
Oxygen-carrying nanoparticle-based chemo-sonodynamic therapy for tumor suppression and autoimmunity activation†
Yanan
Zhang
a,
Na
Qiu
a,
Yu
Zhang
a,
Huixian
Yan
a,
Jianbo
Ji
a,
Yanwei
Xi
a,
Xiaoye
Yang
*a,
Xiaogang
Zhao
*b and
Guangxi
Zhai
 *a
*a
aDepartment of Pharmaceutics, Key Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Shandong University, Jinan, 250012, PR China. E-mail: professorgxzhai@126.com; yangxiaoye@sdu.edu.cn; Tel: +(86) 531-88382015
bDepartment of Thoracic Surgery, The Second Hospital of Shandong University, Jinan, Shandong 250033, China. E-mail: zhaoxiaogang@sdu.edu.cn; Tel: +(86) 531-85875009
First published on 9th April 2021
Abstract
Sonodynamic therapy (SDT) is a promising non-invasive approach for cancer therapy. However, tumor hypoxia, a pathological characteristic of most solid tumor types, poses a major challenge in the application of SDT. In this study, a novel CD44 receptor-targeted and redox/ultrasound-responsive oxygen-carrying nanoplatform was constructed using chondroitin sulfate (CS), reactive oxygen species (ROS)-generating sonosensitizer Rhein (Rh), and perfluorocarbon (PFC). Perfluoroalkyl groups introduced into the structures preserved the oxygen carrying ability of PFC, increasing the oxygen content in B16F10 melanoma cells and enhancing the efficiency of SDT. Controlled nanoparticles without PFC generated lower ROS levels and exerted inferior tumor inhibition effects, both in vitro and in vivo, under ultrasound-treatment. In addition, SDT promoted immunogenic cell death (ICD) by inducing exposure of calreticulin (CRT) after treatment with CS–Rh–PFC nanoparticles (NPs). The immune system was significantly activated by docetaxel (DTX)-loaded NPs after SDT treatment due to the enhanced secretion of IFN-γ, TNF-α, IL-2 and IL-6 cytokines and tumor-infiltrating CD4+ and CD8+ T cell contents. Our findings support the utility of CS–Rh–PFC as an effective anti-tumor nanoplatform that promotes general immunity and accommodates multiple hydrophobic drugs to enhance the beneficial effects of chemo-SDT therapy.
1. Introduction
Melanoma is considered as the most malignant skin tumor type in terms of metastasis and lethality.1,2 Patients experience pain and suffering before and after surgery during the course of traditional therapy. Moreover, survival rates cannot be guaranteed owing to the high metastasis of melanoma cells leading to nearly 90% mortality worldwide.3,4 Non-invasive treatments, such as near-infrared (NIR) light,5 radioactive rays (X-ray),6 and ultrasound (US), are currently gaining recognition as effective ways of improving patient quality of life.7 However, the barriers of skin and deeper tissues limit the penetration of light, and cautious application of infinitely penetrating rays is essential to avoid detrimental effects to normal cells and tissues.8 US-triggered SDT has shown efficacy as an alternative non-invasive treatment for different cancer types with the aid of sonosensitizers to generate ROS, exhibiting good penetration and safety.9For the effective treatment of cancers with SDT, the pivotal step is choice of sonosensitizer. The existing sonosensitizers are classified into two broad categories, specifically, organic and inorganic. The majority of inorganic sonosensitizers, such as TiO2-based molecules or modified-MnWOx,10 possess semiconductor properties and exert good SDT effects. However, organic sonosensitizers, such as porphyrin molecules,11 IR780,12 ICG13 and chlorin e6 (Ce6),14 are more commonly used for therapy. The application of small hydrophobic sonosensitizers is hindered by low tumor targeting ability and poor solubility, which could be addressed by applying different hydrophilic modifications to ensure optimization of SDT effects. However, these two sonosensitizer types only generate cytotoxic ROS under US stimulation, which has a US-time-dependent tumor inhibitory function. Therefore, the development of continual ROS-releasing sonosensitizers is highly desirable for the improvement of therapeutic efficacy.
Hypoxia is a basic pathological characteristic of solid tumors associated with high metastasis and invasion and poor prognosis after therapy.15 However, the low density of oxygen in the tumor micro-environment also restricts the production of ROS by SDT.16 To alleviate the deficiency of oxygen and formulate novel NPs, an oxygen-carrying compound could be synthesized.17 PFC is used as an artificial blood substitute mainly due to its biocompatibility in vivo and oxygen-carrying capacity.18 The combined application of PFC and photodynamic therapy (PDT) has gained considerable success in recent years.17
CS is a non-immunogenic polysaccharide with excellent biocompatibility that effectively targets tumor tissues in association with CD44 receptors on the surface of cancer cells.19 The preparation of CS-based NPs was predominantly synthesized via covalent bond formation. Rh was initially identified as an effective sonosensitizer that can generate ROS radicals spontaneously in cancer cells and suppress tumor growth by activating JNK/Jun/caspase-3 signaling.20 Tumor suppression by Rh was further enhanced after US treatment owing to the generation of more ROS molecules.21 Therefore, the Rh-based nanoplatform could produce ROS slowly but continuously in tumor tissues following intravenous injection, overcoming the limitation of ROS generation only within a few minutes of US treatment. In addition, the inherent anti-tumor and anti-metastasis properties of Rh contribute to the therapeutic activity of SDT.22 To improve anti-tumor effects, a model antineoplastic drug, DTX, with the ability to block the cell cycle was incorporated into NPs, with a view to induce combined therapeutic actions.23
Here, we report for the first time the self-assembled CS–Rh–PFC NPs generated by amide reactions using the disulfide bond. The newly generated NPs were certified as a novel oxygen and DTX carrier with ROS-generating ability. Upon application to B16F10 cells, our NPs could actively target tumor cells and induce eversion of CRT protein, leading to ICD.20 The addition of DTX augmented the ICD effect in keeping with recent reports.24 Moreover, NPs effectively activated the immune system upon injection into mice and DTX-loaded NPs significantly suppressed the growth of B16F10 tumor tissues.
2. Materials and methods
2.1. Materials
A medical ultrasonic instrument (DM-300B; Shenzhen Dimip Technology Co., Ltd, Shenzhen, China) was employed for therapy using the following parameters: ultrasound stimulation (1 MHz), duty cycle (10–90%, 40% applied) and pulse repetition cycle (10 ms). Acoustic intensity generally varies within the range of 0.3–3.0 W cm−2. Considering clinical application and safety, an acoustic intensity of 1.2 W cm−2 was applied in this study. All parameters were consistently maintained during SDT.
C57BL/6N mice (male, 5–8 weeks) and Wistar rats (male, 250 ± 10 g) were acquired from Beijing Vital River Laboratory Animal Technology Co. Ltd (Beijing, China). The blood samples were collected from the rabbit bought from the Xiling Angle Breeding Center (Jinan, China). All animal experimental operation complied with the requirements of the National Act on the treatment of experimental animals (People's Republic of China). The animal experiments were also approved by the Animal Ethics Committee of Shandong University, in accordance with the “Guideline for the Care and Use of Laboratory Animals”.
2.2. Synthesis of CS–Rh–PFC
The final synthesis of CS–Rh–PFC polymer was conducted in three steps. Firstly, the carboxyl terminals of CS were activated and linked to amino groups in cys to synthesize CS–cys polymers.25 In brief, 0.2 g of CS was dissolved in 50 mL of distilled water (DW), then EDC and NHS (5 equiv./CS) were added and stirred for 15 min. Subsequently, 2.8 g of cys was added and the reaction was conducted for 48 h at 25 °C with stirring. The product was dialyzed (MWCO 3500) against DW to remove residual agents, followed by lyophilization. Quantification of cys was performed by comparing the integration proportions of cys and CS in the 1H-NMR spectra.Rh was grafted into CS–cys using the reported method with several modifications.26 Firstly, 0.35 g of Rh was completely dissolved in 500 mL of NaHCO3 solution (10 g L−1 in DW) via heating and stirring. After cooling of Rh solution to room temperature, EDC (5 equiv./Rh) was added and stirred for 20 min, followed by NHS (5 equiv./Rh), and the reaction was maintained for 24 h away from light at 25 °C. The acquired solution was dialyzed against DW (MWCO 3500), followed by lyophilization. The CS–cys–Rh polymers were synthesized with different Rh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CS–cys ratios (1:10 to 5:10). The Rh degree of substitution (DS) was detected with UV-Visible spectrophotometry (UV-vis) at 443 nm.
:CS–cys ratios (1:10 to 5:10). The Rh degree of substitution (DS) was detected with UV-Visible spectrophotometry (UV-vis) at 443 nm.
PFC was combined with CS–Rh conjugates through amidation coupling reactions according to an established protocol with little modification.17 To activate carboxylic groups, PFC (2 g) was dissolved in 150 mL of dimethyl sulfoxide (DMSO) and mixed with similar amounts (5 equiv./PFC) of EDC and NHS under nitrogen protection, with stirring for 12 h away from light at room temperature. The mixture was purified by dialysis against a mixed solution (DMSO:H2O = 6:1, MWCO 3500) followed by DW before lyophilization. Finally, polymers with different ratios of PFC to CS–Rh (2:1, 1:1 and 1:2) were synthesized.
2.3. Characterization
NMR and Fourier transform infrared (FT-IR) spectroscopy were applied for the evaluation of conjugation. CS or CS–cys was dissolved in D2O, and CS–cys or CS–cys–Rh was dissolved in a mixed solution of DMSO-d6/D2O (6:1, v/v) for confirming the CS–cys–Rh structure by 1H-NMR spectra. For 19F-NMR spectra, CS–cys–Rh or CS–Rh–PFC copolymers were fully dissolved in the mixed solution of DMSO-d6/D2O (6:1, v/v). Malvern laser granulometry (Zeta Nano ZS, UK) was applied for examining the size changes in NPs. The morphology was imaged via transmission electron microscopy (TEM, Japan).
2.4. Measurement of critical aggregation concentration (CAC)
Pyrene fluorescence probe detection was performed to determine CAC values of CS–Rh–PFC polymers.27 Briefly, 10.2 mg of pyrene was dissolved in 2 mL of acetone and diluted 100-fold to acquire a final pyrene concentration of 51 μg mL−1. Next, the CS–Rh–PFC solutions were added to 20 μL of diluted pyrene solution at different concentrations (1 ng mL−1 to 1 μg mL−1) after the evaporation of acetone. To examine fluorescence intensity, a fluorospectrometer instrument (Hitachi, Japan) using an emission wavelength of 380 nm was employed. A plot of I373/I384 against logarithmic concentrations was generated to obtain CAC values.2.5. Detection of dissolved oxygen
The content of dissolved oxygen was determined using a portable dissolved oxygen meter (JPBJ-608, INESA Scientific Instrument Co., Ltd).28 Firstly, the polymers of CS–Rh and CS–Rh–PFC were dissolved in ultrapure water with an Rh concentration of 2 mg mL−1, using ultrapure water alone as a control. After stirring for 5 min at room temperature, O2 at a constant pressure of 5 MPa was applied to flush all samples continuously for 5 min. The O2 content values were measured every 2.5 min within the first 2 h as well as at different time-points (3, 4, 6, 8, 12 and 24 h). Different concentrations of CS–Rh–PFC solutions were additionally examined for their O2 content.2.6. Detection of singlet oxygen
Singlet oxygen was measured using the fluorescent probe 9,10-dimethylanthracene (DMA) with few modifications.29 Briefly, DMA in DMF was added to solutions of DW, CS–Rh–PFC, and CS–Rh–PFC pre-treated with 20 mM DTT, CS–Rh, and CS–Rh–PFC under N2 conditions. The Rh concentration was 5 μg mL−1 in all samples, and the DMA content was maintained at 20 μM. All samples were subjected to US at 1.2 W cm−2 for 3 min, incubated for 4 h, and scanned via fluorescence spectrophotometer (Ex of 360 nm and Em of 380–550 nm). The fluorescence intensity was recorded for further comparative analyses.2.7. Preparation of NPs and DTX-loaded NPs
NPs were self-assembled using the probe ultrasonic method. Briefly, CS–Rh–PFC conjugates were added to DW or PBS, followed by sonication for 15 min with an ultrasonic probe (Ningbo Scientz Biotechnology Co., Ltd, China, 150 W, 4 s off/2 s on) in an ice bath. The solution was purified through a 0.8 μm membrane filter. All samples were stored at 4 °C.DTX-loaded NPs (DTX/NPs) were prepared using a simple modified dialysis method. DTX (100 μL) in methanol solution was added dropwise to solutions of CS–Rh–PFC in DW (1 mL) under stirring for 5 h at 25 °C. The resulting solution was sonicated for 15 min and dialyzed (MWCO 3500) against DW to remove methanol. Following centrifugation at 4000 rpm for 15 min, the supernatant was passed through a 0.8 μm membrane filter. Importantly, the whole reaction was conducted in the dark.
The DTX content was determined via high-performance liquid chromatography (HPLC) on a Hypersil-BDS C18 column (5 μm, 250 mm × 4.6 mm) at a detection wavelength of 230 nm. The mobile phase comprised a mixture of H2O and CH3CN (52:48 (v/v)) at a constant speed of 1.0 mL min−1. Drug loading (DL) and entrapment efficiency (EE) were calculated according to the following equations:
 | (1) |
 | (2) |
2.8. DTX and Rh release assays
In vitro drug release was examined in PBS (0.2% Tween-80, 7.4, 0.1 M) containing different concentrations of DTT (0, 20 mM and 20 μM). The DTX/NPs were placed within dialysis bags (MW 3500) and incubated in a shaking bed (37 °C, 100 rpm) for 0.5 h to 72 h. At the desired time-points, 1 mL of different media containing DTX and Rh were retrieved for HPLC and UV-vis detection, respectively. Isopyknic blank medium was added to replenish the original volume. Each step was repeated three times.2.9. Hemolysis assay
Rabbit blood was collected and stored in heparinized tubes, followed by centrifugation (3000 rpm, 10 min). The lower layer of red blood cells was washed with saline and centrifuged three times (3000 rpm, 10 min). Cells were re-suspended in saline to prepare 2% concentrations. Erythrocyte solution was mixed with NPs solutions (0.1–1.0 mg mL−1) and incubated for 1 h at 37 °C, followed by centrifugation (3000 rpm, 10 min). Then the supernatant was separated and scanned via UV-vis at 541 nm. Assays were conducted in triplicate.2.10. In vivo stability
In vivo stability was assessed in PBS (pH 7.4, 0.1 M), 0.1 M NaCl and 10% FBS after culturing for 24 h, and anti-dilution ability (against 1000-fold H2O) was also evaluated. The stability of NPs in complete 1640 medium with 10% FBS was analyzed within 48 h. All samples were incubated in a 37 °C bath with stirring at 100 rpm. Reductive stability was additionally evaluated with PBS (pH 7.4, 0.1 M) containing DTT (0, 20 μM and 20 mM) at different time-points. The size was measured by the dynamic light scattering (DLS) method, and relative morphology images were obtained using transmission electron microscopy (TEM, Japan).2.11. Cellular uptake and distribution assay
Coumarin 6 (C6) was used to observe intracellular uptake following encapsulation into NPs with the same method of DTX/NPs.30 Free C6 was used as a control. B16F10 cells were initially seeded in 12-well plates at a density of 1.6 × 104 cells per well and cultured overnight. Cell medium was replaced with 2.412 μg mL−1 C6, C6-loaded NPs (C6/NPs, 2.412 μg mL−1 C6) or blank medium, with incubation periods of 1, 2 and 4 h, respectively. Following incubation, the cells were washed three times with PBS. Next, cells were digested and collected for flow cytometry (FCM) measurements. For observation of the internalization mechanism of NPs, an inverted fluorescence microscope (NIKON Ti-U, Japan) was used. Similarly, B16F10 cells were seeded on 12-well plates at a density of 8 × 103 cells per well until 70% confluence. Cells were washed with PBS three times after incubation with free C6 and C6/NPs solutions at different time-points. Subsequently, cells were fixed with 4% paraformaldehyde and the nuclei were stained with DAPI, followed by washing with PBS. For the determination of the intracellular distribution of C6/NPs, incubation was terminated at 1 h and cells were washed with PBS, followed by staining with Golgi-Tracker Red (Beyotime Biotech, China) for 0.5 h. Cells were washed, fixed and stained with DAPI for observation.2.12. Cytotoxicity study
The in vitro toxicity of NPs and DTX/NPs without or with US over different time-periods was investigated in B16F10 cells via the CCK8 assay. In brief, cells were seeded in a 96-well plate (5 × 103 cells per well) with 100 μL of complete medium. After culturing for 12 h, the medium was substituted with NPs or DTX/NPs solution containing different Rh concentrations. All cells were incubated for 24 h before rinsing with PBS and replenished with a new medium. For the dark toxicity assay, cells were further incubated for 24 h. Cells in the SDT treatment groups were incubated for another 24 h after exposure to US at 1.2 W cm−2 for 1, 3 and 5 min. The cell viability of free Rh, CS–Rh + SDT and NPs + SDT groups was additionally evaluated at the same Rh concentrations and US times. All plates were treated with CCK8 and their optical density (OD) values were recorded on a microplate reader at 450 nm. Cell viability was calculated as follows:| Cell viability = (ODs − ODb)/(ODc − ODb) × 100% | (3) |
2.13. Live/dead cell assay
B16F10 cells were seeded in a 12-well plate (1.0 × 104 cells per well) and incubated for 24 h. The medium was replaced with blank medium (control group), CS–Rh (4 μg mL−1 Rh), NPs (4 μg mL−1 Rh) and DTX/NPs (4 μg mL−1 Rh, 6.22 μg mL−1 DTX) for 8 h. After washing with PBS, cells were treated with or without US and incubated with fresh medium for 4 h. The Calcein-AM/PI Kit was employed for the visualization of live and dead cells under an inverted fluorescence microscope following the manufacturer's instructions.312.14. In vitro ROS detection
The DCFH-DA kit (Beyotime Biotech, China) was applied for the measurement of ROS. After seeding on 14 mm Petri dishes (1.0 × 104 cells per well), B16F10 cells were cultured overnight and further immersed in fresh medium or media containing CS–Rh (4 μg mL−1 Rh) or NPs (4 μg mL−1 Rh). Cells were cultured for 4 h, and a 10 μM DCFH–DA probe prepared with fresh medium was added before the cells were exposed to US (or not) (1.2 W cm−2, 3 min). After 30 min of incubation, cells were washed and fixed, and nuclei were stained with DAPI, followed by examination via confocal laser scanning microscopy (CLSM, Zeiss LSM 780, Switzerland).2.15. In vitro calreticulin evaluation
Immunofluorescence staining was applied for the evaluation of CRT exposure induced by DTX/NPs.32 Briefly, B16F10 cells (2 × 104 cells per well) were seeded in 14 mm Petri dishes and cultured overnight. The cell medium was discarded and replenished with fresh medium or media containing CS–Rh (4 μg mL−1 Rh), NPs (4 μg mL−1 Rh) and DTX/NPs (4 μg mL−1 Rh, 6.22 μg mL−1 DTX) after rinsing with PBS. After 6 h incubation, cells were washed and treated with US (1.2 W cm−2, 3 min) or left untreated. Cells were continuously cultured for 24 h, washed three times with PBS and subsequently fixed with 4% paraformaldehyde. CRT rabbit monoclonal antibody (1:200 dilution) was added to dishes and cultured for 1 h at 37 °C, followed by staining and incubation with Dylight 488-goat anti-rabbit IgG (1:200 dilution) for further 1 h. DAPI dye was applied to stain the cell nuclei, followed by recording and examination of CRT exposure using CLSM.
2.16. Western blot analysis
B16F10 cells were seeded on 75 cm2 flasks and cultured with fresh medium, CS–Rh (4 μg mL−1 Rh), NPs (4 μg mL−1 Rh) and DTX/NPs (4 μg mL−1 Rh, 6.22 μg mL−1 DTX), respectively. After 12 h of incubation, cells in SDT groups were washed twice with PBS and subjected to US (1.2 W cm−2 for 3 min). The other groups were incubated in the dark during the course of the experiment. All cells were cultured for 12 h before collection for protein extraction. Briefly, cells were initially lysed in RIPA buffer with 1% phenylmethanesulfonyl fluoride (PMSF). Following sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), protein bands were transferred to polyvinylidene fluoride membranes and incubated with primary antibodies specific for cleaved caspase-3, cleaved caspase-9, MMP9, BAX and Bcl-2 (1000-fold dilution) using β-actin as the internal reference, followed by horseradish peroxidase-labeled secondary antibodies. Protein bands were visualized and gray values were measured.2.17. Analysis of cellular apoptosis
Dysregulation of cellular apoptosis, essential in the lost homeostasis state during cell life,33 was examined using an apoptosis detection kit (Solarbio, China). Briefly, B16F10 cells were seeded on a 12-well plate (1.8 × 104 cells per well) until 75% confluence. Cells were incubated with fresh medium, CS–Rh (4 μg mL−1 Rh), NPs (4 μg mL−1 Rh) or DTX/NPs (4 μg mL−1 Rh and 6.22 μg mL−1 DTX) for 12 h, respectively. Subsequently, cells were washed with PBS, and replenished with fresh medium. US treatment groups underwent persistent ultrasonic therapy (1.2 W cm−2, 3 min). After 4 h, cells were stained using the kit according to the manufacturer's instructions and measured using FCM. The results were analyzed using Cyt Expert 2.3 software (n = 3).2.18. Cell cycle evaluation
Cell cycle analysis was performed via FCM using a specific cell cycle detection kit (Keygen Biotech, China).34 B16F10 cells were initially plated on a 12-well plate (1.8 × 104 cells per well) and cultured for 12 h. Cells were washed and re-immersed in fresh medium, CS–Rh (4 μg mL−1 Rh), NPs (4 μg mL−1 Rh), free DTX (6.22 μg mL−1) or DTX/NPs (4 μg mL−1 Rh and 6.22 μg mL−1 DTX) for 12 h, respectively. US-treated groups were rinsed and incubated in the fresh medium, followed by SDT treatment (1.2 W cm−2, 3 min) and a further 12 h incubation period. Finally, cells were collected and treated with diluted PI/RNase A before culturing for 60 min away from light. FCM and ModFit LT software were used for recording and analysis of DNA changes, respectively.2.19. Microtubule regrowth study
As one of the substructures of the cytoskeletal filament, microtubules are related to critical cellular activities, such as endocytosis and intracellular transport.35 CLSM using Tubulin Tracker Red (Beyotime Biotech, China) was applied for the examination of microtubule forms. Briefly, cells were seeded in 14 mm Petri dishes (1.0 × 104 cells per well) and cultured overnight. After discarding the old medium, cells were washed and co-cultured with fresh medium, NPs (4 μg mL−1 Rh), DTX/NPs (4 μg mL−1 Rh and 6.22 μg mL−1 DTX) or free DTX (6.22 μg mL−1) for 12 h. SDT groups were subjected to US and all cells were incubated for 4 h. Subsequently, cells were washed, fixed with 4% paraformaldehyde, and stained according to the manufacturer's instructions. Cell nuclei were labeled via DAPI staining.2.20. Construction of tumor-bearing mice
To investigate the biodistribution and tumor inhibition effects of DTX/NPs, the B16F10 tumor-bearing C57BL/6N mice models were constructed. Generally, B16F10 cells were collected and diluted at a density of 1 × 107 cells per mL in PBS, and 100 μL of the cell suspension was injected into the right flanks of the male mice (4–6 weeks). When the tumor volume reached 30–80 mm3, mice were intravenously injected with different formulations, and the tumor volume was calculated according to the equation below:| Tumor volume (mm3) = La × Lb2/2 | (4) |
2.21. In vivo ROS generation
To detect the in vivo ROS level, the singlet oxygen sensor green probe (SOSG, Invitrogen™, USA) was applied according to the report of Xing with slight modifications.36 Briefly, B16F10 tumor-bearing mice were divided into three groups before intravenous injection with 100 μL of NS, CS–Rh or NPs solutions containing 25 μM SOSG probe. Mice were exposed to US (1.2 W cm−2, 3 min) for 1 h after the injection. Following the SDT treatment for 1 h, mice were sacrificed and the tumor tissues were frozen and sectioned. Images of sections were obtained via fluorescence scanning microscopy.2.22. In vivo hypoxia assay
The in vivo hypoxia assay was conducted according to a previous protocol with limited modification.36 In brief, B16F10 tumor-bearing mice were intravenously injected with normal saline (NS), CS–Rh and NPs solutions, and SDT was conducted after 1 h of injection. Hypoxyprobe™-1 solution dissolved in 0.9% NaCl at a concentration of 60 mg kg−1 was intraperitoneally injected into mice. After 1.5 h, mice were euthanized and tumors were collected. Later, the tumors were sectioned and the slices were stained with relative secondary antibody in the kit for further imaging via fluorescence scanning microscopy.2.23. In vivo biodistribution
To observe the biodistribution in mice after intravenous injection, 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR) was encapsulated into the NPs, and free DiR as well as DiR/NPs were separately intravenously injected into B16F10 tumor-bearing mice at a dose of 200 μg mL−1. The bioluminescence imaging system (IVIS Kinetic, USA) was applied to capture the fluorochrome signal after 1, 4, 8 and 24 h. In addition, mice were sacrificed at a 24 h time-point and major tissues were collected and imaged. The DiR intensity was recorded for further quantification of accumulated NPs. All data were obtained with Living Image 4.1 software.2.24. Evaluation of efficacy and systemic immune response
After successful establishment of the B16F10 tumor-bearing mice model, animals were randomly divided into 7 groups. All mice were intravenously injected with different solutions and treated with or without SDT as follows: normal saline (NS, 1), CS–Rh + SDT (2), NPs + SDT (3), DTX/NPs (4), Taxotere® (5), and DTX/NPs + SDT (6) (n = 5). The dose of DTX and Rh in all groups was 10 mg kg−1 and 3 mg kg−1, respectively. Mice were injected every three days from Day 0 to Day 9, and were subjected to US for 3 min at 1.2 W cm−2 at 1 h after injection. Every 2 days, the tumor volume and body weight of mice were measured and recorded.On day 14, blood was collected from all mice through enucleation of eye after fasting for 12 h, and the cytokines of helper T cells (Th, IFN-γ, TNF-α, IL-2, IL-6, Multi Sciences) were detected in serum. Tumor tissues from a number of mice were removed and ground for analysis of Th (CD4+ T cells) and cytotoxic T cells (Tc, CD8+ T cells) via FCM. Additionally, the tumor sections were stained with CD3, CD4 and CD8 antibodies for the observation of Th and Tc cells. Additionally, the tumor draining lymph nodes were collected from the mice body, followed by grinding and staining with anti-CD11c-FITC, anti-CD80-APC and anti-CD86-PE antibodies. The dendritic cells (DCs) were thereby detected using FCM.
Subsequently, mice were sacrificed and tumors of all groups were obtained for imaging. Besides tumor tissues were collected and sliced for TdT-mediated d UTP Nick-End Labeling (TUNEL) assay with brown staining of nuclei. For safety evaluation of these preparations, all major tissues of heart, liver, lungs, spleen and kidneys were collected and sectioned for hematoxylin–eosin (H&E) along with tumor staining.
2.25. Pharmacokinetic analysis
Wistar rats were fasted for 12 h and subjected to femoral vein injection after random allocation into two groups (n = 3). The commercially available drug Taxotere® and self-assembled DTX/NPs were injected at a dose of 15 mg kg−1 DTX. At the specified time-points, 0.3 mL of blood was extracted into heparinized tubes from sinus jugularis of rats, and samples were centrifuged at 3000 rpm for 10 min after incubation for about 2 h at room temperature. Subsequently, 150 μL of supernatant plasma was mixed with 2 mL of methyl-tert-butyl ether for further extraction of DTX, and the mixtures were further centrifuged at 3000 rpm for 10 min to acquire the supernatant. After evaporation of the supernatant under nitrogen flow, DTX was re-dissolved in acetonitrile and detected by the HPLC method in all samples.2.26. Statistical analysis
All data were expressed as mean ± standard deviation (SD). Statistical analysis was conducted via one-way analysis of variance (ANOVA) at confidence levels of 95% and 99%. Data were regarded as ‘highly significant’ and ‘significant’ at p < 0.01 and p < 0.05, respectively.3. Results and discussion
3.1. Synthesis and characterization of polymers
Based on the EDC/NHS activation chemistry, polymers were successively fabricated by introducing Rh and PFC groups for generating ROS and carrying oxygen, respectively, as shown in Scheme 1 and Fig. 1A. 1H-NMR, 19F-NMR and FT-IR spectroscopy were applied to validate the polymer synthesis. First, CS and CS–cys were dissolved in D2O. Peak a (δ = 1.97 ppm) represented the characteristic group of CS, while the other four peaks (b, c, d, and e) at 2.67–3.10 ppm represented the successful linkage of cys groups. Based on the integration ratio of peak a [3H, –CH3] to peaks of b–e [4H, –CH2–CH–S–S–CH–CH2–], the DS of cys was 76.9 (Fig. 1B). After the introduction of Rh to the conjugate, 1H-NMR spectra of mixed solutions were acquired. Peaks at 7.0–9.0 ppm were characteristic signals of [3H, –OH] in the Rh group (Fig. 1C). Since the fluorine atom in PFC only emerged after the synthesis process, CS–Rh–PFC and CS–Rh were measured using 19F-NMR in mixed solutions. Emerging f, g, and h peaks at around −80, −117 and −126 ppm provided strong evidence of the introduction of PFC (Fig. 1D). FT-IR spectra of all polymers were subsequently generated. The peak at 3294.67 cm−1 represented a characteristic signal of –OH stretch vibration and –NH symmetrical vibration, and peaks at 1604.71 and 1222.08 cm−1 represented –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or –SO stretching vibrations, respectively, while the CS–cys spectrum displayed similar features as CS, except for a small shift in the peak at 1217.40 cm−1 due to the –NH vibration of cys. In CS–Rh, two peaks at 1652.77 and 1629.80 cm−1 were observed for the –CO stretch vibration, supporting the novel introduction of –CO bonds. Moreover, the peak at 1450.22 cm−1 represented a –C–C stretch vibration in the benzene ring of CS–Rh, indicating the successful linkage of Rh. In the spectrum of CS–Rh–PFC, the new peaks at 2360.80 and 1037.75 cm−1 represented –NO and –C–F stretch vibrations, respectively, supporting the successful generation of this polymer.
O or –SO stretching vibrations, respectively, while the CS–cys spectrum displayed similar features as CS, except for a small shift in the peak at 1217.40 cm−1 due to the –NH vibration of cys. In CS–Rh, two peaks at 1652.77 and 1629.80 cm−1 were observed for the –CO stretch vibration, supporting the novel introduction of –CO bonds. Moreover, the peak at 1450.22 cm−1 represented a –C–C stretch vibration in the benzene ring of CS–Rh, indicating the successful linkage of Rh. In the spectrum of CS–Rh–PFC, the new peaks at 2360.80 and 1037.75 cm−1 represented –NO and –C–F stretch vibrations, respectively, supporting the successful generation of this polymer.
 | ||
| Scheme 1 (A) Synthesis of the CS–cys–Rh–PFC polymer and self-assembly process of DTX-loaded NPs. (B) Accumulation of injected DTX-loaded NPs and SDT-induced immunotherapy in tumor cells. (C) Intracellular hydrolysis of DTX-loaded NPs and release of drugs and oxygen molecules after US treatment. | ||
 | ||
| Fig. 1 Synthesis scheme of CS–cys–Rh–PFC (A). 1H-NMR spectra of CS and CS–cys in D2O, (B) and CS–cys and CS–cys–Rh in d6-DMSO/D2O (C). 19F-NMR spectra of CS–cys–Rh and CS–cys–Rh–PFC in d6-DMSO/D2O (D). FT-IR spectra of CS, Cs–cys, CS–cys–Rh and CS–cys–Rh–PFC (E). | ||
3.2. Preparation and characterization of NPs
During the synthesis process, the weight ratio of PFC to CS–Rh changed from 2:1 to 1:2 for the formulation of different final conjugates. After the polymers were lyophilized, re-dissolved in PBS and placed under the ultrasonic probe for 15 min in an ice-bath, the self-assembled NPs were successfully prepared. The size of NPs increased from 131.1 to 149.6 nm with similar low PDI values based on different CS–Rh–PFC conjugates, with the achievement of homogeneous distribution in PBS (Table 1). Upon increasing the PFC:CS–Rh ratio, the diameter of NPs decreased mainly for the introduction of a more dense hydrophobic core, resulting in a strong hydrophobic interaction among the NPs, while the content of Rh decreased correspondingly. To ensure a benign SDT effect along with the enhanced permeation and retention (EPR) effect, CS–Rh–PFC (1:2) with the smallest size distribution was employed for subsequent investigation, and the zeta potential of the corresponding NPs was −33.8 mV. In addition, the morphology of the NPs was further evaluated via TEM (depicted in Fig. 2A), and the self-assembled NPs appeared as uniform and well-dispersed spheres in PBS solution.
 | ||
| Fig. 2 Characterization of CS–Rh–PFC NPs. TEM images of NPs incubated in PBS (A), 20 μM DTT (B), and 20 mM DTT (C) solutions after 24 h. (D) Hemolysis rate of NPs (1–7 represent 0.1, 0.25, 0.5, 0.75, 1 mg mL−1 NPs, blank control and positive control, respectively). (E) Size distribution of NPs in 20 μM or 20 mM DTT solution at 0, 8 or 24 h. (F) Stability of NPs in PBS, 0.1 M NaCl, 10% FBS solutions and 1000-fold dilution with PBS. (G) and (H) represent the cumulative release rate curves of DTX and Rh from NPs upon incubation in PBS or reductive solutions, respectively. (I) Curves of the intensity ratio (I373/I384) excited with pyrene against logarithms of CS–Rh–PFC concentrations. (J) The curves of dissolved oxygen over time with H2O, CS–Rh and CS–Rh–PFC at an Rh concentration of 2 mg mL−1. (K) Curves of dissolved oxygen of H2O and CS–Rh–PFC at different concentrations. **p < 0.01. | ||
| Sample | Rh (%) | Diameter (nm) | PDI | ζ (mV) | CMC (μg mL−1) |
|---|---|---|---|---|---|
| CS–Rh–PFC (2:1) |
11.68 | 149.6 ± 1.30 | 0.228 ± 0.015 | −32.8 | 35.12 |
| CS–Rh–PFC (1:1) |
10.88 | 136.3 ± 1.78 | 0.272 ± 0.012 | −35.4 | 33.83 |
| CS–Rh–PFC (1:2) |
10.78 | 131.1 ± 4.30 | 0.270 ± 0.020 | −33.8 | 27.51 |
The hemolysis ratio of NPs is shown in Fig. 2D. All values were <2%, even at the highest concentration of 1 mg mL−1. The benign safety of intravenous administration might be attributed to the addition of PFC groups, which served as an artificial blood substitute with good biocompatibility.
3.3. Stability of NPs
Initially, the redox-responsive stability of NPs was analyzed by incubation with 20 mM or 20 μM DTT solution for 24 h. Size and morphological variations were recorded using DLS and TEM. Upon addition of 20 mM DTT solution, NPs changed from stable spheres to large clusters owing to the breakage of disulfide bonds, while the spheres remained stable in 20 μM DTT solution (Fig. 2B and C). Rupture of NPs promoted the inner drug release into the hydrophobic core, and could realize accurate dosing to tumor tissues after intravenously injection into the human body.34 The DLS results revealed an increase in size from 129.7 nm to 331.1 nm and 766.0 nm after incubation in 20 mM DTT at 0 h, 8 h and 24 h (Fig. 2E), while the NPs size in 20 μM DTT showed negligible changes after 24 h incubation, indicative of favorable redox-stability in non-tumor regions.Stability of NPs in different solutions was evaluated via DLS after incubation with PBS, 10% FBS and 0.1 M NaCl solutions, respectively, for 24 h. As shown in Fig. 2F, the CS backbone was slightly hydrolyzed in PBS as previously reported,14 but remained stable in the presence of 10% FBS and 0.1 M NaCl solutions, which favored the maintenance of morphology and the precise drug delivery of NPs after drug administration. Furthermore, NPs maintained stability within two days when placed in 10% FBS (Fig. S1†). In addition, the NPs exhibited favorable anti-dilution stability.
3.4. Preparation and DTX release of DTX/NPs
To achieve effective combination therapy, the model drug DTX was loaded into the inner core of NPs with different feed ratios of DTX to CS–Rh–PFC polymers (1:10 to 5:10). After a simple ultrasound-dialysis method, the NPs were prepared and DL% and EE% were calculated (shown in Table 2). Obviously, the DL% increased significantly from 5.18% to 18.33% with increasing ratio, while EE% increased from 37.84% to 67.73% when the ratio reached 3:10, but showed a decrease at a ratio of 5:10. This phenomenon might be attributed to hydrophobic/hydrophilic imbalance with further addition of DTX, resulting in the rupture of hydrophobic interaction between DTX and polymers. Accordingly, the 3:10 ratio was selected for further experiments.
| Sample | DTX/polymer | DL (%) | EE (%) |
|---|---|---|---|
| CS–Rh–PFC (1:2) |
1:10 |
5.18 ± 0.06 | 37.84 ± 0.12 |
| 2:10 |
7.06 ± 0.02 | 54.60 ± 0.69 | |
| 3:10 |
16.89 ± 0.07 | 67.73 ± 0.34 | |
| 5:10 |
18.33 ± 0.13 | 44.89 ± 0.39 |
Subsequently, in vitro drug release was investigated in the presence of DTT solutions. The cumulative DTX release rate from NPs into PBS medium reached 30.69% in the first 8 h and 54.21% by 72 h (Fig. 2G). A similar release behavior was observed for NPs in 20 μM DTT solution (34.49% and 63.12% at 8 h and 72 h, respectively). Notably, the release rate was highly accelerated in 20 mM DTT solution, reaching 96.94% at the 72 h time-point, owing to rapid release after the breakage of inner disulfide bonds. Solutions were subsequently examined using UV-vis at 443 nm for Rh detection. The cumulative release rate curve of Rh was similar to that of DTX (Fig. 2H). However, the maximum value of Rh acquired in 20 mM DTT solution at 72 h was lower than that from DTX curves (only 61.11%), which could be attributed to the incomplete fracture of disulfide bonds within the estimated time.
3.5. CAC determination
As shown in Fig. 2I, the CAC was determined using the pyrene probe method. The CAC was 0.034 and 0.035 mg mL−1 for CS–Rh–PFC (1:1) and CS–Rh–PFC (1:2), respectively, indicating the slight effect of the hydrophilic CS groups. At a PFC to CS–Rh ratio of 2:1, the CAC significantly reduced to 0.027 mg mL−1, facilitating the formation and morphology retention of NPs. Accordingly, CS–Rh–PFC (2:1) was used for subsequent analyses.
3.6. Detection of O2 release
The final constructed fluoropolymer possessed the ability to dissolve oxygen via van der Waals's forces between oxygen and CS–Rh–PFC molecules, while interactions among the polymers were impaired.37 Here, oxygen contents were measured at different time-points after the addition of polymers (2 mg mL−1) and saturation with oxygen gas for 5 min. As shown in Fig. 2J, CS–Rh–PFC initially had the greatest oxygen content (17.68 mg L−1) compared to CS–Rh (15.39 mg L−1) and water (11 mg L−1) (p < 0.001), and the degree of reduction with time was minimal. Interestingly, the oxygen content of CS–Rh was higher than that of water, mainly due to its amphiphilic structure that could self-assemble into NPs and maintain oxygen molecules within the inner core. The dissolved oxygen content decreased in a time-dependent manner, which might be attributed to the release of oxygen and the ROS-generating effect of Rh.38 Variable concentrations of CS–Rh–PFC were additionally evaluated (Fig. 2K). The oxygen content decreased with the reduced concentrations and prolonged time-periods, supporting the potential of CS–Rh–PFC to overcome hypoxia and enhance the effect of SDT in tumor cells and tissues.3.7. Cellular uptake and in vitro distribution
After measurement of the fluorescence intensity of C6 in C6/NPs, intracellular uptake of NPs was investigated at time-points of 1, 2 and 4 h using FCM and fluorescence microscopy. As shown in Fig. 3A and B, C6 intensity in cells increased with the passage of incubation time for both NPs and free drug groups, with better performance of the NPs than the free C6 group at every time-point. Data were additionally quantified using FCM. The fluorescence intensity in the NPs group was nearly an order of magnitude stronger than that of free C6 at 1 h, indicating enhanced cellular uptake after loading of the drug in NPs. The synthesized NPs thus showed good targeting of tumor cells through CD44 receptors. | ||
| Fig. 3 (A) Fluorescence images of B16F10 cells after culturing with free C6 and C6/NPs for 1, 2 and 4 h, respectively (scale bar, 50 μm). (B) FCM histograms of C6 in B16F10 cells after culturing with free C6 and C6/NPs for 1, 2 and 4 h. (C) Images of B16F10 cells cultured with free C6 and C6/NPs for 1 h, whereby Golgi apparatus was stained red (scale bar, 50 μm). (D) Cell viability curves of NPs and (E) DTX/NPs treated under dark conditions or with SDT for 1, 3 and 5 min (F) B16F10 cells incubated with blank medium, CS–Rh, NPs and DTX/NPs with/without SDT treatment, followed by staining with the Calcein-AM/PI kit. Red and green cells represent dead and live cells, respectively (scale bar, 100 μm). (G) ROS detection in B16F10 cells incubated with different preparations with/without SDT treatment via CLSM (scale bar, 20 μm). **p < 0.01, ***p < 0.001. | ||
Considering the important roles of Golgi apparatus in the synthesis and processing of essential proteins, the distribution of free C6 and C6/NPs in Golgi apparatus was examined. After 1 h of incubation, Golgi apparatus and cell nuclei were stained with Golgi-Tracker Red marker and DAPI, respectively, and the overlap of C6 and the Golgi-Tracker Red staining part was recorded (Fig. 3C). The data showed favorable accumulation and targeting ability of CS-based NPs to Golgi apparatus, similar to previous reports.39,40 Golgi structures could be easily destroyed after SDT treatment, leading to the inhibition of multiple metastasis-associated proteins.41
3.8. Chemo-SDT cytotoxicity of NPs
The CCK-8 assay was employed for the assessment of cytotoxicity. First, the cytotoxicity of CS–Rh–PFC NPs was investigated using B16F10 cells in the dark without or with SDT treatment for 1, 3 and 5 min (1.2 W cm−2) under variable Rh concentrations (0.1 to 32 μg mL−1). As shown in Fig. 3D, the cytotoxicity was significantly higher in the SDT groups (p < 0.001). The IC50 values were determined as 15.11, 4.885, 1.604 and 1.245 μg mL−1 for darkness, SDT 1 min, SDT 3 min and SDT 5 min groups, respectively. Thus, SDT induced lower cell viability owing to the generation of ROS by Rh, and the cytotoxicity increased with the extension of US time. The cytotoxicity of DTX/NPs for chemo-SDT was additionally evaluated. DTX/NPs displayed remarkable cell killing ability after SDT treatment, even at a low concentration of 0.1 μg mL−1, with a cell viability of ∼50% determined for the SDT 3 min treatment group (Fig. 3E). In all groups, the cell viability was decreased with increasing Rh concentration, with no significant difference between SDT 3 min and SDT 5 min groups. Considering the effectiveness and short US time, DTX/NPs (4 μg mL−1 Rh) treated with SDT for 3 min were employed for subsequent experiments. Additionally, the cytotoxicity of NPs, free Rh and CS–Rh groups was compared after US treatment for 3 min (Fig. S2†). The results exhibited that the cell viability for the NPs group decreased significantly than that for the CS–Rh group, reflecting the successful O2 carrying and ROS-generating ability of NPs.The chemo-SDT cytotoxicity of different preparations was further investigated via Calcein-AM and PI staining. The number of red cells (dead) and green cells (live) in control and CS–Rh groups was similar, indicating negligible cytotoxicity under CS–Rh culture (Fig. 3F). The number of dead cells increased slightly after SDT treatment. However, the overall cell number was reduced for NPs and DTX/NPs groups, and the number of dead cells was significantly increased in NPs + SDT and DTX/NPs + SDT groups compared with NPs and DTX/NPs groups, respectively. Consequently, all the results described the better anti-tumor property for NPs than CS–Rh, and the NPs could gain better chemo-SDT effect after encapsulating DTX.
3.9. SDT for ROS generation
The production of ROS using a sonosensitizer under US treatment is considered an essential step for the induction of apoptosis.42 The 2′-7′dichlorofluorescin diacetate (DCFH–DA) kit was employed for ROS detection based on alteration of DCFH to fluorescent DCF after reaction with ROS, and the images were obtained using CLSM. After 4 h of incubation, the DCF fluorescence was brighter and stronger in order as shown in Fig. 3G. Furthermore, the NPs + SDT group showed a stronger ROS signal than the CS–Rh + SDT group, which was potentially attributed to the oxygen-carrying capacity of PFC and consequently improved ROS production.Singlet oxygen generation in solutions was measured with DMA after US treatment for 4 h (Fig. S3†). Contrary to the DCF probe, DMA fluorescence intensity decreased after reaction with singlet oxygen. Therefore, the DW with 20 μM DMA showed the highest peaks in the result, while DW without DMA only appeared as a baseline. According to the curves obtained, the NPs group generated more singlet oxygen than CS–Rh and NPs + N2 groups, supporting an essential role of oxygen content in SDT-mediated cancer cell destruction,43 and NPs could generate more singlet oxygen than the NPs + 20 mM DTT group, which was probably induced by the disassembly of PFC groups from NPs structures under a redox environment that resulted in deficient oxygen supplement for generating ROS.
3.10. Cell cycle assay
The cell cycle in cancer cells is not disrupted owing to various reasons, leading to uncontrolled proliferation.44 In this study, the B16F10 cell cycle was evaluated after incubating with different preparations for 12 h with or without SDT treatment. In the DNA content-frequency histograms (Fig. 4A (a and b)), the left red peak refers to the G0/G1 phase and the right red peak represents the G2/M phase. Moreover, the mesh regions belong to the S phase of cells. Histograms of control + SDT and CS–Rh ± SDT groups were similar to that of the control group, suggesting no effect of the sonosensitizer on the cell cycle. However, the NPs ± SDT groups displayed increased G2/M and reduced G0/G1 phases compared with CS–Rh and control groups, indicating that the addition of PFC regulated the cell cycle and promoted apoptosis. Finally, all DTX-based groups (free DTX, DTX/NPs and DTX/NPs + SDT) showed an obvious increase in the G2/M phase (p < 0.01). Accordingly, it was concluded that the addition of DTX could significantly restrict the cell cycle to anaphase mitosis, preventing the chaotic proliferation of cancer cells, and PFC exerted the same effect, but to a lower extent. | ||
| Fig. 4 (A) (a) DNA content frequency histograms of B16F10 cells treated with blank medium, CS–Rh, NPs, DTX and DTX/NPs with or without SDT treatment. (b) Cell cycle distribution of B16F10 cells. (B) Morphology of microtubulin in B16F10 cells after incubation with blank medium, NPs, DTX/NPs and DTX with or without SDT treatment. Scale bar: 10 μm. (C) CLSM images of CRT exposure in B16F10 cells treated with free medium, CS–Rh, NPs and DTX/NPs and SDT. Scale bar: 10 μm. (D) Western blot results of changes in cleaved caspase-3, cleaved caspase-9, MMP9, BAX, and Bcl-2 proteins in B16F10 cells treated with different preparations with/without SDT. (E) Apoptosis of B16F10 cells subjected to different treatments. | ||
3.11. Microtubule morphology assay
DTX was regarded to target microtubulin and could result in the double-strand DNA breaks and subsequent apoptosis.6,45 B16F10 cells were stained with Tubulin-Tracker Red after incubation with different preparations with/without SDT treatment (Fig. 4B). Compared with the free DTX group which showed condensed microtubule distribution and brighter fluorescence, the control group exhibited negligible tubulin polymerization. However, tubulin polymerization in the DTX/NPs group was significantly enhanced. Tubulin showed greater aggregation in all SDT groups than non-SDT ones except the DTX/NPs + SDT group due to rapid apoptosis of tumor cells during incubation. Microtubule morphology changes were consistent with cell cycle results, supporting the favorable anti-proliferative effects of DTX/NPs and SDT.3.12. CRT assay
CRT, one of the damage-associated molecular patterns, was exposed outside the cell membrane after ICD.1 Here, CRT was detected via the immunofluorescence staining method after cultivation by different preparations. Greater CRT exposure on B16F10 cells was recorded in the CS–Rh + SDT group compared with control + SDT groups, which was more significant for NPs + SDT and DTX/NPs + SDT groups (Fig. 4C). The degree of CRT exposure was similar among the non-SDT treated groups, except for the DTX/NPs group (shown in Fig. S4†), mainly for the encapsulation of DTX. Therefore, SDT treatment could enhance the degree of CRT exposure and generate greater ‘eat me’ signals to stimulate the antigen presentation of dendritic cells.46 As a result, the DTX/NPs + SDT group may activate a battery of immunological responses, such as CD8+ T cell proliferation and secretion of Th-related cytokines (Fig. 6).3.13. Western blot analysis
To investigate the effects on apoptosis, related proteins of the caspase and Bcl-2 families were detected via western blot (Fig. 4D). The gray value analysis result could be seen in Fig. S5.† As one of the famous markers of apoptosis, cleaved caspase-3 was deemed as a new therapeutic target in cancer nowadays, which could increase the therapy efficacy and decrease spontaneous tumor development.47 Compared with the control group, the expression of cleaved caspase-3 protein was significantly increased, particularly in NPs + SDT and DTX/NPs + SDT groups (p < 0.01), manifesting the favorable anti-proliferative effect on tumor cells. Expression of cleaved caspase-9, the upstream inducer of cleaved caspase-3, was also reduced (p < 0.01). In terms of BAX and Bcl-2 protein expression, BAX/Bcl-2 ratios were significantly increased, especially in NPs + SDT and DTX/NPs + SDT groups, compared with the control group (p < 0.001) (Fig. S6†). Therefore, the CS–Rh–PFC NPs and DTX-loaded NPs exerted a favorable cellular apoptosis effect with SDT treatment. Additionally, the expression of the MMP9 enzyme that degrades the extracellular matrix and promotes invasion of cancer cells was detected.14 Also, the MMP9 plays a crucial role in tumor invasion and metastasis.48 According to the results, the MMP9 levels were significantly decreased in all treated groups compared with the control group, and the NPs + SDT group possessed a lower MMP9 protein content than the CS–Rh + SDT group. The expression of MMP9 was significantly decreased after encapsulating DTX into the core of NPs. The findings suggested that DTX/NPs significantly inhibited cell invasion and NPs performed better than CS–Rh, which might be attributed to their oxygen-carrying capacity that would alleviate the hypoxia environment of tumor cells. Therefore, Rh-based polymers with SDT treatment show promising anti-tumor effects, and DTX/NPs serve as effective therapy in combination with chemo-SDT.3.14. Apoptosis assay
In view of the finding that SDT treatment enhances cellular apoptosis and necrosis, the extent of cellular apoptosis with or without SDT treatment was examined using an Annexin V-FITC/PI kit.49 As shown in Fig. 4E, SDT treatment induced limited apoptosis and necrosis effects on B16F10 cancer cells (<7%), but the rate of apoptosis was significantly increased after culturing with CS–Rh, NPs and DTX/NPs (p < 0.01). Upon application of SDT, apoptotic and necrotic rates were enhanced, and the rate of necrosis reached 31.71% for DTX/NPs with SDT. Therefore, NPs and DTX/NPs exerted favorable anti-tumor effects with the aid of SDT, consistent with western blot results (Fig. 4D).3.15. Drug distribution and elimination assay
To examine the in vivo drug distribution of NPs or free drug, the fluorescent probe of DiR was encapsulated into NPs.50 After the two preparations were intravenously injected into B16F10 tumor-bearing mice, animals were scanned and images were obtained after administration for 1, 4, 8 and 24 h using the Caliper IVIS Lumina II system. As shown in Fig. 5A, DiR was distributed mainly in tumor sites and lung tissues, and was additionally detected in kidney tissues. Fluorescence of DiR was reduced with increasing time, and signals disappeared at the 24 h time-point for the free DiR group owing to the rapid metabolism and excretion by liver and kidney tissues. However, signals in DiR/NPs were significantly strong. Owing to the excellent stability of NPs (Fig. 2 and Fig. S1†), the in vivo blood circulation time was prolonged and anti-tumor efficiency enhanced without hydrolysis in normal cells. Mice were euthanized and dissected after 24 h to examine fluorescence intensity in major organs (Fig. 5B and C). The DiR contents in major tissues, except heart, in the DiR/NPs group, were significantly higher than those of the free DiR group, indicating longer retention times in the blood and greater stability in the non-reductive environment of normal tissues.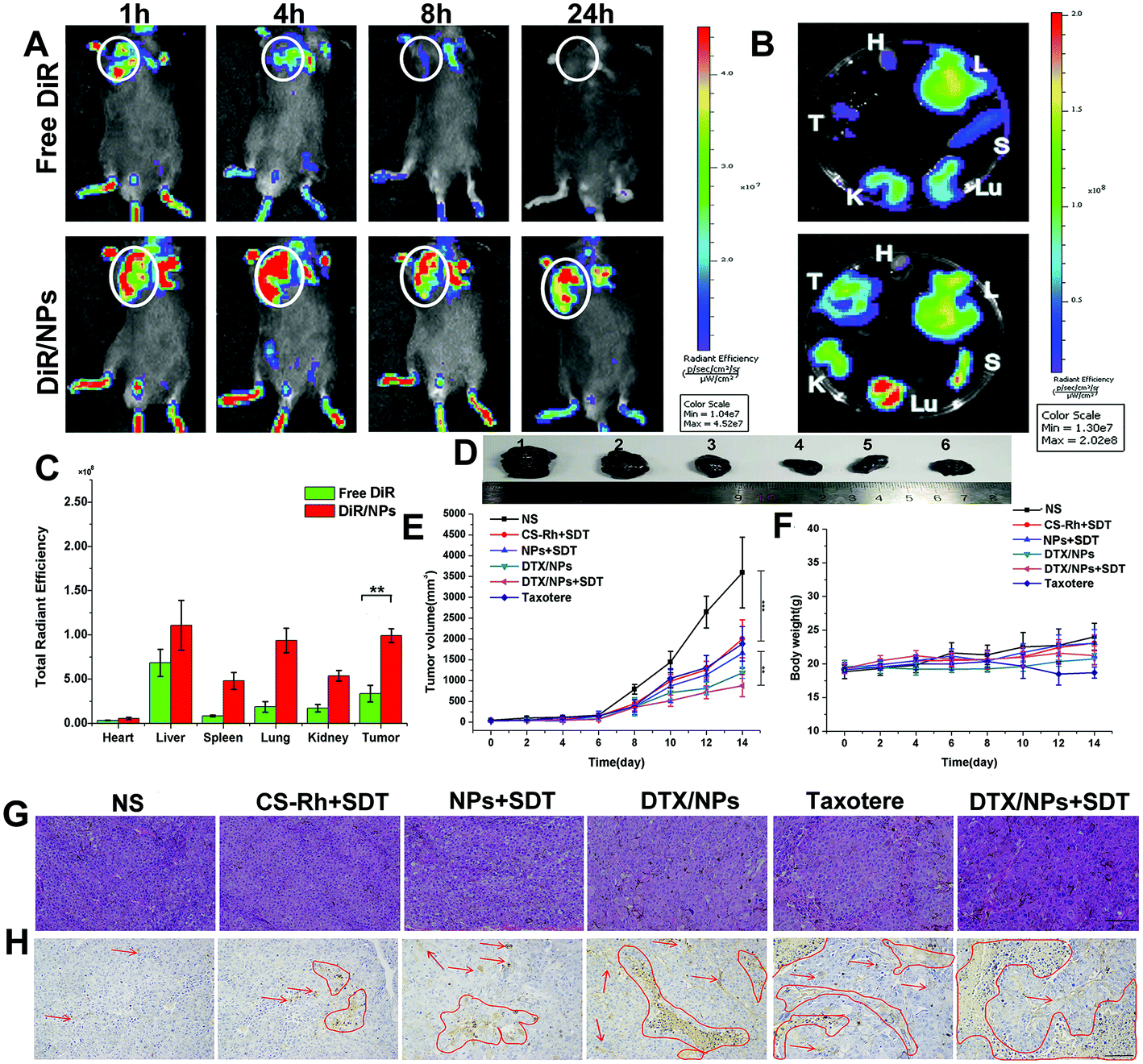 | ||
| Fig. 5 (A) In vivo fluorescence distribution in B16F10 tumor-bearing mice at 1, 4, 8 and 12 h after intravenous injection of DiR/NPs or free DiR solutions. The tumor foci were marked by white circles. (B) Ex vivo fluorescence distribution in heart (H), liver (L), spleen (S), lungs (Lu), kidneys (K), and tumors (T) after 24 h injection of DiR/NPs or free DiR solutions. (C) Total radiant efficiency values of major organs after 24 h of injection (n = 3). **p < 0.01. (D) Tumor images for control, CS–Rh + SDT, NPs + SDT, DTX/NPs, Taxotere® and DTX/NPs + SDT groups (1 to 7), respectively. (E) Curves of tumor volume changes and (F) body weight changes of B16F10 tumor-bearing mice subjected to different treatments. (G) H&E staining and (H) TUNEL images of tumor tissues collected from B16F10 tumor-bearing mice after the administration of different preparations. | ||
Elimination of drugs in blood was additionally examined after intravenous injection of 15 mg kg−1 DTX in Taxotere® and NPs. The results are listed in Table 3. The maximum concentration (Cmax) value of DTX/NPs was nearly four times than that of Taxotere®, and t1/2 was increased nearly 4.75-fold. The area under curve (AUC) and mean residence time (MRT) for DTX/NPs were greater than those of the commercially available drug Taxotere®, but the clearance (CL) decreased more than 5.7-fold. Therefore, the long-term circulation capacity of NPs was beneficial for in vivo drug administration.
| Parameter | Taxotere® | DTX/NPs |
|---|---|---|
| AUC0–36 (mg L−1 h−1) | 6.177 ± 0.30 | 30.86 ± 0.30 |
| AUC0–∞ (mg L−1 h−1) | 6.465 ± 0.36 | 32.12 ± 0.30 |
| MRT0–36 (h) | 1.557 ± 0.03 | 4.526 ± 1.03 |
| MRT0–∞ (h) | 1.820 ± 0.10 | 6.756 ± 1.36 |
| t 1/2z (h) | 2.048 ± 0.12 | 9.518 ± 1.91 |
| CL [L (h kg)−1] | 2.325 ± 0.13 | 0.4670 ± 0.02 |
| C max (mg L−1) | 6.247 ± 1.01 | 25.09 ± 1.85 |
3.16. In vivo anti-tumor effect and safety evaluation
The in vivo anti-tumor therapeutic effect of DTX/NPs in B16F10 tumor-bearing mouse models was additionally evaluated. After 14 days, mice were dissected and tumors were removed, as shown in Fig. 5D. In combination with volume change curves, our data showed that tumors in CS–Rh + SDT, NPs + SDT and Taxotere® groups were significantly decreased relative to the control group (p < 0.001), while the volume was further decreased in the DTX/NPs and DTX/NPs + SDT groups compared with the Taxotere® group (p < 0.01) (Fig. 5E). However, the body weight was not significantly changed, except for a slight decrease in the Taxotere® group, on Day 10 and Day 12 (Fig. 5F), suggesting a mild virulent property of Taxotere® compared with NPs and DTX/NPs. The relative tumor volume of mice and tumor inhibition rate are presented in Fig. S7 and S8.† H&E staining of tumors showed significantly enhanced necrotic areas in the DTX/NPs and DTX/NPs + SDT groups (Fig. 5G). The area of apoptosis in tumor tissues was additionally detected (marked with red arrows and circles in Fig. 5H). Apoptotic regions were significantly increased in the DTX/NPs ± SDT relative to the control group. Additionally, the maximum number of apoptotic bodies was detected in the DTX/NPs + SDT group.3.17. In vivo hypoxia attenuation and ROS production capacity
To establish the hypoxia-relieving ability of DTX/NPs in B16F10 tumor-bearing mice, tumor tissues were collected and sectioned for further immunohistochemical staining after intravenous injection of mice with the different preparations.36 Hypoxic regions and cell nuclei were stained red and blue, respectively (Fig. 6B). The NS group displayed significant and maximal hypoxia areas, illustrating the inherent anoxic nature of tumor tissues. Notably, NPs significantly ameliorated the hypoxic environment in B16F10 solid tumors under conditions of SDT treatment, while tumor tissues exhibited large hypoxic areas in the non-PFC-containing group of CS–Rh. The in vivo ROS production was additionally evaluated. As a fluorescent probe that effectively penetrates living cells, the ROS indicator SOSG was injected after treatment of different preparations and SDT. Tumors were sliced and stained with DAPI for the observation of cell nuclei (blue). As shown in Fig. 6C, SOSG (green) fluorescence was significantly increased in CS–Rh + SDT and NPs + SDT groups relative to the control group, clearly indicating strong ROS generation capacity. US treatment could accelerate movement of NPs and promote release of oxygen loaded in the nanoplatform core that was carried by PFC.51 Therefore, NPs combined with US generated more ROS molecules than CS–Rh and could overcome the hypoxia environment. | ||
| Fig. 6 (A) Overall schedule of in vivo tumor inhibition analysis. (B) Immunofluorescence staining of tumor slices for the detection of hypoxia in vivo. Cell nuclei and anoxic regions were stained blue and red, respectively. (C) Generation of singlet oxygen of NS, CS–Rh + SDT and NPs + SDT groups in vivo. Singlet oxygen production was measured with SOSG, detected as green fluorescence, while the cell nuclei were stained blue. (D) Proportion of CD4+ and (E) CD8+ T cells in tumor tissues determined using FCM. (F) Quantitative analysis of various cytokines in serum samples of B16F10 tumor-bearing mice after different treatments. (G) and (H) represent the CD3+ CD8+ and CD3+ CD4+ immunofluorescence staining of B16F10 tumor tissues incubated with different preparations, respectively. The CD3+ and cell nuclei were all marked in red and blue, respectively, and CD4+ and CD8+ cells were separately marked in green. Scale bar, 100 μm. | ||
3.18. Anti-tumor immunity assay
Mice were injected with different preparations four times and sacrificed, followed by acquisition of tumor tissues on Day 14, and the CD4+ and CD8+ T cells were measured to assay immune responses (Fig. 6A). As shown in Fig. 6D and E, SDT treatment groups could recruit more CD4+ and CD8+ T cells after US therapy compared with the NS group, while the CD4+ T cell content in Taxotere® was similar to that of the DTX/NPs group owing to the inherent ICD-inducing effect of DTX.52 However, DTX/NPs recruited significantly more CD8+ T cells in tumor tissues compared with Taxotere®, probably owing to the enhanced cytotoxicity of cancer cells after the formulation of NPs.53 Furthermore, DTX/NPs + SDT recruited the maximum number of CD4+ and CD8+ T cells in tumor tissues, indicating an immunologic enhancement effect of SDT (Fig. S9 and S10†). Mouse serum in different groups was collected for the detection of IL-2, IL-6, TNF-α and IFN-γ via ELISA. A heat map of quantitative analysis was sketched (Fig. 6F). Therefore, circulating cytokine levels of IL-6, TNF-α and IFN-γ were significantly higher in the DTX/NPs + SDT group relative to the control group (p < 0.01), while concentrations of IL-2 cytokines showed limited differences (Fig. S11†). Therefore, the DTX/NPs + SDT group showed an increased extent of T cell infiltration as well as cytokine secretion capacity, which was beneficial for in vivo immune response activation. Furthermore, immunofluorescence results revealed the increased expression of CD4+ and CD8+ cells in DTX/NPs and DTX/NPs + SDT groups (Fig. 6G, H and Fig. S12 and S13†). Compared with the NS group, the DTX/NPs + SDT group displayed elevated CD3+ CD4+ and CD3+ CD8+ T cell expression, consistent with the results shown in Fig. 6D and E. Additionally, the maturity of DCs was also detected in tumor draining lymph nodes via FCM after staining with CD11+, CD80+ and CD86+ antibodies. As shown in Fig. S14,† the results showed that the DTX/NPs + SDT boosted the maturation of DCs (23.86%) more powerfully than the DTX/NPs (21.37%) and Taxotere® (18.31%) groups, while the maturity degree of DCs was very low in the NS group (11.67%). Therefore, the combined therapy could induce strong immune response in the mice body.4. Conclusion
In this study, an oxygen-carrying amphiphilic nanoplatform containing a novel sonosensitizer, Rh, was constructed, which exerted an enhanced therapeutic effect in combination with chemo-SDT. A CS–Rh conjugate was synthesized by grafting of Rh on to the CS backbone. The conjugate was endowed with anti-tumor properties of Rh and showed better performance than free Rh, which could be attributable to the formulation of CS–Rh NPs. Linking to PFC molecules generated CS–Rh–PFC NPs that could dissolve and transport oxygen molecules to tumor tissues. The SDT effect of CS–Rh was enhanced by oxygen molecules carried by PFC and greater levels of ROS were produced, which further inhibited tumor growth. Furthermore, CS–Rh–PFC NPs could alleviate the hypoxia situation around tumor tissues, which was beneficial for the suppression of tumor metastasis and invasion. Our observation of reduced MMP9 expression, a crucial contributor to metastasis and invasion, further validated this finding. After hydrolysis of disulfide bonds in the presence of glutathione in tumor cells, DTX was released into the cytoplasm. Compared with the commercially available drug, Taxotere®, DTX/NPs exhibited a longer circulation time and half-life in vivo, which were beneficial for anti-tumor therapy. Following administration into B16F10 tumor-bearing mice, the DTX/NPs + SDT group showed a marked increase in CD8+ and CD4+ T cells in infiltrating tumor tissues compared with the NS group. However, there was no significant difference in tumor infiltrate lymphocytes and peripheral blood between CS–Rh + SDT and NPs + SDT groups, implying the critical role of Rh in activating tumor immunity. Also, the DC maturation after the combined chemo-SDT could initiate and direct the immune responses. In summary, we synthesized a novel Rh-based nanoplatform that exerted enhanced therapeutic effects in combined chemo-SDT, which relieved hypoxia pressure and realized precise tumor-targeted drug release. In the future, the anti-tumor effects of the novel nanoplatform could be further improved through simultaneous encapsulation of other drugs or immune adjuvants and multifunctional sonosensitizers.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This work was supported by the National Science and Technology Major Special Project-Major New Drug Creation (2019ZX09301-112) and Major Basic Research Projects of Shandong Natural Science Foundation, P.R. China (No. ZR2018ZC0232), Shandong Provincial Major Science & Technology Innovation Project, P.R. China (2018CXGC1411) and Shandong Provincial Program of Taishan Industrial Experts (No. TSCY 20190331).References
- Y. Li, H. Zhang, Q. Li, P. Zou, X. Huang, C. Wu and L. Tan, Cancer Lett., 2020, 495, 12–21 CrossRef CAS PubMed.
- X. Kong, T. Kuilman, A. Shahrabi, J. Boshuizen, K. Kemper, J. Y. Song, H. W. M. Niessen, E. A. Rozeman, M. H. Geukes Foppen, C. U. Blank and D. S. Peeper, Nature, 2017, 550, 270–274 CrossRef PubMed.
- D. L. Bellan, S. M. P. Biscaia, G. R. Rossi, A. M. Cristal, J. P. Goncalves, C. C. Oliveira, F. F. Simas, D. A. Sabry, H. A. O. Rocha, C. R. C. Franco, R. Chammas, R. J. Gillies and E. S. Trindade, Carbohydr. Polym., 2020, 250, 116869 CrossRef CAS PubMed.
- M. Artomov, eLife, 2019, 8, e48145 CrossRef CAS PubMed.
- X. Ge, Q. Fu, L. Su, Z. Li, W. Zhang, T. Chen, H. Yang and J. Song, Theranostics, 2020, 10, 4809–4821 CrossRef CAS PubMed.
- Z. Wang, N. Hauser, G. Singer, M. Trippel, R. A. Kubik-Huch, C. W. Schneider and M. Stampanoni, Nat. Commun., 2014, 5, 3797–3805 CrossRef CAS PubMed.
- R. M. S. Sigrist, J. Liau, A. E. Kaffas, M. C. Chammas and J. K. Willmann, Theranostics, 2017, 7, 1303–1329 CrossRef PubMed.
- H. Xu, N. Yu, J. Zhang, Z. Wang, P. Geng, M. Wen, M. Li, H. Zhang and Z. Chen, Biomaterials, 2020, 257, 120239 CrossRef CAS PubMed.
- J. Zhu, C. Chu, D. Li, X. Pang, H. Zheng, J. Wang, Y. Shi, Y. Zhang, Y. Cheng, E. Ren, J. Cheng, X. Chen and G. Liu, Adv. Funct. Mater., 2019, 29, 1904056 CrossRef.
- F. Gong, L. Cheng, N. Yang, O. Betzer, L. Feng, Q. Zhou, Y. Li, R. Chen, R. Popovtzer and Z. Liu, Adv. Mater., 2019, 31, e1900730 CrossRef PubMed.
- S. Liang, X. Deng, Y. Chang, C. Sun, S. Shao, Z. Xie, X. Xiao, P. Ma, H. Zhang, Z. Cheng and J. Lin, Nano Lett., 2019, 19, 4134–4145 CrossRef CAS PubMed.
- R. Zhang, L. Zhang, H. Ran, P. Li, J. Huang, M. Tan, Y. Yang and Z. Wang, Biomater. Sci., 2020, 8, 4581–4594 RSC.
- Z. Liu, J. Li, W. Chen, L. Liu and F. Yu, Biomaterials, 2020, 232, 119685 CrossRef CAS PubMed.
- M. Liu, A. R. Khan, J. Ji, G. Lin, X. Zhao and G. Zhai, J. Controlled Release, 2018, 290, 150–164 CrossRef CAS PubMed.
- M. Yashiro, H. Kinoshita, G. Tsujio, T. Fukuoka, Y. Yamamoto, T. Sera, A. Sugimoto, S. Nishimura, S. Kushiyama, S. Togano, K. Kuroda, T. Toyokawa and M. Ohira, Oncol. Lett., 2021, 21, 38 CAS.
- C. Li, X. Q. Yang, J. An, K. Cheng, X. L. Hou, X. S. Zhang, Y. G. Hu, B. Liu and Y. D. Zhao, Theranostics, 2020, 10, 867–879 CrossRef CAS PubMed.
- Q. Wang, J. M. Li, H. Yu, K. Deng, W. Zhou, C. X. Wang, Y. Zhang, K. H. Li, R. X. Zhuo and S. W. Huang, Biomater. Sci., 2018, 6, 3096–3107 RSC.
- Q. Zeng, L. Qiao, L. Cheng, C. Li, Z. Cao, Z. Chen, Y. Wang and J. Liu, ACS Biomater. Sci. Eng., 2020, 6, 2956–2969 CrossRef CAS PubMed.
- J. Luo, T. Gong and L. Ma, Carbohydr. Polym., 2020, 249, 116887 CrossRef CAS PubMed.
- A. Wang, H. Jiang, Y. Liu, J. Chen, X. Zhou, C. Zhao, X. Chen and M. Lin, J. Cancer, 2020, 11, 500–507 CrossRef CAS PubMed.
- J. An, Y. G. Hu, K. Cheng, C. Li, X. L. Hou, G. L. Wang, X. S. Zhang, B. Liu, Y. D. Zhao and M. Z. Zhang, Biomaterials, 2020, 234, 119761 CrossRef CAS PubMed.
- L. Yang, S. Lin, Y. Kang, Y. Xiang, L. Xu, J. Li, X. Dai, G. Liang, X. Huang and C. Zhao, J. Exp. Clin. Cancer Res., 2019, 38, 31–43 CrossRef PubMed.
- J. Kim, J. Lee, J. Lee, H. Keum, Y. Kim, Y. Kim, B. Yu, S. Y. Lee, J. Tanaka, S. Jon and M. C. Choi, Adv. Mater., 2020, 32, e2002902 CrossRef PubMed.
- H. Qiao, X. Chen, E. Chen, J. Zhang, D. Huang, D. Yang, Y. Ding, H. Qian, J. Feijen and W. Chen, Biomater. Sci., 2019, 7, 2749–2758 RSC.
- M. Liu, H. Du, A. R. Khan, J. Ji, A. Yu and G. Zhai, Carbohydr. Polym., 2018, 184, 82–93 CrossRef CAS PubMed.
- X. Wang, Y. Guo, L. Qiu, X. Wang, T. Li, L. Han, H. Ouyang, W. Xu and K. Chu, Carbohydr. Polym., 2019, 206, 121–131 CrossRef CAS PubMed.
- X. Yang, X. Shi, Y. Zhang, J. Xu, J. Ji, L. Ye, F. Yi and G. Zhai, J. Controlled Release, 2020, 323, 333–349 CrossRef CAS PubMed.
- Y. Wang, F. Zhang, H. Lin and F. Qu, ACS Appl. Mater. Interfaces, 2019, 11, 43964–43975 CrossRef CAS PubMed.
- A. Hollmann, M. A. Castanho, B. Lee and N. C. Santos, Biochem. J., 2014, 459, 161–170 CrossRef CAS PubMed.
- D. Zhang, J. Zhang, Q. Li, A. Song, Z. Li and Y. Luan, ACS Appl. Mater. Interfaces, 2019, 11, 32633–32646 CrossRef CAS PubMed.
- T. Zhou, X. Liang, P. Wang, Y. Hu, Y. Qi, Y. Jin, Y. Du, C. Fang and J. Tian, ACS Nano, 2020, 14, 12679–12696 CrossRef CAS PubMed.
- Q. Li, D. Zhang, J. Zhang, Y. Jiang, A. Song, Z. Li and Y. Luan, Nano Lett., 2019, 19, 6647–6657 CrossRef CAS PubMed.
- Y. Chen and J. S. Lin, Biochimie, 2017, 132, 1–8 CrossRef CAS PubMed.
- X. Shi, X. Yang, M. Liu, R. Wang, N. Qiu, Y. Liu, H. Yang, J. Ji and G. Zhai, Carbohydr. Polym., 2021, 254, 117459 CrossRef CAS PubMed.
- T. Hohmann and F. Dehghani, Cells, 2019, 8, 362–416 CrossRef CAS PubMed.
- L. Xing, J.-H. Gong, Y. Wang, Y. Zhu, Z.-J. Huang, J. Zhao, F. Li, J.-H. Wang, H. Wen and H.-L. Jiang, Biomaterials, 2019, 206, 170–182 CrossRef CAS PubMed.
- D. L. Tao, L. Z. Feng, Y. Chao, C. Liang, X. J. Song, H. R. Wang, K. Yang and Z. Liu, Adv. Funct. Mater., 2018, 28, 11–21 Search PubMed.
- G. Zhou, F. Peng, Y. Zhong, Y. Chen, M. Tang and D. Li, Int. J. Oncol., 2017, 50, 933–941 CrossRef CAS PubMed.
- H. Liewen, N. Markuly, H. Laubli, Y. Liu, M. S. Matter, N. Liewen, C. Renner, A. Zippelius and F. Stenner, Targeted Oncol., 2019, 14, 577–590 CrossRef PubMed.
- H. O'Farrell, B. Harbourne, Z. Kurlawala, Y. Inoue, A. L. Nagelberg, V. D. Martinez, D. Lu, M. H. Oh, B. P. Coe, K. L. Thu, R. Somwar, S. Lam, W. L. Lam, A. M. Unni, L. Beverly and W. W. Lockwood, J. Thorac. Oncol., 2019, 14, 656–671 CrossRef PubMed.
- H. Li, P. Zhang, J. Luo, D. Hu, Y. Huang, Z. R. Zhang, Y. Fu and T. Gong, ACS Nano, 2019, 13, 9386–9396 CrossRef CAS PubMed.
- R. Hou, X. Liang, X. Li, X. Zhang, X. Ma and F. Wang, Biomater. Sci., 2020, 8, 2526–2536 RSC.
- J. Chen, H. Luo, Y. Liu, W. Zhang, H. Li, T. Luo, K. Zhang, Y. Zhao and J. Liu, ACS Nano, 2017, 12849–12862 CrossRef CAS PubMed.
- S. Kar, Cell Syst., 2016, 2, 8–10 CrossRef CAS PubMed.
- M. C. O. da Rocha, P. B. da Silva, M. A. Radicchi, B. Y. G. Andrade, J. V. de Oliveira, T. Venus, C. Merker, I. Estrela-Lopis, J. P. F. Longo and S. N. Bao, J. Nanobiotechnol., 2020, 18, 43 CrossRef PubMed.
- W. Li, J. Yang, L. Luo, M. Jiang, B. Qin, H. Yin, C. Zhu, X. Yuan, J. Zhang, Z. Luo, Y. Du, Q. Li, Y. Lou, Y. Qiu and J. You, Nat. Commun., 2019, 10, 3349–3354 CrossRef PubMed.
- A. Bernard, S. Chevrier, F. Beltjens, M. Dosset, E. Viltard, A. Lagrange, V. Derangere, A. Oudot, F. Ghiringhelli, B. Collin, L. Apetoh, O. Feron, S. Chen, L. Arnould, F. Vegran and R. Boidot, Cancer Res., 2019, 79, 5958–5970 CrossRef CAS PubMed.
- L. Meng, Y. Cheng, X. Tong, S. Gan, Y. Ding, Y. Zhang, C. Wang, L. Xu, Y. Zhu, J. Wu, Y. Hu and A. Yuan, ACS Nano, 2018, 12, 8308–8322 CrossRef CAS PubMed.
- H. Wang, Y. Yang, X. Sun, F. Tian, S. Guo, W. Wang, Z. Tian, H. Jin, Z. Zhang and Y. Tian, Theranostics, 2018, 8, 4969–4984 CrossRef CAS PubMed.
- Y. Fan, Q. Wang, G. Lin, Y. Shi, Z. Gu and T. Ding, Acta Biomater., 2017, 62, 257–272 CrossRef CAS PubMed.
- F. Zhang, J. Zhuang, B. Esteban Fernández de Ávila, S. Tang, Q. Zhang, R. H. Fang, L. Zhang and J. Wang, ACS Nano, 2019, 13, 11996–12005 CrossRef CAS PubMed.
- T. Flieswasser, J. Van Loenhout, L. Freire Boullosa, A. Van den Eynde, J. De Waele, J. Van Audenaerde, F. Lardon, E. Smits, P. Pauwels and J. Jacobs, Cells, 2020, 9, 1474–1490 CrossRef CAS PubMed.
- A. Krol, P. Pomastowski, K. Rafinska, V. Railean-Plugaru and B. Buszewski, Adv. Colloid Interface Sci., 2017, 249, 37–52 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1bm00198a |
| This journal is © The Royal Society of Chemistry 2021 |