
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of poly(2-alkyl-2-oxazoline)s in hydrogels and biofabrication
Lucca
Trachsel
a,
Marcy
Zenobi-Wong
 *a and
Edmondo M.
Benetti
*bc
*a and
Edmondo M.
Benetti
*bc
aTissue Engineering + Biofabrication Laboratory, Department of Health Sciences and Technology, ETH Zürich, 8093 Zürich, Switzerland
bLaboratory for Surface Science and Technology, Department of Materials, ETH Zürich, Vladimir-Prelog-Weg 5, 8093 Zürich, Switzerland. E-mail: edmondo.benetti@mat.ethz.ch
cBiointerfaces, Swiss Federal Laboratories for Materials Science and Technology (Empa), Lerchenfeldstrasse 5, CH-9014, St. Gallen, Switzerland
First published on 22nd February 2021
Abstract
Poly(2-alkyl-2-oxazoline)s (PAOXAs) have been rapidly emerging as starting materials in the design of tissue engineering supports and for the generation of platforms for cell cultures, especially in the form of hydrogels. Thanks to their biocompatibility, chemical versatility and robustness, PAOXAs now represent a valid alternative to poly(ethylene glycol)s (PEGs) and their derivatives in these applications, and in the formulation of bioinks for three-dimensional (3D) bioprinting. In this review, we summarize the recent literature where PAOXAs have been used as main components for hydrogels and biofabrication mixtures, especially highlighting how their easily tunable composition could be exploited to fabricate multifunctional biomaterials with an extremely broad spectrum of properties.
Introduction
During the last decade, poly(2-alkyl-2-oxazoline)s (PAOXAs) have emerged as starting materials for a variety of different biomedical applications,1–4 including the generation of drug delivery systems,5 tissue engineering constructs, biofabrication, and the design of synthetic biointerfaces.6–14 In particular, water-soluble PAOXAs such as poly(2-methyl-2-oxazoline) (PMOXA) and poly(2-ethyl-2-oxazoline) (PEOXA) have been increasingly applied in different biomedical formulations. This is due to their in vitro cytocompatibility15–18 and excellent biocompatibility in vivo,19–21 coupled with their antifouling and stealth properties,13,22 which significantly reduce unspecific interactions of PAOXA-functionalized supports with biomolecules and cells.6,12,13Besides these positive aspects, PMOXA, PEOXA, and their derivatives applied in biomaterials also appear to be advantageous compared to the use of poly(ethylene glycol) (PEG)-based analogues. Increasing evidence suggests that PEGs are potentially immunogenic in patients,23–26 while PEG itself is susceptible to oxidative degradation within physiological environments, leading to the generation of toxic by-products.27,28 In contrast, although PMOXA and PEOXA feature similar physicochemical properties to PEGs, they present tertiary amide groups along their backbone, which are less susceptible to fast hydrolytic degradation within physiological media with respect to ether functions.13,29,30
By exploiting cationic ring-opening polymerization (CROP), multifunctional PAOXAs can be synthesized in a controlled fashion and under relatively mild and accessible conditions, providing polymers with different functionalities, or copolymers with diverse structural properties. The chemical versatility and synthetic accessibility of PAOXAs thus represent major advantages over PEGs, whose availability in the form of functional (co)polymers is intrinsically limited.31,32
The unique physicochemical properties of PAOXAs, coupled to their biocompatibility, chemical robustness and multifunctional character have recently triggered their increasing use as precursors for the generation of hydrogels. Especially in the field of tissue engineering, PAOXA-based networks were applied as supports for cell cultures, as multifunctional matrices for the encapsulation of mammalian cells and as synthetic components within bio-inks in three-dimensional (3D) bioprinting. In this short, comprehensive review paper, we aim to highlight the most prominent and recent examples of PAOXA-based hydrogel formulations, paying particular attention to how the compositional versatility of this emerging class of polymers has enabled a variety of cross-linking strategies and the design of bioactive supports.
PAOXA hydrogels for cell cultures and tissue engineering
In 2013, Farrugia and coworkers reported the first example of PMOXA-based hydrogels applied as supports for 3D cell cultures.33 The starting polymers for generating PMOXA networks were based on statistical copolymers including 2-methyl-2-oxazoline (MOXA) and 2-(dec-9-enyl)-2-oxazoline (DecEnOXA), which contained a long aliphatic side chain with a terminal alkene group (Fig. 1a). Using the UV-mediated thiol–ene reaction, RGD peptides featuring a thiol group were subsequently coupled to the alkene groups of the P(MOXA-ran-DecEnOXA) precursors. A second thiol–ene reaction using dithiothreitol (DTT) as cross-linking agent and Irgacure 2959 as photoinitiator enabled the formation of P(MOXA-ran-DecEnOXA)-RGD hydrogels. Interestingly, an increment in RGD loading within the networks did not translate to a significant increase in the number of cells adhering to their surface. In contrast, an increase in the concentration of adhesive peptides markedly influenced cell morphology and orientation of actin fibres, which became more aligned (Fig. 1b). Besides seeding cells onto the P(MOXA-ran-DecEnOXA)-RGD supports, fibroblasts were encapsulated within the network structure by including them in the precursor solutions, which were subsequently cured using bis-cysteine RGD peptide and DTT as cross-linking agents. High cell viability was finally demonstrated, providing the first evidence of the excellent cytocompatibility of PMOXA-based networks.2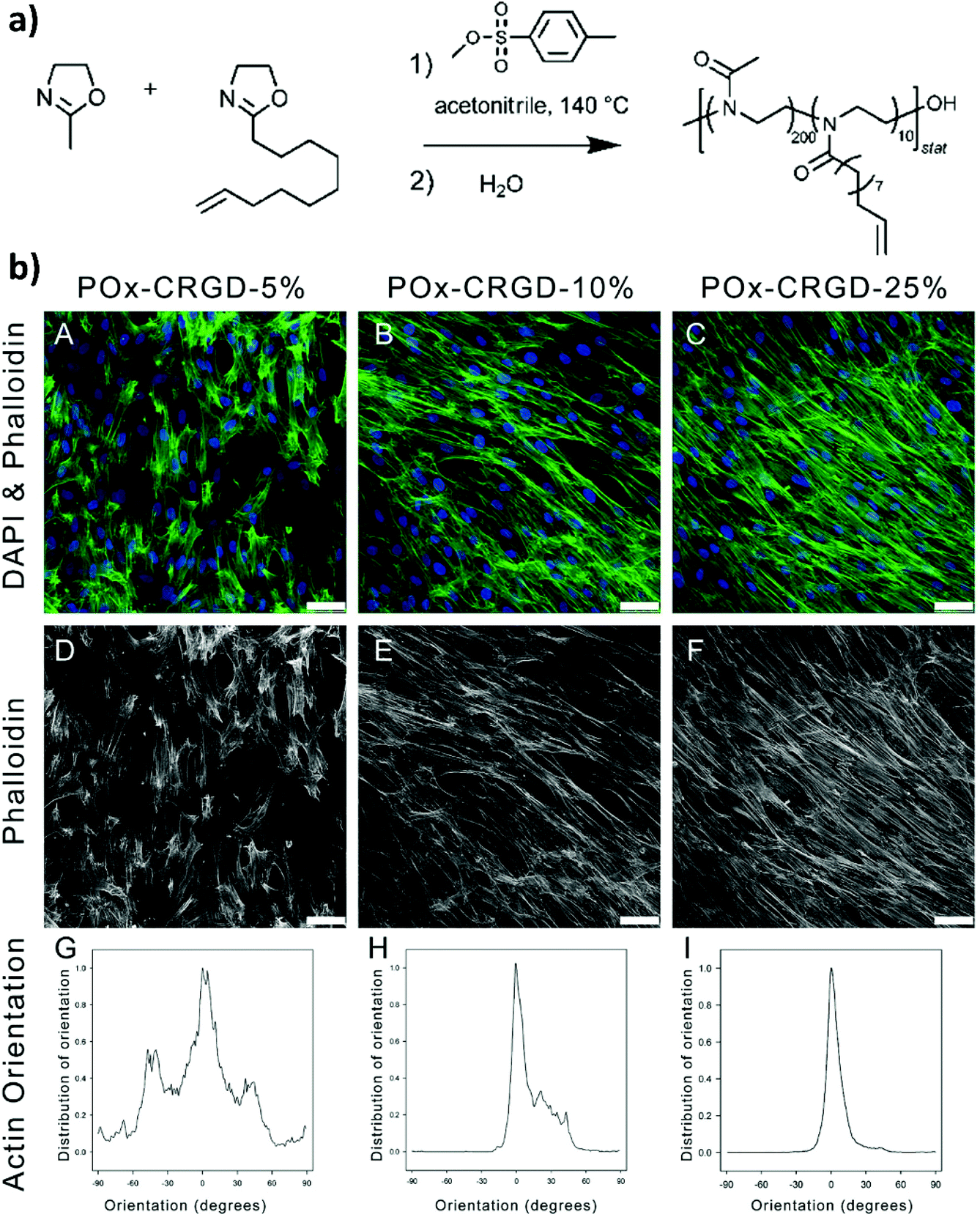 | ||
| Fig. 1 (a) CROP of MOXA and DecEnOXA, using methyl para-toluenesulfonate as the initiator and water as the terminating agent to yield P(MOXA-ran-DecEnOXA). (b) Effect of an increasing amount of the incorporated RGD peptide into PMOXA hydrogels on fibroblast attachment and morphology; (A, D) 5% CRGDSG, (B, E) 10% CRGDSG, and (C, F) 25% CRGDSG. (A–C) Immunofluorescence micrographs showing the cells seeded onto PMOXA hydrogels, following staining with phalloidin and DAPI to visualize actin cytoskeleton fibres and cell nuclei, respectively. (D–F) Immunofluorescence micrographs showing the actin fibres. Scale bar = 50 μm. (G–I) Quantification of the actin orientation of fibroblasts adhered to the surface of PAOXA hydrogels with an increasing amount of RGD. Reproduced with permission from Farrugia et al.33 Copyright 2013 American Chemical Society. | ||
Analogous cross-linking strategies were applied in several other studies aiming to fabricate PAOXA hydrogels from copolymer precursors including alkene groups in their side chains.34–37 For example, Hickey et al. encapsulated cells within porous PAOXA hydrogels to create artificial female reproductive tract tissue, which was intended as a model system for studying infection.38 Specifically, hydrogels composed of cross-linked P(MOXA-ran-DecEnOXA) were frozen and lyophilized to introduce pores with typical areas in the range 200–3200 μm2. The porous character of the hydrogels enabled the cells that were seeded on top of the supports to infiltrate into their bulk structure, thus avoiding exposure to UV irradiation necessary for cross-linking, which could lead to cell damage.
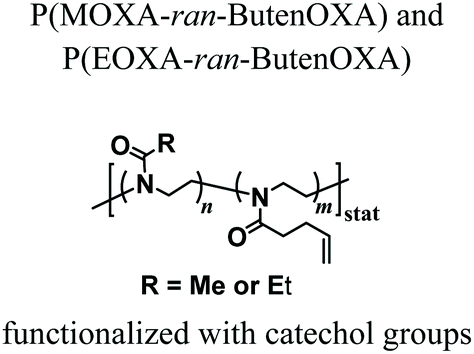
Groll and coworkers recently proposed alternative strategies to covalently cross-link PAOXA precursors, while focusing on the development of a biosynthetic hydrogel adhesive for the treatment of articular cartilage defects.39 In particular, catechol-bearing PAOXA precursors readily formed tissue-adhesive hydrogels through the oxidation of catechols to quinones followed by their polymerization or reaction with fibrinogen, which was added to the pre-hydrogel mixtures. Interestingly, push-out tests revealed strong adhesion by the PAOXA hydrogels on the native cartilage tissue, with adhesive strength values of up to 20 kPa, significantly higher than the values commonly recorded with the sealant fibrin or even surgical glue. In addition, when degradable ester bonds were incorporated within the polymer networks, lateral cartilage integration studied using an in vitro disc/ring model showed augmented cell invasion and stronger deposition of the cartilaginous matrix at the defect site, without compromising the bond strength to the cartilage rings.
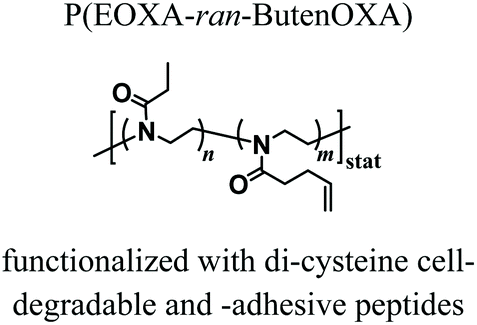
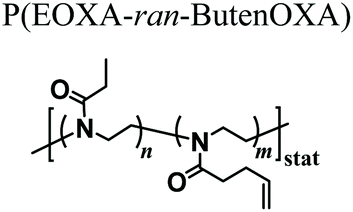
The first in vivo biocompatibility data on PAOXA-based hydrogels came from a very recent study by You et al.,40 who fabricated cell-degradable hydrogels based on P(EOXA-ran-2-butenyl-2-oxazoline) (P(EOXA-ran-ButenOXA)) cross-linked via thiol–ene chemistry. The authors first found that by tuning the mechanical and cell-degradable properties of PAOXA hydrogels, the morphology of encapsulated mesenchymal stromal cells (MSCs) could be regulated, subsequently influencing the expression and secretion of pro-angiogenic cytokines and growth factors. In addition, exploiting their excellent tissue adhesiveness, pre-hydrogel mixtures including MSCs were applied directly onto the epicardium of the heart in a rat myocardial infarction (MI) model, and subsequently photo-cross-linked yielding cell-loaded hydrogels. Immunostaining of the epicardium for micro-capillary-specific markers (isolectin B4) indicated that the MSC-loaded PAOXA hydrogels promoted neovascularization in the infarcted cardiac tissue and at the epicardium–hydrogel interface, confirming the pro-angiogenic phenotype observed in vitro (Fig. 2a–c). Gene expression analysis revealed a significant upregulation of cardiac reparative factors (VCAM-1 and VEGF) in the infarcted epicardium treated with the MSC-loaded PAOXAs, compared to an untreated infarcted cardiac tissue (Fig. 2d). Moreover, echocardiographic imaging of the rat hearts 28 days after inducing MI demonstrated that the epicardial placement of the MSC-loaded PAOXA hydrogels enhanced the cardiac function, while histological analysis showed reduced collagen deposition and interstitial fibrosis, both associated with scar formation in the infarcted area.
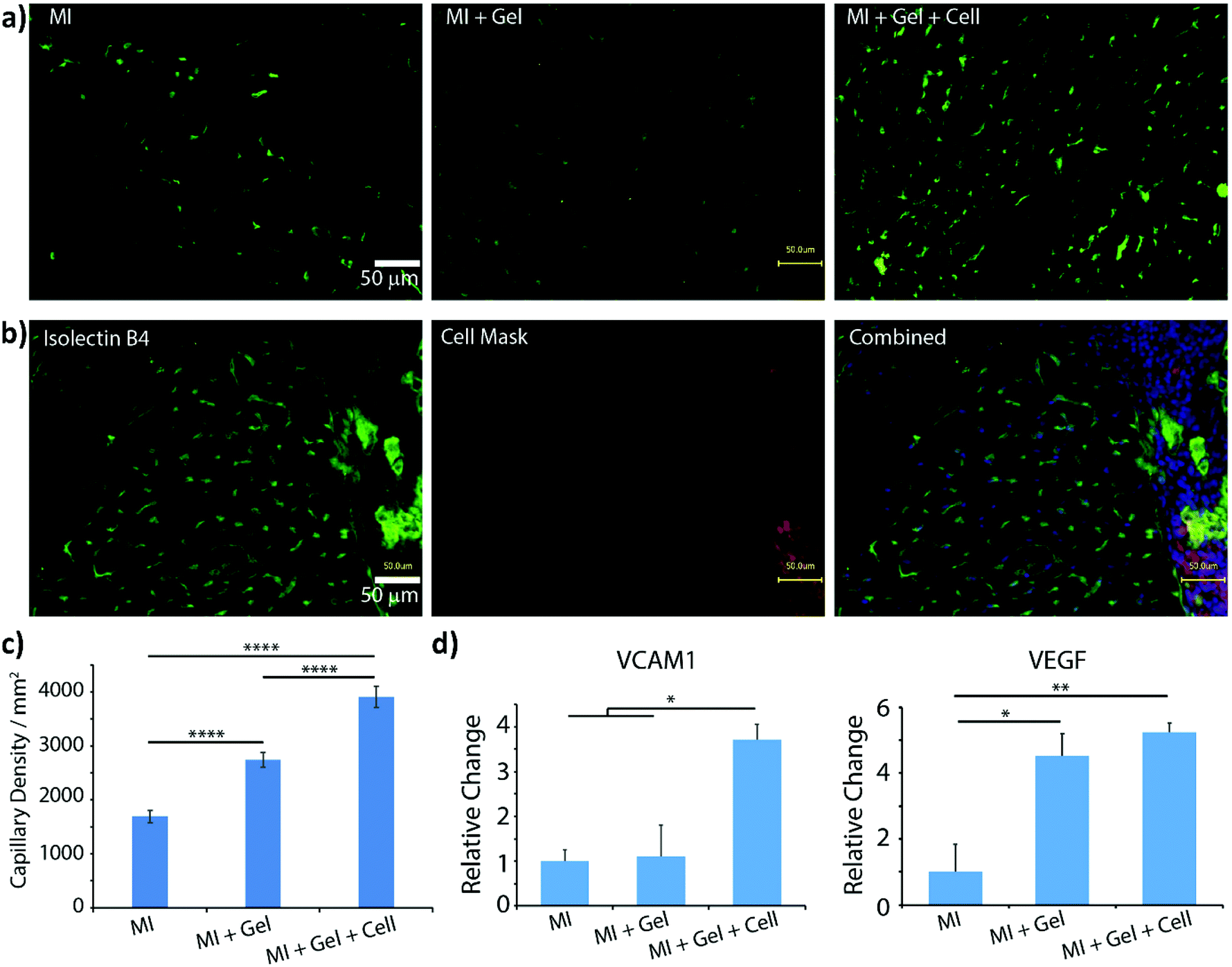 | ||
| Fig. 2 (a) Isolectin B4 staining demonstrated increased capillary density in the border area of myocardial infarction (MI) induction followed by PAOXA hydrogel placement (MI + Gel) and MI induction followed by MSC-loaded PAOXA hydrogel (MI + Gel + cell) treatment groups compared to the MI only group; scale bar = 50 μm. (b) Capillaries formed inside the PAOXA hydrogel layer. Red, CM-Dil labelled MSCs; blue, DAPI; green, isolectin B4; scale bar = 50 μm. (c) Corresponding quantification of capillary densities. MI group (n = 3), MI + Gel group (n = 4) and MI + Gel + cell group (n = 4). (d) Quantification of VCAM-1 and VEGF gene expression in different MI groups (n = 3 for each group). *p < 0.05; **P < 0.01; ****p < 0.0001. Reproduced with permission from You et al.40 Copyright 2021 Elsevier. | ||
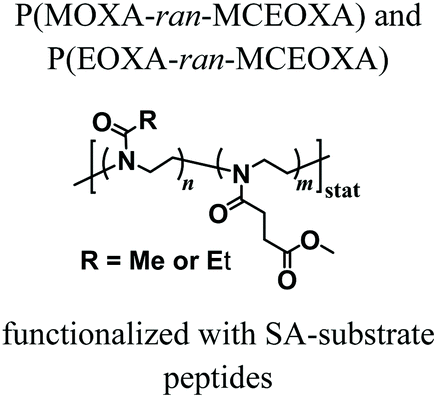
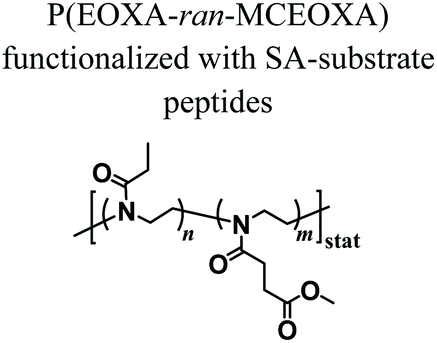
Despite the variety of efficient methods for the generation of hydrogels from PAOXA-based precursors, the application of strong nucleophiles or electrophiles, such as in Michael addition-based crosslinking reactions,41 coupled with free-radical processes involving UV light and photoinitiators (which often lack chemospecificity and bio-orthogonality42) can be detrimental to cell viability.43,44 To overcome these limitations, enzymatic cross-linking has emerged as a particularly suitable method for forming hydrogels in the presence of cells, due to its fast kinetics, excellent specificity in aqueous environments, and tolerance to oxygen and biocompatibility.45 Following this strategy, Trachsel and coworkers fabricated PMOXA and PEOXA-based hydrogels using Sortase A (SA),46 a bacterial transpeptidase which enables bio-orthogonal cross-linking reactions.47,48 Statistical copolymers based on PMOXA and PEOXA presenting carboxylic acid side chains were synthesized using CROP, and derivatised with peptide sequences acting as substrates for SA-mediated cross-linking. Enzymatic ligation reactions could be subsequently performed under physiological conditions (Fig. 3), yielding hydrogels with storage moduli (G′) between a few tens of Pa to ∼1 kPa, depending on the concentration of SA employed. Relevantly, the encapsulation of human articular chondrocytes (hACs) during network formation produced hydrogels with cell viability higher than 90% after three weeks of culturing in the absence of any cell-adhesive cues.
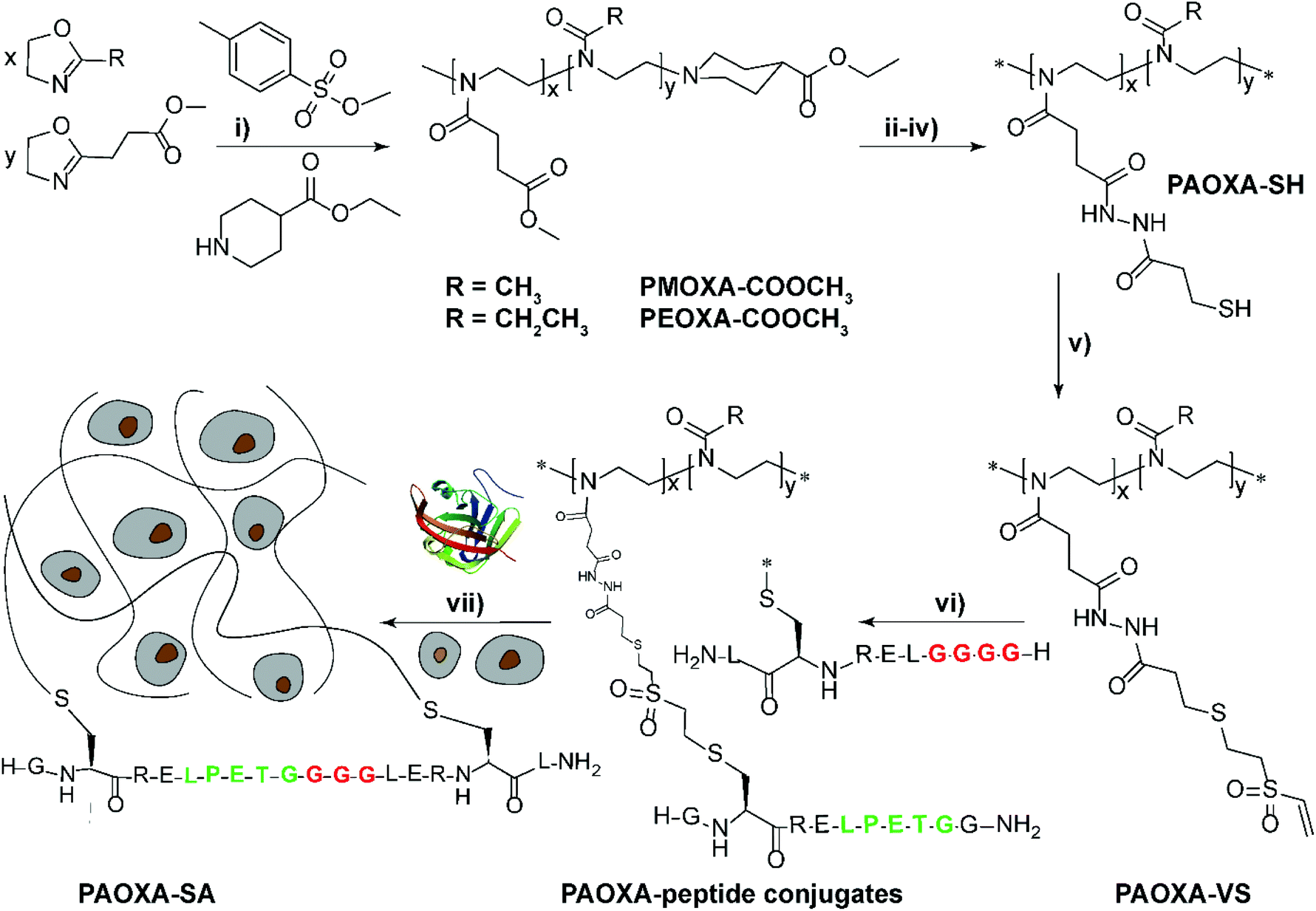 | ||
Fig. 3 Synthesis of PAOXA–peptide conjugates and their cross-linking via SA in the presence of hACs. (i) CROP of MOXA (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH3) or EOXA (RCH2CH3) with a methyl–ester containing a comonomer, 2-methoxycarbonylethyl-2-oxazoline (MCEOXA), using methyl tosylate as an initiator and ethyl isonipecotate as a terminating agent, yielding statistical PAOXA copolymers, containing 12–15 mol% methyl ester side chains (x). (ii–iv) First, hydrolysis of the methyl ester side chains, then coupling of the carboxylic acid side chains with a disulfide-containing compound, and subsequent reduction to thiol groups, yielding PAOXA–SH. (v) Vinylsulfonation (VS) via the thiol-Michael addition of thiol moieties on the side chains with divinyl sulfone, yielding PAOXA–VS. (vi) Coupling of SA-substrate peptides via thiol-Michael addition, yielding PAOXA–peptide conjugates (vii) SA-mediated ligation of the coupled peptides resulting in the cross-linking of the polymers (PAOXA–SA) in the presence of hACs under physiological pH conditions and at 37° C. Reproduced with permission from Trachsel et al.46 Copyright 2018 Royal Society of Chemistry. CH3) or EOXA (RCH2CH3) with a methyl–ester containing a comonomer, 2-methoxycarbonylethyl-2-oxazoline (MCEOXA), using methyl tosylate as an initiator and ethyl isonipecotate as a terminating agent, yielding statistical PAOXA copolymers, containing 12–15 mol% methyl ester side chains (x). (ii–iv) First, hydrolysis of the methyl ester side chains, then coupling of the carboxylic acid side chains with a disulfide-containing compound, and subsequent reduction to thiol groups, yielding PAOXA–SH. (v) Vinylsulfonation (VS) via the thiol-Michael addition of thiol moieties on the side chains with divinyl sulfone, yielding PAOXA–VS. (vi) Coupling of SA-substrate peptides via thiol-Michael addition, yielding PAOXA–peptide conjugates (vii) SA-mediated ligation of the coupled peptides resulting in the cross-linking of the polymers (PAOXA–SA) in the presence of hACs under physiological pH conditions and at 37° C. Reproduced with permission from Trachsel et al.46 Copyright 2018 Royal Society of Chemistry. | ||
PAOXA hydrogels for the incorporation and release of bioactive compounds
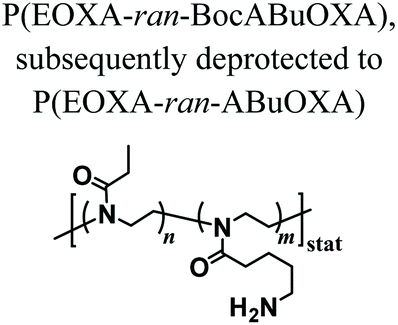
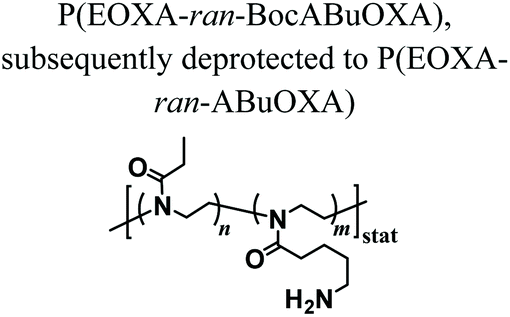
PAOXA hydrogels have also been applied by Hartlieb et al. for the reversible binding of DNA and its subsequent release. In particular, amine-containing PAOXAs were first obtained by the random co-polymerization of 2-ethyl-2-oxazoline (EOXA) with 2-(4-((tert-butoxycarbonyl)amino)butyl)-2-oxazoline (BocABuOXA), which bears tert-butyloxycarbonyl (Boc)-protected amino groups at its side chains, followed by deprotection.49 Hydrogels were subsequently formed by reacting a fraction of the amino groups with epichlorohydrin in aqueous sodium hydroxide solution, whereas the remaining NH2 groups on the PAOXA segments were used to immobilize negatively charged DNA within the network. DNA release was then triggered by the addition of polyanionic heparin.Interestingly, similar PAOXA hydrogels were formed within porous polyethylene (PE) and polypropylene (PP) matrices, generating hydrogel microstructures with tunable pore sizes.50 DNA was bound to the generated hybrid materials and subsequently released by exploiting different stimuli including temperature and pH.
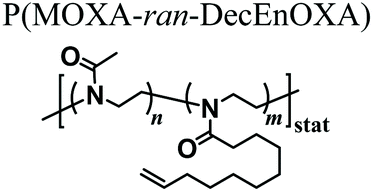
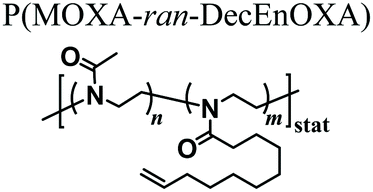
As an alternative starting polymer, poly(ethylene imine) (PEI) obtained from the complete hydrolysis of PEOXA has been recently used by Englert and coworkers to generate precursor mixtures for the fabrication of PAOXA hydrogels. Specifically, the PEI segments were derivatized with 3-butenyl side chains, yielding random copolymers poly(2-(3-butenyl)-2-oxazoline-ran-ethylene imine) that feature reactive double-bonds at their side chains. Hydrogel formation was achieved by thiol–ene photo-addition reaction, using difunctional thiols as cross-linkers and by applying UV-irradiation, yielding networks with swelling properties that could be finely tuned by varying the relative content of double bond-bearing butenyl functions and ethylene imine units along the copolymer backbone.51 The PEI-PAOXA hydrogels were finally applied for binding and releasing DNA, a process which could be modulated by varying the concentration of free amines along the copolymer segments and their protonation.
Biofabrication using PAOXA-based hydrogels
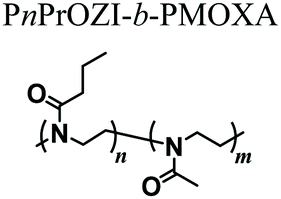
While an increasing interest has been shown to developing PAOXA-based formulations for biomaterials, the application of this class of polymers has simultaneously become significant for several biofabrication methods. The fully tunable composition of PAOXAs and their biocompatible character have especially triggered their use in extrusion-based 3D bioprinting, an additive manufacturing technique that uses a printable bioink composed of polymer precursors and cells to generate 3D cellularized tissue-like hydrogels with a predefined architecture.In 2017, Lorsen and coworkers proposed the first example of extrusion-based 3D bioprinting of PAOXAs, also introducing poly(2-alkyl-2-oxazine)s (PAOZIs) in their formulation—polymers strictly related to PAOXAs, although obtained from the CROP of six-membered cyclic imino ethers.6,52–55 In particular, the bioink was based on amphiphilic diblock copolymers, including a hydrophilic PMOXA and a thermoresponsive poly(2-n-propyl-2-oxazine) (PnPrOZI) block, yielding PnPrOZI-b-PMOXA.56 PnPrOZI shows a lower critical solution temperature (LCST) at approximately 12 °C,57 and thus its relative content within PnPrOZI-b-PMOXA species could be adjusted to modulate the thermogelling properties of the entire bioink, which featured LCST values between 15 °C and 35 °C. In particular, at 37 °C the thermogelling bioink showed isothermal shear thinning and rapid shear recovery behaviors, two rheological properties that are crucial for extrusion-based 3D printing (Fig. 4a and b). The reduction of the temperature to 25 °C triggered the gelation of the copolymer mixture, providing transparent hydrogels with G′ values more than 1 kPa. Relevantly, bioinks based on PnPrOZI-b-PMOXA enabled printing in the presence of fibroblasts, obtaining 3D cell-loaded hydrogel grids that showed exceptional cell viability 24 hours after printing (Fig. 4c–e), although with relatively modest shape fidelity and low printing resolution.
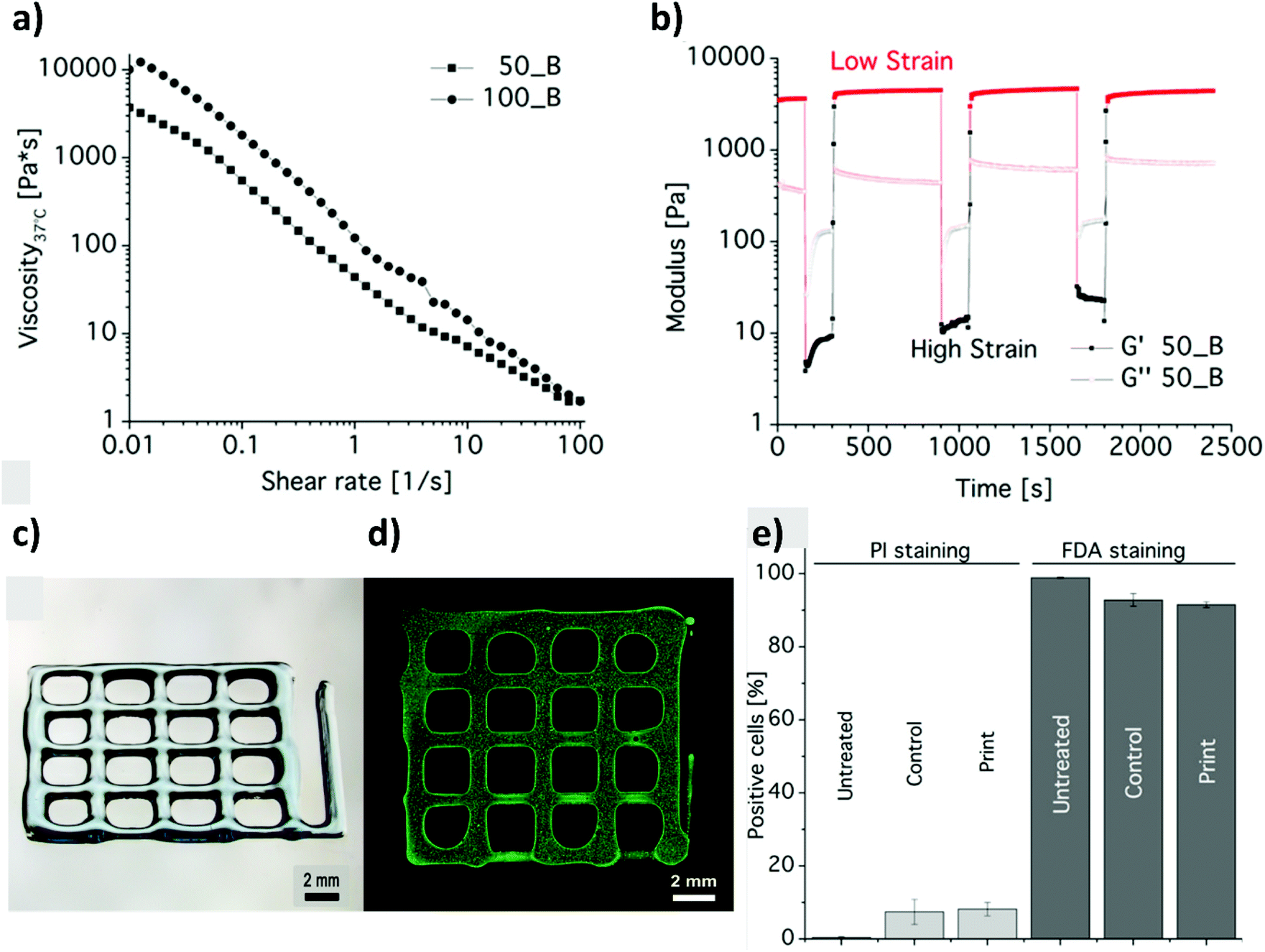 | ||
| Fig. 4 (a) Flow curves obtained from shear rate sweep tests of 20 wt% diblock copolymer solutions at 37 °C, revealing the shear thinning nature of PnPrOZI50-b-PMOXA50 (50_B) and PnPrOZI100-b-PMOXA100 (100_B). (b) Oscillatory time sweep tests of 20 wt% PnPrOZI50-b-PMOXA50 (50_B) solution at 37 °C and at an angular frequency of 10 rad s−1 under alternating strains (a low strain of 0.5% and a high strain of 150% that simulate the large shear deformation during the extrusion process), showing a rapid and complete shear recovery. (c) Light microscopy image of a 3D printed grid composed of orthogonal stacks of hydrogel strands with a base area of 12 × 12 mm2. (d) Cell-loaded constructs. (e) Quantification of cell viability by FACS analysis to reveal the influence of the printing process on the viability of fibroblasts. “Untreated” represents the cells just in the medium, and “control” corresponds to the cells that were re-dispersed in the bioink but not printed. “Print” refers to the cell viability measured 24 h post printing. Reproduced with permission from Lorson et al.56 Copyright 2017 American Chemical Society. | ||
Despite the relatively high PnPrOZI-b-PMOXA concentration (20 wt%) used during printing, complex 3D shapes could not be obtained, primarily due to the relatively low yield point of the bioink, which caused the deposited material to flow after the extrusion process and compromised the shape fidelity of the resulting printed grids (Fig. 4c and d). It is also important to emphasize that a high polymer content within bioinks might induce clogging of the nozzle tip during extrusion, leading to a more confined environment for the encapsulated cells as a result of macromolecular crowding.58,59 Relevantly, this phenomenon could adversely affect cell viability, proliferation and migration.
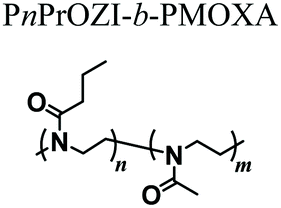
In order to overcome this inconvenience, in a subsequent work the same authors complemented the PnPrOZI-b-PMOXA with clay minerals with the aim of improving the printability and viscoelastic properties of the entire bioink.60 The addition of LAPONITE® XLG in the form of disk-shaped platelets did not critically affect the thermoresponsive properties and cytocompatibility of the copolymer mixture. However, it significantly improved its rheological properties, including shear thinning, zero-shear viscosity, G′ and yield stress, finally promoting the printability of the bioink.
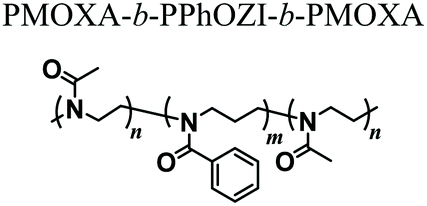
Interestingly, upon replacing PnPrOZI-b-PMOXA with an ABA triblock copolymer including PMOXA (A) and poly(2-phenyl-2-oxazine) (PPhOZI) (B), the bioink underwent an inverse and reversible gelation below a critical temperature, forming a reversible nanoscale wormlike network.54 The resulting hydrogel could reach G′ values of up to 110 kPa, while retaining shear thinning and rapid self-healing properties, and it could be efficiently 3D printed. The reversible gelation of PMOXA-b-PPhOZI-b-PMOXA was exploited to generate sacrificial supports to assist the 3D printing of polymeric matrices that are intrinsically difficult to create with sufficient resolution, such as alginate structures. A mixture of PMOXA-b-PPhOZI-b-PMOXA and alginate underwent gelation when subjected to relatively low temperature, enabling 3D printing with excellent shape fidelity. The subsequent incubation of the printed hybrid structure in CaCl2 led to alginate cross-linking. Final heating up to 37 °C resulted in the dissolution of the sacrificial triblock-copolymer matrix, and the formation of an intact alginate scaffold with the desired structure.
Alternative bioinks characterized by superior mechanical properties relied on the formation of double-network (DN) hydrogels, comprising an enzymatically cross-linked PEOXA network and an alginate hydrogel, which is cross-linked through reversible, ionic interactions (Fig. 5a and c).61 In particular, the primary, PEOXA-based network was densely and covalently cross-linked, providing elasticity to the entire material, while the secondary, alginate-based hydrogel was embedded in the former, and contained sparsely non-covalent cross-links that granted reversible and dissipative properties.62
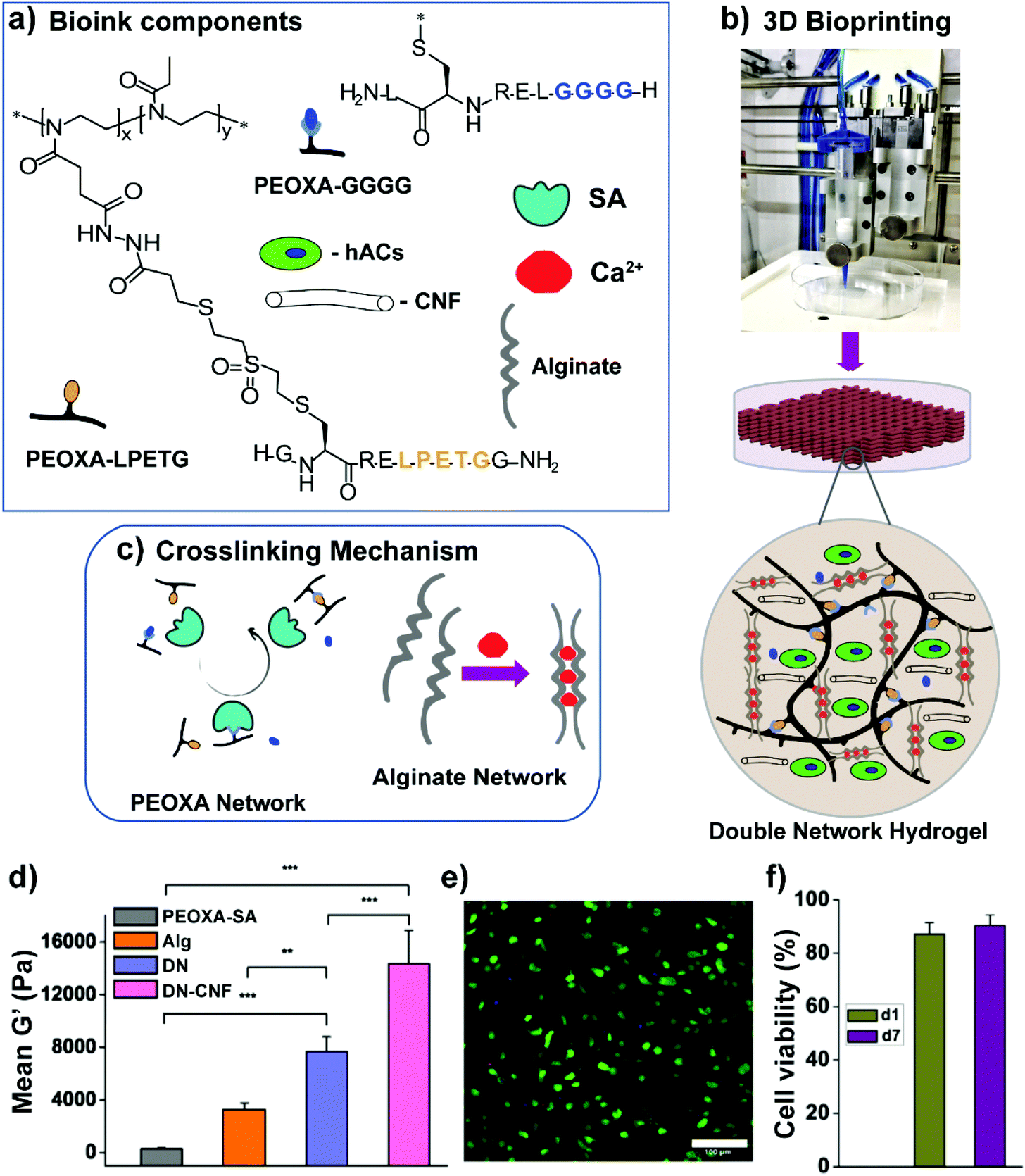 | ||
| Fig. 5 (a) Bioink components, including PEOXA-peptide conjugates, alginate, Sortase A (SA), Ca2+ ions, and hACs. (b) Extrusion-based 3D bioprinting process. (c) SA-mediated and ionic crosslinking mechanism to form PEOXA and alginate networks, respectively. (d) Mean G′ at equilibrium (recorded after 60 min) for different hydrogels. (e) Two-photon microscopy image showing hACs stained with calcein AM (live cells in green) and propidium iodide (dead cells in red) encapsulated in 3D bioprinted constructs 1 day post-bioprinting, as maximum intensity projections over 100 μm. Scale bar = 100 μm. (f) Quantification of hAC viability within 3D bioprinted grids 1 day (d1) and 7 days (d7) post-printing; SD n = 3 for all conditions. Reproduced with permission from Trachsel et al.61 Copyright 2019 American Chemical Society. | ||
PEOXA/alginate DNs showed G′ values of up to 8 kPa, reaching values more than twofold higher than the combined values of G′ obtained on the single constituting networks (Fig. 5d). Following the addition of a relatively low content of cellulose nanofibres (CNF), the DN-hydrogel formulations became shear thinning and shear-recoverable bioinks, gaining the rheological properties necessary for bioprinting. The resulting 3D printed constructs featured dimensionally stable grids with good shape fidelity, while human auricular chondrocytes loaded into the bioink showed excellent viability (90 ± 2%) both 1 and 7 days after printing (Fig. 5e and f).
In addition to extrusion-based 3D bioprinting, PAOXA-based formulations have recently been employed as inks in other 3D-printing techniques. In 2014, Groll and coworkers applied PEOXA as a starting material to fabricate 3D fibrous scaffolds by melt electrowriting (MEW).63 This technique has recently gained increasing attention, and represents a hybrid fabrication technology, having features in common with solution electrospinning and extrusion-based 3D printing.64,65 MEW is a solvent-free technique that enables the fabrication of 3D fibrous structures with typical fibre diameters ranging from 800 nm to 150 μm, which are much lower than those achievable by extrusion-based bioprinting. Unlike bioprinting, MEW is a cell-free manufacturing process, where cells are typically seeded onto the hydrated fibrous material following manufacturing.
Groll et al. systematically investigated the key parameters for MEW, such as heating temperature, feeding pressure, acceleration voltage, and collector distance (Fig. 6b). By heating PEOXA to 220 °C (well above its Tg), and by applying an acceleration voltage of up to 7 kV, a sufficient flow of the molten polymer was attained, generating fibres with diameters ranging from 8 to ∼140 μm. Digital control over the translating collector finally enabled the precise positioning of the fibres, generating microstructured 3D scaffolds with excellent resolution.
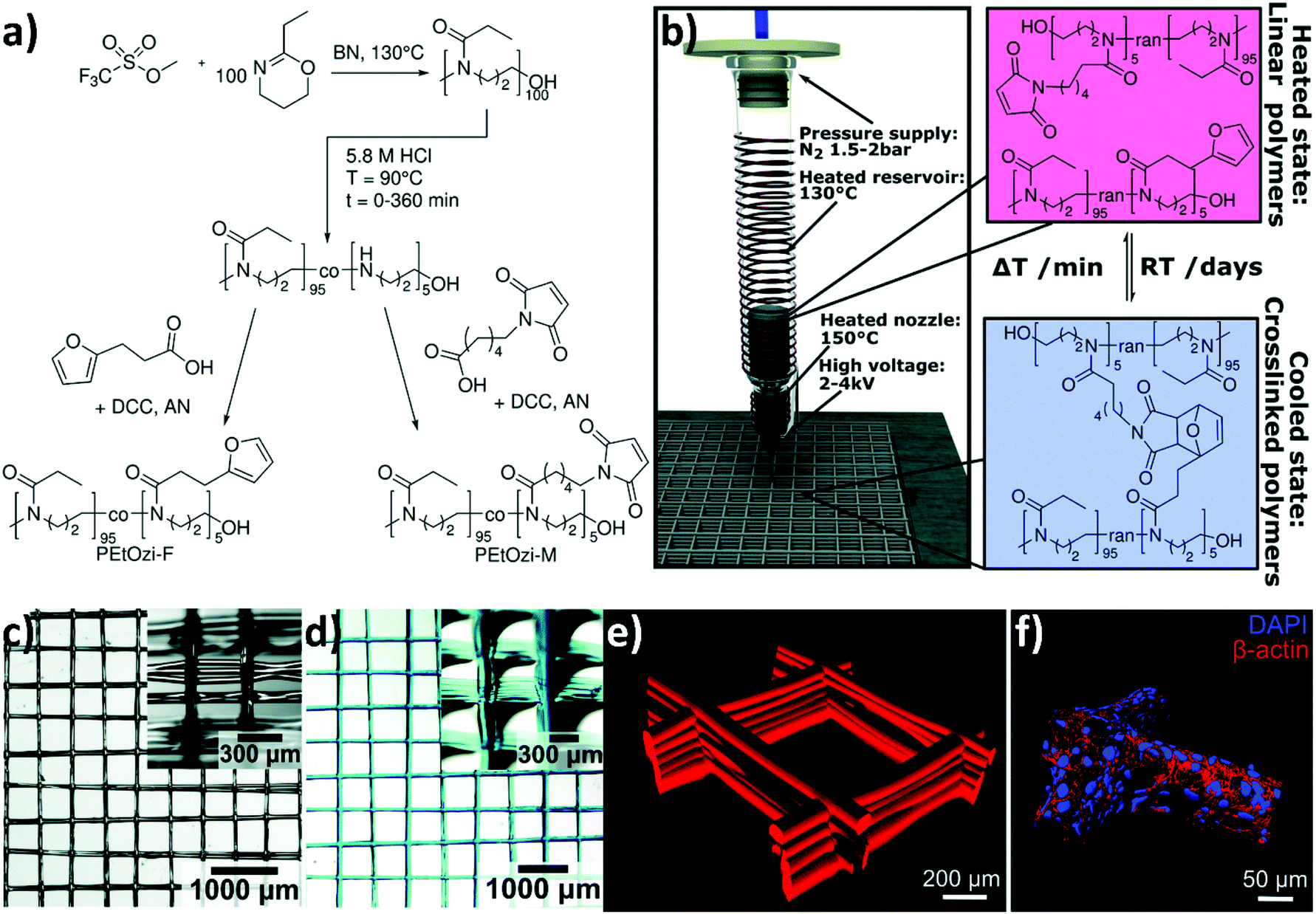 | ||
Fig. 6 (a) The CROP of 2-ethyl-2-oxazine initiated with methyl trifluoromethanesulfonate and terminated with an excess of KOH in methanol to yield poly(2-ethyl-2-oxazine) (PEOzi) homopolymers. Subsequently, the partial acid hydrolysis of PEOzi affords statistical poly(2-ethyl-2-oxazine-co-propylene imine) copolymers followed by the reaction of furan or maleimide moieties with the secondary amines yielding furan and maleimide functionalized PEOzi, i.e., PEOzi-F and PEOzi-M, respectively. (DCC: dicyclohexylcarbodiimide, AN: acetonitrile, and BN: benzonitrile). (b) Schematic illustration of the MEW setup, processing conditions and the reversible DA equilibrium, allowing liquefaction and melt processing at elevated temperatures followed by chemical crosslinking upon cooling. (c) Stereomicroscopic image of the MEW printed scaffold in the dry state and (d) swollen state, which was hydrated and stained with DY-647P1-maleimide for improved visualization. Insets show the scaffolds from a tilted angle to better visualize the fibre morphology and stacking. (e) Confocal microscopy image of a fluorescently labelled hydrogel scaffold with 500 μm fibre spacing. (f) Human embryonic kidney cells were grown on hydrogel scaffolds functionalized with the RGD adhesion peptide (NH2–CGGG![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif) ![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) ![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) S–COOH). Following four days of growth at the scaffold, the cells were stained with cytoskeletal marker protein β-actin (red) and DAPI (nucleus in blue), which allowed the 3D surface-reconstruction of human embryonic kidney cells attached to the scaffold. Reproduced with permission from Nahm et al.66 Copyright 2020 Royal Society of Chemistry. S–COOH). Following four days of growth at the scaffold, the cells were stained with cytoskeletal marker protein β-actin (red) and DAPI (nucleus in blue), which allowed the 3D surface-reconstruction of human embryonic kidney cells attached to the scaffold. Reproduced with permission from Nahm et al.66 Copyright 2020 Royal Society of Chemistry. | ||
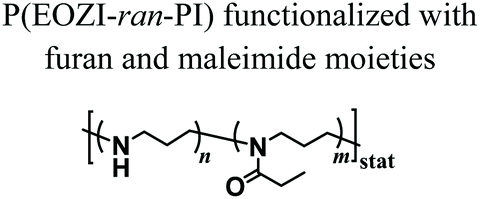
Nahm and coworkers recently expanded the functional character of the materials fabricated using MEW by exploiting the cross-linking of the polymers that constitute the fibres, yielding porous hydrogel-based structures.66 Poly(2-ethyl-2-oxazine) PEOZI—a hydrophilic polymer with a relatively low Tg—was partially hydrolyzed to obtain statistical copolymers poly(2-ethyl-2-oxazine-ran-propylene imine) (P(EOZI-ran-PI)) presenting functionalisable propylene imine units, which were derivatised with furan and maleimide moieties prior to the MEW process (Fig. 6a). The molten, electrified jet was liquid at high temperature but spontaneously cross-linked via Diels–Alder (DA) reaction between the functional side chains during cooling while leaving the nozzle (Fig. 6b). This MEW process enabled the fabrication of micro-periodic fibrous structures with a well-defined diameter, spacing, and number of layers. The DA click reactions enabled cross-linking between fibres from different layers at their junctions, obtaining dimensionally stable scaffolds that could be hydrated in aqueous or cell culture media without dissolving or distorting the entire architecture (Fig. 6c and d). Moreover, the obtained hydrogels retained unreacted furan and maleimide groups available for further functionalization, as was demonstrated by linking different fluorophores and/or peptide units. Preliminary experiments to assess the biological compatibility of arginine–glycine–aspartic acid (RGD)-bearing PEOZI constructs revealed a rapid settlement by human embryonic kidney cells, and an overall low cytotoxicity of the polymeric support (Fig. 6e and f).
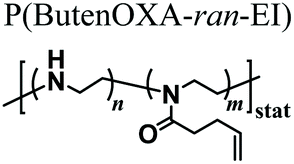
MEW could be additionally applied to fabricate a sacrificial template based on structured fibres, which could be exploited to generate micro-channels with a controlled structure within a hydrogel. This approach was exploited by the group of Dargaville, who used 3D printed poly(ε-caprolactone) (PCL) fibres obtained by MEW to fabricate micro-sized channels within the hydrogels based on P(EOXA-ran-ButenOXA).67 Relevantly, this strategy could be exploited to fabricate microfluidic devices showing a precisely tunable structure and morphology, or generate hydrogel supports with interconnected channels to mimic tissues with organized vasculature.
Following simpler although still robust approaches, fibrous supports based on the PAOXA hydrogels were fabricated by Sanyal and coworkers, exploiting solution electrospinning.68 In particular, the P(EOXA-ran-ButenOXA) mixed with a tetra-functional thiol cross-linker and photoinitiator was electrospun and simultaneously irradiated with UV light. This produced cross-linked fibrous structures that could retain their shapes when swollen within aqueous environments.
When compared to MEW, solution electrospinning only enables the fabrication of structures with randomly distributed fibres. Stubbe et al. performed electrospinning of PEOXA fibres starting from aqueous solutions, and systematically investigated the effect of molar mass, dispersity (Đ) and concentration of the polymer on the morphology and dimensions of the obtained constructs.69 The cross-linking of the electrospun PEOXA fibres could be achieved by applying selenol-modified PEOXA, and triggering the formation of dynamic diselenide bonds.70 Alternatively, the application of benzophenone-modified PEOXA enabled temporal control over the cross-linking process by exploiting UV irradiation.71 In another report, random copolymers poly(EOXA-ran-ethylenimine), obtained by the partial hydrolysis of PEOXA, were used for aqueous electrospinning, subsequently functionalised with glutaraldehyde vapor, and finally cross-linked to yield structurally stable fibrous constructs.72
Finally, it is worth mentioning two-photon polymerization (2PP) as an emerging additive manufacturing method which has recently enabled the precise fabrication of structured PAOXA hydrogels. 2PP exploits a near-infrared ultrashort-pulsed laser to excite photoinitiators in a precise way and in a confined space to a two-photon state that triggers the polymerization of monomers in solution. This enables spatiotemporal control over the 3D structuring of scaffolds, also in the presence of cells.73–75
Wloka et al. have recently performed 2PP of PEOXA bis(acrylates) (PEOXA-DA) combined with a newly synthesized water-soluble photoinitiator, successfully achieving the formation of 3D-structured PEOXA hydrogels with very high spatial resolution and accuracy.73 These preliminary experiments revealed the great potential of 2PP in the fabrication of supports for tissue engineering.
Conclusions
During the past two decades, the application of PAOXAs has been extended to a variety of bio-related processes, including the design of drug carriers,5,52,76 polymer–protein conjugates,77 polymer–drug conjugates,78 and the fabrication of coatings for biomaterials and sensors.37,79–81 More recently, PAOZIs have been successfully introduced in similar applications, especially as components for drug delivery formulations53 and as surface modifiers with excellent bioinert properties,6 emerging as alternative or complementary materials to PAOXAs. However, the use of these polymers in the formulations of hydrogels and carrier materials for biofabrication is still limited, although the pioneering examples reported in this short review and summarized in Table 1 prove that it holds great potential.| Polymer composition | Type of crosslinking | Fabrication/processing | Application | Ref. |
|---|---|---|---|---|
| Abbreviations: MOXA: 2-methyl-2-oxazoline, DecEnOXA: 2-(dec-9-enyl)-2-oxazoline, DTT: dithiothreitol, ButenOXA: 2-butenyl-2-oxazoline, BocABuOXA: 2-(4-((tert-butoxycarbonyl)amino)butyl)-2-oxazoline, ABuOXA: 2-aminobutyl-2-oxazoline, PE: polyethylene, PP: polypropylene, EI: ethylene imine, MCEOXA: 2-methylcarboxyethyl-2-oxazoline, SA: sortase A, PnPrOZI: poly(2-n-propyl-2-oxazine), PPhOZI: poly(2-phenyl-2-oxazine), EOZI: 2-ethyl-2-oxazine, PI: propylene imine, MEW: melt electrowriting, DA: Diels–Alder, and PCL: poly(ε-caprolactone). | ||||
|
Thiol–ene reaction using DTT and CRGDSCG for cross-linking | UV irradiation | Human dermal fibroblasts 3D culture | 33 |
|
Thiol–ene reaction using DTT for cross-linking and to couple CRGDSG peptide | UV irradiation | Human primary stromal cell 3D culture | 38 |
|
NaIO4-mediated oxidation of catechols to quinones that react with each other forming crosslinks | — | Injectable tissue adhesive hydrogel for articular cartilage defect treatment | 39 |
|
The addition of epichlorohydrin that reacts with the amines on the polymer resulting in crosslinking | — | Cationic hydrogels for reversible DNA complexing | 49 |
|
Supported structures were generated by soaking porous PE or PP filter materials in a copolymer/epichlorhydrin solution that cross-linked upon heating | Thermal treatment | Matrix supported hydrogels for DNA binding and release upon change in the temperature or pH | 50 |
|
Hydrogels formed by thiol–ene cross-linking using dithiols | UV irradiation | DNA binding and release | 51 |
|
Hydrogels formed by thiol–ene cross-linking | UV irradiation | Epicardial placement of mesenchymal stem cells for myocardial repair | 40 |
|
Hydrogels formed by enzyme (SA)-mediated cross-linking | — | Injectable hydrogel for 3D cell culture of human articular chondrocytes | 46 |
|
Thermogelation after extrusion yields hydrogels | Extrusion-based 3D printing | 3D bioprinting of fibroblast-loaded hydrogel grids | 56 |
|
Thermogelation after extrusion yields hydrogels | Extrusion-based 3D printing | Improving the printability of the thermoresponsive ink by the addition of clay minerals (LAPONITE® XLG) | 60 |
|
Inverse thermogelation of the amphiphilic triblock copolymers | Extrusion-based 3D printing | Sacrificial support material by 3D printing of alginate | 54 |
|
SA-mediated and ionic cross-linking of PEOXA and alginate, respectively, post extrusion forming hydrogels | Extrusion-based 3D printing | 3D bioprinting of human auricular chondrocytes-loaded constructs | 61 |
|
Spontaneous cross-linking via dynamic DA reaction post processing | MEW | Fabrication of micro-periodic fibrous scaffolds for human embryonic kidney cells | 66 |
|
MEW of PCL fibres acting as a sacrificial template within photo-cross-linked PEOXA hydrogels | UV irradiation | Fabrication of hydrogel supports with interconnected channels that mimic organized vasculature | 67 |
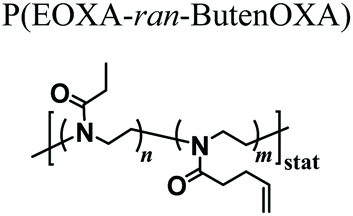
|
Thiol–ene cross-linking during electrospinning | Solution electrospinning and UV irradiation | Hydrogels from cross-linked fibrous structures | 68 |
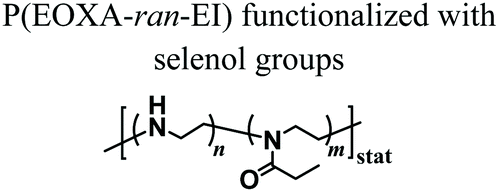
|
Formation of diselenide bonds between selenol-modified P(EOXA-ran-EI) | Solution electrospinning | Degradable diselenide-cross-linked nanofibres | 70 |
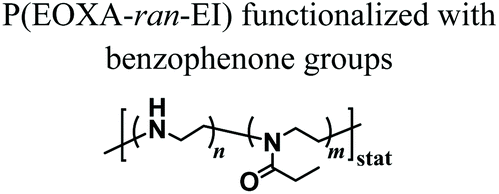
|
Radical addition of benzophenone to neighboring polymer chains forming cross-links | Solution electrospinning UV irradiation | Fabrication of water-stable nanofibres using temporally controlled UV crosslinking | 71 |
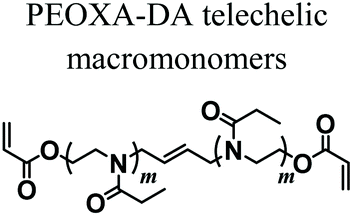
|
2PP | Near-infrared laser irradiation | 3D micro-structured PEOXA hydrogels | 73 |
In all of these applications, PAOXAs and PAOZIs show several advantages in relation to PEG and its derivatives, such as an easily tunable chemical composition and an improved stability within physiological environments. In addition, according to a variety of data obtained from in vivo experiments, PAOXAs show good biocompatibility when dissolved in solutions and in the form of nanoparticles (NPs),82,83 while in vitro biocompatibility was demonstrated for polymer networks and coatings.84,85
Despite increasing interest and promising studies, the successful translation of PAOXA (or PAOZI)-based formulations into clinical therapies has not yet been accomplished. We believe that the access to PAOXA-based materials in clinics would certainly boost their application in materials science and would trigger the interest of more researchers in including this class of polymers in their formulations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Prof. Katharina Maniura (EMPA) for scientific support.References
- T. Lorson, M. M. Lübtow, E. Wegener, M. S. Haider, S. Borova, D. Nahm, R. Jordan, M. Sokolski-Papkov, A. V. Kabanov and R. Luxenhofer, Biomaterials, 2018, 178, 204–280 CrossRef CAS PubMed.
- T. R. Dargaville, B. G. Hollier, A. Shokoohmand and R. Hoogenboom, Cell Adhes. Migr., 2014, 8, 88 CrossRef PubMed.
- V. R. De La Rosa, J. Mater. Sci. Mater. Med., 2014, 25, 1211–1225 CrossRef CAS PubMed.
- O. Sedlacek, B. D. Monnery, S. K. Filippov, R. Hoogenboom and M. Hruby, Macromol. Rapid Commun., 2012, 33, 1648–1662 CrossRef CAS PubMed.
- R. Luxenhofer, Y. Han, A. Schulz, J. Tong, Z. He, A. V. Kabanov and R. Jordan, Macromol. Rapid Commun., 2012, 33, 1613–1631 CrossRef CAS PubMed.
- G. Morgese, B. Verbraeken, S. N. Ramakrishna, Y. Gombert, E. Cavalli, J.-G. Rosenboom, M. Zenobi-Wong, N. D. Spencer, R. Hoogenboom and E. M. Benetti, Angew. Chem., Int. Ed., 2018, 57, 11667–11672 CrossRef CAS PubMed.
- S. N. Ramakrishna, G. Morgese, M. Zenobi-Wong and E. M. Benetti, Macromolecules, 2019, 52, 1632–1641 CrossRef CAS.
- G. Morgese, E. Cavalli, J. G. Rosenboom, M. Zenobi-Wong and E. M. Benetti, Angew. Chem., Int. Ed., 2018, 57, 1621–1626 CrossRef CAS PubMed.
- G. Morgese, S. N. Ramakrishna, R. Simic, M. Zenobi-Wong and E. M. Benetti, Biomacromolecules, 2018, 19, 680–690 CrossRef CAS PubMed.
- G. Morgese, Y. Gombert, S. N. Ramakrishna and E. M. Benetti, ACS Appl. Mater. Interfaces, 2018, 10, 41839–41848 CrossRef CAS PubMed.
- W. Yan, M. Divandari, J.-G. Rosenboom, S. Ramakrishna, N. D. Spencer, G. Morgese, E. M. Benetti and L. Trachsel, Polym. Chem., 2018, 9, 2580–2589 RSC.
- B. Pidhatika, J. Möller, E. M. Benetti, R. Konradi, E. Rakhmatullina, A. Mühlebach, R. Zimmermann, C. Werner, V. Vogel and M. Textor, Biomaterials, 2010, 31, 9462–9472 CrossRef CAS PubMed.
- R. Konradi, C. Acikgoz and M. Textor, Macromol. Rapid Commun., 2012, 33, 1663–1676 CrossRef CAS PubMed.
- R. Konradi, B. Pidhatika, A. Mühlebach and M. Textor, Langmuir, 2008, 24, 613–616 CrossRef CAS PubMed.
- M. Bauer, C. Lautenschlaeger, K. Kempe, L. Tauhardt, U. S. Schubert and D. Fischer, Macromol. Biosci., 2012, 12, 986–998 CrossRef CAS PubMed.
- M. Bauer, S. Schroeder, L. Tauhardt, K. Kempe, U. S. Schubert and D. Fischer, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1816–1821 CrossRef CAS.
- M. N. Leiske, A.-K. Trützschler, S. Armoneit, P. Sungur, S. Hoeppener, M. Lehmann, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2017, 5, 9102–9113 RSC.
- J. Kronek, Z. Kroneková, J. Lustoň, E. Paulovičová, L. Paulovičová and B. Mendrek, J. Mater. Sci. Mater. Med., 2011, 22, 1725–1734 CrossRef CAS PubMed.
- P. Goddard, L. E. Hutchinson, J. Brown and L. J. Brookman, J. Controlled Release, 1989, 10, 5–16 CrossRef CAS.
- F. C. Gaertner, R. Luxenhofer, B. Blechert, R. Jordan and M. Essler, J. Controlled Release, 2007, 119, 291–300 CrossRef CAS PubMed.
- M. C. Woodle, C. M. Engbers and S. Zalipsky, Bioconjugate Chem., 1994, 5, 493–496 CrossRef CAS PubMed.
- B. L. Tardy, J. J. Richardson, V. Nithipipat, K. Kempe, J. Guo, K. L. Cho, M. A. Rahim, H. Ejima and F. Caruso, Biomacromolecules, 2019, 20, 1421–1428 CrossRef CAS PubMed.
- T.-L. Cheng, P.-Y. Wu, M.-F. Wu, J.-W. Chern and S. R. Roffler, Bioconjugate Chem., 1999, 10, 520–528 CrossRef CAS PubMed.
- R. P. Garay, R. El-Gewely, J. K. Armstrong, G. Garratty and P. Richette, Expert Opin. Drug Delivery, 2012, 9, 1319–1323 CrossRef CAS PubMed.
- H. Schellekens, W. E. Hennink and V. Brinks, Pharm. Res., 2013, 30, 1729–1734 CrossRef CAS PubMed.
- C.-J. Chang, C.-H. Chen, B.-M. Chen, Y.-C. Su, Y.-T. Chen, M. S. Hershfield, M.-T. M. Lee, T.-L. Cheng, Y.-T. Chen, S. R. Roffler and J.-Y. Wu, Nat. Commun., 2017, 8, 522 CrossRef PubMed.
- G. Pasut and F. M. Veronese, Adv. Drug Delivery Rev., 2009, 61, 1177–1188 CrossRef CAS PubMed.
- R. Webster, V. Elliott, B. K. Park, D. Walker, M. Hankin and P. Taupin, in PEGylated Protein Drugs: Basic Science and Clinical Applications, ed. F. M. Veronese, Birkhäuser Basel, Basel, 2009, pp. 127–146 Search PubMed.
- B. Pidhatika, M. Rodenstein, Y. Chen, E. Rakhmatullina, A. Mühlebach, C. Acikgöz, M. Textor and R. Konradi, Biointerphases, 2012, 7, 1 CrossRef CAS PubMed.
- Y. Chen, B. Pidhatika, T. von Erlach, R. Konradi, M. Textor, H. Hall and T. Lühmann, Biointerphases, 2014, 9, 031003 CrossRef CAS PubMed.
- B. Obermeier, F. Wurm, C. Mangold and H. Frey, Angew. Chem., Int. Ed., 2011, 50, 7988–7997 CrossRef CAS PubMed.
- C. Mangold, F. Wurm and H. Frey, Polym. Chem., 2012, 3, 1714–1721 RSC.
- B. L. Farrugia, K. Kempe, U. S. Schubert, R. Hoogenboom and T. R. Dargaville, Biomacromolecules, 2013, 14, 2724–2732 CrossRef CAS PubMed.
- A. M. Kelly, A. Hecke, B. Wirnsberger and F. Wiesbrock, Macromol. Rapid Commun., 2011, 32, 1815–1819 CrossRef CAS PubMed.
- A. M. Kelly and F. Wiesbrock, Macromol. Rapid Commun., 2012, 33, 1632–1647 CrossRef CAS PubMed.
- K. P. Luef, C. Petit, B. Ottersbock, G. Oreski, F. Ehrenfeld, B. Grassl, S. Reynaud and F. Wiesbrock, Eur. Polym. J., 2017, 88, 701–712 CrossRef CAS PubMed.
- N. Ahmad, B. Colak, M. J. Gibbs, D. W. Zhang, J. E. Gautrot, M. Watkinson, C. R. Becer and S. Krause, Biomacromolecules, 2019, 20, 2506–2514 CrossRef CAS PubMed.
- D. J. Van Der Heide, B. Verbraeken, R. Hoogenboom, T. R. Dargaville and D. K. Hickey, Biomater. Tissue Technol., 2017, 1, 1–5 Search PubMed.
- O. Berberich, J. Blöhbaum, S. Hölscher-Doht, R. H. Meffert, J. Teßmar, T. Blunk and J. Groll, J. Ind. Eng. Chem., 2019, 80, 757–769 CrossRef CAS.
- Y. You, K. Kobayashi, B. Colak, P. Luo, E. Cozens, L. Fields, K. Suzuki and J. Gautrot, Biomaterials, 2020, 120356 Search PubMed.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- S. A. Fisher, A. E. G. Baker and M. S. Shoichet, J. Am. Chem. Soc., 2017, 139, 7416–7427 CrossRef CAS PubMed.
- S. J. Stohs, J. Basic Clin. Physiol. Pharmacol., 1995, 6, 205 CAS.
- S. J. Bryant, C. R. Nuttelman and K. S. Anseth, J. Biomater. Sci., Polym. Ed., 2000, 11, 439 CrossRef CAS PubMed.
- L. S. Moreira Teixeira, J. Feijen, C. A. van Blitterswijk, P. J. Dijkstra and M. Karperien, Biomaterials, 2012, 33, 1281–1290 CrossRef CAS PubMed.
- L. Trachsel, N. Broguiere, J.-G. Rosenboom, M. Zenobi-Wong and E. M. Benetti, J. Mater. Chem. B, 2018, 6, 7568–7572 RSC.
- N. Broguiere, F. A. Formica, G. Barreto and M. Zenobi-Wong, Acta Biomater., 2018, 77, 182–190 CrossRef CAS PubMed.
- C. S. Theile, G. Bozkurt, L. Kundrat, A. E. M. Blom and H. L. Ploegh, Nat. Protoc., 2013, 8, 1787 CrossRef PubMed.
- M. Hartlieb, D. Pretzel, K. Kempe, C. Fritzsche, R. M. Paulus, M. Gottschaldt and U. S. Schubert, Soft Matter, 2013, 9, 4693–4704 RSC.
- M. Hartlieb, D. Pretzel, C. Englert, M. Hentschel, K. Kempe, M. Gottschaldt and U. S. Schubert, Biomacromolecules, 2014, 15, 1970–1978 CrossRef CAS PubMed.
- C. Englert, L. Tauhardt, M. Hartlieb, K. Kempe, M. Gottschaldt and U. S. Schubert, Biomacromolecules, 2014, 15, 1124–1131 CrossRef CAS PubMed.
- O. Sedlacek and R. Hoogenboom, Adv. Ther., 2020, 3, 1900168 CrossRef.
- M. M. Lübtow, L. Hahn, M. S. Haider and R. Luxenhofer, J. Am. Chem. Soc., 2017, 139, 10980–10983 CrossRef PubMed.
- L. Hahn, M. Maier, P. Stahlhut, M. Beudert, V. Flegler, S. Forster, A. Altmann, F. Töppke, K. Fischer, S. Seiffert, B. Böttcher, T. Lühmann and R. Luxenhofer, ACS Appl. Mater. Interfaces, 2020, 12, 12445–12456 CrossRef PubMed.
- Z. Kroneková, T. Lorson, J. Kronek and R. Luxenhofer, ChemRxiv, 2018 DOI:10.26434/chemrxiv.5793990.v1.
- T. Lorson, S. Jaksch, M. M. Lübtow, T. Jüngst, J. Groll, T. Lühmann and R. Luxenhofer, Biomacromolecules, 2017, 18, 2161 CrossRef CAS PubMed.
- M. M. Bloksma, R. M. Paulus, H. P. C. van Kuringen, F. van der Woerdt, H. M. L. Lambermont-Thijs, U. S. Schubert and R. Hoogenboom, Macromol. Rapid Commun., 2012, 33, 92–96 CrossRef CAS PubMed.
- D. Chimene, R. Kaunas and A. K. Gaharwar, Adv. Mater., 2020, 32, 1902026 CrossRef CAS PubMed.
- A. Parak, P. Pradeep, L. C. du Toit, P. Kumar, Y. E. Choonara and V. Pillay, Drug Discovery Today, 2019, 24, 198–205 CrossRef CAS PubMed.
- C. Hu, L. Hahn, M. Yang, A. Altmann, P. Stahlhut, J. Groll and R. Luxenhofer, J. Mater. Sci., 2021, 56, 691–705 CrossRef CAS.
- L. Trachsel, C. Johnbosco, T. Lang, E. M. Benetti and M. Zenobi-Wong, Biomacromolecules, 2019, 20, 4502–4511 CrossRef CAS PubMed.
- K. Yasuda, J. P. Gong, Y. Katsuyama, A. Nakayama, Y. Tanabe, E. Kondo, M. Ueno and Y. Osada, Biomaterials, 2005, 26, 4468 CrossRef CAS PubMed.
- G. Hochleitner, J. F. Hümmer, R. Luxenhofer and J. Groll, Polymer, 2014, 55, 5017–5023 CrossRef CAS.
- P. D. Dalton, Curr. Opin. Biomed. Eng., 2017, 2, 49–57 CrossRef.
- A. Hrynevich, B. Ş. Elçi, J. N. Haigh, R. McMaster, A. Youssef, C. Blum, T. Blunk, G. Hochleitner, J. Groll and P. D. Dalton, Small, 2018, 14, 1800232 CrossRef PubMed.
- D. Nahm, F. Weigl, N. Schaefer, A. Sancho, A. Frank, J. Groll, C. Villmann, H. W. Schmidt, P. D. Dalton and R. Luxenhofer, Mater. Horiz., 2020, 7, 928–933 RSC.
- J. N. Haigh, Y. M. Chuang, B. Farrugia, R. Hoogenboom, P. D. Dalton and T. R. Dargaville, Macromol. Rapid Commun., 2016, 37, 93–99 CrossRef CAS PubMed.
- O. I. Kalaoglu-Altan, B. Verbraeken, K. Lava, T. N. Gevrek, R. Sanyal, T. Dargaville, K. De Clerck, R. Hoogenboom and A. Sanyal, ACS Macro Lett., 2016, 5, 676–681 CrossRef CAS.
- B. Stubbe, Y. Li, M. Vergaelen, S. Van Vlierberghe, P. Dubruel, K. De Clerck and R. Hoogenboom, Eur. Polym. J., 2017, 88, 724–732 CrossRef CAS.
- Y. Li, M. Vergaelen, X. Pan, F. E. Du Prez, R. Hoogenboom and K. De Clerck, Macromolecules, 2018, 51, 6149–6156 CrossRef CAS.
- Y. Li, M. Vergaelen, E. Schoolaert, R. Hoogenboom and K. De Clerck, Eur. Polym. J., 2019, 112, 24–30 CrossRef CAS.
- O. I. Kalaoglu-Altan, Y. Li, R. McMaster, A. Shaw, Z. Hou, M. Vergaelen, R. Hoogenboom, T. R. Dargaville and K. De Clerck, Eur. Polym. J., 2019, 120, 109218 CrossRef CAS.
- T. Wloka, S. Czich, M. Kleinsteuber, E. Moek, C. Weber, M. Gottschaldt, K. Liefeith and U. S. Schubert, Eur. Polym. J., 2020, 122, 109295 CrossRef CAS.
- A. Marino, C. Filippeschi, V. Mattoli, B. Mazzolai and G. Ciofani, Nanoscale, 2015, 7, 2841–2850 RSC.
- J.-F. Xing, M.-L. Zheng and X.-M. Duan, Chem. Soc. Rev., 2015, 44, 5031–5039 RSC.
- T. X. Viegas, M. D. Bentley, J. M. Harris, Z. Fang, K. Yoon, B. Dizman, R. Weimer, A. Mero, G. Pasut and F. M. Veronese, Bioconjugate Chem., 2011, 22, 976–986 CrossRef CAS PubMed.
- O. Sedlacek, V. R. de la Rosa and R. Hoogenboom, Eur. Polym. J., 2019, 120, 109246 CrossRef CAS.
- M. Schmidt, L. K. Bast, F. Lanfer, L. Richter, E. Hennes, R. Seymen, C. Krumm and J. C. Tiller, Bioconjugate Chem., 2017, 28, 2440–2451 CrossRef CAS PubMed.
- G. Morgese and E. M. Benetti, Eur. Polym. J., 2017, 88, 470–485 CrossRef CAS.
- L. Trachsel, M. Romio, S. N. Ramakrishna and E. M. Benetti, Adv. Mater. Interfaces, 2020, 2000943 CrossRef CAS.
- L. Tauhardt, K. Kempe, M. Gottschaldt and U. S. Schubert, Chem. Soc. Rev., 2013, 42, 7998–8011 RSC.
- R. Tavano, L. Gabrielli, E. Lubian, C. Fedeli, S. Visentin, P. Polverino De Laureto, G. Arrigoni, A. Geffner-Smith, F. Chen, D. Simberg, G. Morgese, E. M. Benetti, L. Wu, S. M. Moghimi, F. Mancin and E. Papini, ACS Nano, 2018, 12, 5834–5847 CrossRef CAS PubMed.
- D. Le, F. Wagner, M. Takamiya, I.-L. Hsiao, G. Gil Alvaradejo, U. Strähle, C. Weiss and G. Delaittre, Chem. Commun., 2019, 55, 3741–3744 RSC.
- M. Ryma, J. Blöhbaum, R. Singh, A. Sancho, J. Matuszak, I. Cicha and J. Groll, ACS Biomater. Sci. Eng., 2019, 5, 1509–1517 CrossRef CAS PubMed.
- F. Wang, P. Ren, K. V. Bernaerts, Y. Fu, W. Hu, N. Zhou and T. Zhang, ACS Appl. Bio Mater., 2020, 3, 5428–5437 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |