI†). Among the PAC variants, NgPAC1 photoactivation which exhibited the highest deviation for HCN2 mobility from the control was opted for optical stimulation. The instantaneous diffusion coefficient of mEos::HCN2ΔCNBD (Dm = 0.085 ± 0.014 μm2 s−1) remained similar upon optical elevation of cAMP by photoactivation of NgPAC1 (Fig. S5A, ESI†). The cumulative distribution of the instantaneous diffusion coefficient and the mean square displacement curve of mEos::HCN2ΔCNBD also reflected no deviation in mobility upon stimulation (Fig. S5B and C, ESI†). Consistently, the diffusion characteristics of mEos::HCN2ΔCNBD including the diffusion coefficient (Dm), cumulative distribution of instantaneous diffusion coefficient and mean square displacement remained unaltered upon application of 8-Br-cAMP, in accordance with optical studies (Fig. S5D–F, ESI†). A comparative analysis of the mobility kinetics of HCN2 channels and of the deletion construct lacking CNBD upon optical (Fig. S6A, C and E, ESI†) and pharmacological (Fig. S6B, D and F, ESI†) stimulation was performed. The mobility of mEos::HCN2 was significantly reduced on elevating the intracellular cAMP levels by stimulation. On the contrary, the diffusion dynamics of mEos::HCN2ΔCNBD remained unaltered in the presence of elevated cAMP. Together, these results confirmed the cAMP dependent alterations in HCN2 lateral diffusion to be mediated by its cyclic nucleotide binding domain. We confirmed our observations by generating two point mutants of HCN2, namely R591A and R591E, reported to have reduced sensitivity to changes in intracellular cAMP (Fig. S6G and H, ESI†).31,32 In contrast to wild type HCN2 whose mobility was significantly reduced to half by elevated cAMP levels, the point mutants (R591A and R591E) displayed slight alteration in their mobility upon stimulation, similar to the deletion construct (HCN2ΔCNBD) (Fig. S6G and H, ESI†). Our results confirmed that the deviations in HCN2 lateral diffusion was indeed specific and predominantly mediated by direct binding of cAMP.
To understand if the cAMP mediated regulation was specific to HCN2, we extended this study to assess the dynamics of the AMPA receptor subunit GluA1, another type of ion channel which is not known to directly bind to cAMP. Interestingly, upon application of 8-Br-cAMP, a slight regulation of GluA1 dynamics and increase in mobility was observed, contrary to HCN2 (Fig. S5G–I, ESI†). Previous reports have shown indirect regulation of GluA1 function by cAMP dependent PKA, which phosphorylates GluA1 subunits within their C terminus,33 which could explain this deviation in mobility. The results signified the importance of cAMP dependent differential regulation of diverse ion channels, indicating a universal mechanism for controlling membrane excitability.
Alteration in nanoscale segregation of HCN2 channels via optogenetic modulation of cAMP
To assess the effect of altering cAMP on the nanoscale organization of HCN2, super-resolution PALM and STORM (Stochastic Optical Reconstruction Microscopy) imaging were performed to determine the morphology and distribution of HCN2 at a spatial resolution of 30–50 nm. The experimental details have been provided in the ESI,† Section 1.3 and 1.4. The reconstructed super-resolved intensity localization maps from PALM experiments revealed mEos::HCN2 to be organized as discrete nanoclusters (enclosed within black regions) with a cluster area >0.0025 μm2 (Fig. 4A). The morphological characteristics including the area, average intensity, and total intensity of HCN2 clusters were extracted and analyzed between the control and stimulated conditions. The total area of the HCN2 clusters increased upon optical stimulation of bPAC, TpPAC, and NgPAC1 with reference to the control (Fig. 4B). Comparing the histogram of the distribution area of HCN2 clusters also indicated a significant increase in the cluster area as well as in the number of clusters upon stimulation of PACs (Fig. 4C and D). Concurrently, an increase in the total intensity of mEos::HCN2 nanoclusters was observed upon optical induction of cAMP (Fig. 4H). The total intensity distribution per cluster showed a right shift towards higher values upon cAMP induction by PAC variants, compared to the control (Fig. 4I and J). However, no significant change in the average intensity of HCN2 clusters or their distribution was observed with reference to the control upon stimulation (Fig. 4E–G). Between the different PACs, the morphological characteristics of the HCN2 nanodomains remained similar upon photoactivation (Table S2, ESI†).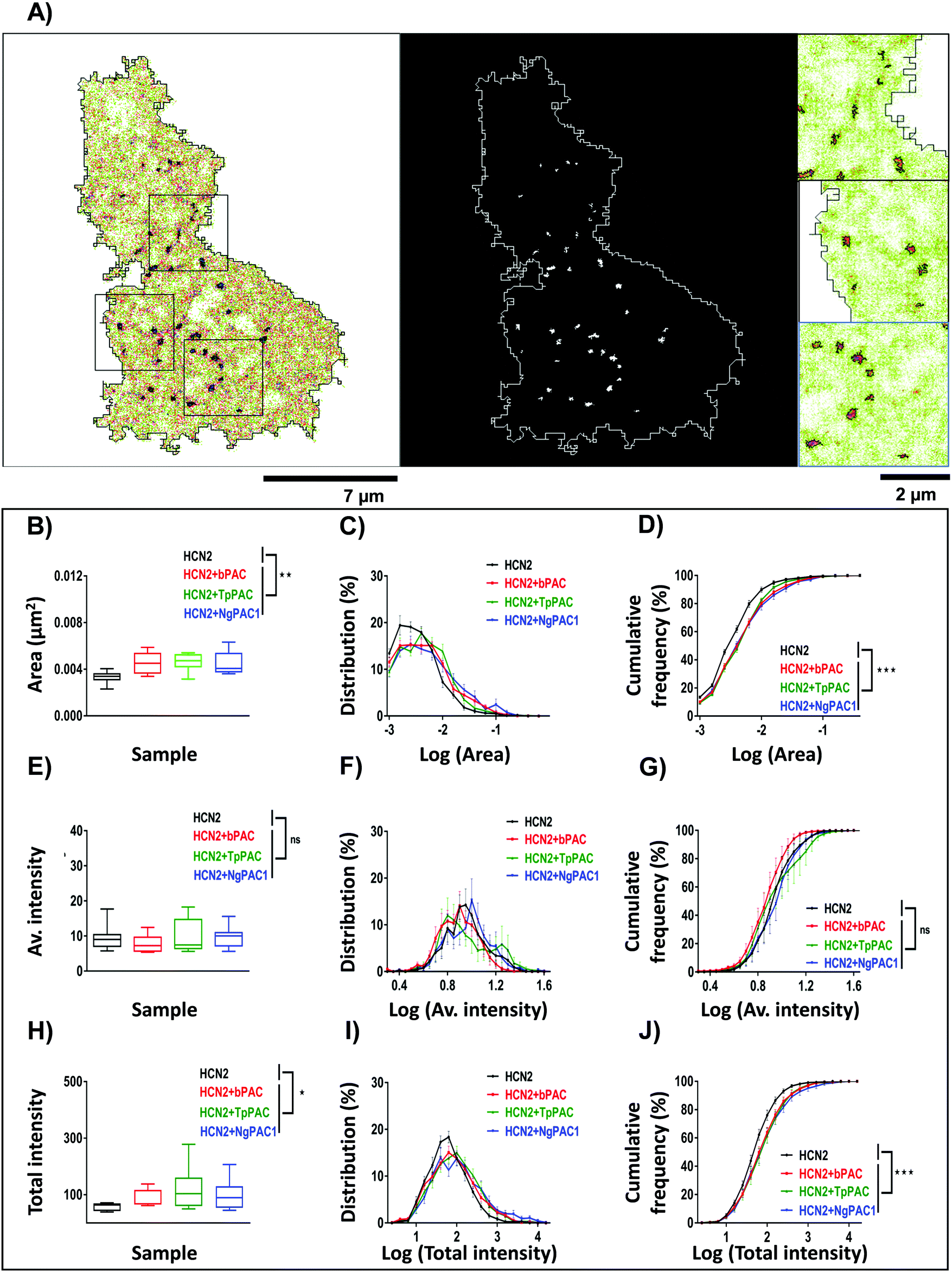 | ||
| Fig. 4 Modulation of nanoscale aggregation of HCN2 clusters upon light regulated elevation of cAMP. (A) Super-resolution intensity image (left panel) and segmented intensity map (middle panel) representing the clusters of mEos::HCN2 (outlined in black in the left panel, and white in the middle panel). Scale bar: 7 μm. Zoomed regions (right panel) from a representative cell marked by black boxes in the intensity image. Scale bar: 2 μm. (B, E, and H) Quantification of the area, average and total intensity of HCN2 clusters upon elevating the cAMP level compared to the control condition, respectively (n = 9, median, *p < 0.05, **p < 0.01, Mann–Whitney test). (C, F, and I) Probability distribution of area, average and total intensity of HCN2 clusters for the control and elevated cAMP condition, respectively. (D, G, and J) Cumulative frequency distribution plotted for the area, average and total intensity of HCN2 clusters for the control and upon increasing the cAMP level, respectively (n = 9, mean ± s.e.m from 2 biological replicates, ***p < 0.001, 2-way Anova). A significant increase in the cluster area, number and total intensity of mEos::HCN2 nanodomains was observed upon cAMP induction by PAC variants, compared to the control, in contrast to the average intensity of domains which remained unaltered. | ||
To understand if the nano-organization and mobility changes observed for HCN2 channels upon altering the cAMP levels were due to over-expression defects, the nanocluster organization for ectopically expressed and endogenous HCN2 clusters were assessed by STORM imaging. STORM imaging was performed on Neuro-2a cells over-expressing mEos::HCN2 (Fig. S7, ESI†) and on endogenous HCN2 (Fig. S8, ESI†) in the presence and absence of pharmacological application of forskolin to modulate cAMP. For STORM imaging, both untransfected and cells over-expressing mEos::HCN2 were labelled using HCN2 antibodies marked by Alexa-647. Quantitatively comparable results of morphological parameters of clusters namely, area, average and total intensity from control and forskolin treated cells were obtained. Comparison of HCN2 nanocluster sizes between control and forskolin treated cells for ectopically expressed mEos::HCN2 (Fig. S7A–C, ESI†) and endogenous HCN2 (Fig. S8A–C, ESI†) reflected an increase in size upon stimulation, in accordance with the PALM data. The total intensity of nanoclusters also increased for both ectopically expressed mEos::HCN2 (Fig. S7G–I, ESI†) and endogenous HCN2 (Fig. S8G–I, ESI†) after forskolin treatment, supporting the previous data. No significant change in the average intensity of domains was observed upon stimulation for mEos::HCN2 (Fig. S7D, ESI†) or endogenous HCN2 (Fig. S8D, ESI†). Interestingly, the histogram of average intensity distribution per nanocluster for ectopically expressed mEos::HCN2 (Fig. S7E and F, ESI†) and endogenous HCN2 (Fig. S8E and F, ESI†) shifted towards higher values upon stimulation. In summary, an increase in the area and intensity of HCN2 clusters was observed upon both optical and pharmacological stimulation of ectopically expressed as well as endogenous protein, overruling any over-expression artefacts and confirming that HCN2 channels responded to altering cAMP levels by modulating their localization and kinetics.
All these results emphasize a novel behavior of HCN2 i.e. its tendency to get immobilized with increasing levels of cAMP and self-organize into sub 100 nm sized nanoclusters. Though such an organization has not been reported previously for HCN2, ligand gated and voltage gated ion channels and several trans-membrane molecules including cell adhesion molecules, scaffolding molecules and molecules involved in degenerative diseases have been known to exist in similar functional membrane nanodomains.34–39 Similar to ligand gated receptors and voltage gated ion channels, HCN2 was also observed to undergo Brownian motion alternating between confined and free diffusional states, allowing us to observe the physiochemical map of molecular behavior in real-time with a precision of few tens of nanometers. It is very well known that the gating kinetics of HCN2 is altered upon binding to cAMP. It is thought that binding of cAMP could alter the conformation of HCN2 molecules.40 It has been proposed that the signalling state of the channel with zero, two or four ligands is much more stable than with one or three ligands.41 Thus, a transient increase in the ligand would prefer an increase in the number of channels which are in the stabilized state, which would also increase the immobilization kinetics of the channel. It is still unclear if there are complementary molecular mechanisms involving scaffolding molecules that could be involved in the cAMP dependent alteration of mobility of HCN2 channels. Akin to scaffolding slot molecules, these interacting molecules could modulate the strength of immobilization of these channels depending on the intracellular cAMP levels. However, our studies indicate the alteration of HCN2 channel mobility to be predominantly dependent on direct cAMP binding rather than indirect mechanisms including PKA signalling pathways.
In short, we demonstrate a paradigm to successfully use optogenetic probes to observe different physiochemical properties of a molecule of interest at nanometer resolution in living cells. We demonstrate that both acute and chronic stimulation of PACs can have differential effects on the HCN2 channel kinetics and clustering. Most of the molecular movements on the membrane are described by lateral diffusion governed by the rules of Brownian diffusion. Here, with the help of optogenetic approaches, we were able to control this random movement and modulate both the nanoscale localization and real-time mobility of HCN2 channels. We believe that similar paradigms to alter the chemical map of a cell with high spatio-temporal precision in combination with nanoscale imaging holds great potential in understanding the directionality of molecular signalling and the pre-requisites for this kind of signalling to occur. Though these mechanisms are vague at present, our studies open up the possibility to use a genetically encodable optical approach to study real-time molecular changes in nano-organization which could be extended to different molecular complexes, giving a higher control on manipulating their localization and trafficking dynamics.
Conclusions
Ability to manipulate biochemical pathways in living cells with molecular precision in real-time has been a challenge for cellular biologists. The multiple signalling cascades that are modulated and amplified by second messengers make this an orchestra of finely regulated molecular events. In recent years, with the advent of optogenetics, there has been a surge towards improving the spatial and temporal precision for controlling the local availability of second messengers to manipulate signalling cascades within a cell. However, due to the ensemble nature of photoactivation and reporting paradigms based on conventional microscopy, most of the previous attempts have been limited to understanding the changes in an ensemble of molecules that are involved in a signal cascade. Here, using novel genetic encoders that elevate intracellular cAMP levels upon photoactivation combined with multiple modes of microscopy, we illustrate the potential to assess both the ensemble and single molecule behavior of molecules of interest whose physiochemical properties could be altered by optogenetic activation of second messengers. In the present work using optogenetic variants of photoactivated adenylyl cyclases, we have optically modulated cAMP and shown that HCN2 channels are segregated into nanoclusters controlled by lateral diffusion and transient trapping. By optogenetic and pharmacological manipulation of intracellular cAMP, we show that the molecular properties which regulate the spatio-temporal behavior of HCN2 channels are altered and that these changes are mediated through the cyclic nucleotide binding domain of HCN2. Using HCN2 point mutants displaying low sensitivity to cAMP and truncated HCN2 lacking CNBD, we confirmed the alterations in lateral organization and mobility of these channels to be specific to direct binding of cAMP. Though it is well understood that HCN channels react differentially to cAMP levels, such changes in immobilization and lateral organization of these channels have not yet been reported. Elucidating the spatial organization of HCN channels makes us comprehend the role of alteration of ionic conductivity at the molecular scale, an important step towards understanding how the plasma membrane maintains its instantaneous permeability to different ions. The functional relevance of nanoscale clustering of HCN channels remains vague at this point; but it opens interesting questions on their regulation in response to varying cAMP levels and how this could contribute to the diverse functions involved. Our observation of altering GluA1 kinetics to intracellular cAMP also signifies the relevance as to whether the dynamic regulation of these biomolecules in response to cAMP would be a universal mechanism for controlling membrane excitability. In summary, merging super-resolution microscopy with optogenetics has enabled us to bring deeper insight into the real-time molecular basis of localization and trafficking of HCN channels and their response to dynamic cAMP signal transduction at the nanoscale level. We believe that a similar combination of nanoscopy with optogenetic modulation will provide unprecedented spatio-temporal information on the local biochemical maps that modulate molecular function and behavior in the native biological environment.Experimental section
Materials and methods
:1000, Sigma, T8328) and rabbit anti-HCN2 (1:800, Alomone, APC-030) marked with Alexa 488-conjugated anti-mouse IgG (1:500; Invitrogen, A11029) and Alexa 647-conjugated anti-rabbit IgG (1:500; Invitrogen, A21245), respectively. For imaging of ectopically expressed HCN2, cells were transfected with mEos::HCN2 using Turbofect reagent prior to fixation and labelling with anti-HCN2 antibodies.
Imaging and analysis
Details of ensemble and single molecule imaging and analysis is provided in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Bioimaging Facility for allowing us to use their microscopy resources and the Central Animal Facility at IISc, Bangalore for the animals for the experiments conducted. We are thankful to Dr Jean-Baptistse Sibarita, University of Bordeaux, for allowing us to use the custom-made plugin for single particle tracking analysis. We are grateful to the Science and Engineering Research Board (DST-SERB) for the Early Career Research Award to M. J., the Department of Biotechnology for Ramalingaswami Fellowships to M. J., and D. N., the Har Gobind Khorana Innovative Young Biotechnologist Award to M. J., the DBT-RA program fellowship in Biotechnology and Life Sciences to M. T., the DBT-IISc Partnership Program (D. N.), the DBT-Genetic Engineering Taskforce (D. N.), the MHRD-IISc-STAR program grant (S. K., D. N.) and the Tata Program Grant for generous funding given to execute the project, to create the infrastructure and for human resource management.Notes and references
- K. M. Walton and R. P. Rehfuss, Mol. Neurobiol., 1990, 4, 197–210 CrossRef CAS PubMed.
- E. R. Kandel, Mol. Brain, 2012, 5, 14 CrossRef CAS PubMed.
- J. A. Beavo and L. L. Brunton, Nat. Rev. Mol. Cell Biol., 2002, 3, 710–718 CrossRef CAS PubMed.
- P. A. Insel, L. Zhang, F. Murray, H. Yokouchi and A. C. Zambon, Acta Physiol., 2012, 204, 277–287 CrossRef CAS.
- H. Rehmann, A. Wittinghofer and J. L. Bos, Nat. Rev. Mol. Cell Biol., 2007, 8, 63–73 CrossRef CAS PubMed.
- S. S. Taylor, R. Ilouz, P. Zhang and A. P. Kornev, Nat. Rev. Mol. Cell Biol., 2012, 13, 646–658 CrossRef CAS PubMed.
- J. L. Bos, Trends Biochem. Sci., 2006, 31, 680–686 CrossRef CAS PubMed.
- U. B. Kaupp and R. Seifert, Physiol. Rev., 2002, 82, 769–824 CrossRef CAS.
- R. B. Robinson and S. A. Siegelbaum, Annu. Rev. Physiol., 2003, 65, 453–480 CrossRef CAS PubMed.
- C. Wahl-Schott and M. Biel, Cell. Mol. Life Sci., 2009, 66, 470–494 CrossRef CAS PubMed.
- M. Iseki, S. Matsunaga, A. Murakami, K. Ohno, K. Shiga, K. Yoshida, M. Sugai, T. Takahashi, T. Hori and M. Watanabe, Nature, 2002, 415, 1047–1051 CrossRef CAS PubMed.
- M. Stierl, P. Stumpf, D. Udwari, R. Gueta, R. Hagedorn, A. Losi, W. Gartner, L. Petereit, M. Efetova, M. Schwarzel, T. G. Oertner, G. Nagel and P. Hegemann, J. Biol. Chem., 2011, 286, 1181–1188 CrossRef CAS PubMed.
- A. Penzkofer, M. Stierl, P. Hegemann and S. Kateriya, Chem. Phys., 2011, 387, 25–38 CrossRef CAS.
- A. Penzkofer, M. Tanwar, S. K. Veetil, S. Kateriya, M. Stierl and P. Hegemann, Chem. Phys., 2013, 423, 192–201 CrossRef CAS.
- A. Penzkofer, M. Tanwar, S. K. Veetil, S. Kateriya, M. Stierl and P. Hegemann, J. Photochem. Photobiol., A, 2014, 287, 19–29 CrossRef CAS.
- A. Penzkofer, M. Tanwar, S. K. Veetil and S. Kateriya, J. Photochem. Photobiol., B, 2015, 153, 90–102 CrossRef CAS PubMed.
- A. Penzkofer, M. Tanwar, S. K. Veetil and S. Kateriya, Trends Appl. Spectrosc., 2014, 11, 39–62 CAS.
- M. Ohki, K. Sugiyama, F. Kawai, H. Tanaka, Y. Nihei, S. Unzai, M. Takebe, S. Matsunaga, S. Adachi, N. Shibayama, Z. Zhou, R. Koyama, Y. Ikegaya, T. Takahashi, J. R. Tame, M. Iseki and S. Y. Park, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6659–6664 CrossRef CAS PubMed.
- S. Schroder-Lang, M. Schwarzel, R. Seifert, T. Strunker, S. Kateriya, J. Looser, M. Watanabe, U. B. Kaupp, P. Hegemann and G. Nagel, Nat. Methods, 2007, 4, 39–42 CrossRef PubMed.
- S. Weissenberger, C. Schultheis, J. F. Liewald, K. Erbguth, G. Nagel and A. Gottschalk, J. Neurochem., 2011, 116, 616–625 CrossRef CAS PubMed.
- A. Hartmann, R. D. Arroyo-Olarte, K. Imkeller, P. Hegemann, R. Lucius and N. Gupta, J. Biol. Chem., 2013, 288, 13705–13717 CrossRef CAS PubMed.
- Z. Zhou, K. F. Tanaka, S. Matsunaga, M. Iseki, M. Watanabe, N. Matsuki, Y. Ikegaya and R. Koyama, Sci. Rep., 2016, 5, 19679 CrossRef PubMed.
- M. Tanwar, L. Khera, N. Haokip, R. Kaul, A. Naorem and S. Kateriya, Sci. Rep., 2017, 7, 12048 CrossRef PubMed.
- F. Zhang and E. S. Tzanakakis, Sci. Rep., 2017, 7, 9357 CrossRef PubMed.
- W. Steuer Costa, S. C. Yu, J. F. Liewald and A. Gottschalk, Curr. Biol., 2017, 27, 495–507 CrossRef CAS PubMed.
- K. Harada, M. Ito, X. Wang, M. Tanaka, D. Wongso, A. Konno, H. Hirai, H. Hirase, T. Tsuboi and T. Kitaguchi, Sci. Rep., 2017, 7, 7351 CrossRef PubMed.
- A. Ludwig, X. Zong, M. Jeglitsch, F. Hofmann and M. Biel, Nature, 1998, 393, 587–591 CrossRef CAS PubMed.
- V. Beaumont and R. S. Zucker, Nat. Neurosci., 2000, 3, 133–141 CrossRef CAS PubMed.
- K. B. Seamon, W. Padgett and J. W. Daly, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 3363–3367 CrossRef CAS.
- R. H. Alasbahi and M. F. Melzig, Pharmazie, 2012, 67, 5–13 CAS.
- S. Chen, J. Wang and S. A. Siegelbaum, J. Gen. Physiol., 2001, 117, 491–504 CrossRef CAS PubMed.
- L. Zhou and S. A. Siegelbaum, Structure, 2007, 15, 655–670 CrossRef CAS PubMed.
- T. G. Banke, D. Bowie, H. Lee, R. L. Huganir, A. Schousboe and S. F. Traynelis, J. Neurosci., 2000, 20, 89–102 CrossRef CAS PubMed.
- D. Nair, E. Hosy, J. D. Petersen, A. Constals, G. Giannone, D. Choquet and J. B. Sibarita, J. Neurosci., 2013, 33, 13204–13224 CrossRef CAS PubMed.
- S. Kedia, P. Ramakrishna, P. R. Netrakanti, M. Jose, J. B. Sibarita, S. Nadkarni and D. Nair, Nanoscale, 2020, 12, 8200–8215 RSC.
- O. Rossier, V. Octeau, J. B. Sibarita, C. Leduc, B. Tessier, D. Nair, V. Gatterdam, O. Destaing, C. Albiges-Rizo, R. Tampe, L. Cognet, D. Choquet, B. Lounis and G. Giannone, Nat. Cell Biol., 2012, 14, 1057–1067 CrossRef CAS PubMed.
- S. Venkatesan, S. Subramaniam, P. Rajeev, Y. Chopra, M. Jose and D. Nair, eNeuro, 2020, 7 Search PubMed.
- J. Heck, P. Parutto, A. Ciuraszkiewicz, A. Bikbaev, R. Freund, J. Mitlohner, M. Andres-Alonso, A. Fejtova, D. Holcman and M. Heine, Neuron, 2019, 103, 66–79 e12 CrossRef CAS PubMed.
- M. Heine, J. Heck, A. Ciuraszkiewicz and A. Bikbaev, Neuropharmacology, 2020, 169, 107556 CrossRef CAS PubMed.
- W. N. Zagotta, N. B. Olivier, K. D. Black, E. C. Young, R. Olson and E. Gouaux, Nature, 2003, 425, 200–205 CrossRef CAS PubMed.
- J. Kusch, S. Thon, E. Schulz, C. Biskup, V. Nache, T. Zimmer, R. Seifert, F. Schwede and K. Benndorf, Nat. Chem. Biol., 2011, 8, 162–169 CrossRef PubMed.
- E. Lou, S. Fujisawa, A. Morozov, A. Barlas, Y. Romin, Y. Dogan, S. Gholami, A. L. Moreira, K. Manova-Todorova and M. A. S. Moore, PLoS One, 2012, 7(3), e33093 CrossRef CAS.
- K. Harada, M. Ito, X. Wang, M. Tanaka, D. Wongso, A. Konno, H. Hirai, H. Hirase, T. Tsuboi and T. Kitaguchi, Sci. Rep., 2017, 7(1), 7351 CrossRef.
- G.-J. Kremers, K. L. Hazelwood, C. S. Murphy, M. W. Davidson and D. W. Piston, Nat. Methods, 2009, 6(5), 355–358 CrossRef CAS PubMed.
- J. Chen, J. S. Mitcheson, M. Lin and M. C. Sanguinetti, J. Biol. Chem., 2000, 275(46), 36465–36471 CrossRef CAS PubMed.
- M. Zhang, H. Chang, Y. Zhang, J. Yu, L. Wu, W. Ji, J. Chen, B. Liu, J. Lu, Y. Liu, J. Zhang, P. Xu and T. Xu, Nat. Methods, 2012, 9(7), 727–729 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Figures and Tables, Supplementary Experimental section, Abbreviations. See DOI: 10.1039/d0cb00124d |
| This journal is © The Royal Society of Chemistry 2021 |