
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox responsive Pluronic micelle mediated delivery of functional siRNA: a modular nano-assembly for targeted delivery†
Sandeep
Kadekar‡
a,
Ganesh N.
Nawale‡
 a,
Vignesh K.
Rangasami
ab,
Vadim
Le Joncour
c,
Pirjo
Laakkonen
c,
Jöns
Hilborn
d,
Oommen P.
Varghese
*a and
Oommen P.
Oommen
*b
a,
Vignesh K.
Rangasami
ab,
Vadim
Le Joncour
c,
Pirjo
Laakkonen
c,
Jöns
Hilborn
d,
Oommen P.
Varghese
*a and
Oommen P.
Oommen
*b
aTranslational Chemical Biology Laboratory, Polymer Chemistry Division, Department of Chemistry – Ångström Laboratory, Uppsala University, 751 21, Uppsala, Sweden. E-mail: oommen.varghese@kemi.uu.se
bBioengineering and Nanomedicine Group, Faculty of Medicine and Health Technologies, Tampere University, 33720, Tampere, Finland. E-mail: oomen.oommen@tuni.fi
cTranslational Cancer Medicine Research Program, Faculty of Medicine, University of Helsinki, Helsinki, Finland
dPolymer Chemistry, Department of Chemistry – Ångström Laboratory, Uppsala University, 751 21, Uppsala, Sweden
First published on 4th May 2021
Abstract
There is an unmet need to develop strategies that allow site-specific delivery of short interfering RNA (siRNA) without any associated toxicity. To address this challenge, we have developed a novel siRNA delivery platform using chemically modified pluronic F108 as an amphiphilic polymer with a releasable bioactive disulfide functionality. The micelles exhibited thermoresponsive properties and showed a hydrodynamic size of ∼291 nm in DLS and ∼200–250 nm in SEM at 37 °C. The grafting of free disulfide pyridyl groups enhanced the transfection efficiency and was successfully demonstrated in human colon carcinoma (HCT116; 88%) and glioma cell lines (U87; 90%), non-cancerous human dermal fibroblast (HDF; 90%) cells as well as in mouse embryonic stem (mES; 54%) cells. To demonstrate the versatility of our modular nanocarrier design, we conjugated the MDGI receptor targeting COOP peptide on the particle surface that allowed the targeted delivery of the cargo molecules to human patent-derived primary BT-13 gliospheres. Transfection experiments with this design resulted in ∼65% silencing of STAT3 mRNA in BT-13 gliospheres, while only ∼20% of gene silencing was observed in the absence of the peptide. We believe that our delivery method solves current problems related to the targeted delivery of RNAi drugs for potential in vivo applications.
Introduction
Efficient intracellular delivery of oligonucleotides to target cells without premature loss in the endosome is one of the key challenges for the successful clinical translation of nucleic acid therapy. Several nanoparticle-based delivery systems are developed based on positively charged lipids,1 polymers,2,3 lipid-polymer hybrids4 or inorganic materials5 that assist in the translocation of siRNA across the cellular barrier and escape the endosomal trap for effective gene silencing. The inherent inflammatory response elicited by cationic transfection reagents and a lack of a tissue-specific delivery strategy for non-liver targets limit the use of such technologies.6,7 Such toxic effects could be partially eliminated by the removal of the excess reagents from the cell-culture media.8,9 We have recently shown that by employing anionic delivery systems such as hyaluronic acid10 or by neutralizing the charge of the cationic polymer by coating with an anionic glycosaminoglycan such as chondroitin sulfate, we could not only improve the transfection efficacy by enabling controlled endosomal release of the cargo molecules but also allow the CD44 receptor-mediated uptake of nanoparticles.11 Another strategy that has been reported includes delivery systems having disulfide-bonds on the surface which improve intracellular trafficking by targeting the cell-surface thiols.12 This includes siRNA modifications having disulfide-linkages that allow the carrier-free delivery of siRNA depending on the accessibility of disulfide bonds for interacting with cell-surface receptors.13,14 The redox state of the intracellular compartment also facilitates the release of the drug molecule from the carrier, enabling efficient gene silencing capability. We hypothesized that nanoparticles decorated with disulfide groups could offer a multimodal supramolecular assembly that could facilitate the conjugation of the drug molecule (siRNA) and targeting ligand as well as allow improved intracellular trafficking capability with a redox-sensitive linkage. Attempts to design siRNA delivery systems by conjugating a cell-surface receptor targeting ligand have achieved limited success as most of the cargo molecules remain trapped within the endosomal compartment. A few exceptions are the molecular conjugate of RNA with N-acetyl-D-galactosamine (GalNAc) that targets the asialoglycoprotein receptor (ASGPR) in the liver15 and the folate conjugate that targets the folate receptor overexpressed in breast cancer cells.16 Of these, GalNAc-conjugates are most efficient as this ligand enables quick uptake and release of siRNA from the endosomal compartment due to the high turnover rate (15 minutes) of the ASGPR receptor.17 Validation of other siRNA-targeting ligand conjugates (e.g., antibody-siRNA) demonstrated that only a small subset of drug conjugates showed functional siRNA internalization.18,19For designing the modular micellar nanocarrier, we utilized Pluronic F108, a well-known tri-block polymer possessing a hydrophobic poly(propylene glycol) (PPG) segment flanked by two hydrophilic poly(ethylene glycol) units or PEGs. This amphiphilic polymer self-assembles under aqueous conditions to micellar structures and can be used as a drug delivery vehicle.20 A Pluronic-based formulation encapsulating the chemotherapeutic agent doxorubicin, SP1049C from Supratek Pharma Inc., has been shown to passively target cancer stem cells in vivo21 and is currently in Phase-III clinical trials for treating esophagus and gastric cancers. Pluronic F108 also exhibits adjuvant activity that improves the gene transfection efficiency for lentiviral vectors.22 Such micelles are also successfully utilized to stabilize siRNA-PEI complexes and facilitate intracellular transport.23,24 However, shortcomings with these strategies include the variabilities in micellar self-assembly, poor siRNA loading efficiency, variability in the knockdown efficiency and a lack of active targeting ability. Other key challenges in the field of siRNA delivery involve the rescuing of functional nucleic acid from endosomal degradation and enhancing serum stability after infusion. We have recently engineered Pluronic-based micelles for delivering Tissue Factor siRNA for improving stem cell survivability and function.25 To address the stability issues we have also designed 4′-guanidinium modified siRNA that significantly enhances the serum stability and bioactivity of the native siRNA sequence.26 Other strategies include the use of calcium ions (Ca2+) that are known to form nanocomplexes with siRNA and plasmid DNA and help in endosomal escape as well as providing the serum stability.27 However, the colloidal stability of calcium phosphate-nucleic acid nanoprecipitates is difficult to control, resulting in high variability in gene and siRNA delivery efficiency.28 Attempts to circumvent these issues were made by physically coating these nanocomplexes with block polymers such as Pluronic-F127![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 or poly(ethylene glycol)–block–poly(aspartic acid).28 These methods are not proficient as the coating of ionic complexes with amphiphilic polymers is not very stable, resulting in inefficient transfection efficiency and high variability. We, therefore, envisaged that the covalent conjugation of siRNA with Pluronic F108 would provide a stable nanoparticle formulation with efficient siRNA loading, which would yield a consistent target-specific gene knockdown.
29 or poly(ethylene glycol)–block–poly(aspartic acid).28 These methods are not proficient as the coating of ionic complexes with amphiphilic polymers is not very stable, resulting in inefficient transfection efficiency and high variability. We, therefore, envisaged that the covalent conjugation of siRNA with Pluronic F108 would provide a stable nanoparticle formulation with efficient siRNA loading, which would yield a consistent target-specific gene knockdown.
Results and discussion
To begin with, we first synthesized Pluronic F108 functionalized with pyridyl disulfide groups (Scheme 1). For this purpose, the amino-disulfide pyridyl linker 2 was synthesized following the reported procedure.30 Thereafter, the terminal hydroxyl groups were activated using p-nitro-phenyl chloroformate to afford intermediate 4 with quantitative yield.31 The nucleophilic substitution of phenyl chloroformate functionalized Pluronic F108 4 with linker 2 afforded a disulfide pyridyl derivative of Pluronic F108 5 with nearly 92% substitution. It is important to note that, hitherto there are no reports on complete modification of Pluronic and only 60% of modification has been reported previously.31 The product 5 was purified by dialysis, and the degree of pyridyl disulfide incorporation was quantified by UV spectroscopy.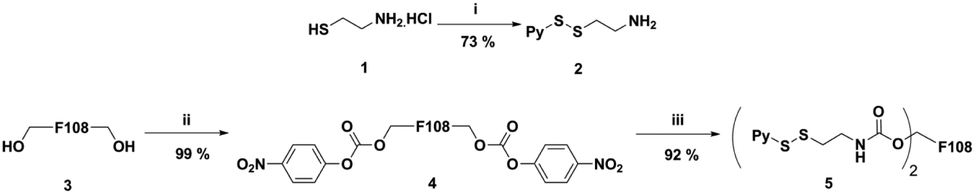 | ||
| Scheme 1 Synthesis of pyridyl disulfide of Pluronic F108 (P-SS) (5). (i) 2,2′-Dipyridyl disulfide, MeOH, AcOH, rt, 48 h; (ii) 4-nitrophenyl chloroformate, DCM, rt, overnight; and (iii) 2, DCM, reflux, overnight. | ||
In the next step, we conjugated siRNA targeting STAT3 as it is one of the key transcription factors that play an important role in tumor growth and survival and aid in tumor immune evasion.32,33 Effective silencing of the STAT3 gene by siRNA has been reported to suppress tumor growth and the antisense drug targeting this gene is being clinically validated for treating lymphoma and lung cancer.34–36 To perform the siRNA conjugation reaction, the disulfide protecting groups of siRNA at the 3′ end of the sense strand were reduced with dithiothreitol (DTT) and the purified material was treated with an excess (100 eq.) of pyridyl disulfide modified Pluronic 5 in PBS buffer (pH 8), at room temperature overnight.37 (Scheme 2).
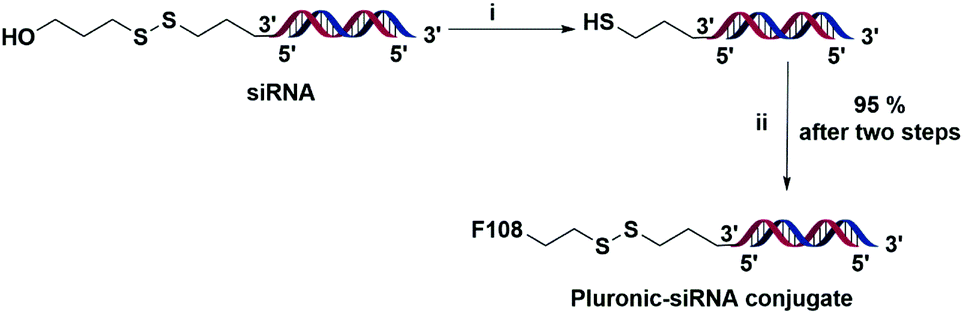 | ||
| Scheme 2 Synthesis of the Pluronic F108-siRNA conjugate through a disulfide linkage. The red strand is the sense strand and the blue strand is the antisense strand. (i) Dithiothreitol, H2O, 37 °C, 2 h; and (ii) 5, PBS buffer (pH 8), rt, overnight. | ||
The ratio of siRNA to SS-pyridyl groups was carefully optimized, and 8 mol% of siRNA to SS-pyridyl was chosen as they displayed excellent coupling efficiency. The native PAGE analysis of the reaction mixture indicated ∼95% of conjugation efficiency (Fig. 1C). Further analysis of the Pluronic F108-siRNA conjugate (P-SS-STAT3) indicated nanoparticle formation with a hydrodynamic size to ∼276 nm and a zeta potential of −12.6 mV as determined by dynamic light scattering (DLS) (Fig. 1A and Fig. S2 in the ESI†). For transfection studies, Ca2+ was complexed with the Pluronic siRNA particles (P-SS-STAT3/Ca), which resulted in an increase in the hydrodynamic size to ∼343 nm. The zeta potential also changed from −12.6 mV to −0.153 mV upon the addition of Ca2+ to the complexes (Fig. 1B and S2 in the ESI†). This indicates that the addition of Ca2+ ions neutralized the net negative charge of the particle to yield a neutral nanocarrier. As Pluronic-derived micelles are known to display thermo-responsive properties,38 we investigated the effect of temperature on the size distribution of P-SS-STAT3/Ca by performing DLS at 37 °C and scanning electron microscopy (SEM) where the samples were dried at 37 °C. Gratifyingly, the hydrodynamic size of the P-SS-STAT3/Ca complexes after 30 minutes of incubation at 37 °C displayed a decrease in hydrodynamic size to ∼291 nm (Fig. S2†), whereas the SEM analysis showed a size of ∼200–250 nm (Fig. 1D). For conducting biological studies, we evaluated four types of siRNA nanocomplexes namely the Ca2+-siRNA ionic complex (STAT3/Ca); Ca2+-siRNA ionic complex coated with unmodified Pluronic F108 (P/STAT3/Ca); Ca2+-siRNA ionic complex physically coated with dithio-pyridyl functionalized Pluronic F108 (P-SS/STAT3/Ca) where STAT3 siRNA without 3′-thiol modification was used; and Ca2+-siRNA ionic complex covalently conjugated with Pluronic F108 by employing the thiol-exchange reaction using thiolated siRNA and dithio-pyridyl modified Pluronic (P-SS-STAT3/Ca). The ‘-’ in the abbreviation indicates a covalent linkage while ‘/’ indicates physical association. The concentration of Ca2+ was fixed at 5 mM, as it was reported to give the best transfection efficiency.27
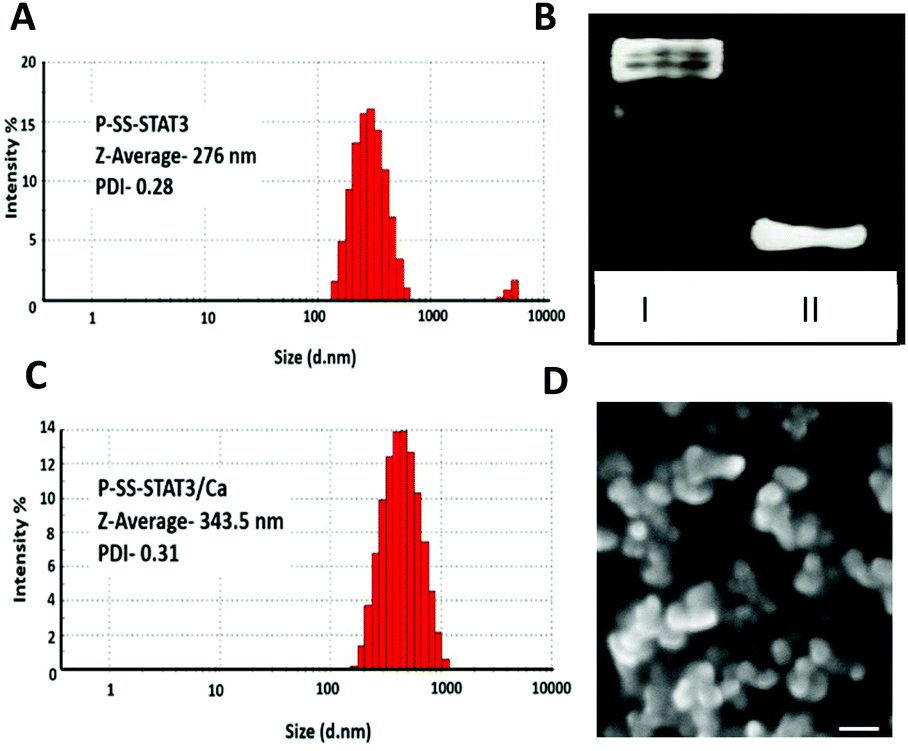 | ||
| Fig. 1 Hydrodynamic size of (A) P-SS-STAT3 and (B) P-SS-STAT3/Ca as determined by the dynamic light scattering method at 25 °C. (C) Native PAGE (15%) to visualize the conjugation of siRNA and P-SS (5). Lane 1 shows P-SS-STAT3 and lane 2 shows unmodified STAT3 siRNA. (D) SEM micrograph image depicting Plu-SS/STAT3/Ca after incubation at 37 °C. Scale bar: 200 nm. | ||
In this study, we used the human colon carcinoma cell line HCT116 to perform STAT3 knockdown studies as colon cancer cells are known to constitutively express this gene and are known to play a crucial role in tumor invasion and nodal metastasis.39 Our results from transfection experiments indicate that both STAT3/Ca and physically coated P/STAT3/Ca gave a modest 35% STAT3 knockdown in 24 h when treated with a 50 nM siRNA concentration. The transfection experiment using a commercial transfection reagent RNAiMAX which was utilized as a positive control displayed a 94% gene silencing under similar conditions. Next, we tested the gene silencing efficacy of physically coated P-SS/STAT3/Ca and covalently conjugated P-SS-STAT3/Ca under identical experimental conditions. Interestingly, P-SS/STAT3/Ca displayed an increase in transfection efficiency from 35% to 61.5% when compared with P/STAT3/Ca that lacked the pyridyl disulfide group on the Pluronic backbone. We believe that the enhanced gene knockdown in the presence of disulfide groups on the nanoparticle surface is predominantly due to the enhanced cytosolic internalization of the nanoparticles by targeting the cell-surface free thiols, as reported recently with other disulfide containing reagents.13 In order to understand the impact of the covalent conjugation of siRNA on the transfection efficiency, we tested P-SS-STAT3/Ca under these conditions. Gratifyingly, P-SS-STAT3/Ca displayed 88% STAT3 knockdown, which is comparable with that of the RNAiMAX group (92%). The gene knockdown was further substantiated at the protein level by western blot assay that showed a lower expression of STAT3 protein levels after 48 h of treatment with P-SS-STAT3/Ca (Fig. 2B). The positive control groups with RNAiMAX showed an efficient gene knockdown with both STAT3 and P-SS-STAT3. We further evaluated the effect of the nanoformulation on cellular toxicity. For this purpose, we performed the cytotoxicity assay with alamarBlue in HCT116 cells. These experiments demonstrated that all nanoformulations were biocompatible and did not show any cytotoxicity even after 48 h treatment of P-SS-STAT3/Ca.
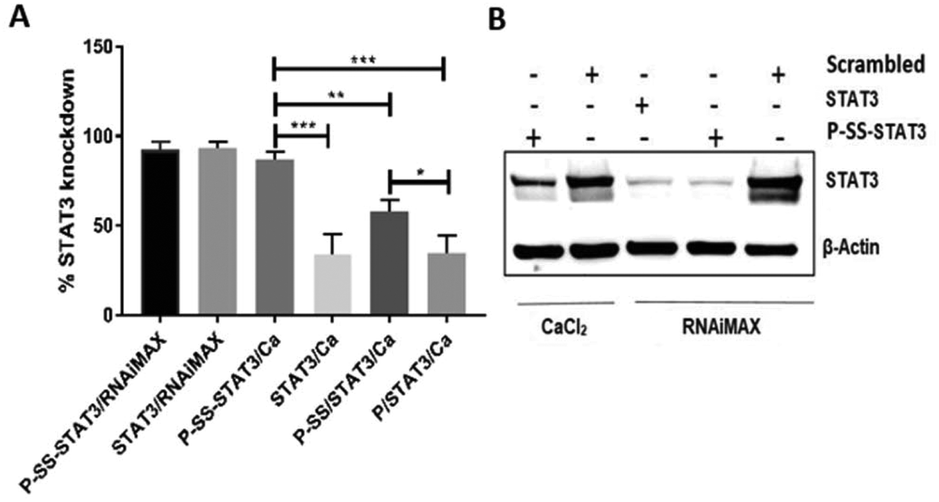 | ||
| Fig. 2 Knockdown of STAT3 in HCT116 cells by (A) qRT-PCR. Statistics for the groups done by one-way ANOVA (***P < 0.001 **P < 0.01). Individual comparison is done by the T-test (*P < 0.05) and (B) western blot analysis with CaCl2 and RNAiMAX. | ||
In order to understand the role of the dithiol conjugation in siRNA delivery and cellular uptake, we performed flow cytometry and confocal microscopy studies using the Cy5.5 tagged siRNA nanocomplex using HCT116 cells. The FACS analysis clearly showed enhanced cellular uptake of the P-SS-STAT3-Cy5.5/Ca nanocomplex as compared to P-SS-STAT3-Cy5.5/RNAiMax suggesting that SS-conjugation enhanced cellular delivery (Fig. S3A in the ESI†). The confocal images of the transfected cells displayed comparable intracellular distribution and perinuclear localization of Cy5.5 labelled siRNA and flow cytometry confirmed the uptake of Cy5.5 labelled siRNA (Fig. S3B in the ESI†). Next, we tested the versatility of our delivery system in different cell types and with different target sequences. For this purpose, we performed STAT3 knockdown experiments in a non-cancerous human dermal fibroblast (HDF) cell line and human glioblastoma cell line U87-MG. Interestingly, we observed over 90% gene silencing in both these cell lines with P-SS-STAT3/Ca similar to the RNAiMAX control. To verify the generality of the approach we tested another sequence using the nanoformulation with siRNA that targets OCT4 (P-SS-OCT4/Ca), a transcription factor that is key to maintaining the stemness of mouse (mES) and human embryonic stem cells.
The transfection experiments with mES cells resulted in ∼54% silencing of OCT4 mRNA when transfected with a 100 nM P-SS-OCT4/Ca complex. We also observed ∼83% and ∼75% knockdown when P-SS-OCT4 and free OCT4 siRNA were transfected with RNAiMAX (Fig. 3B).
 | ||
| Fig. 3 Versatility of the P-SS-Delivery system: (A) STAT3 gene knockdown in U87-MG and human dermal fibroblast (HDF) cell lines. (B) Knockdown of the OCT4 gene in mES cells. (C) Expression level of MDGI when BT13 cells were cultured under 2D conditions and 3D spheroid conditions. (D) Transfection efficiency of the P-SS-STAT3-COOP/Ca and P-SS-STAT3/Ca nanoparticles in patient-derived gliospheres (BT-13). **P < 0.05 statistical analysis is done by ANOVA. | ||
Finally, to develop a clinically relevant system, we envisioned to utilize the excess disulfide pyridyl groups on the nanocarrier surface to engineer a targeted drug delivery system. For this purpose, we conjugated a glioma targeting peptide (COOP) via disulfide-exchange reaction with the N-terminal cysteine modified peptide and SS-pyridyl modified Pluronic as described earlier. The COOP peptide was chosen as the targeting ligand as it is known to target mammary derived growth inhibitor (MDGI) receptors that are overexpressed in human glioblastoma cells, specifically the invasive tumor satellite cells and the vessels produced by these glioblastomas.40 To test our targeted siRNA delivery system, we utilized patient-derived primary BT-13 gliospheres (cells grown as spheroids) that are known to express MDGI receptors under serum free conditions41 and compared with BT-13 cells cultured under 2D conditions. We first analyzed the MDGI mRNA levels in BT-13 cells when cultured under serum-free (3D spheroids) and 10% serum (2D) conditions. As anticipated, we observed a ∼2.5-fold increase in the MDGI mRNA levels in the 3D spheroid cultures, when compared to the BT-13 cells grown in 2D in the presence of serum (Fig. 3C). One of the advantages of using spheroid cultures is that it recapitulates the in vivo conditions having a tissue penetration barrier with higher extracellular matrix composition as in a natural tissue. The conjugation of the COOP peptide on the P-SS-STAT3 micelle (P-SS-STAT3-COOP) resulted in the formation of self-assembled micelles with a hydrodynamic size of ∼230 nm which upon complexation with Ca+2 increased to ∼337 nm (data not shown). Interestingly, the transfection experiments with the COOP peptide conjugated nanoparticles (P-SS-STAT3-COOP/Ca) displayed a significantly higher STAT3 knockdown (∼60%) as compared to the non-targeting variant of the nanoparticles (P-SS-STAT3/Ca) (∼22%); however, with RNAiMax we observed a knockdown efficiency of only 33% (Fig. 3D). Interestingly, the effect of the targeting peptide was not observed when the cells were cultured in 2D in the presence of serum (Fig. S4 in the ESI†). This result demonstrates the significance of cell-specificity and targeted delivery of RNAi drugs to tumor cells.
Conclusions
In summary, we have designed a Pluronic-based thermoresponsive siRNA delivery formulation that allows efficient intracellular delivery of RNA in cancer cell lines, embryonic stem cells and primary glioma cells. Our results also highlight the significance of a modular nanocarrier design that allows covalent conjugation of siRNA and the targeting peptide to promote cell-specific delivery. The mild transfection conditions also demonstrated minimal toxicity, which is important for any siRNA delivery systems for potential in vivo applications.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Notes and references
- S. Ramishetti, I. Hazan-Halevy, R. Palakuri, S. Chatterjee, S. Naidu Gonna, N. Dammes, I. Freilich, L. Kolik Shmuel, D. Danino and D. Peer, Adv. Mater., 2020, 32, 1906128 CrossRef CAS PubMed.
- Y. Zou, M. Zheng, W. Yang, F. Meng, K. Miyata, H. J. Kim, K. Kataoka and Z. Zhong, Adv. Mater., 2017, 29, 1703285 CrossRef PubMed.
- S. Watanabe, K. Hayashi, K. Toh, H. J. Kim, X. Liu, H. Chaya, S. Fukushima, K. Katsushima, Y. Kondo, S. Uchida, S. Ogura, T. Nomoto, H. Takemoto, H. Cabral, H. Kinoh, H. Y. Tanaka, M. R. Kano, Y. Matsumoto, H. Fukuhara, S. Uchida, M. Nangaku, K. Osada, N. Nishiyama, K. Miyata and K. Kataoka, Nat. Commun., 2019, 10, 1–13 CrossRef CAS PubMed.
- K. Thanki, D. van Eetvelde, A. Geyer, J. Fraire, R. Hendrix, H. Van Eygen, E. Putteman, H. Sami, C. de Souza Carvalho-Wodarz, H. Franzyk, H. M. Nielsen, K. Braeckmans, C. M. Lehr, M. Ogris and C. Foged, J. Controlled Release, 2019, 310, 82–93 CrossRef CAS PubMed.
- Y. Liu, X. Ji, W. W. L. Tong, D. Askhatova, T. Yang, H. Cheng, Y. Wang and J. Shi, Angew. Chem., Int. Ed., 2018, 57, 1510–1513 CrossRef CAS PubMed.
- A. Wittrup and J. Lieberman, Nat. Rev. Genet., 2015, 16, 543–552 CrossRef CAS PubMed.
- S. Akhtar, Expert Opin. Drug Metab. Toxicol., 2010, 6, 1347–1362 CrossRef CAS PubMed.
- M. Paidikondala, S. Kadekar and O. Varghese, Int. J. Mol. Sci., 2018, 20, 56 CrossRef PubMed.
- M. Paidikondala, G. N. Nawale and O. P. Varghese, Biomacromolecules, 2019, 20, 1317–1324 CrossRef CAS PubMed.
- M. Paidikondala, V. K. Rangasami, G. N. Nawale, T. Casalini, G. Perale, S. Kadekar, G. Mohanty, T. Salminen, O. P. Oommen and O. P. Varghese, Angew. Chem., Int. Ed., 2019, 58, 2815–2819 CrossRef CAS PubMed.
- H. Yan, O. P. Oommen, D. Yu, J. Hilborn, H. Qian and O. P. Varghese, Adv. Funct. Mater., 2015, 25, 3907–3915 CrossRef CAS.
- E. K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088–2091 CrossRef CAS PubMed.
- Z. Shu, I. Tanaka, A. Ota, D. Fushihara, N. Abe, S. Kawaguchi, K. Nakamoto, F. Tomoike, S. Tada, Y. Ito, Y. Kimura and H. Abe, Angew. Chem., Int. Ed., 2019, 58, 6611–6615 CrossRef CAS PubMed.
- F. Gauthier, S. Claveau, J. R. Bertrand, J. J. Vasseur, C. Dupouy and F. Debart, Bioorg. Med. Chem., 2018, 26, 4635–4643 CrossRef CAS PubMed.
- J. M. Brown, J. E. Dahlman, K. K. Neuman, C. A. H. Prata, M. C. Krampert, P. M. Hadwiger and H.-P. Vornlocher, Nucleic Acid Ther., 2019, 29, 231–244 CrossRef CAS PubMed.
- E. A. Orellana, S. Tenneti, L. Rangasamy, L. T. Lyle, P. S. Low and A. L. Kasinski, Sci. Transl. Med., 2017, 9, eaam9327 CrossRef PubMed.
- A. L. Schwartz, S. E. Fridovich and H. F. Lodish, J. Biol. Chem., 1982, 257, 4230–4237 CrossRef CAS.
- G. E. Rhizobium, Nucleic Acids Res., 2013, 1, 13–14 Search PubMed.
- T. Sugo, M. Terada, T. Oikawa, K. Miyata, S. Nishimura, E. Kenjo, M. Ogasawara-Shimizu, Y. Makita, S. Imaichi, S. Murata, K. Otake, K. Kikuchi, M. Teratani, Y. Masuda, T. Kamei, S. Takagahara, S. Ikeda, T. Ohtaki and H. Matsumoto, J. Controlled Release, 2016, 237, 1–13 CrossRef CAS PubMed.
- A. Pitto-Barry and N. P. E. Barry, Polym. Chem., 2014, 5, 3291–3297 RSC.
- D. Y. Alakhova, Y. Zhao, S. Li and A. V. Kabanov, PLoS One, 2013, 8, e72238 CrossRef CAS PubMed.
- I. Höfig, M. J. Atkinson, S. Mall, A. M. Krackhardt, C. Thirion and N. Anastasov, J. Gene Med., 2012, 14, 549–560 CrossRef PubMed.
- S. H. Lee, S. H. Choi, S. H. Kim and T. G. Park, J. Controlled Release, 2008, 125, 25–32 CrossRef CAS PubMed.
- D. Rafael, P. Gener, F. Andrade, J. Seras-Franzoso, S. Montero, Y. Fernandez, M. Hidalgo, D. Arango, J. Sayos, H. F. Florindo, I. Abasolo, S. Schwartz and M. Videira, Drug Deliv., 2018, 25, 961–972 CrossRef CAS PubMed.
- V. K. Rangasami, G. Nawale, K. Asawa, S. Kadekar, S. Samanta, B. Nilsson, K. N. Ekdahl, S. Miettinen, J. Hilborn, Y. Teramura, O. P. Varghese and O. P. Oommen, Biomacromolecules, 2021, 22(5), 1980–1989 CrossRef CAS PubMed.
- G. N. Nawale, S. Bahadorikhalili, P. Sengupta, S. Kadekar, S. Chatterjee and O. P. Varghese, Chem. Commun., 2019, 55, 9112–9115 RSC.
- E. Ruvinov, O. Kryukov, E. Forti, E. Korin, M. Goldstein and S. Cohen, J. Controlled Release, 2015, 203, 150–160 CrossRef CAS PubMed.
- Y. Kakizawa, K. Miyata, S. Furukawa and K. Kataoka, Adv. Mater., 2004, 16, 699–702 CrossRef CAS.
- L. Qin, Y. Sun, P. Liu, Q. Wang, B. Han and Y. Duan, J. Biomater. Sci. Polym. Ed., 2013, 24, 1757–1766 CrossRef CAS PubMed.
- G. T. Zugates, D. G. Anderson, S. R. Little, I. E. B. Lawhorn and R. Langer, J. Am. Chem. Soc., 2006, 128, 12726–12734 CrossRef CAS PubMed.
- J. T. Li, J. Carlsson, J. N. Lin and K. D. Caldwell, Bioconjugate Chem., 1996, 7, 592–599 CrossRef CAS PubMed.
- H. Yu, D. Pardoll and R. Jove, Nat. Rev. Cancer, 2009, 9, 798–809 CrossRef CAS PubMed.
- M. Demaria, A. Camporeale and V. Poli, Int. J. Cancer, 2014, 135, 1997–2003 CrossRef CAS PubMed.
- M. Kortylewski, P. Swiderski, A. Herrmann, L. Wang, C. Kowolik, M. Kujawski, H. Lee, A. Scuto, Y. Liu, C. Yang, J. Deng, H. S. Soifer, A. Raubitschek, S. Forman, J. J. Rossi, D. M. Pardoll, R. Jove and H. Yu, Nat. Biotechnol., 2009, 27, 925–932 CrossRef CAS PubMed.
- A. Alshamsan, S. Hamdy, J. Samuel, A. O. S. El-Kadi, A. Lavasanifar and H. Uludaǧ, Biomaterials, 2010, 31, 1420–1428 CrossRef CAS PubMed.
- D. Hong, R. Kurzrock, Y. Kim, R. Woessner, A. Younes, J. Nemunaitis, N. Fowler, T. Zhou, J. Schmidt, M. Jo, S. J. Lee, M. Yamashita, S. G. Hughes, L. Fayad, S. Piha-Paul, M. V. P. Nadella, M. Mohseni, D. Lawson, C. Reimer, D. C. Blakey, X. Xiao, J. Hsu, A. Revenko, B. P. Monia and A. R. MacLeod, Sci. Transl. Med., 2015, 7, 314ra185 CrossRef PubMed.
- O. Y. Wong, P. I. Pradeepkumar and S. K. Silverman, Biochemistry, 2011, 50, 4741–4749 CrossRef CAS PubMed.
- K. H. Bae, S. H. Choi, S. Y. Park, Y. Lee and T. G. Park, Langmuir, 2006, 22, 6380–6384 CrossRef CAS PubMed.
- L. Lin, A. Liu, Z. Peng, H. J. Lin, P. K. Li, C. Li and J. Lin, Cancer Res., 2011, 71, 7226–7237 CrossRef CAS PubMed.
- M. Hyvönen, J. Enbäck, T. Huhtala, J. Lammi, H. Sihto, J. Weisell, H. Joensuu, K. Rosenthal-Aizman, S. El-Andaloussi, U. Langel, A. Närvänen, G. Bergers and P. Laakkonen, Mol. Cancer Ther., 2014, 13, 996–1007 CrossRef PubMed.
- V. Le Joncour, P. Filppu, M. Hyvönen, M. Holopainen, S. P. Turunen, H. Sihto, I. Burghardt, H. Joensuu, O. Tynninen, J. Jääskeläinen, M. Weller, K. Lehti, R. Käkelä and P. Laakkonen, EMBO Mol. Med., 2019, 11(6), e9034 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1bm00428j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |