
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiposomes loaded with polyphenol-rich grape pomace extracts protect from neurodegeneration in a rotenone-based in vitro model of Parkinson's disease†
Attilio
Marino‡
 *a,
Matteo
Battaglini‡
a,
Andrea
Desii
a,
Chiara
Lavarello
bc,
Giada
Genchi
a,
Andrea
Petretto
b and
Gianni
Ciofani
*a
*a,
Matteo
Battaglini‡
a,
Andrea
Desii
a,
Chiara
Lavarello
bc,
Giada
Genchi
a,
Andrea
Petretto
b and
Gianni
Ciofani
*a
aIstituto Italiano di Tecnologia, Smart Bio-Interfaces, Viale Rinaldo Piaggio 34, 56025 Pontedera, Italy. E-mail: attilio.marino@iit.it; gianni.ciofani@iit.it
bIRCCS Istituto Giannina Gaslini, Core Facilities-Clinical Proteomics and Metabolomics, Via Gerolamo Gaslini 5, 16147 Genova, Italy
cUniversity of Genoa, Department of Chemistry and Industrial Chemistry, Via Dodecaneso 31, 16146 Genova, Italy
First published on 30th September 2021
Abstract
Parkinson's disease (PD) is a progressive neurodegenerative disease with no satisfactory therapy options. Similar to other neurodegenerative conditions, such as Alzheimer's and Huntington's diseases, oxidative stress plays a key factor in the neurodegeneration process. To counteract the uncontrolled increase of reactive oxygen species (ROS) and oxidative stress-dependent cell death, several preclinical and clinical tests exploit natural-derived organic antioxidants, such as polyphenols. Despite some promising results, free antioxidants show scarce brain accumulation and may exhaust their scavenging activity before reaching the brain. In this work, we developed an antioxidant therapeutic nanoplatform consisting of nano-sized functionalized liposomes loaded with selected polyphenol-rich vegetal extracts with high blood–brain barrier crossing capabilities. The antioxidant extracts were obtained from the grape seeds and skins as a byproduct of wine production (i.e., pomace), following a sustainable circular approach with reduced environmental impact. The antioxidant nanoplatform was successfully tested in a relevant in vitro model of PD, where it completely rescued the ROS levels, prevented the aggregation of α-synuclein fibrils, and restored cell viability, paving the way for preclinical translation of the approach.
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disease after Alzheimer's Disease (AD), affecting between 0.1% and 0.2% of the world population.1 PD is characterized at the cellular level by the degeneration of the dopaminergic neurons of the substantia nigra, and this phenomenon is at the basis of the movement impairments commonly associated with the disease, such as resting tremor, postural instability, and bradykinesia.2 The main hallmarks of the disease include aggregation and accumulation of the α-synuclein (α-syn) protein, mitochondrial dysfunctions, and impairments in protein clearance pathways such as ubiquitin-proteasome.3–5 A high level of reactive oxygen species (ROS) has been identified as a key element of PD.4 ROS are highly reactive molecules at the basis of several biological functions and under physiological conditions, they are regulated by mechanisms such as antioxidant enzymes (e.g., catalase, superoxide dismutase) and ROS scavenging molecules (e.g., glutathione).6–8 However, when antioxidant defense mechanisms fail to keep ROS levels under control, their overproduction can induce damage to biological macromolecules, including proteins, lipids, or nucleic acids.7,9 High levels of ROS are commonly associated with neurodegenerative disease due to the high oxygen consumption occurring in the central nervous system (CNS) and the relatively low concentration of antioxidant molecules present in neurons.10,11 In the case of PD, it has been hypothesized that the auto-oxidation of dopamine present in the dopaminergic neurons of the substantia nigra could be one of the key factors leading to PD pathogenesis. ROS induce the misfolding of α-syn monomers, which in turn causes their aggregation and, consequently, mitochondrial dysfunctions. In turn, α-syn aggregates and mitochondrial dysfunctions can induce the generation of ROS in a continuous cycle leading to neuronal cell death.4,12–15 Currently, there is no cure for PD, and the main treatment of the disease involves the use of 3,4-dihydroxyl-phenylalanine (L-DOPA) to counteract the motor symptoms typical of PD.16 Antioxidant molecules such as melatonin, vitamin E, coenzyme Q10, and creatine have been proposed as a potential treatment of the disease, and some antioxidant molecules like polyphenols (e.g., resveratrol), glutathione, and N-acetylcysteine have also been investigated in clinical trials for the treatment of PD.15,17In this regard, grapes, and especially their seeds and skin, represent one of the richest natural sources of antioxidant phenolic compounds.18–20 Grape pomace, a mixture of seeds and skins obtained by pressing grapes, represents a by-product of wine production characterized by a relatively high concentration of phenolics,21 being the skins rich in anthocyanins and the seeds in non-anthocyanin molecules.22 The cost-effective and sustainable conversion of the grape pomace into valuable antioxidant extracts is a “green” paradigm adopted in food, cosmetic, and pharmaceutical industries to reduce the environmental impact of waste disposal in winemaking.23 It is well-known that polyphenols possess ROS scavenging properties in cells. The work of Brezoiu et al. recently demonstrated enhanced scavenger activity through the encapsulation of phenolic extract in silica matrices with respect to the free extract alone.24 The encapsulation of phenolic compounds is also considered an efficient strategy to increase their bioavailability25 and promote the crossing of the blood–brain barrier (BBB),26 a biological barrier that hinders the ability of many antioxidant therapeutic molecules to reach the brain.
The term BBB refers to a continuous barrier mainly formed by brain endothelial cells and other cells such as astrocytes and pericytes that envelopes the brain separating the CNS from the bloodstream.27,28 This separation is achieved mainly by the presence of molecular structures called tight junctions present in the contact region between brain endothelial cells and limiting the paracellular transport of substances.27,28 The BBB is a pivotal structure due to its ability to prevent potentially dangerous substances from reaching the brain. However, BBB is also one of the main obstacles for the delivery of potentially therapeutic moieties limiting the passage of all large molecules drugs and of approximately 98% of small molecules drugs.29
The use of nanostructures has been proposed as a means to improve the brain targeting efficiency of antioxidant molecules. Thanks to their physiochemical properties, nanostructures can be used as drug carriers able to encapsulate therapeutic moieties improving their bioavailability and pharmacokinetics, while reducing possible adverse effects. Moreover, nanostructure surfaces can be functionalized with specific molecules able to increase their targeting efficiency.30,31 In particular lipid-based nanostructures represent one of the main studied classes of nanovectors in brain targeting.15,32 Among these, liposomes are the most common lipid-based carriers exploited in human healthcare, being the first nanostructures approved by the FDA for cancer treatment and with several examples of liposomes being currently investigated for the treatment of different neurological disorders.15,33,34 Liposomes are normally composed of a mixture of amphiphilic phospholipids, cholesterol, and other lipid molecules assembled into one or more lipid bilayers and characterized by an aqueous core. Due to the presence of a water-based core, liposomes are commonly used to encapsulate hydrophilic molecules.15,31,32 Examples of liposomes loaded with antioxidant molecules as potential candidates for the treatment of brain disorders include quercetin-loaded and curcumin-modified liposomes for the treatment of AD, quercetin-loaded unilamellar liposomes for the treatment of ischemia, and ascorbic acid- and α-tocopherol-loaded liposomes as a protective agent against ischemic stroke.15,35–37
In this work, we developed a novel liposome formulation loaded with antioxidant molecules able to specifically target and cross the BBB and to revert some of the ROS-induced damage present in an in vitro toxin-induced model of PD. The liposome surface was functionalized to increase the BBB targeting and crossing efficiency of our formulation. Liposomes were loaded with antioxidant-rich grape pomace extracts obtained from Cinque Terre land, where the distinctive features of soils are known to generate wines with high antioxidant content.38 The use of grape pomace, a byproduct of wine production rich in polyphenols, as a source of antioxidants represents an example of a sustainable and circular process in which the resulting solid wastes can be finally composted for producing fertilizers. The BBB crossing efficiency of the extract-loaded liposomes (ext-LSs) was assessed on a multicellular in vitro model of the BBB. Moreover, the potential efficacy of ext-LSs for the treatment of PD was assessed on a rotenone-induced in vitro PD model. Rotenone is an inhibitor of the mitochondrial electron transport chain complex I commonly used to induce PD-like features both in vivo and in vitro.39–41 In particular, we assessed the ability of ext-LSs to reduce ROS, to improve cell viability, and to hinder α-syn aggregation in the in vitro PD model.
Experimental section
Extract preparation and characterization
Pomaces from white grape (WG) and from black grape (BG) varieties (Vitis vinifera) cultured in the Cinque Terre territories (La Spezia, Liguria, Italy) were used as a natural source of polyphenols. Specifically, the mixture of the WG varieties included 60% of Albarola, 30% of Vermentino, and 10% of Bosco, while the BG mixture 50% Cigliegiolo, and 50% Cannaiolo. The hydroalcoholic extraction was carried out as indicated elsewhere, with some modifications.42 Specifically, the pomaces were blended by using a mincer (Imetech CH 3000; 1000 W), ground with mortar and pestle, and dispersed in a 50% v/v water–ethanol solution (25% w/v). The extraction solution was shaken with a vortex for 15 min (200 rpm) and stirred for 16 h. The extraction solution was subsequently filtered with filter papers (nominal basis weight 75 ± 5 g m−2; Forlab), centrifuged (5000 rpm; Universal 320 R Hettich), and the supernatant was collected with a serological pipette.For the analysis of the total phenol content (TPC) and the trolox equivalent antioxidant capacity (TEAC), the hydroalcoholic solutions from WG and BG pomace were freeze-dried to quantify the solid extract and then suspended in water. The TPC was determined by using the Folin–Ciocalteu colorimetric assay (Sigma). Folin–Ciocalteu phenol quantification43 was carried out on 3 concentrations (0.375, 0.750, and 1.500 mg mL−1) of each extract (from WG and BG pomaces), absorbance was measured at λ = 800 nm by using a multiplate reader (Victor3, PerkinElmer), and the polyphenol concentration of each sample was expressed as resveratrol equivalent per g of dry weight by using a calibration curve (Fig. S1a†). The TEAC of WG and BG pomace extracts was measured by using a colorimetric TEAC assay kit based on the Cu2+ reduction (Sigma) as described in a previous work.44 The absorbance was measured at λ = 570 nm through a microplate reader (Victor3, PerkinElmer), values were corrected by subtracting the blank, and TEAC results were expressed as μmol of trolox equivalent per mg of dry weight by using a calibration curve (Fig. S1b†).
High-performance liquid chromatography (HPLC) was performed as previously described in the literature45 to quantify the presence of individual phenolic compounds (i.e., gallic acid, catechins, and epicatechins) in the extracted solutions. Briefly, an LC-20 Shimadzu system equipped with a C-18 column (150 mm × 4.6 mm i.d., 5 μm particle size) was used. The mobile phases were 2.0% acetic acid in ddH2O (phase A) and acetonitrile (phase B). The column was eluted at a 1.0 mL min−1 rate by using a linear gradient from 5% to 75% of B in 20 min, to 100% in 5 min, isocratic for 5 min, and to 5% in 10 min. 20 μl of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 dilution samples were injected and compounds were detected at λ = 254 nm. The concentrations of gallic acid, catechin, and epicatechin were quantified using purified compounds (Sigma) as standards.
:10 dilution samples were injected and compounds were detected at λ = 254 nm. The concentrations of gallic acid, catechin, and epicatechin were quantified using purified compounds (Sigma) as standards.
The molecular composition of the extracts was analyzed by using a Vanquish Horizon UHPLC coupled to Q-Exactive Orbitrap mass spectrometer. The extracted molecules were diluted (1:10 dilution) in methanol and 5 μl samples were directly injected into the reverse phase (RP) column. The molecular separation was carried out at 40 °C with an ACQUITY C18 BEH 1.7 μm 2.1 × 100 mm column (Waters S.p.A.). The linear gradient started from 1% B phase (acetonitrile, 0.1% formic acid) and 99% A (H2O, 0.1% formic acid) to 100% B in 15 min with a 250 μl min−1 flow rate, then the columns were stabilized for 5 minutes with 1% phase B. The experiments were performed in data-dependent acquisition mode alternating full MS and MS/MS scans. The precursors were ionized using an electrospray at −3.5 kV and the inlet capillary temperature was held at 300 °C. Nitrogen sheath gas and nitrogen auxiliary gas were set at a flow rate of 30 and 10 arbitrary units (AU), respectively. Single MS survey scans were performed in the Orbitrap, recording a mass window between 70 and 1000 m/z with automatic gain control (AGC) target of 106, at the maximum injection time of 100 ms and a resolution of 35000 at 200 m/z. Data-dependent MS/MS analysis was performed in top speed mode with a 2 s cycle-time with an isolation window of 1.2 m/z and an exclusion list for 2 s. The intensity threshold was set at 1.6 × 105 using an isolation window of 1.4 Da. 17500 resolution, 105 AgC, and 50 ms maximum injection time were used for MS2 scan. Raw data files were processed by Compound Discoverer™ 3.1 software. Briefly, raw files were aligned with adaptive curve setting with 5 ppm mass tolerance and 0.8 min retention time shift. Unknown compounds were detected with a 5 ppm mass tolerance, 3 signal to noise ratio, 30% of relative intensity tolerance for isotope search, 300000 minimum peak intensity, and then grouped with 5 ppm mass and 0.2 min retention time tolerances. A procedural blank sample was used for background subtraction. Peak areas, across all samples, were subsequently normalized to the total area of the corresponding samples and then for the extract concentration. Molecules were identified by using the mzCloud spectral library. Only the best match higher than 85 were considered.
Liposome fabrication, functionalization, and characterization
Extract-loaded liposomes (ext-LSs) were fabricated by using the thin-film evaporation method similarly as described in a previous work.46 Specifically, 5 mg of brain lipids (Avanti Polar Lipids, Inc.) and 10 mg of a biotin-conjugated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(polyethylene glycol)-5000 (biotin-PEG-DSPE; Avanti Polar Lipids, Inc.) were dissolved in 2 mL of chloroform and then transferred to an orbital shaker for evaporation (overnight at room temperature). The obtained dried lipids were subsequently re-hydrated in 2 mL of a water solution with 4 mg mL−1 extracts, extruded through a 100 nm pore polycarbonate membrane (Avanti® Mini Extruder; 11 passages through the membrane were performed), and finally dialyzed (300 kDa molecular weight cut off; Spectrum Laboratories, Inc.). Non-loaded liposomes (LSs) were used as a control for biocompatibility studies, ROS level measurement, and for the assessment of apoptosis/necrosis.Liposome functionalization with an anti-transferrin receptor antibody was achieved through the biotin–streptavidin interaction between the biotin-PEG-DSPE and the streptavidin-conjugated Ab (anti-TfR Ab; 25 μg mL−1; Abcore) as described and characterized in previous works.46,47 A fluorescence staining of the liposomes with the lipophilic DiO dye (1:25 v/v dilution in a 5 mg mL−1 liposome dispersion; Vybrant™) was performed for the cell up-take analysis and the blood–brain barrier (BBB) crossing investigations. After staining, liposomes were purified as described above.
For scanning electron microscopy (SEM) imaging, A 20 μl drop of a diluted dispersion (30 μg mL−1) of ext-LSs and plain LSs was deposited and let dry on a glass coverslip under a sterile hood. Then, the sample was gold-sputtered (60 nA for 20 s), and SEM imaging was performed with a Helios NanoLab 600i FIB/SEM, FEI.
Transmission electron microscopy (TEM) imaging of ext-LSs and plain LSs was carried out with a JEOL 1011. For sample preparation, a drop of the liposome dispersion was deposited on an amorphous carbon film-coated plasma-treated Cu grid. The grid was then treated with 1% uranyl acetate solution in H2O.
The hydrodynamic size and the Z-potential of ext-LSs and plain LSs (80 μg mL−1 dispersion) were measured with a Zetasizer Nano ZSP (Malvern Instrument).
To investigate the concentration of polyphenols loaded in ext-LSs, the polyphenol concentration of the dialyzed solution was analyzed by following the Folin–Ciocalteu method above described; the concentration of the liposome-loaded polyphenols was calculated by subtraction.
The polyphenol release profile of 3 mg ml−1 ext-LSs in PBS was instead investigated at 37 °C in different experimental conditions: pH 7.4, pH 4.5, pH 7.4 with 50 μM H2O2, and pH 4.5 with 50 μM H2O2. The concentration of the released polyphenols was assessed at different time points (4, 8, 24, and 48 h) by performing the Folin–Ciocalteu assay on the dialysate solution (Amicon® centrifuge filters; Ultra-4 Centrifugal Filter Unit [MWCO 100 kDa], Sigma-Aldrich). The cumulative release was calculated as the sum of the polyphenol amount at a specific time point plus the amount of polyphenols that was removed and replaced with new buffer and finally expressed as % of the initially loaded polyphenols.
Cell culture and treatment
Human neuroblastoma-derived cells (SH-SY5Y; ATCC CRL-2266) were cultured at 30–90% confluence in T75 flasks by using Dulbecco's Modified Eagle's Medium/F12 (DMEM/F12; Life Technologies) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 μg mL−1 streptomycin and 100 U mL−1 penicillin (Gibco). SH-SY5Y-derived neurons were obtained by following a previously described low-confluence differentiation protocol.48,49 Specifically, SH-SY5Y cells were seeded at a density of 15000 cells per cm2 and, at 24 h from seeding, treated for 4 days with a low-serum medium (DMEM with 1% FBS) supplemented with 10 μM all-trans-retinoic acid, 100 μg mL−1 streptomycin, and 100 U mL−1 penicillin.
At day 4 of differentiation, the biocompatibility of LSs and ext-LSs was tested. Cells were treated for 72 h with 0, 25, 100, 400, or 800 μg mL−1 nanoparticles, and then cell viability was evaluated through the 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4disulfophenyl)-2H-tetrazolium (WST-1) cell viability assay (BioVision), following manufacturer's instruction. WST-1 was diluted in 1:11 in phenol red-free complete medium (DMEM with 10% FBS) and incubated for 40 min before reading the absorbance at λ = 450 nm with a microplate reader (Victor3; PerkinElmer). Blank was subtracted and results were expressed as % of non-treated controls.
The above mentioned WST-1 assay protocol was also applied for the assessment of the cell viability in response to the plain rotenone (rot) treatment (1.00 μM; 72 h incubation), to the 72 h co-treatment with rot (1.00 μM) and ext-LS (400 μg mL−1), and to the 24 h rot pre-treatment (1.00 μM) followed by 48 h of co-treatment with rot and ext-LSs (400 μg mL−1). Furthermore, the cell viability was assessed in response to the 24 h rot pre-treatment (1.00 μM) followed by 48 h of co-treatment with rot and free extracts (ext with 8.06 μg mL−1 polyphenols, corresponding to the concentration loaded in 400 μg mL−1 ext-LSs). The effects of the rot vehicle (dimethyl sulfoxide – DMSO – 1:1000 dilution) on cell viability were also investigated.
hCMEC/D3 human brain microvascular endothelial cell line represents a well-established endothelial cell line for modeling the in vitro BBB function,50 and suitable for investigating the BBB crossing capability of different nanomaterials.51–53 hCMEC/D3 cells were cultured at 30–90% confluence in T75 flasks by using DMEM with 10% FBS, 100 μg mL−1 streptomycin, and 100 U mL−1 penicillin. WST-1 assay was performed for testing the biocompatibility of plain LSs and ext-LSs in hCMEC/D3; for this experiment, cells were seeded at a density of 15000 cells per cm2 and, at 24 h from seeding, were treated for 72 h with 0, 25, 100, 400, and 800 μg mL−1 of either empty LSs or ext-LSs.
As described in detail in the following section, human astrocytes were used with hCMEC/D3 cells for the preparation of the multicellular BBB model. Human astrocytes derived from the cerebral cortex (Innoprot) were cultured in T-75 flasks at 30–90% confluence with DMEM supplemented with 10% FBS, 100 IU mL−1 penicillin (Gibco), and 100 μg mL−1 streptomycin (Gibco).
Multicellular BBB model and nanoparticle crossing analysis
The in vitro multicellular BBB model was obtained following the double seeding approach presented in a previous work.54 Human astrocytes were seeded on the bottom surface (i.e., abluminal side) of 3 μm pore transwell inserts (Corning Incorporated) at a density of 10000 cells per cm2 and left for 15 h to adhere to the porous membranes in the humid chamber of the incubator. Subsequently, inserts were placed into a 24 multi-well plate and hCMEC/D3 cells were seeded at a high density (80000 cells per cm2) on the upper surface (luminal side) of the inserts. The co-cultures were incubated for 4 days with phenol red-free complete medium (DMEM with 10% FBS and antibiotics).
At day 4, the BBB crossing of DiO-stained ext-LSs was investigated: 400 μg mL−1 of ext-LSs (200 μl volume) were incubated in the upper (i.e., luminal) compartment, and the fluorescence in the bottom (i.e., abluminal) compartment (700 μl volume) was measured at different time points (8, 24, 48, and 72 h) by using a microplate reader (Victor3, PerkinElmer; λex = 485 nm, λem = 535 nm). The measured fluorescence intensities were finally converted to liposome concentrations by using a calibration curve (Fig. S2†). The barrier functionality was evaluated before and after the liposome treatment in terms of transendothelial electrical resistance (TEER) between the luminal and abluminal compartments by using a Millicell ERS-2 Volt-Ohmmeter (Millipore). The electrodes were placed in the two compartments and the electric resistance was read after 10 s. The resistance values of 6 barriers were measured before and after incubation with ext-LSs (72 h treatment). Finally, the resistance values were multiplied by the surface of the barrier (0.33 cm2) and reported in the graph as average ± standard deviation.
Liposome internalization analysis
The cell up-take analysis was performed in SH-SY5Y-derived neurons treated with 400 μg mL−1 of DiO-stained ext-LSs. The analysis of ext-LS-positive cells (ext-LSs+ cells) was quantitatively assessed by flow cytometry at 4, 24, and 48 h from liposome incubation. Cells were washed twice, detached by trypsin, centrifuged (300 rcf for 6 min), and resuspended in PBS. Subsequently, the fluorescence of the single cells was read by using a flow cytometer (Beckman Coulter CytoFLEX; λex = 488 nm, λem = 525 nm). Fluorescence emission distributions were collected and the higher fluorescence intensity detected in the non-treated control cells was used to define the fluorescence threshold. The cells displaying fluorescence intensity higher than the threshold were considered ext-LSs+ cells. Values are expressed as the percentage of the total number of cells.Internalization pathways in pinosomes and lysosomes were investigated at different time points (4, 24, and 48 h) by confocal laser scanning microscopy (CLSM) imaging (C2s system; Nikon). For pinosomes, cells were co-incubated with the DiO-stained ext-LSs and the cell-impermeant water-soluble Cascade Blue hydrazide (30 μM; Molecular Probes), washed with PBS, and then imaged by CLSM at the different time points. For lysosome imaging, cells incubated with DiO-labeled ext-LSs were stained with the Lysotracker acidotropic probe (1:1500 dilution, 1 h incubation; Invitrogen) 1 h before performing CLSM imaging. Finally, image analysis of signal co-localization (Pearson's correlation coefficients) of ext-LSs vs. pinosomes and ext-LSs vs. lysosomes was carried out by using NIS Elements software (Nikon).
Measurement of the reactive oxygen species in living cells
The levels of reactive oxygen species (ROS) in SH-SY5Y-derived neurons were assessed in different experimental conditions: in response to the rot treatment (1 μM, 72 h incubation; “Rot” experimental class), to the rot pre-treatment (1 μM, 24 h incubation) followed by the combined rot (1 μM) and ext-LS (400 μg mL−1) co-treatment (48 h incubation; “Rot + ext-LSs” experimental class), and to the 24 h rot the pre-treatment (1.00 μM) followed by 48 h of co-treatment with rot and free extracts (“Rot + ext” experimental class). Cells were detached by trypsin, centrifuged (300 rcf for 6 min), resuspended in PBS, and stained with the CellRox Green Reagent (1:2000 dilution, 30 min incubation; Invitrogen). Subsequently, the fluorescence intensity of the single cells was read by using a flow cytometer (Beckman Coulter CytoFLEX; λex = 488 nm, λem = 525 nm). The fluorescence emission distributions were collected and compared to the non-treated control cells (“Control” experimental class). The higher fluorescence intensity in non-stained cells was used to define the fluorescence threshold. The cells displaying a fluorescence intensity superior to the threshold were considered ROS+ cells. Values are expressed as the percentage of the total number of cells.
Apoptosis/necrosis investigations
The amount of healthy, early apoptotic, late apoptotic, and necrotic cells was investigated by flow cytometry in the “Control”, “Rot”, “Rot + ext-LSs”, and “Rot + ext” experimental classes following a previously described protocol.55 Before flow cytometry, the cells were detached by using trypsin, centrifuged (300 rcf for 6 min), and stained with FITC-annexin V (1:20 dilution; Thermo Fisher) and propidium iodide (PI, 1:1000 dilution; Thermo Fisher) in annexin V binding buffer (1×) for 15 min at 37 °C. After staining, the fluorescence intensity of the single cells was read by using a flow cytometer (Beckman Coulter CytoFLEX; FITC-A channel: λex = 488 nm, λem = 525 nm; ECD-A channel: λex = 488 nm, λem = 610 nm). The percentages of healthy, early apoptotic, late apoptotic, and necrotic cells were analyzed using the CytoFLEX software and finally reported on histograms.
Real-time reverse-transcription polymerase chain reaction
The transcription levels of genes encoding for antioxidant enzymes (catalase – Cat, superoxide dismutase 1 – SOD1, superoxide dismutase 2 – SOD2, and glutathione synthetase – GSS) and apoptosis-related proteins (p21 and p53) were investigated in the “Control”, “Rot”, and “Rot + ext-LSs” experimental classes. For the transcription level analysis, quantitative real-time reverse-transcription polymerase chain reaction (qRT-PCR) was performed similarly as previously described.56 The cells were detached by using trypsin, centrifuged (300 rcf for 6 min), and the mRNA was extracted by using a High Pure RNA Isolation Kit (Roche) following the manufacturer's protocol. mRNA was quantified through spectrophotometric analysis (NanoDrop; Thermo Scientific) and 100 ng of mRNA from each sample was processed with the iScriptTM Reverse Transcription Supermix (Bio-Rad) to obtain the cDNA (the temperature cycle used for the reverse transcription was 25 °C for 5 min, 42 °C for 45 min, 48 °C for 15 min, and 85 °C for 5 min; CFX Connect™ Real-Time PCR Detection System thermocycler, Bio-Rad). The amplification of the obtained cDNA was carried out with the SsoAdvanced SYBR Green Supermix (Bio-Rad) by using the following temperature protocol: 1 cycle at 98 °C (30 s duration), 40 cycles at 98 °C (3 s duration), and 60 °C (7 s duration), and a temperature ramp from 65 to 95 °C with 0.5 °C s−1 increments for obtaining the melting curve. Hprt and Rpl32 were used as reference genes. The cycle threshold (Ct) of the control (non-treated cultures) was used to calculate the ΔΔCt of the treatments. The forward and reverse primers of the investigate genes have been reported in Table S1.†Immunofluorescence analysis
Immunofluorescence analyses were carried out to investigate the intracellular levels of α-syn and of phosphorylated α-syn (phospho-α-syn) aggregates, as well as the β3-tubulin differentiation marker expression in the “Control”, “Rot”, and “Rot + ext-LSs” experimental classes. After treatments, cells were fixed with 4% paraformaldehyde (PFA) in PBS at 4 °C for 25 min, membrane permeabilization was performed with 0.1% Triton X-100 for 25 min, and blocking was carried out with a 10% goat serum solution in PBS for 70 min. Samples were then treated with a primary rabbit anti-α-syn antibody (1:200 dilution; Abcam), a primary rabbit anti-α-syn (phospho S129) antibody (1:200 dilution; Abcam), or a primary anti-β3-tubulin antibody produced in rabbit (0.3 μg mL−1; Sigma), in 10% goat serum solution in PBS at 37 °C for 1 h. After primary antibody incubation, samples were washed 4 times and then treated for 1 h with a staining solution including 10 μg mL−1 of Alexa Fluor 488 goat anti-Rabbit IgG (Invitrogen) secondary antibody, TRITC-conjugated phalloidin (100 μM; Millipore) for f-actin staining, and Hoechst 33342 (1 μg mL−1; ThermoFisher) for nucleus counterstaining. After washing the cultures with PBS, CLSM imaging was performed with a C2s system (Nikon). The automatic detection of the α-syn and of the phospho-α-syn aggregates was carried out with the NIS Elements software (Nikon) by imposing both signal intensity and size thresholds. Aggregates >3 μm2 were considered for the analysis. The number of α-syn/phospho-α-syn aggregates was then normalized for the number of cells and reported in bar graphs. Similarly, the number of cells positive to β3-tubulin was detected by NIS Elements software (Nikon) upon signal thresholding and finally reported in the graph.
Statistical analysis
Statistical analysis was carried out by using the R software (https://www.r-project.org/). The two-tailed Student's t-test was used as a two-sample parametric test. For multiple comparisons, analysis of variance (ANOVA) parametric test was carried out, and, in the case of p < 0.05, Tukey's honestly significant difference (HSD) post-hoc test was performed. Concerning qRT-PCR data, statistical analysis was performed by using the Bio-Rad CFX Manager software. For both qRT-PCR and mass spectrometry data, p < 0.05 and at least 3-fold regulation was considered as the significance threshold. The * symbol was used to indicate in the graphs the experimental treatments that resulted significantly different (p < 0.05) with respect to controls.Results and discussion
Preparation of a highly antioxidant catechins-rich extract from grape-pomace
The extracts obtained from WG and BG pomaces were characterized in terms of total phenol content (TPC), trolox equivalent antioxidant capacity (TEAC), and concentration of individual phenolic compounds (i.e., gallic acid, catechins, and epicatechins). Results are reported in Fig. 1. In Fig. 1a, the polyphenol concentrations were reported for 0.4, 0.8, and 1.5 mg mL−1 extract concentrations from WG (black rhombus symbol in the graph) and BG (gray square in the graph) grape pomaces. The extract from WG pomace was characterized by a significantly higher polyphenol concentration (145.0 ± 1.5 μg mL−1 resveratrol equivalent for 1.5 mg mL−1 of extract) with respect to the extract obtained from BG pomace (107.7 ± 4.6 μg mL−1 resveratrol equivalent for 1.5 mg mL−1 of extract; p < 0.05). The resulting TPC of the extracts from WG and BG pomace were 29.0 ± 0.3 and 18.9 ± 0.8 mg resveratrol equivalent per g dry matter, respectively (Table 1). The higher TPC value of the WG pomace-derived extract was associated with a higher TEAC (Fig. 1b). The trolox equivalent of 6 μg of WG pomace-derived extract (33.9 ± 0.7 nmol) was significantly higher compared to the trolox equivalent of 6 μg BG pomace-derived extract (16.8 ± 2.4 nmol; p < 0.05). The resulting TEAC of the extracts from WG and BG pomace were 1.5 ± 0.3 and 0.7 ± 0.2 μmol trolox equivalent per mg dry matter, respectively (Table 1). HPLC analyses (Fig. 1c) suggested a similar concentration of gallic acid in the two extracts (0.7 mg g−1 dry matter for WG and 0.6 mg g−1 dry matter for BG pomace-derived extract). However, a higher amount of catechins (6.2 mg g−1 dry matter), and epicatechins (8.0 mg g−1 dry matter) was detected in WG pomace-derived extract with respect to the BG one (2.5 mg g−1 dry matter catechins and 4.6 mg g−1 dry matter epicatechins; Table 1). Altogether, the characterization highlighted a relatively high concentration of TEAC associated with a superior catechin and epicatechin concentration. Table 1 also provides a comparison of TPC, TEAC, gallic acid, catechin, and epicatechin contents described in the literature in extracts from different types of grape varieties, including different sources such as grape skin, seeds, and pomace;57–61 the extract from WG pomace from Cinque Terre presented in this work is characterized by superior TEAC values.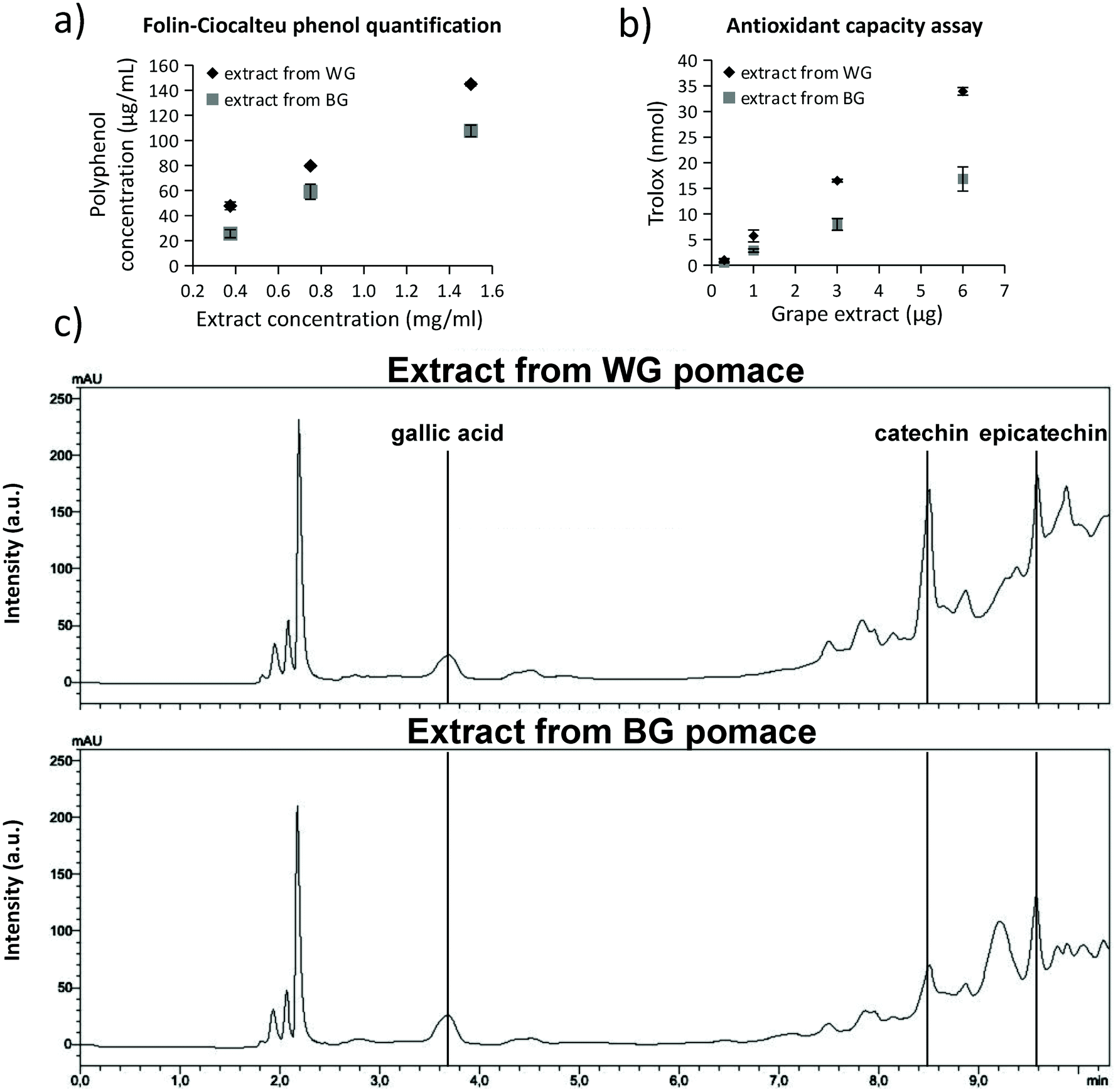 | ||
| Fig. 1 Characterization of the antioxidant extracts from white grapes (WG) with respect to the black grapes (BG) pomace from Cinque Terre. (a) Folin–Ciocalteu phenol quantification for 0.4, 0.8 and 1.5 mg mL−1 extracts from WG (black rhombus symbol in the graph) and BG (gray square in the graph) grape pomaces. The extract from WG pomace was characterized by a significantly higher polyphenol concentration; (b) the trolox-equivalent antioxidant capacity (TEAC) of 1, 3, and 6 μg of the WG pomace-derived extract (black rhombus) were significantly higher with respect to the TEAC of the BG pomace-derived extract; (c) HPLC measurements showed a similar concentration of gallic acid in the two extracts. Higher content of catechins and epicatechins was detected in WG pomace-derived extract. | ||
| Reference | GC | Grape variety | GA | CAT | EPIC | TPC | TEAC |
|---|---|---|---|---|---|---|---|
| Extract from WG pomace included in liposomes | Pomace | Albarola (60%) | 0.7 | 6.2 | 8.0 | 29.0 | 1.5 |
| Vermentino (30%) | |||||||
| Bosco (10%) | |||||||
| Extract from BG pomace non-included in liposomes | Pomace | Ciliegiolo (50%) | 0.6 | 2.5 | 4.6 | 18.9 | 0.7 |
| Cannaiolo (50%) | |||||||
| Pastrana-Bonilla et al. (2003)57 | Seeds | Muscadine (V. rotundifolia) | 0.03–0.09 | 0.3–14 | 0.4–18 | 15–32 | 0.2–0.48 |
| Pastrana-Bonilla et al. (2003)57 | Skin | Muscadine (V. rotundifolia) | n.d | n.d | n.d. | 2.6–5.4 | 0.011–0.014 |
| Yilmaz & Toledo (2004)58 | Seeds | Muscadine (V. rotundifolia) | 0.99 | 0.12 | 0.96 | n.d. | n.d. |
| Yilmaz & Toledo (2004)58 | Seeds | Chardonnay | 0.15 | 3.58 | 4.21 | n.d. | n.d. |
| Yilmaz & Toledo (2004)58 | Seeds | Merlot | 0.1 | 1.27 | 1.15 | n.d. | n.d. |
| Yilmaz & Toledo (2004)58 | Skin | Chardonnay | 0.05 | 0.6 | 0.44 | n.d. | n.d. |
| Yilmaz & Toledo (2004)58 | Skin | Merlot | 0.03 | 0.16 | 0.13 | n.d. | n.d. |
| Anastasiadi et al. (2010)59 | Seeds | Mandilaria | 1.61 | 6.61 | 2.03 | 26.16 | n.d. |
| Anastasiadi et al. (2010)59 | Seeds | Voidomatis | 0.19 | 4.78 | 3.71 | 27.32 | n.d. |
| Anastasiadi et al. (2010)59 | Seeds | Asyrtiko | 0.07 | 10.67 | 2.2 | 33.13 | n.d. |
| Anastasiadi et al. (2010)59 | Seeds | Aidani | 0.09 | 1,43 | 0.89 | 8.25 | n.d. |
| Anastasiadi et al. (2010)59 | Skin | Mandilaria | 0.0017 | n.d. | n.d. | 3.52 | n.d. |
| Anastasiadi et al. (2010)59 | Skin | Voidomatis | 0.0005 | n.d. | n.d. | 0.64 | n.d. |
| Anastasiadi et al. (2010)59 | Skin | Asyrtiko | 0.0018 | n.d. | n.d. | 1.19 | n.d. |
| Anastasiadi et al. (2010)59 | Skin | Aidani | 0.0002 | n.d. | n.d. | 0.9 | n.d. |
| Maluf et al. (2018)60 | Pomace | Vitis labrusca L. | n.d. | n.d. | n.d. | 35.5 | n.d. |
| Weidner et al. (2013)61 | Seeds | Vitis Coignetiae | 0.252 | 0.32 | 0.29 | 8.99 | 0.071 |
| Weidner et al. (2013)61 | Seeds | Vitis Coignetiae | 0.439 | 0.38 | 0.36 | 12.81 | 0.094 |
| Weidner et al. (2013)61 | Seeds | Pinot noir | 0.774 | 0.53 | 0.45 | 21.21 | 0.152 |
| Weidner et al. (2013)61 | Seeds | Pinot noir | 1.057 | 0.6 | 0.47 | 30.02 | 0.196 |
Mass spectrometry (MS) analyses of the WG and BG pomace-derived extracts were carried out to compare the relative amount of the different compounds detected by using a C18 column in negative ion mode. The relative compound amounts in WG, normalized to BG, are shown in Fig. 2 (green color for data > 1; red color for data < 1). Out of 29 molecules detected, 5 were found in significantly higher amount in WG pomace-derived extract: catechin (6.3-fold increase; p < 0.05), corchorifatty acid F (4.7-fold increase; p < 0.05), dodecanedioic acid (22.6-fold increase; p < 0.05), quercetin-3β-D-glucoside (5.2-fold increase; p < 0.05), and rutin (17.5-fold increase; p < 0.05). Furthermore, 5 molecules were found in significantly lower amount in WG pomace-derived extract: 2,4,6-trihydroxy-2-(4-hydroxybenzyl)-1-benzofuran-3(2H)-one (3.3-fold decrease; p < 0.05), astilbin (3.1-fold decrease; p < 0.05), isorhamnetin (5.5-fold decrease; p < 0.05), NP-000308 (4.6-fold decrease; p < 0.05), and quercetin (5.2-fold decrease; p < 0.05).
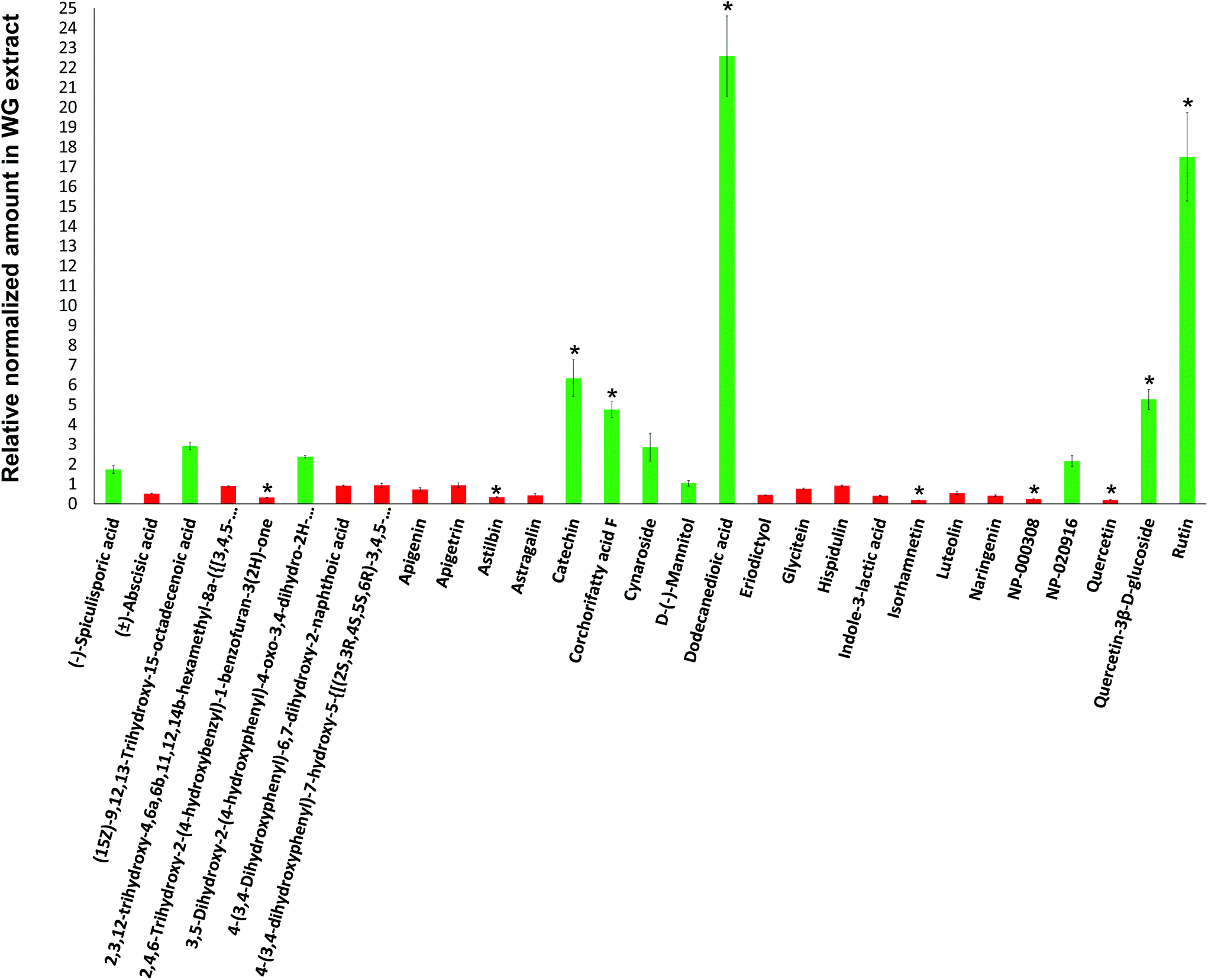 | ||
| Fig. 2 Mass spectrometry (MS) analysis of the WG and BG pomace-derived extracts. The relative amount of the compounds in WG were normalized for BG (green color for data > 1; red color for data < 1). Catechin, corchorifatty acid F, dodecanedioic acid, quercetin-3β-D-glucoside, and rutin were found in higher amounts in WG pomace-derived extract. 2,4,6-trihydroxy-2-(4-hydroxybenzyl)-1-benzofuran-3(2H)-one, astilbin, isorhamnetin, NP-000308, and quercetin were found in higher amounts in BG pomace-derived extract. | ||
Characterization of ext-LSs
Due to the superior TPC, TEAC and catechin/epicatechin content, the extract from WG pomace was used for the fabrication of the extract-loaded liposomes (ext-LSs; Fig. 3). The schematic of the ext-LSs functionalized with an antibody against the transferrin receptor (anti-TfR antibody) is shown in Fig. 3a. The SEM and TEM imaging is reported in Fig. 3b and c, respectively, while the hydrodynamic size (2r) of the nanoparticles is shown in Fig. 3d. As expected, the hydrodynamic size of the liposomes (133.0 ± 27.0 nm) is slightly higher than 100 nm, which corresponds to the size of the membrane pores used for the liposome extrusion. This can be attributed to the elastic deformation of the liposomes;62 the Z-potential was −21.5 ± 11.9 mV. Hydrodynamic size and Z-potential were comparable to other anti-TfR antibody functionalized brain lipid-based liposomes obtained in a previous work.46 The round-shaped morphology, the hydrodynamic size and the Z-potential of the ext-LSs are comparable to those ones of empty LSs (Fig. S3;† 2r = 139.2 ± 32.72 nm; Z-potential = −31.1 ± 7.54 mV). The loading assessment highlighted a concentration of 20.15 μg mL−1 of polyphenols in 1 mg mL−1 of ext-LSs. The % polyphenol release at different time points (4, 8, 24, and 48 h) in PBS is reported in the graph of Fig. 3e. The % of released polyphenols at 48 h was 31.70 ± 1.46% in buffer at pH 7.4 (light blue) and 35.11 ± 0.94% at pH 7.4 with 50 μM H2O2 (gray). A significant increase in polyphenol release was obtained at pH 4.5 (37.26 ± 1.64% at pH 4.5; p < 0.05; orange) compared to pH 7.4. The highest % release was observed with pH 4.5 with 50 μM H2O2 (39.35 ± 2.20% at pH 4.5 with 50 μM H2O2). This result is interesting since the ext-LSs are expected to interact both with acidic microenvironment (i.e., when internalized in acidic organelles such as late endosomes and lysosomes) and ROS (i.e., in rot-treated cells and dopaminergic neurons of PD). Different works in the literature reported as the membrane destabilization induced by the acidic environment and lipid peroxidation promote drug release from liposomes.63–65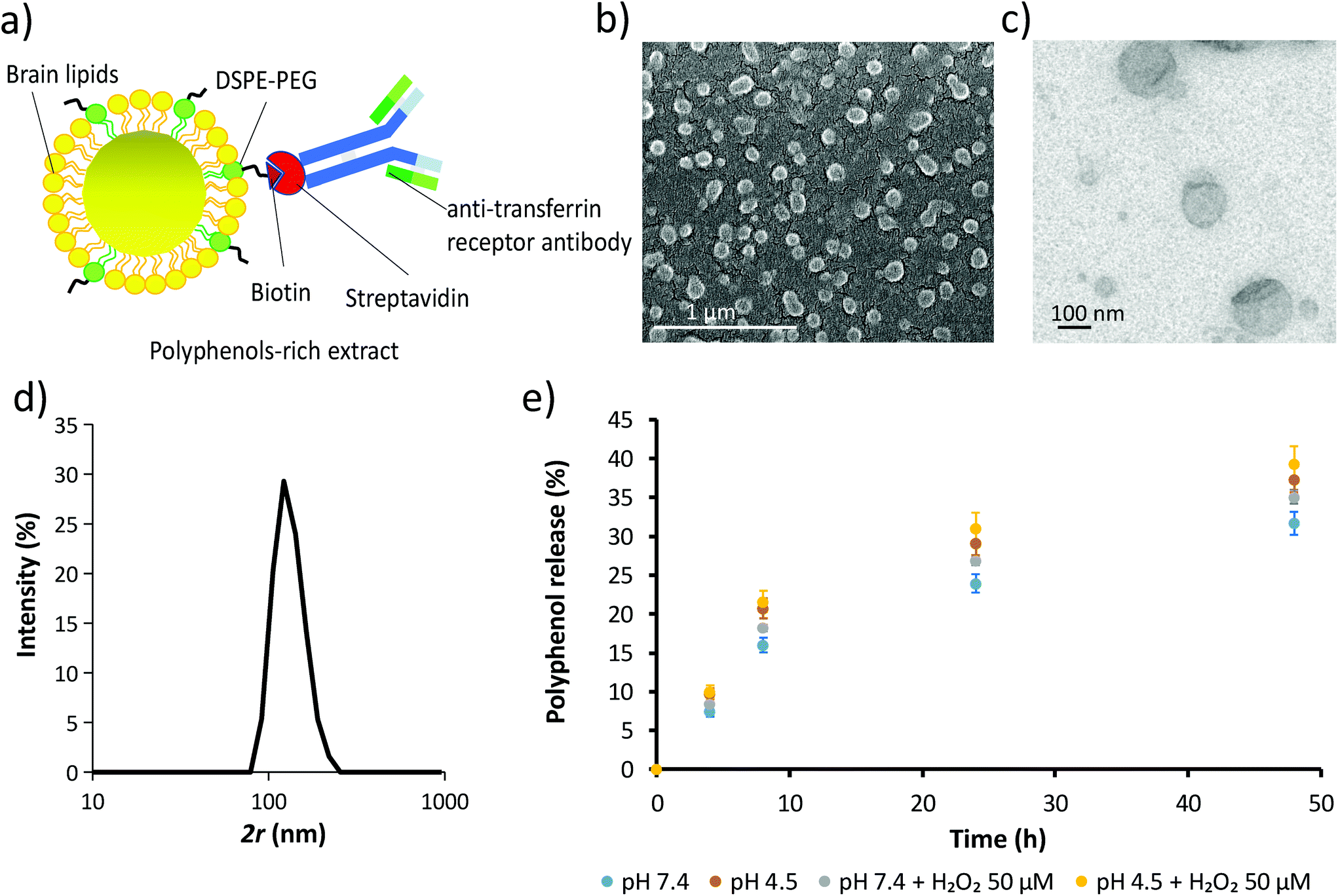 | ||
| Fig. 3 Extract-loaded liposomes (ext-LSs) functionalized with an antibody against the transferrin receptor (anti-TfR antibody). (a) The schematic of the ext-LSs; (b) representative scanning electron microscopy (SEM) and (c) representative transmission electron microscopy (TEM) image; (d) hydrodynamic size (2r); (e) cumulative polyphenol release at pH 7.4 (light blue), pH 4.5 (orange), pH 7.4 with 50 μM H2O2 (gray), and pH 4.5 with 50 μM H2O2 (yellow). | ||
The liposome biocompatibility was tested on both SH-SY5Y-derived neurons and on human brain microvascular endothelial hCMEC/D3 cells. Cell viability was investigated in response to a 72 h treatment with non-loaded LSs or ext-LSs (0, 25, 100, 400, or 800 μg mL−1, corresponding to 0, 0.50, 2.15, 8.06 and 16.12 μg mL−1 of polyphenols in ext-LSs). Results are reported as ESI (Fig. S4†), and highlight relatively high biocompatibility of these nanovectors compared to other examples reported in the literature:66 no significant effects have been observed on both cell lines with the exception of the highest evaluated concentration (800 μg mL−1), that conversely produced a significant decrement of cell metabolic activity (p < 0.05). A significant difference between “Ext-LSs” and “LSs” was found at 800 μg mL−1 (p < 0.05). Specifically, the treatment with ext-LSs was less cytotoxic than empty LSs. This can be attributed to the presence of the protective antioxidant polyphenols in ext-LSs, which reduced the cytotoxicity of the nanoplatform at this high concentration. According to these data, the following experiments have been performed by using the highest non-toxic liposome concentration (400 μg mL−1, corresponding to 8.06 μg mL−1 of loaded polyphenols).
Nanoparticle up-take in SH-SY5Y-derived neurons
Before evaluating the potential protective effects of ext-LSs on the in vitro Parkinson's disease (PD) model, cellular uptake analysis was carried out on SH-SY5Y-derived neurons treated with 400 μg mL−1 of DiO-stained ext-LSs (Fig. 4). In Fig. 4a, the fluorescence emission distributions of the cells incubated for 4, 24, and 48 h are shown; the graphs in Fig. 4b show the % of the ext-LSs+ cells. A progressive internalization of the ext-LSs was observed at 4 h (22.1 ± 1.4%), 24 h (46.2 ± 1.6%), and 48 h (77.6 ± 1.3%) of incubation. The CLSM imaging in Fig. 4c shows the progressive internalization of the ext-LSs (in green) in the subcellular compartments involved in pinocytosis (pinosomes: top images in red) and in digestive organelles (lysosomes: bottom images in red). Qualitatively, a higher signal co-localization (in yellow) can be appreciated for both pinosomes and lysosomes after 48 h of incubation. The Pearson's correlation was used to quantitatively assess the co-localization of pinosomes vs. ext-LS (Fig. 4d) and lysosomes vs. ext-LS (Fig. 4e) fluorescence signals. For both pinosomes and lysosomes, the increase of the Pearson's correlation from 4 to 48 h incubation (for pinosomes, 0.24 ± 0.06 at 4 h, 0.34 ± 0.03 at 24 h, 0.50 ± 0.06 at 48 h; for lysosomes, 0.04 ± 0.02 at 4 h, 0.11 ± 0.03 at 24 h, 0.43 ± 0.04 at 48 h) reveals the continuous nanoparticle internalization in SH-SY5Y-derived neurons, being the progressive internalization through active cell uptake mechanisms like pinocytosis typical of negatively charged-liposomes.67 A relation between incubation duration and internalization extent has been also observed by other works in the literature using liposomes with similar properties in terms of size and surface charge. As an example, Cauzzo et al. investigated in vitro the fate of dye-loaded negatively charged liposomes with ∼150 nm size and analyzed the fluorescence intensity of RAW 264.7 macrophages during uptake. A continuous and gradual increase of cell fluorescence was found during the 24 h of liposome internalization, therefore indicating a progressive nanoparticle uptake.68 Similarly, Kang et al. observed a remarkable increase in internalization of ∼120 nm-sized negatively charged liposomes conjugated with a αvβ3 ligand from 3 to 16 h of treatment in HUVECs and U87 cells.69 The mechanism can be attributed to the interface of new particles with the cells along the incubation.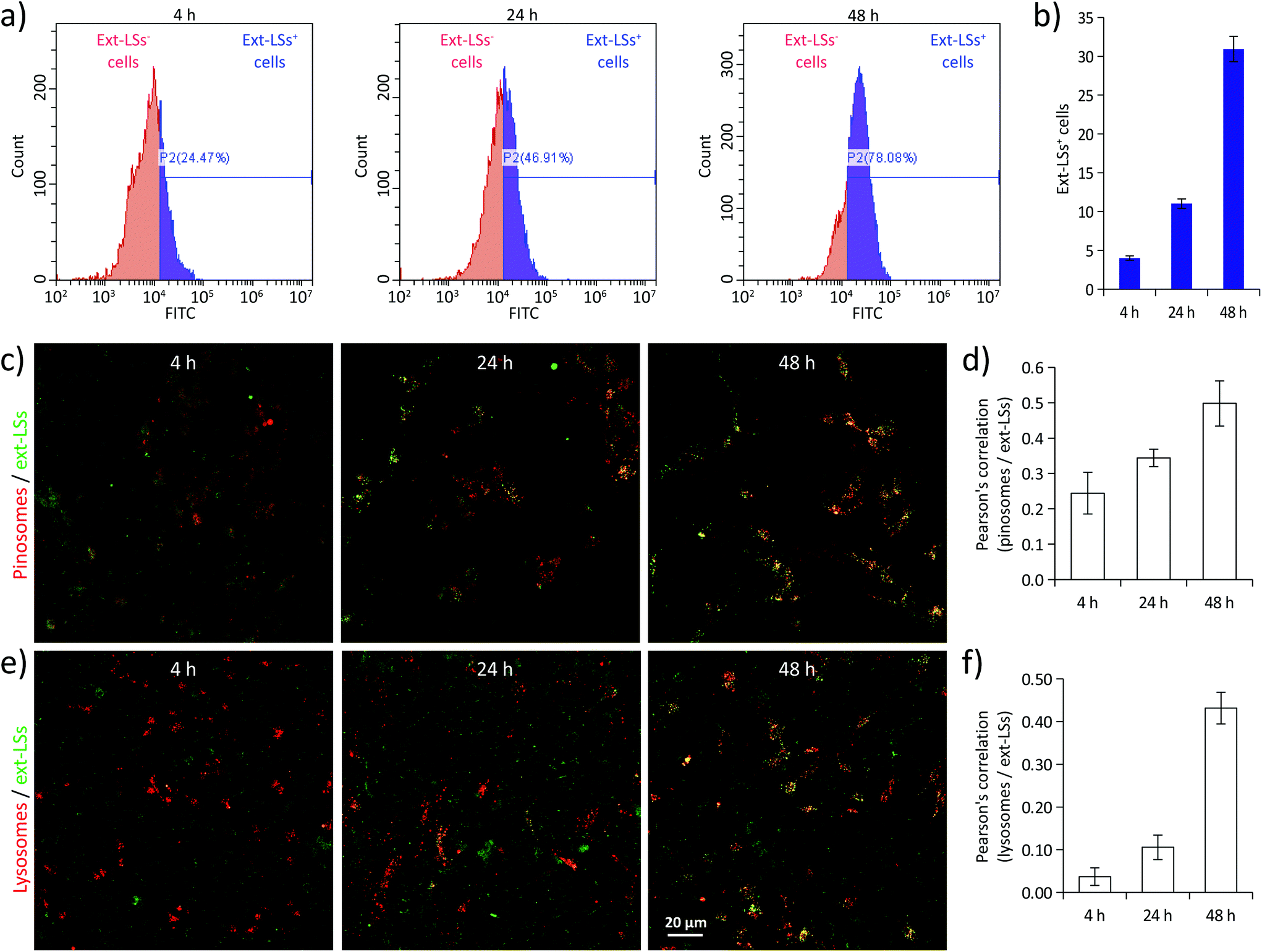 | ||
| Fig. 4 Cell up-take analysis in SH-SY5Y-derived neurons treated with 400 μg mL−1 of fluorescently-stained ext-LSs. (a) Fluorescence emission distributions of the cells incubated for 4, 24, and 48 h; (b) percentage (%) of ext-LSs+ cells; (c) confocal laser scanning microscopy (CLSM) imaging of ext-LSs (in green) in the subcellular compartments involved in pinocytosis (pinosomes: top images, in red) and in digestive organelles (lysosomes: bottom images, in red); Pearson's correlation coefficient of pinosomes vs. ext-LS (d) and of lysosomes vs. ext-LS (e) fluorescence signals. | ||
Ext-LSs rescue the cell viability in the rotenone model of PD
The in vitro rotenone (rot) PD model was used to test the protective effect of ext-LSs in SH-SY5Y-derived neurons (Fig. 5). The vehicle (DMSO, 1:1000 dilution) was not able to affect the cell viability (107 ± 42%; p > 0.05). A significant decrease of cell viability compared to controls (100 ± 29%) was observed for the 1.00 μM rot treatment (50.72 ± 11.4% viability; “Rot” experimental class; p < 0.05). This result is in line with other works in literature, where a cell survival ranging between 35–60%70,71 was observed in SH-SY5Y cells treated with 1.00 μM in similar culture conditions (1% FBS). Interestingly, both the synchronous co-treatment with 1.00 μM rot and ext-LSs for 72 h (107 ± 38%; Fig. S5†), as well as the pre-treatment with 1.00 μM rot for 24 h followed by the combined 1.00 μM rot + ext-LS treatment for 48 h (99 ± 37%; “Rot + ext-LSs”; Fig. 5) were able to completely rescue the cell viability (no significant difference compared to controls; p > 0.05). This result is particularly interesting since the polyphenol-rich ext-LSs demonstrated to protect cells from the neurodegenerative effects of the rot, even when applied 24 h after the rot pre-treatment. Only a partial rescue of the cell viability was instead obtained with 1.00 μM rot for 24 h followed by the combined 1.00 μM rot + free extracts incubation for 48 h (65 ± 10%; “Rot + ext”; p < 0.05; Fig. 5). These findings are in line with the literature, where other antioxidant compounds have been reported to protect cells from rot-induced cytotoxicity. However, some of these antioxidant compounds were used in previous works as pre-treatment to reduce rot neurotoxicity and were not able to completely rescue the cell viability. For example, genistein showed protective activity in A53T mutant α-syn overexpressing SH-SY5Y cells. Pre-treatments with both 20 and 40 μM genistein restored cell viability to about 80% of the control.72 Similarly, the pre-treatment with 20 μM resveratrol has been shown to partially protect SH-SY5Y cells from rot-induced cytotoxicity by acting on autophagy.73 In our work, the complete viability rescue obtained in cells pre-treated for 24 h with rot and subsequently co-treated with rot and ext-LSs for 48 h represents an excellent result, since it demonstrates the restorative potential of ext-LSs in insulted cells, such as in the case of dopaminergic neurons in PD. Similarly, a complete recovery of cell viability was observed by Rey et al. in SH-SY5Y cells pre-incubated for 48 h with the 1-methyl-4-phenylpyridinium (MPP+) toxin and subsequently co-treated with MPP+ and 4 U mL−1 erythropoietin (EPO), a type I cytokine with well known neuroprotective and neurotrophic activity.74 Specifically, EPO exerted the restorative action on MPP+ pre-treated neurons by recovering the mitochondrial dysfunction and promoting the recovery of the dopaminergic phenotype.
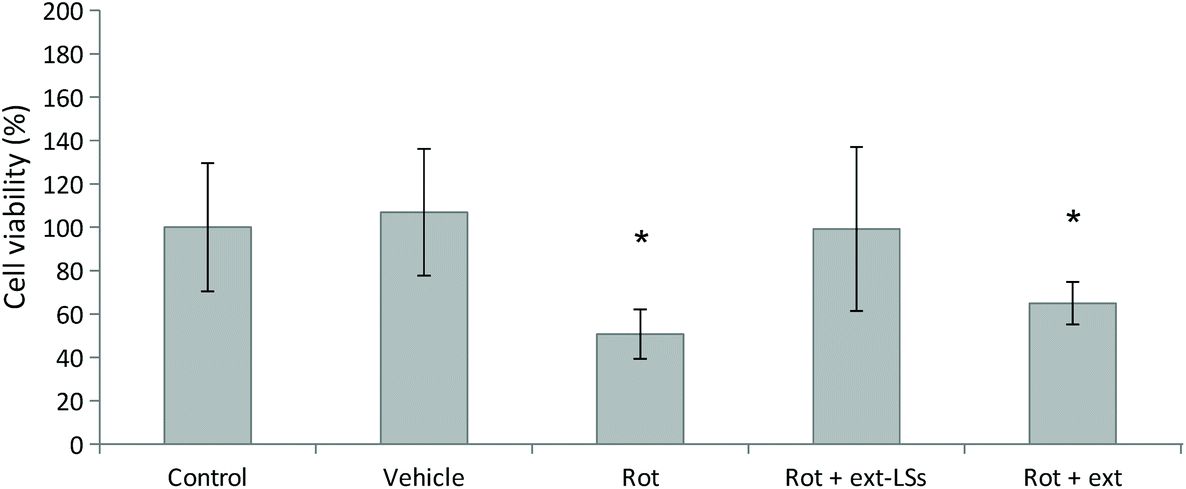 | ||
| Fig. 5 Protective effect of ext-LSs in the in vitro rotenone-derived PD model. WST-1 cell viability assay of SH-SY5Y-derived neurons treated with rot (1.00 μM) for 72 h (“Rot)”, with 1.00 μM rot for 24 h followed by the combined 1.00 μM rot + ext-LS incubation for 48 h (“Rot + ext-LSs”), or with 1.00 μM rot for 24 h followed by the combined 1.00 μM rot + free extracts incubation for 48 h (“Rot + ext”). The vehicle is DMSO (1:1000 dilution). *p < 0.05. | ||
The following experiments were focused on deepening the protective mechanisms of ext-LSs after rot-pretreatment (“Rot + ext-LSs” experimental group corresponding to 1.00 μM rot pre-treatment for 24 h followed by the combined 1.00 μM rot + 400 μg mL−1 ext-LS treatment for 48 h). Results were compared to the “Rot” experimental group (72 h treatment with 1.00 μM rot),“Control” (non-treated control cultures), and “Rot + ext” (24 h pre-treatment with 1.00 μM rot followed by 48 h of co-treatment with rot and free extracts).
Ext-LSs reduce oxidative stress, apoptosis and necrosis in the PD model
The levels of reactive oxygen species (ROS), apoptosis, and necrosis were investigated in “Control”, “Rot”, “Rot + ext-LSs”, and “Rot + ext” experimental classes (Fig. 6). The intracellular ROS levels were measured by flow cytometry upon CellRox fluorescence staining (Fig. 6a and b). The graphs in Fig. 6a show representative fluorescence intensity distributions. The shift to the right of the “Rot” distribution indicates an increase in the intracellular ROS levels. Instead, the distribution shift cannot be appreciated in “Rot + ext-LSs”. A partial shift to the right of the fluorescence distribution can be appreciated in “Rot + ext”. Quantitatively, ROS+ cells (Fig. 6b) were 3.6 ± 2.4% in “Control”, remarkably higher in “Rot” (15.9 ± 0.5%, corresponding to ∼4.4 fold higher compared to controls; p < 0.05), and in “Rot + ext” (8.0 ± 0.5%; ∼2.2 fold higher compared to controls). The ROS+ cells in “Rot + ext-LSs” were just 3.3 ± 3.8%, thus non-significantly different to “Control” (p > 0.05). This result indicates a complete rescue of the ROS levels in “Rot + ext-LSs” and a partial rescue in “Rot + ext”.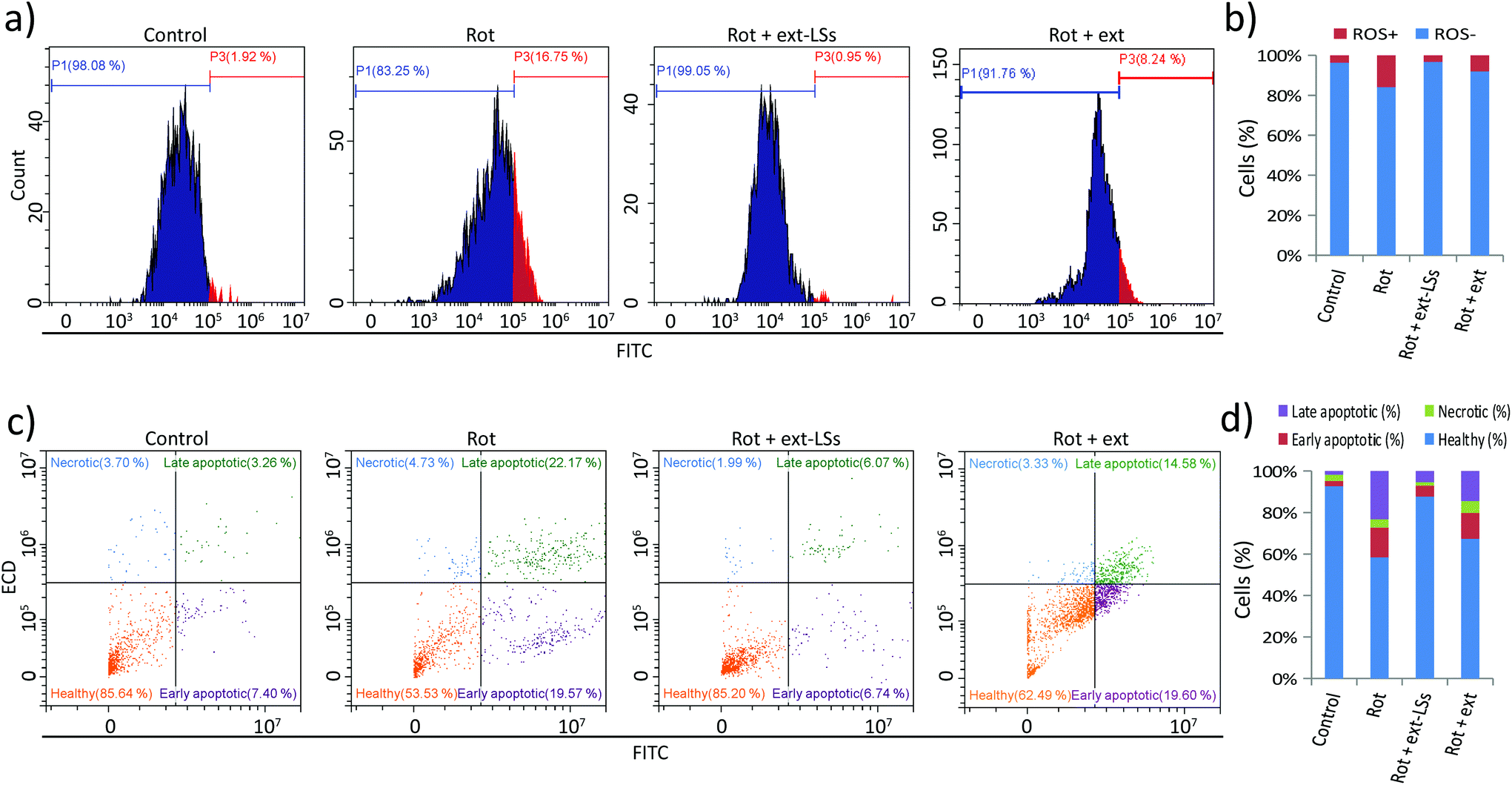 | ||
| Fig. 6 Levels of reactive oxygen species (ROS), apoptosis, and necrosis in “Control”, “Rot”, “Rot + ext-LSs”, and “Rot + ext” experimental classes. Intracellular ROS levels investigated by flow cytometry upon CellRox fluorescence staining: (a) representative fluorescence intensity distributions; (b) graph reporting the % of ROS+ cells. Analysis of apoptotic and necrotic effects: (c) representative fluorescence distributions of FITC-annexin V and PI stained cells, with subpopulations of healthy (in orange), early apoptotic (in purple), necrotic (in light blue), and late apoptotic (in green) cells; (d) graph reporting the percentage of healthy, early apoptotic, necrotic, and late apoptotic cells. * p < 0.05. | ||
Apoptotic and necrotic effects were also investigated. Results are shown in Fig. 6c and d. Representative fluorescence distributions of FITC-annexin V and PI stained cells in “Control”, “Rot”, “Rot + ext-LSs”, and “Rot + ext” experimental groups are reported in Fig. 6c. The subpopulation of healthy (in orange), early apoptotic (in purple), necrotic (in light blue) and late apoptotic (in green) cells have been highlighted in representative scatter plots. In Fig. 6d, the graph reports the percentage of healthy (92.7 ± 8.7% in “Control”, 58.4 ± 4.3% in “Rot”, 87.7 ± 2.8% in “Rot + ext-LSs”, and 67.3 ± 9.9% in “Rot + ext”), early apoptotic (2.5 ± 4.3% in “Control”, 14.4 ± 4.8% in “Rot”, 5.3 ± 1.9% in “Rot + ext-LSs”, and 12.5 ± 6.6% in “Rot + ext”), necrotic (3.1 ± 1.6% in “Control”, 4.0 ± 1.9% in “Rot”, 1.6 ± 0.4% in “Rot + ext-LSs”, and 5.8 ± 3.3% in “Rot + ext”), and late apoptotic cells (1.8 ± 2.8% in “Control”, 23.3 ± 2.8% in “Rot”, 5.4 ± 0.6% in “Rot + ext-LSs”, and 14.4 ± 4.6% in “Rot + ext”) in the 4 experimental groups. The “Rot” treatment was able to significantly affect cell viability (34.3% decrease of healthy cells compared to controls) mostly by increasing apoptosis (11.9% increase of early apoptotic cells and 21.5% increase of late apoptotic cells). This result is in line with the literature: indeed, rot has been demonstrated to be involved in the impairment of mitochondrial complex I, with the consequent apoptotic induction through ROS formation.75 However, thanks to the antioxidant ext-LSs, the complete rescue of healthy cells was obtained in “Rot + ext-LSs”. Indeed, the % of healthy cells in “Rot + ext-LSs” (87.7 ± 2.8%) was remarkably higher than in “Rot” treatment (58.4 ± 4.3%; p < 0.05), and non-significantly different with respect to the “Control” group (92.7 ± 8.7%; p > 0.05). This result highlights the high protective potential of the ext-LSs against apoptosis induction in PD models, and opens new perspectives for a green nanomedicine in the treatment of neurodegenerative diseases associated with uncontrolled ROS increase.
The analysis of apoptotic levels demonstrated as the free extract is able to only partially inhibit the toxic effects of rot. This finding is in line with the obtained results on cell viability and ROS level. In this regard, independent works demonstrated as the encapsulation of phenolic extracts enhances the intracellular scavenger activity with respect to the free extract alone.24,76 The improved scavenging activity is attributed to the nanoparticle-dependent protection from oxidation in the extracellular microenvironment (i.e., prolongation of lifetime and effectiveness of encapsulated molecules) as well as the enhanced intracellular delivery of the preserved antioxidants. Differently form the ext-LSs, the free extracts were not able to completely rescue the ROS, cell viability and apoptotic levels. Therefore, the following experiments have been carried out to characterize the gene expression profile and the α-syn aggregation only in “Control”, “Rot”, and “Rot + ext-LSs”.
Gene expression analysis in response to rotenone and ext-LSs
The mRNA expression of genes encoding for antioxidant enzymes (Cat, SOD1, SOD2, and GSS) and apoptosis-related proteins (p21 and p53) were investigated in the “Control”, “Rot”, and “Rot + ext-LSs” experimental classes (Fig. 7). The bar graph in Fig. 7a reports the normalized expression (ΔΔCq) of the investigated genes, while Fig. 7b shows the associated clustergram. The volcano plots showing the p-value versus the fold change of the different comparisons are reported as ESI (Fig. S6†). The “Rot” treatment was able to significantly upregulate the expression of SOD2 (3.8-fold regulation; p < 0.05) and of p53 (5.3-fold regulation; p < 0.05). The upregulation of the antioxidant enzymes is an intrinsic protection mechanism of the cells to counteract oxidative stress conditions. Specifically, SOD2 encodes for the mitochondria-targeted Mn-dependent superoxide dismutase (Mn-SOD), which is known to be upregulated in response to increased ROS levels via activation of multiple mitogen-activated protein kinase MAPK (i.e., ERK, JNK, and p38) pathways.77 The same pathways are also known to upregulate the p53 expression.77 Mn-SOD is expressed only in the mitochondrial matrix, where exerts its antioxidant protective activity by converting the superoxide to H2O2, which is then metabolized to H2O by the peroxiredoxin 3.78 The physiologic relevance of Mn-SOD has been demonstrated in knockout mice, which are characterized by neonatal lethality (differently from the knockout of the other isoforms).79 Many independent studies highlighted as rot increases ROS by acting on the complex I of the mitochondrial respiratory chain;80,81 at this regard, Li et al. observed as Mn-SOD overexpressing cells were more resistant to rotenone-induced apoptosis compared to control cells.82 Therefore, it is not surprising that the mitochondria-specific SOD2 gene is overexpressed by the cells as a regulatory response to the rot treatment. In “Rot + ext-LSs”, the SOD2 expression is instead comparable to “Control” (0.75-fold regulation; p > 0.05). The rescued expression of SOD2 in “Rot + ext-LSs” cultures can be associated with their basal ROS levels (values comparable to “Control” and significantly lower than “Rot”). Also, the p53 upregulation observed in “Rot” is completely rescued by the ext-LS (0.75-fold regulation in “Rot + ext-LSs”; p > 0.05). These results are in line with the flow cytometry studies, where the early and late apoptotic cells were comparable in “Control” and “Rot + ext-LSs”. Despite the increase of p21 and Cat expression in the “Rot” experimental class compared to “Control” is not statistically significant (i.e., the combination of p < 0.05 and >3-fold regulation requirements is not satisfied), a clear trend of expression can be observed, with a significant downregulation in “Rot + ext-LSs” compared to “Rot”.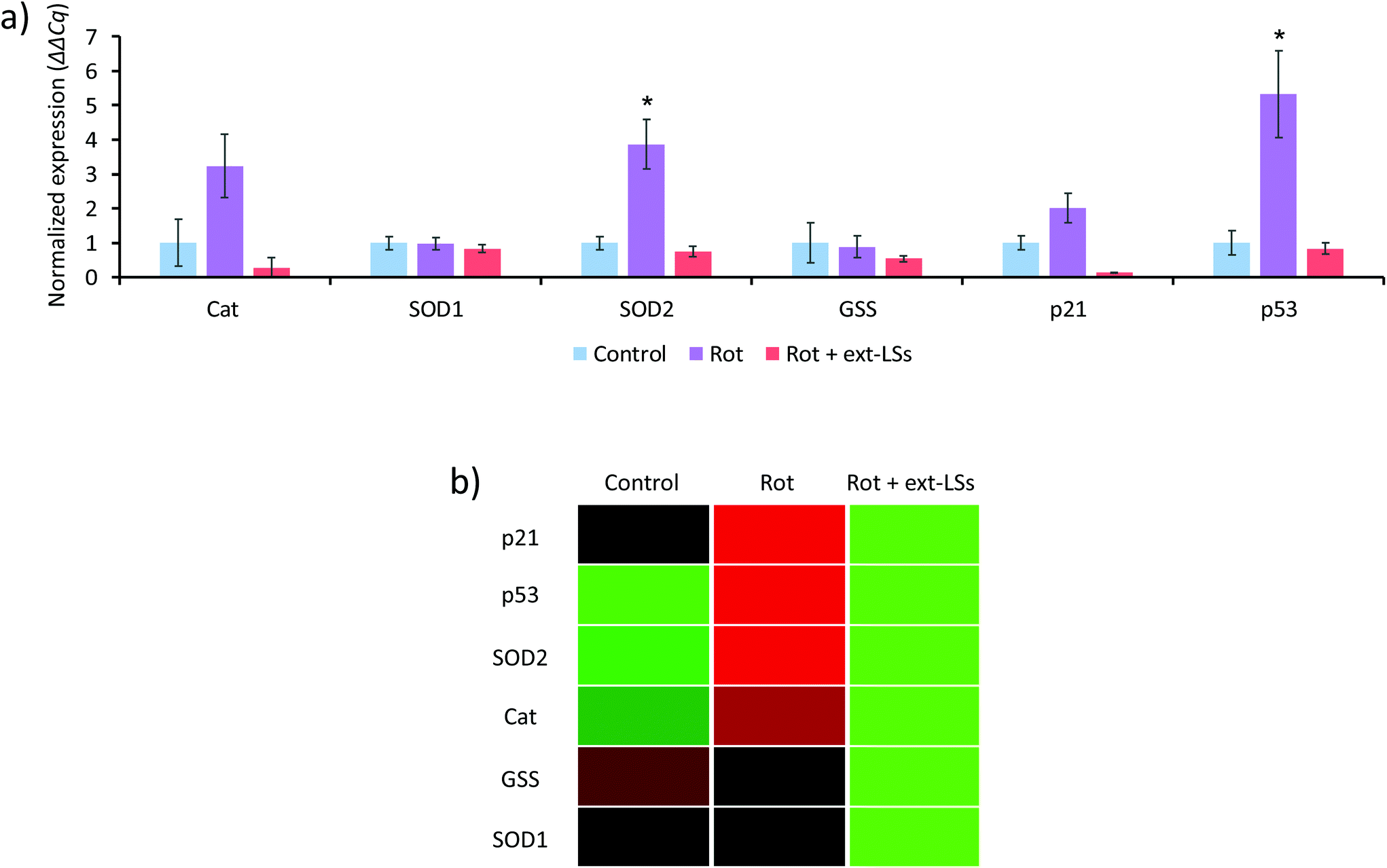 | ||
| Fig. 7 mRNA expression of genes encoding for antioxidant enzymes (Cat, SOD1, SOD2, and GSS) and for apoptosis-related proteins (p21 and p53): (a) bar graph reporting the normalized expression (ΔΔCq) of the investigated genes and (b) clustergram (upregulation in red; downregulation in green). *p < 0.05 and regulation >3 folds. | ||
Complete rescue of the α-syn levels and partial rescue of β3-tubulin
α-Syn is a presynaptic protein that is neuropathologically associated with PD. α-Syn and its phosphorylated form (phospho-α-syn) misfold and aggregate upon ROS damage. The aggregated forms are highly toxic, can affect different neuronal functions (e.g., the synaptic activity), and promote neural cell death. Both α-syn and phospho-α-syn are highly expressed within Lewy bodies (LBs), which represent the gold standard markers in PD and in dementia with Lewy bodies (DLB). Similarly, the impairment of mitochondrial complex I induced by rot treatment induces the aggregation of α-syn and phospho-α-syn in neural cells.83 In this work, the expression of these PD markers was investigated in “Control”, “Rot”, and “Rot + ext-LSs” experimental classes by immunofluorescence followed by CLSM imaging (Fig. 8). In Fig. 8a, the panel reports the expression of α-syn (top) and phospho-α-syn (bottom) in the 3 experimental classes. For each panel, f-acting (in red), α-syn/phospho-α-syn (in green), nuclei (in blue), and the merged signal (“merge”) are shown. As expected, a clear increase in the number of α-syn and phospho-α-syn aggregates can be observed in “Rot”. Instead, the presence of ext-LSs inhibits the rot-dependent formation of these intracellular protein aggregates. Indeed, the elevated number of aggregates observed in “Rot” cannot be detected in “Rot + ext-LSs”. In Fig. 8b, the graphs report the number of protein aggregates (α-syn in the top, phospho-α-syn in the bottom) per cell in “Control” (0.02 ± 0.02 α-syn aggregates per cell; 0.03 ± 0.03 phospho-α-syn aggregates per cell), “Rot” (0.18 ± 0.04 α-syn aggregates per cell; 0.44 ± 0.13 phospho-α-syn aggregates per cell), and “Rot + ext-LSs” (0.01 ± 0.02 α-syn aggregates per cell; 0.01 ± 0.02 phospho-α-syn aggregates per cell). The number of both α-syn and phospho-α-syn aggregates significantly increase in “Rot” (p < 0.05) with respect to “Control”, as described in other works.84 The incubation with ext-LSs allowed the complete rescue of the “Control” condition in terms of both α-syn and phospho-α-syn aggregates (p > 0.05 with respect to control). Since the α-syn and the phospho-α-syn aggregation is mediated by their misfolded conformation upon ROS damage, the inhibition of the intracellular protein accumulation in “Rot + ext-LSs” can be attributed to the protective effects of ext-LSs against the intracellular ROS. In the literature, other antioxidant compounds were proposed as inhibitors of the α-syn aggregation in PD models. In this regard, a seaweed polyphenol, dieckol, has shown retarding effects in rot-induced neurotoxicity and α-syn aggregation in SH-SY5Y cells thanks to its protecting antioxidant activity. In this cited work, a partial rescue of the α-syn aggregation was observed when treating SH-SY5Y cells with rot (2 μM in proliferative conditions −10% FBS).85 Moreover, recent investigations also showed as phenolic moieties, such as the hydroxytyrosol (HT), can inhibit the formation of proteolysis-resistant fibrils of α-syn through their stabilizing effects.86 However, free antioxidants show scarce BBB crossing capabilities and may exhaust their scavenging activity before reaching the brain.15 For this reason, the encapsulation of the antioxidants in nanocarriers for brain delivery, such as liposomes, is fundamental to exert their protective function. Nanoparticle-mediated brain drug delivery is also known to be more efficient in neuropathologies (such as PD, Alzheimer's disease, CNS cancers, etc.), where BBB is dysfunctional and disrupted.87 Intending to exploit the delivery potential of nanoparticles, Piersimoni et al. recently fabricated lipoic acid ligand-shell Au nanoconjugates to protect neural cells from the oxidative stress associated with α-syn aggregates.88 In addition, antioxidant nanoparticles can also protect from chronic activation of microglia; as an example, antioxidant shell-core nanoparticles composed of ferulic acid diacid molecule and tannic acid were exploited to reduce the intracellular α-syn aggregation in microglia, a process sustaining neurotoxicity and inflammation.89 A reduction in pro-inflammatory cytokine release was found (both TNF-α and IL-6), indicating the potential of antioxidant nanoparticles in reducing the neuroinflammation processes in PD.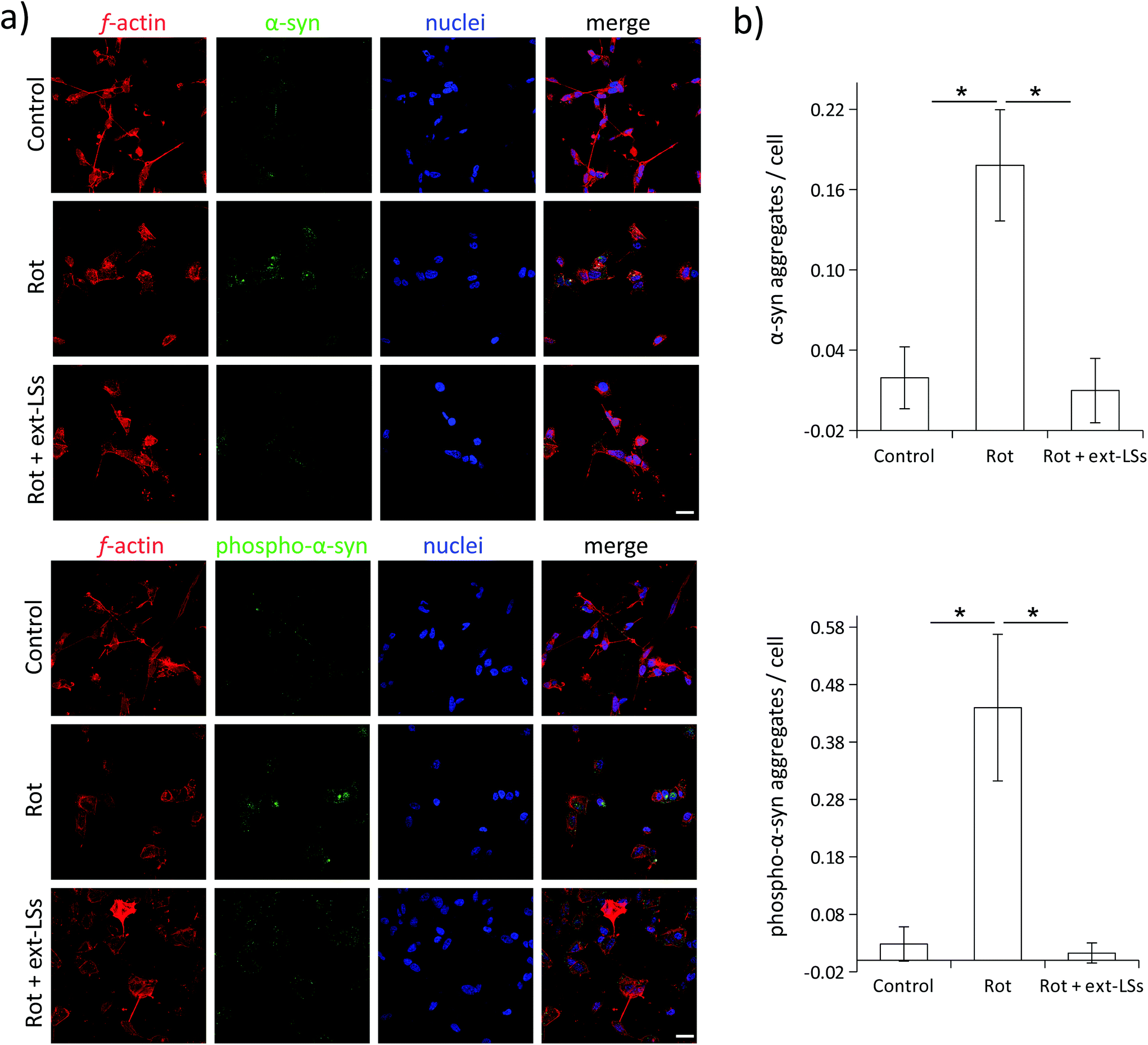 | ||
| Fig. 8 Immunofluorescence of α-syn and phospho-α-syn. (a) Panel reporting the expression of α-syn (top) and phospho-α-syn (bottom): f-acting (in red), α-syn/phospho-α-syn (in green), nuclei (in blue), and merging; (b) graph reporting quantitative analysis of protein aggregates (α-syn in the top; phospho-α-syn in the bottom) per cell. *p < 0.05. Scale bar: 20 μm. | ||
β3-Tubulin is a neuronal-specific microtubule element of the tubulin family with a fundamental role in axon guidance, maintenance, and synaptic vesicle transport to the presynaptic termini. The expression of this marker is remarkably downregulated in PD.90 The β3-tubulin expression is highly sensitive to the rot treatment,91 and levels of this neuronal marker can already decrease after 2 h of treatment with this drug.92 The expression of the β3-tubulin neuronal marker was investigated in “Control”, “Rot”, and “Rot + ext-LSs” experimental groups by immunofluorescence followed by CLSM imaging (Fig. 9). The panel in Fig. 9a shows the expression of f-actin (in red), β3-tubulin (in green), and nuclei (in blue). The graph in Fig. 9b reports the % of β3-tubulin+ cells. A significant decrease of the β3-tubulin+ cells was found in “Rot” experimental condition (64.4 ± 7.2%) with respect to “Control” (91.9 ± 2.7%; p > 0.05). Partial rescue of the “Control” condition was obtained in “Rot + ext-LSs”, where 78.2 ± 2.1% of β3-tubulin+ cells were detected.
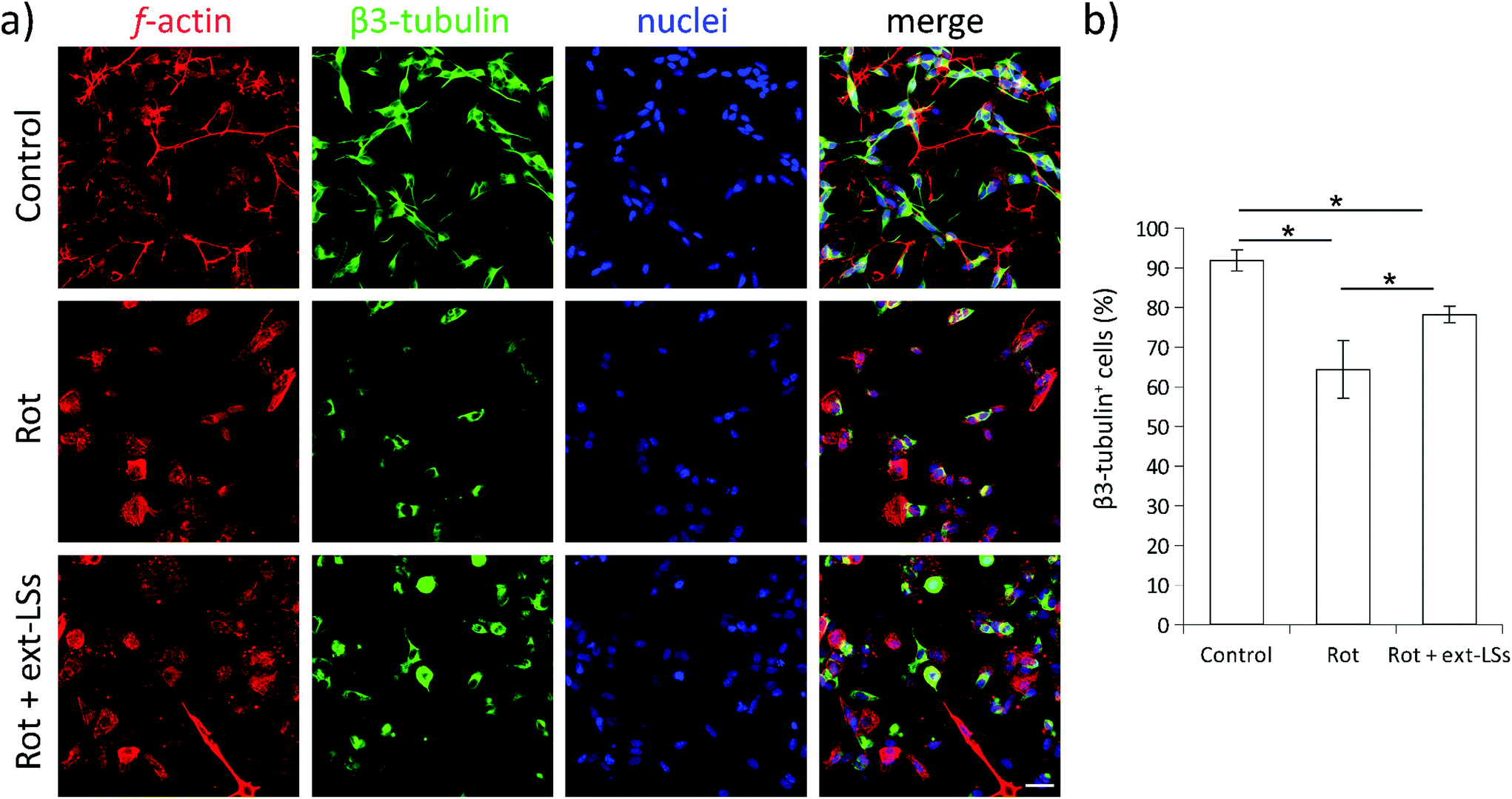 | ||
| Fig. 9 Immunofluorescence of the β3-tubulin neuronal marker. (a) Panel showing f-actin (in red), β3-tubulin (in green), and nuclei (in blue); (b) graph reporting percentage (%) of β3-tubulin+ cells. *p < 0.05. Scale bar: 20 μm. | ||
Altogether, these results indicate that ext-LSs counteract the rot-induced ROS in the cells, completely prevent the α-syn and phospho-α-syn aggregation, and partially protect the neural cells from the β3-tubulin depletion in PD models.
Finally, the BBB crossing capability of the ext-LSs was assessed in a multicellular in vitro model (results available as ESI; Fig. S7†) similarly as previously described.54,93 In Fig. S7a,† the schematic representation of the in vitro BBB system with luminal (in blue) and abluminal (in red) compartments separated by astrocytes and by an endothelial cell layer is shown. A progressive increase of the ext-LS concentration was detected in the abluminal compartment (18.2 ± 7.6 μg mL−1 at 8 h, 24.8 ± 2.5 μg mL−1 at 24 h, 43.9 ± 3.8 μg mL−1 at 48 h, 49.2 ± 4.1 μg mL−1 at 72 h) after incubating the luminal compartment with 400 μg mL−1 DiO-stained ext-LSs (Fig. S7b†). Considering the result at 72 h, the concentration of the ext-LSs in the abluminal compartment represented 55% of the concentration at the equilibrium (88.9 μg mL−1). The majority (6%) of the ext-LSs crossed the BBB system in 72 h, demonstrating a satisfactory BBB penetrating capability. Moreover, the TEER was measured before (115.0 ± 7.6 Ω cm2) and after (117.7 ± 11.2 Ω cm2) ext-LS incubation (Fig. S7c†): no significant decrease of the TEER was observed after ext-LS treatment, suggesting as ext-LSs do not affect BBB functionality. This observation, together with the biocompatibility results observed by using the same liposome concentrations, supports the safety of the approach and paves the way for preclinical investigations.
Conclusions
Summarizing, functionalized liposomes loaded with extracts rich in catechins and epicatechins from selected grape pomace completely restored the ROS levels, prevented the α-syn aggregation, rescued the cell viability, and inhibited the β3-tubulin depletion in an in vitro rot-induced PD model. Furthermore, the alteration of the gene expression profile in these cells was successfully rescued by ext-LSs. This antioxidant nanoplatform demonstrated excellent BBB crossing capacity in a multicellular BBB in vitro model, paving the way for future preclinical studies. Considering the potential role of antioxidant nanoparticles in decreasing the neuroinflammation in PD, future studies will be also focused on elucidating the potential anti-inflammatory properties of ext-LSs, including the analysis of cytokine expression and release in microglia.Conflicts of interest
A. M., M. B., A. D., and G. C. declare a patent filing related to some of the technologies presented in this article (Italian patent application IT102020000015493, filed on 26/06/2020). The authors declare no other conflicts of interest.Acknowledgements
We acknowledge Mr Mirco Busco, who kindly donated the grape pomaces.References
- O. B. Tysnes and A. Storstein, J. Neural Transm., 2017, 124(8), 901–905 CrossRef PubMed.
- P. M. A. Antony, N. J. Diederich, R. Krüger and R. Balling, FEBS J., 2013, 280, 5981–5993 CrossRef CAS PubMed.
- W. S. Kim, K. Kagedal and G. M. Halliday, Alzheimer's Res. Ther., 2014, 6(5), 73 CrossRef PubMed.
- J. D. Guo, X. Zhao, Y. Li, G. R. Li and X. L. Liu, Int. J. Mol. Med., 2018, 41, 1817–1825 CAS.
- R. Betarbet, T. B. Sherer and J. T. Greenamyre, Exp. Neurol., 2005, 191, S17–S27 CrossRef CAS PubMed.
- C. Tapeinos and A. Pandit, Adv. Mater., 2016, 30, 2485 Search PubMed.
- G. Pizzino, N. Irrera, M. Cucinotta, G. Pallio, F. Mannino, V. Arcoraci, F. Squadrito, D. Altavilla and A. Bitto, Oxid. Med. Cell. Longevity, 2017, 2017, 8416763 Search PubMed.
- V. Lobo, A. Patil, A. Phatak and N. Chandra, Pharmacogn. Rev., 2010, 118–126 CrossRef CAS PubMed.
- S. Kwiecien, K. Jasnos, M. Magierowski, Z. Sliwowski, R. Pajdo, B. Brzozowski, T. Mach, D. Wojcik and T. Brzozowski, J. Physiol. Pharmacol., 2014, 65(5), 613–622 CAS.
- M. Bélanger, I. Allaman and P. J. Magistretti, Cell Metab., 2011, 14, 724–738 CrossRef PubMed.
- J. Wojsiat, K. M. Zoltowska, K. Laskowska-Kaszub and U. Wojda, Oxid. Med. Cell. Longevity, 2018, 2018, 6435861 Search PubMed.
- S. B. Berman and T. G. Hastings, J. Neurochem., 1999, 73, 1127–1137 CrossRef CAS PubMed.
- J. Bové, D. Prou, C. Perier and S. Przedborski, NeuroRx, 2005, 2, 484–494 CrossRef PubMed.
- S. Manoharan, G. J. Guillemin, R. S. Abiramasundari, M. M. Essa, M. Akbar and M. D. Akbar, Oxid. Med. Cell. Longevity, 2016, 8590578 Search PubMed.
- C. Martinelli, C. Pucci, M. Battaglini, A. Marino and G. Ciofani, Adv. Healthcare Mater., 2020, 9, 1901589 CrossRef CAS PubMed.
- E. L. Lane, Eur. J. Neurosci., 2019, 9, 3 Search PubMed.
- R. Filograna, M. Beltramini, L. Bubacco and M. Bisaglia, Curr. Neuropharmacol., 2016, 14, 2016 Search PubMed.
- H. A. R. Suleria, C. J. Barrow and F. R. Dunshea, Foods, 2020, 9, 1206 CrossRef CAS PubMed.
- E.-Q. Xia, G.-F. Deng, Y.-J. Guo and H.-B. Li, Int. J. Mol. Sci., 2010, 11, 622 CrossRef CAS PubMed.
- L. Sochorova, B. Prusova, M. Cebova, T. Jurikova, J. Mlcek, A. Adamkova, S. Nedomova, M. Baron and J. Sochor, Molecules, 2020, 25, 5311 CrossRef CAS PubMed.
- M. L. Manca, E. Casula, F. Marongiu, G. Bacchetta, G. Sarais, M. Zaru, E. Escribano-Ferrer, J. E. Peris, I. Usach, S. Fais, A. Scano, G. Orrù, R. G. Maroun, A. M. Fadda and M. Manconi, Sci. Rep., 2020, 10, 1–14 CrossRef PubMed.
- C. M. Peixoto, M. I. Dias, M. J. Alves, R. C. Calhelha, L. Barros, S. P. Pinho and I. C. F. R. Ferreira, Food Chem., 2018, 253, 132–138 CrossRef CAS PubMed.
- P. Chowdhary, A. Gupta, E. Gnansounou, A. Pandey and P. Chaturvedi, Environ. Pollut., 2021, 278, 116796 CrossRef CAS PubMed.
- A. M. Brezoiu, C. Matei, M. Deaconu, A. M. Stanciuc, A. Trifan, A. Gaspar-Pintiliescu and D. Berger, Food Chem. Toxicol., 2019, 133, 110787 CrossRef CAS PubMed.
- J. Grgić, G. Šelo, M. Planinić, M. Tišma and A. Bucić-Kojić, Antioxidants, 2020, 9, 1–36 CrossRef PubMed.
- Y. Zhang, C. Lv and G. Zhao, Food Rev. Int., 2021 DOI:10.1080/87559129.2021.1888973.
- N. J. Abbott, A. A. K. Patabendige, D. E. M. Dolman, S. R. Yusof and D. J. Begley, Neurobiol. Dis., 2010, 37(1), 13–25 CrossRef CAS PubMed.
- N. J. Abbott and A. Friedman, Epilepsia, 2012, 53(s6), 1–6 CrossRef PubMed.
- W. M. Pardridge, NeuroRx, 2005, 2, 3–14 CrossRef PubMed.
- J. Nicolas, S. Mura, D. Brambilla, N. MacKiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- C. Martinelli, C. Pucci and G. Ciofani, APL Bioeng., 2019, 3, 011502 CrossRef PubMed.
- C. Tapeinos, M. Battaglini and G. Ciofani, J. Controlled Release, 2017, 264, 306–332 CrossRef CAS PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- Z. E. Suntres, J. Toxicol., 2011, 2011, 152474 Search PubMed.
- S. Mourtas, A. N. Lazar, E. Markoutsa, C. Duyckaerts and S. G. Antimisiaris, Eur. J. Med. Chem., 2014, 80, 175–183 CrossRef CAS PubMed.
- S. Sarkar and N. Das, Mech. Ageing Dev., 2006, 127(4), 391–397 CrossRef CAS PubMed.
- J. Sinha, N. Das and M. K. Basu, Biomed. Pharmacother., 2001, 55(5), 264–271 CrossRef CAS.
- I. Rellini, C. Scopesi, P. Piazza, S. Olivari, M. Perrone and M. Firpo, First Contributions to the DEfinition of the “Cinque Terre” Doc Wines Terroir (NW-Italy), 2019, vol. 1 Search PubMed.
- S. Wen, T. Aki, K. Unuma and K. Uemura, Front. Cell. Neurosci., 2020, 14, 581191 CrossRef CAS PubMed.
- M. Pokusa, D. Hajdúchová, V. Menichová, A. Evinová, Z. Hatoková and A. Kráľová-Trančíková, Physiol. Res., 2021, 70, 89–99 CAS.
- T. B. Sherer, R. Betarbet, A. K. Stout, S. Lund, M. Baptista, A. V. Panov, M. R. Cookson and J. T. Greenamyre, J. Neurosci., 2002, 22, 7006–7015 CrossRef CAS PubMed.
- H. Nawaz, J. Shi, G. S. Mittal and Y. Kakuda, Sep. Purif. Technol., 2006, 48, 176–181 CrossRef CAS.
- K. Zhou, L. Su and L. Yu, J. Agric. Food Chem., 2004, 52, 6108–6114 CrossRef CAS PubMed.
- I. Pezzini, A. Marino, S. Del Turco, C. Nesti, S. Doccini, V. Cappello, M. Gemmi, P. Parlanti, F. M. Santorelli, V. Mattoli and G. Ciofani, Nanomedicine, 2017, 12, 403–416 CrossRef CAS PubMed.
- J. Shi, J. Yu, M. Bryan and Y. Wu, Optimization of the extraction of polyphenols from grape seed meal by aqueous ethanol solution Post-harvest Processing of Peanut and Wheat Products to Reduce Inherent Allergens View project Effects of Enzymatic Allergen Reducing Process on Nutritional Quality, Sensory Attributes and Storage Stability of Peanuts View project, 2002 Search PubMed.
- S. Charmsaz, B. Doherty, S. Cocchiglia, D. Varešlija, A. Marino, N. Cosgrove, R. Marques, N. Priedigkeit, S. Purcell, F. Bane, J. Bolger, C. Byrne, P. J. O'Halloran, F. Brett, K. Sheehan, K. Brennan, A. M. Hopkins, S. Keelan, P. Jagust, S. Madden, C. Martinelli, M. Battaglini, S. Oesterreich, A. V. Lee, G. Ciofani, A. D. K. Hill and L. S. Young, BMC Med., 2020, 18, 349 CrossRef CAS PubMed.
- A. Marino, A. Camponovo, A. Degl'Innocenti, M. Bartolucci, C. Tapeinos, C. Martinelli, D. De Pasquale, F. Santoro, V. Mollo, S. Arai, M. Suzuki, Y. Harada, A. Petretto and G. Ciofani, Nanoscale, 2019, 11, 21227–21248 RSC.
- A. Marino, G. Ciofani, C. Filippeschi, M. Pellegrino, M. Pellegrini, P. Orsini, M. Pasqualetti, V. Mattoli and B. Mazzolai, ACS Appl. Mater. Interfaces, 2013, 5, 13012–13021 CrossRef CAS PubMed.
- A. Marino, S. Arai, Y. Hou, E. Sinibaldi, M. Pellegrino, Y. T. Chang, B. Mazzolai, V. Mattoli, M. Suzuki and G. Ciofani, ACS Nano, 2015, 9, 7678–7689 CrossRef CAS PubMed.
- B. Weksler, I. A. Romero and P. O. Couraud, Fluids Barriers CNS, 2013, 10 Search PubMed.
- T. D. Brown, N. Habibi, D. Wu, J. Lahann and S. Mitragotri, ACS Biomater. Sci. Eng., 2020, 6, 4916–4928 CrossRef CAS PubMed.
- L. Battaglia, M. Gallarate, E. Peira, D. Chirio, I. Solazzi, S. M. A. Giordano, C. L. Gigliotti, C. Riganti and C. Dianzani, Nanotechnology, 2015, 26, 255102 CrossRef PubMed.
- G. Graverini, V. Piazzini, E. Landucci, D. Pantano, P. Nardiello, F. Casamenti, D. E. Pellegrini-Giampietro, A. R. Bilia and M. C. Bergonzi, Colloids Surf., B, 2018, 161, 302–313 CrossRef CAS PubMed.
- A. Marino, M. Baronio, U. Buratti, E. Mele and G. Ciofani, Front. Bioeng. Biotechnol., 2021, 9, 66 Search PubMed.
- C. Tapeinos, A. Marino, M. Battaglini, S. Migliorin, R. Brescia, A. Scarpellini, C. De Julián Fernández, M. Prato, F. Drago and G. Ciofani, Nanoscale, 2019, 11, 72–88 RSC.
- A. Marino, S. Arai, Y. Hou, A. Degl'Innocenti, V. Cappello, B. Mazzolai, Y. T. Chang, V. Mattoli, M. Suzuki and G. Ciofani, ACS Nano, 2017, 11, 2494–2505 CrossRef CAS PubMed.
- E. Pastrana-Bonilla, C. C. Akoh, S. Sellappan and G. Krewer, J. Agric. Food Chem., 2003, 51, 5497–5503 CrossRef CAS PubMed.
- Y. Yilmaz and R. T. Toledo, J. Agric. Food Chem., 2004, 52, 255–260 CrossRef CAS PubMed.
- M. Anastasiadi, H. Pratsinis, D. Kletsas, A. L. Skaltsounis and S. A. Haroutounian, Food Res. Int., 2010, 43, 805–813 CrossRef CAS.
- D. F. Maluf, M. M. Gonçalves, R. W. O. D'Angelo, A. B. Girassol, A. P. Tulio, Y. M. Pupo and P. V. Farago, Cosmetics, 2018, 5, 46 CrossRef.
- S. Weidner, A. Rybarczyk, M. Karamać, A. Król, A. Mostek, J. GrÈ©bosz and R. Amarowicz, Molecules, 2013, 18, 3410–3426 CrossRef CAS PubMed.
- S. G. M. Ong, M. Chitneni, K. S. Lee, L. C. Ming and K. H. Yuen, Pharmaceutics, 2016, 8(4), 36 CrossRef PubMed.
- Y. Lee and D. H. Thompson, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9(5) DOI:10.1002/WNAN.1450.
- H. Yi, W. Lu, F. Liu, G. Zhang, F. Xie, W. Liu, L. Wang, W. Zhou and Z. Cheng, J. Nanobiotechnol., 2021, 19, 1–19 CrossRef PubMed.
- J. E. Smolen and S. B. Shohet, J. Lipid Res., 1974, 15, 273–280 CrossRef CAS.
- R. Vieira, P. Severino, L. A. Nalone, S. B. Souto, A. M. Silva, M. Lucarini, A. Durazzo, A. Santini and E. B. Souto, Molecules, 2020, 25(3), 685 CrossRef CAS PubMed.
- D. Montizaan, K. Yang, C. Reker-Smit and A. Salvati, Nanomedicine, 2020, 30, 102300 CrossRef CAS PubMed.
- J. Cauzzo, M. Nystad, A. M. Holsæter, P. Basnet and N. Škalko-Basnet, Int. J. Mol. Sci., 2020, 21, 4847 CrossRef CAS PubMed.
- W. Kang, D. Svirskis, V. Sarojini, A. L. McGregor, J. Bevitt, Z. Wu, W. Kang, D. Svirskis, V. Sarojini, A. L. McGregor, J. Bevitt and Z. Wu, Oncotarget, 2017, 8, 36614–36627 CrossRef PubMed.
- M. Ramalingam, Y. J. Huh and Y. Il Lee, Front. Neurosci., 2019, 13, 1028 CrossRef PubMed.
- M. Ramalingam, S. Jang and H. S. Jeong, Int. J. Mol. Sci., 2021, 22, 1–21 Search PubMed.
- H.-C. Wu, Q.-L. Hu, S.-J. Zhang, Y.-M. Wang, Z.-K. Jin, L.-F. Lv, S. Zhang, Z.-L. Liu, H.-L. Wu and O.-M. Cheng, Neural Regener. Res., 2018, 13, 1375 CrossRef PubMed.
- T.-K. Lin, S.-D. Chen, Y.-C. Chuang, H.-Y. Lin, C.-R. Huang, J.-H. Chuang, P.-W. Wang, S.-T. Huang, M.-M. Tiao, J.-B. Chen and C.-W. Liou, Int. J. Mol. Sci., 2014, 15, 1625–1646 CrossRef PubMed.
- F. Rey, S. Ottolenghi, T. Giallongo, A. Balsari, C. Martinelli, R. Rey, R. Allevi, A. M. Di Giulio, G. V. Zuccotti, S. Mazzucchelli, R. Foresti, M. Samaja and S. Carelli, Antioxidants, 2021, 10, 121 CrossRef CAS PubMed.
- K. Imamura, T. Takeshima, Y. Kashiwaya, K. Nakaso and K. Nakashima, J. Neurosci. Res., 2006, 84, 1376–1384 CrossRef CAS PubMed.
- T. G. Shutava, S. S. Balkundi and Y. M. Lvov, J. Colloid Interface Sci., 2009, 330, 276–283 CrossRef CAS PubMed.
- C. Espinosa-Diez, V. Miguel, D. Mennerich, T. Kietzmann, P. Sánchez-Pérez, S. Cadenas and S. Lamas, Redox Biol., 2015, 6, 183–197 CrossRef CAS PubMed.
- A. Okado-Matsumoto and I. Fridovich, J. Biol. Chem., 2001, 276, 38388–38393 CrossRef CAS PubMed.
- Y. Li, T. T. Huang, E. J. Carlson, S. Melov, P. C. Ursell, J. L. Olson, L. J. Noble, M. P. Yoshimura, C. Berger, P. H. Chan, D. C. Wallace and C. J. Epstein, Nat. Genet., 1995, 11, 376–381 CrossRef CAS PubMed.
- L. Wang, Q. Duan, T. Wang, M. Ahmed, N. Zhang, Y. Li, L. Li and X. Yao, Oxid. Med. Cell. Longevity, 2015, 2015, 217670 Search PubMed.
- S. Heinz, A. Freyberger, B. Lawrenz, L. Schladt, G. Schmuck and H. Ellinger-Ziegelbauer, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- N. Li, K. Ragheb, G. Lawler, J. Sturgis, B. Rajwa, J. A. Melendez and J. P. Robinson, J. Biol. Chem., 2003, 278, 8516–8525 CrossRef CAS PubMed.
- R. Betarbet, T. B. Sherer, G. MacKenzie, M. Garcia-Osuna, A. V. Panov and J. T. Greenamyre, Nat. Neurosci., 2000, 3, 1301–1306 CrossRef CAS PubMed.
- T. B. Sherer, R. Betarbet, A. K. Stout, S. Lund, M. Baptista, A. V. Panov, M. R. Cookson and J. T. Greenamyre, J. Neurosci., 2002, 22, 7006–7015 CrossRef CAS PubMed.
- S. H. Cha, S. J. Heo, Y. J. Jeon and S. M. Park, RSC Adv., 2016, 6, 110040–110046 RSC.
- L. Palazzi, M. Leri, S. Cesaro, M. Stefani, M. Bucciantini and P. Polverino de Laureto, Biochem. Pharmacol., 2020, 173, 113722 CrossRef CAS PubMed.
- X. Dong, Theranostics, 2018, 8, 1481–1493 CrossRef CAS PubMed.
- M. E. Piersimoni, X. Teng, A. E. G. Cass and L. Ying, Nanoscale Adv., 2020, 2, 5666–5681 RSC.
- N. Zhao, X. Yang, H. R. Calvelli, Y. Cao, N. L. Francis, R. A. Chmielowski, L. B. Joseph, Z. P. Pang, K. E. Uhrich, J. Baum and P. V. Moghe, Front. Bioeng. Biotechnol., 2020, 8, 112 CrossRef PubMed.
- F. Simunovic, M. Yi, Y. Wang, L. Macey, L. T. Brown, A. M. Krichevsky, S. L. Andersen, R. M. Stephens, F. M. Benes and K. C. Sonntag, Brain, 2009, 132, 1795–1809 CrossRef PubMed.
- B. Lassus, S. Magifico, S. Pignon, P. Belenguer, M. C. Miquel and J. M. Peyrin, Sci. Rep., 2016, 6, 1–12 CrossRef PubMed.
- M. D. Neely, C. A. Davison, M. Aschner and A. B. Bowman, Toxicol. Sci., 2017, 159, 366–379 CrossRef CAS PubMed.
- S. Charmsaz, B. Doherty, S. Cocchiglia, D. Varešlija, A. Marino, N. Cosgrove, R. Marques, N. Priedigkeit, S. Purcell, F. Bane, J. Bolger, C. Byrne, P. J. O'Halloran, F. Brett, K. Sheehan, K. Brennan, A. M. Hopkins, S. Keelan, P. Jagust, S. Madden, C. Martinelli, M. Battaglini, S. Oesterreich, A. V. Lee, G. Ciofani, A. D. K. Hill and L. S. Young, BMC Med., 2020, 18, 349 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Five figures and one table. Fig. S1 shows the calibration curves of resveratrol for the Folin–Ciocalteu phenol quantification and of trolox for TEAC assessment. Fig. S2 shows the fluorescence calibration curve of different concentrations of DiO-stained ext-LSs. Fig. S3 reports the representative SEM scan, TEM image and the hydrodynamic size of empty liposomes. Fig. S4 shows the WST-1 viability assay of hCMEC/D3 cells and SH-SY5Y-derived neurons treated at different concentrations of LSs and ext-LSs. Fig. S5 shows the WST-1 cell viability assay of the SH-SY5Y-derived neurons co-treated for 72 h with rot and ext-LSs. Fig. S6 shows the volcano plot of gene expression in different experimental conditions. Fig. S7 reports the BBB crossing investigations. Table S1 reports the forward and reverse primers used to perform the gene expression studies. See DOI: 10.1039/d1bm01202a |
| ‡ These authors equally contributed to this work |
| This journal is © The Royal Society of Chemistry 2021 |