
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModerating cellular inflammation using 2-dimensional titanium carbide MXene and graphene variants†
Tochukwu
Ozulumba
 ab,
Ganesh
Ingavle
ac,
Yury
Gogotsi
d and
Susan
Sandeman
*a
ab,
Ganesh
Ingavle
ac,
Yury
Gogotsi
d and
Susan
Sandeman
*a
aSchool of Pharmacy and Biomolecular Sciences, University of Brighton, Brighton, BN2 4GJ, UK. E-mail: s.sandeman@brighton.ac.uk
bDepartment of Chemistry, University of Virginia, Charlottesville, VA 22904, USA
cSymbiosis Centre for Stem Cell Research, Symbiosis International University, Lavale, Pune-412115, India
dDepartment of Material Science and Engineering, and A. J. Drexel Nanomaterials Institute, Drexel University, Philadelphia, PA 19104, USA
First published on 8th January 2021
Abstract
The effective control of microbial and metabolically derived biological toxins which negatively impact physical health remains a key challenge for the 21st century. 2-Dimensional graphene and MXene nanomaterials are relatively new additions to the field of biomedical materials with superior external surface areas suited to adsorptive remediation of biological toxins. However, relatively little is known about their physiological interactions with biological systems and, to date, no comparative biological studies have been done. This study compares titanium carbide MXene (Ti3C2Tx) in multilayered and delaminated forms with graphene variants to assess the impact of variable physical properties on cellular inflammatory response to endotoxin stimulus. No significant impact on cell metabolism or induction of inflammatory pathways leading to cell death was observed. No significant increase in markers of blood cell activation and haemolysis occurred. Whilst graphene nanoplatelets (GNP), graphene oxide (GO) and Ti3C2Tx showed insignificant antibacterial activity towards Escherichia coli, silver nanoparticle-modified GO (GO-Ag) induced bacterial cell death and at a lower dose than silver nanoparticles. All nanomaterials significantly reduced bacterial endotoxin induced THP-1 monocyte IL-8, IL-6 and TNF-α cytokine production by >99%, >99% and >80% respectively, compared to control groups. This study suggests the utility of these nanomaterials as adsorbents in blood contacting medical device applications for removal of inflammatory cytokines linked to poor outcome in patients with life-threatening infection.
1 Introduction
Inflammation is described as the body's defensive response to infection and tissue injury.1,2 Normally, inflammation is tightly regulated by counteracting processes which ensure quick resolution following elimination of the causative pathogen or insult.1,2 However, dysregulation can occur leading to chronic inflammation, build-up of inflammatory mediators and potentially, tissue damage.1 Cytokines are the main drivers of the innate immune response. They include pro-inflammatory cytokines such as interleukin-6 (IL-6), tumour necrosis factor-α (TNF-α), interleukin-8 (IL-8) and interleukin-1β (IL-1β), and anti-inflammatory cytokines such as interleukin-10 (IL-10).3 The accumulation and/or excessive activity of cytokines (cytokine storm) have been linked to the progression of diseases such as liver failure,4 chronic kidney disease,1 sepsis,5 and non-healing wounds.6 The cytokine storm has also been associated with the mortality of patients suffering from SARS-CoV-2 infection (COVID-19), where elevated plasma levels of inflammatory cytokines such as IL-6 have been detected and linked to disease severity.7Modulating cytokine concentrations in blood has been investigated as a potential therapeutic approach for managing the cytokine overload that occurs in some chronic diseases.8 One of the broad-spectrum strategies used to address cytokine build-up is extracorporeal blood purification (EBP). EBP involves the passage of blood (or plasma) via an external purification device, for removal of toxins, and subsequent re-introduction of blood to the body, all via a circuit.9 In haemoperfusion, a common EBP approach, blood is passed through an extracorporeal system where it comes into contact with adsorbents that remove toxins through van der Waals forces, hydrophobic interactions, ionic, covalent or hydrogen bonds.8,9 Synthetic activated carbons,3 carbide derived carbons10 and polymer-based adsorbents11 with large surface areas and high internal nanoporosities have been developed for extracorporeal removal of endotoxins, cytokines and other toxins from blood. Extracorporeal systems based on magnetic separation12,13 and particle-based fluidised beds14,15 have also been developed. Reliance of adsorbents on internal porosity may limit available adsorptive surface area due to narrow pore necks and pore blockage by protein molecules which can slow kinetics and prolong sorption time.10,16 Nanomaterials, such as the 2-dimensional graphenes and MXenes, where adsorptive surface area is external, may overcome these particular limitations.
Graphene refers to a single layer of sp2 hybridized carbon atoms (the elementary subunit of graphite) which are compactly organised to form a honeycomb pattern.17 Graphene is hydrophobic and has a large surface area (theoretical limit of 2630 m2 g−1).18 Similar to activated carbon, graphene can mediate adsorption of biological toxins via hydrophobic interactions with hydrophobic domains in proteins.19 In addition, functionalisation of graphene with heteroatoms and metals enables selective adsorption of biological toxins20 and antimicrobial activity21 respectively. MXenes are a newer group of two-dimensional transition metal carbides and/or nitrides, discovered in 2011, which are derived from the selective etching of their corresponding MAX phases.22 They are represented by the formula, Mn+1XnTx, where M denotes the transition metal, X is carbon and/or nitrogen, Tx indicates the surface terminations (O, OH, F, etc.), n = 1, 2, 3 or 4 and x is the number of terminating groups.23 Apart from being the first synthesised MXene, Ti3C2Tx is also the most commonly studied one.22 Ti3C2Tx is characterised by a layered morphology,22 large surface area,24 tunable surface chemistry,22 hydrophilicity and a large negative charge on the surface.25
Like graphene, Ti3C2Tx has a significant and externally accessible surface area.24 This suggests faster adsorption kinetics for these materials over other adsorbents which may be limited by dependence on internal porosity. However, MXenes differ from graphene and its derivatives in terms of their surface chemistry and hydrophilicity which suggests that mechanisms other than hydrophobic interactions would drive their adsorption of biological toxins. We have previously reported that hydrophobic GNP demonstrated rapid and efficient adsorption of the pro-inflammatory cytokines, IL-8, TNF-α and IL-6, from spiked human plasma.16 We have also shown that hydrophilic multilayer Ti3C2Tx significantly adsorbed the small, water soluble uraemic toxin urea from spent dialysate.26 However, the impact of graphene and MXene surface chemistry on the biological properties of these materials has yet to be comparatively investigated. Although infinite lateral surface area for adsorption may confer faster kinetics, restacking of 2D layers may hinder it. There are also potential concerns about cytotoxicity since nanomaterials have dimensions similar to those of the cellular machinery and therefore may interact to the detriment of the cell. In this paper, the properties of graphene variants (GNP, GO and GO-Ag) were compared to Ti3C2Tx variants {multilayer Ti3C2Tx (ML-MXene) and delaminated Ti3C2Tx (DL-MXene)} for the first time to examine the impact of variable surface chemistry and sheet structure on cell viability, adsorption of bacterial and cellular biotoxins and modulation of cellular inflammatory response to bacterial endotoxin stimulation (Fig. 1).
 | ||
| Fig. 1 Could graphene and Ti3C2Tx be used to moderate cellular inflammation? Schematic of graphene reproduced with permission from Liu et al.27 Copyright 2008 John Wiley and Sons. Schematic of Ti3C2Tx adapted under a Creative Commons Attribution 3.0 Unported License. Published by the PCCP Owner Societies. | ||
2 Results and discussion
2.1 Physical characterisation
Scanning electron micrographs showed variations in the microscopic properties of the materials. GNP28 were arranged as particulate agglomerates and ML-MXene29 as stacked sheets. In contrast, GO,30 GO-Ag31 and DL-MXene32 formed flat sheets indicative of exfoliation from the corresponding graphite/ML-MXene precursors (Fig. 2a–e). TEM (f) and EDS (Table 1) indicated successful functionalisation of GO-Ag sheets with silver nanoparticles in line with previous studies.31 XPS was used to further confirm synthesis of each nanomaterial through detection of C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO bonds in GO and GO-Ag, Ag metal in GO-Ag as well as Ti–C and Ti–O bonds in ML-MXene (Fig. S2†).
O and O–CO bonds in GO and GO-Ag, Ag metal in GO-Ag as well as Ti–C and Ti–O bonds in ML-MXene (Fig. S2†).
 | ||
| Fig. 2 SEM images showing the morphology of GNP (a), GO (b), GO-Ag (c), ML-MXene (d) and DL-MXene (e). TEM image of GO-Ag (f). | ||
| Nanomaterial | Weight (%) | ||||
|---|---|---|---|---|---|
| C | O | Ag | Ti | F | |
| GNP | 96.8 | 3.2 | — | — | — |
| GO | 62.1 | 34.8 | — | — | — |
| GO-Ag | 55.6 | 31.4 | 7.6 | — | — |
| ML-MXene | 7.7 | 9.5 | — | 75.5 | 6.1 |
| DL-MXene | 9.8 | 7.6 | — | 70 | 8.7 |
Nanomaterial surface area, surface charge and size varied depending on surface functional group and delamination in the case of MXene. GNP had a surface area of 498 m2 g−1 which was close to the manufacturer's quoted value of 500 m2 g−1 and comparatively larger than GO (125 m2 g−1) and GO-Ag (132 m2 g−1) (Table 2). The measured surface areas for GO and GO-Ag were similar to previous studies.33,34 It is important to note that unlike the ML-MXene and DL-MXene used in this study, GNP and GO were not derived from the starting precursor nor was GNP exfoliated to produce GO. Megawati et al.35 showed that GO surface area is influenced by the synthesis method used. As the GNP synthesis method is proprietary information,36 it is difficult to pinpoint exact differences in the synthesis protocol that may have contributed to its larger surface area relative to GO. MXene surface area increased from 142 m2 g−1 to 282 m2 g−1 on exfoliation of ML-MXene to DL-MXene, as a result of sheet separation during delamination.25 Another explanation for the observed increase in surface area following delamination of ML-MXene could be because DL-MXene had smaller sheet diameters (386 nm) compared to ML-MXene (1033 nm) (Fig. 3). Reduction in nanomaterial size has been associated with an increase in surface area.37,38 It is important to mention that this is the surface accessible to nitrogen adsorption and the surface available for molecular adsorption from solution may be larger or smaller than the BET surface.
 | ||
| Fig. 3 Nanomaterial surface charge (a) and sheet size (b) were measured using DLS, data represent mean ± SD (n = 3). | ||
| Nanomaterial | Surface area (m2 g−1) |
|---|---|
| GNP | 498 |
| GO | 125 |
| GO-Ag | 132 |
| ML-MXene | 142 |
| DL-MXene | 282 |
Similarly, delamination of ML-MXene to DL-MXene increased the negative surface charge from −13.7 mV to −30.8 mV (Fig. 3a), in agreement with previous studies.25,39 Reduction of GO to GO-Ag decreased its negative surface charge from −29.6 mV to −20.4 mV. Binding of silver ions to oxygenated groups on GO sheets decreased the number of free anionic moieties and consequently, negative surface charge.40 GNP had a negative surface charge of −25.9 mV which has been attributed to oxygenated groups present at defect sites.41
Although DLS measurements typically assume that particles being measured have a spherical shape, the technique is widely used in literature for size measurements of 2D nanomaterials.42–45 Since graphenes and MXenes have anisotropic structures, the exact size of the two-dimensional sheets may not be accurately calculated using this technique. Nonetheless, comparisons can still be made regarding hydrodynamic size differences between materials. DL-MXene had a smaller lateral flake size of 386 ± 5 nm compared to ML-MXene with a mean lateral flake size of 1033 ± 252 nm (Fig. 3b) as a result of the sonication step employed during delamination. GNP had a mean particle size of 956 ± 109 nm which was within the range quoted by the manufacturer (<2 μm) and was half that of GO with a lateral particle size of 1919 ± 286 nm. GO-Ag particle size (4606 ± 717 nm) was about two times larger than that of GO (1919 nm) which could be attributed to increased aggregation brought about by reduction of GO and subsequent loss of oxygen functionalities during GO-Ag synthesis.43,46 Similar increases in size after reduction of GO have been reported in literature.43
2.2 Cell interaction studies
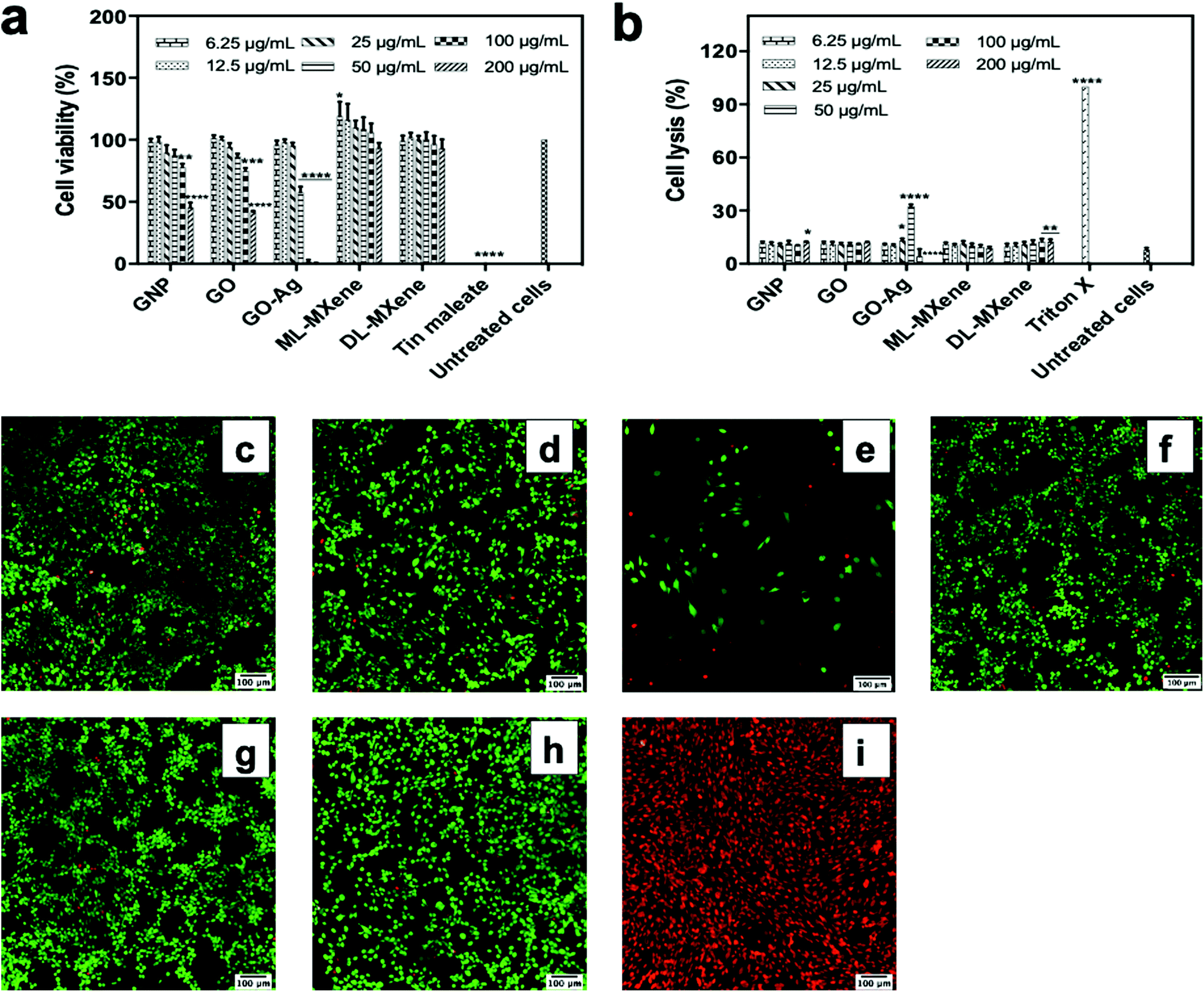 | ||
| Fig. 4 3T3 cell viability from the MTS (a) and LDH (b) assays measured after 24-hour incubation with the nanomaterials (6.25–200 μg mL−1) (n = 3, mean ± SEM). Data were analysed using two-way ANOVA and a Dunnett post hoc test by comparing treated cells to untreated cells (*p < 0.5, **p < 0.01, ***p < 0.001, ****p < 0.0001). Confocal microscopy images showing 3T3 cell viability after 24-hour incubation with GNP (c), GO (d), GO-Ag (e), ML-MXene (f) and DL-MXene (g) at 200 μg mL−1, untreated cells (h) and Triton X (i). Cells were stained with calcein AM and ethidium homodimer-1 – the green and red spots represent live and dead cells respectively. | ||
Although the LDH assay showed that GO-Ag induced significant cell lysis at 25 and 50 μg mL−1, this effect appeared to decrease at higher concentrations in contrast to the membrane damage and reduced cell coverage observed in the Live/Dead assay (Fig. 4e). A probable explanation is that the immobilised silver nanoparticles on the GO-Ag sheet caused assay interference. Silver nanoparticles have been reported to interfere with the LDH assay through enzyme adsorption and inactivation.48–50 Unlike GO-Ag, AgNP alone did not demonstrate a similar trend of decreasing necrosis levels at higher concentrations (Fig. S2c†). This could be due to differences in particle size as the sizes of the immobilised silver nanoparticles on GO-Ag (4 nm) and the silver nanoparticles tested in the cited studies (25–92 nm) were all less than 100 nm. In contrast, the AgNP used in this study was aggregated (Fig. S1†) and had a hydrodynamic size of 507 nm. This likely decreased available surface area for interaction with LDH molecules. The size-dependent interference of silver nanoparticles with the LDH assay has been reported in the scientific literature.50,51 The authors showed that larger silver nanoparticles adsorbed and/or inactivated less LDH compared to smaller silver nanoparticles.
The Live/Dead assay results provided direct confirmation of the LDH assay indicating that GNP (Fig. 4c), GO (Fig. 4d), ML-MXene (Fig. 4f) and DL-MXene (Fig. 4g) had no impact on 3T3 fibroblast viability as the majority of cells stained positive for the viable cell stain calcein AM. Previous studies have also shown that GO52 and DL-MXene39 did not negatively impact cell viability. In contrast, GO-Ag induced membrane damage indicated by the red, ethidium homodimer-1 positive cells in Fig. 4e which were comparable to the dead, ethidium homodimer-positive cells in the positive control (Fig. 4i). Since GO did not impact cell viability, the poor biocompatibility of GO-Ag is ascribed to the immobilised silver nanoparticles. Pristine AgNP did not impact cell viability (Fig. S2†) in contrast to GO-Ag. This could be attributed to variation in nanoparticle size – 507 nm (AgNP) vs. 4 nm (GOAg) as the size-dependent cytotoxicity of silver nanoparticles has been reported in literature.51,53
The LDH assay was a measure of nanomaterial induction of necrotic cell death where lactate dehydrogenases are released from the cells on cell lysis. Induction of apoptotic programmed cell death pathways was measured using the FITC Annexin V/propidium iodide assay (Fig. 5a). The assay indicated that GNP, GO, ML-MXene and DL-MXene did not induce apoptosis in Jurkat T lymphocytes at a concentration of 200 μg mL−1. In contrast, GO-Ag and the positive control camptothecin induced a significant apoptotic response after incubation for 1 and 4 hours respectively (p < 0.0001). Apoptosis induction by GO-Ag could be ascribed to the immobilised silver nanoparticles as GO alone did not induce apoptosis. Previous studies have reported stimulation of cellular apoptosis by silver nanoparticles54 as well as silver-modified GO,55 evidenced by characteristic cellular events such as DNA fragmentation, caspase-3 activation, decrease in membrane mitochondrial potential, elevated expression of proapoptotic genes and reduced expression of antiapoptotic genes.
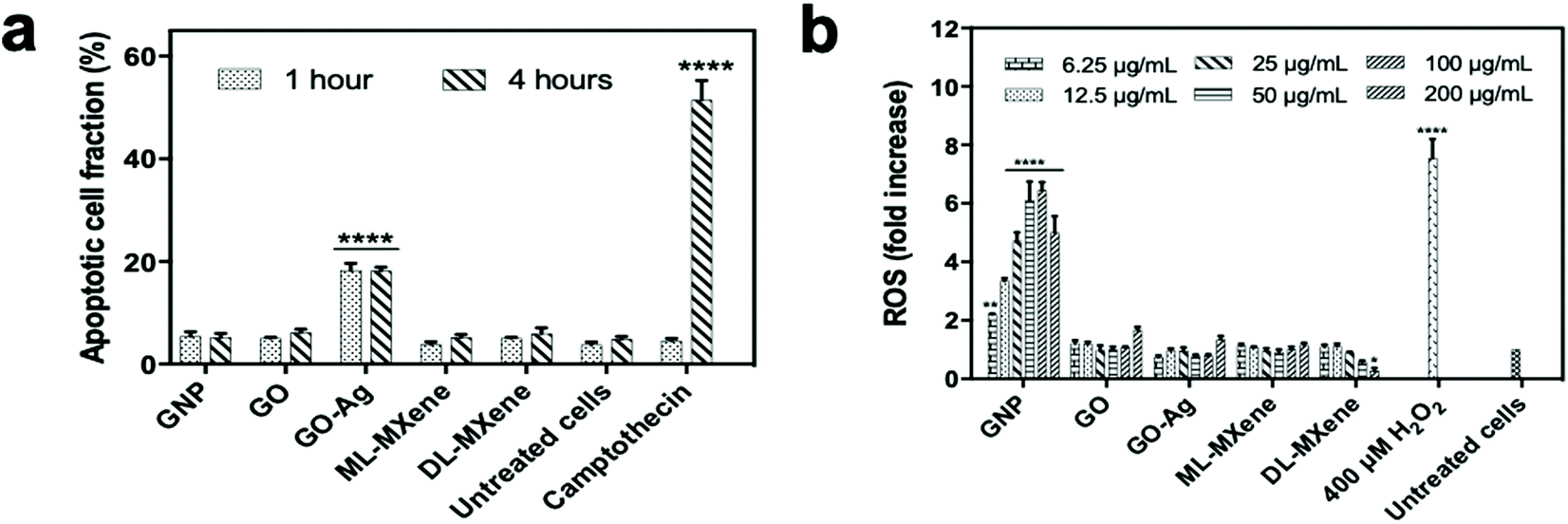 | ||
| Fig. 5 Assessment of nanomaterial biocompatibility with Jurkat T lymphocytes and THP-1 monocytes using the apoptosis (a) and DCFH-DA oxidative stress (b) assays (n = 3, mean ± SEM). The nanomaterial concentration used for the apoptosis assay was 200 μg mL−1. Data were analysed using two-way ANOVA and a Dunnett post hoc test by comparing treated cells to untreated cells (*p < 0.5, ****p < 0.0001). | ||
DCH-DA assay results in Fig. 5b showed that GO, GO-Ag, ML-MXene and DL-MXene did not stimulate ROS production in THP-1 monocytes in contrast to the H2O2 stimulated positive control. This finding disagreed with other studies which reported that GO56 and DL-MXene57 significantly increased ROS production in THP-1 cells and induced oxidative stress. Differences between the findings in the study and prior studies could be attributed to variations in GO synthesis methods, physicochemical properties such as sheet size, surface area and oxygen content, and experimental conditions which are known to affect the outcome of cytotoxicity assays.58,59
Using variations in experimental conditions as an example, studies in the literature which reported induction of oxidative stress by GO56,60–63 assessed GO impact on reactive oxygen species (ROS) levels by adding the probe directly to cells after nanomaterial incubation. In contrast, this study took a different approach. To investigate nanomaterial impact on ROS levels, the THP-1 cells were first labelled with the DCFH-DA probe – the probe passively diffused into cells and underwent deacetylation to the non-fluorescent DCFH by cellular esterases. Next, the nanomaterial suspensions were introduced to the labelled cells for 24-hour incubation. This setup was chosen to limit direct interaction of the nanomaterials with DCFH, the active substrate, and hence minimise the risk of assay interference. Assay interference experiments in Fig. S4† where nanomaterials were in direct contact with the DCFH-DA probe showed that the graphenes (GNP, GO and GO-Ag) induced oxidation of DCFH to DCF to varying extents.
The finding that DL-MXene did not induce ROS production in THP-1 cells (Fig. 5b) disagreed with Jastrzębska et al.57 who reported that DL-MXene induced oxidative stress. A likely explanation for this difference would be variations in synthesis methods and test parameters. The degree of post-synthesis purification may also have affected the purity of the DL-MXene product. In this study, washing and delamination of the MXene product was carried out in deionised water only. Also, the pH of the ML-MXene and DL-MXene suspensions was confirmed to be ∼7 after washing before conducting subsequent experiments. In Jastrzębska et al.,57 the MXene product was first washed with deionised water and then ethanol followed by delamination with DMSO. The last wash was done in isopropyl alcohol. Hence, the induction of oxidative stress may have been caused by chemical residues in the final DL-MXene product. Product purity can affect the biological impact of nanomaterials.64,65
 | ||
| Fig. 6 Assessment of nanomaterial haemocompatibility following 1-hour incubation with healthy donor blood at a mass loading of 50 mg using the haemolysis (a), APTT (b), prothrombin time (c) and platelet activation (d) assays. Data represent n = 3, mean ± SEM. Data were analysed using one-way ANOVA and a Dunnett post hoc test by comparing treated samples to untreated blood (a and d) or normal plasma (b and c) (*p < 0.5, **p < 0.01, ***p < 0.001, ****p < 0.0001). | ||
GNP and ML-MXene did not significantly impact the intrinsic coagulation pathway (p > 0.05) as APTT values were within the range of the normal plasma control (34.4 seconds) (Fig. 6b) and in agreement with an earlier study on GNP.68 In contrast, GO (p < 0.01) and DL-MXene (p < 0.05) significantly extended APTT values in line with other studies for GO.66,69 GNP, GO, ML-MXene and DL-MXene did not significantly impact the extrinsic coagulation pathway (p > 0.05) as all prothrombin clotting time values remained within the normal range of 15.3 seconds (Fig. 6c), in agreement with previous studies on GNP68 and GO.70 In contrast to PMA, the positive control, GNP, GO, ML-MXene and DL-MXene did not induce platelet activation (p > 0.05) as indicated by the percentage of platelets positive for the PAC-1 activation marker (Fig. 6d) and in line with other studies for GO.71
2.3 Antibacterial studies
GO-Ag and AgNP, the positive control, induced significant reductions in E. coli viability (p < 0.0001) at a concentration of 200 μg mL−1 in contrast to GNP, GO, ML-MXene and DL-MXene which did not significantly impact bacterial viability (p > 0.05) (Fig. 7a). This finding disagreed with another study which reported that DL-MXene demonstrated antibacterial activity against E. coli through physical interactions between sharp edges of DL-MXene sheets and bacterial membranes.72 GO-Ag induced a significantly greater reduction in E. coli viability (p < 0.05) compared to AgNP at a concentration of 50 μg mL−1 (Fig. 7b). Similar results were found using a bacterial ATP assay to quantify viable E. coli following incubation with nanomaterials (Fig. S5†). Live/Dead assay results confirmed that GO had no impact on E. coli viability (Fig. 7c) as the majority of bacteria stained for SYTO 9 and fluoresced green which indicated membrane integrity. In contrast, GO-Ag and AgNP induced membrane damage (Fig. 7d and e) evidenced by positive staining of bacteria for propidium iodide and emission of red fluorescence. This indicated that GO-Ag exerted antibacterial activity via membrane damage. The greater antibacterial performance of GO-Ag over AgNP could be attributed to synergistic interactions between GO and the immobilised silver nanoparticles resulting in enhanced bacterial contact.40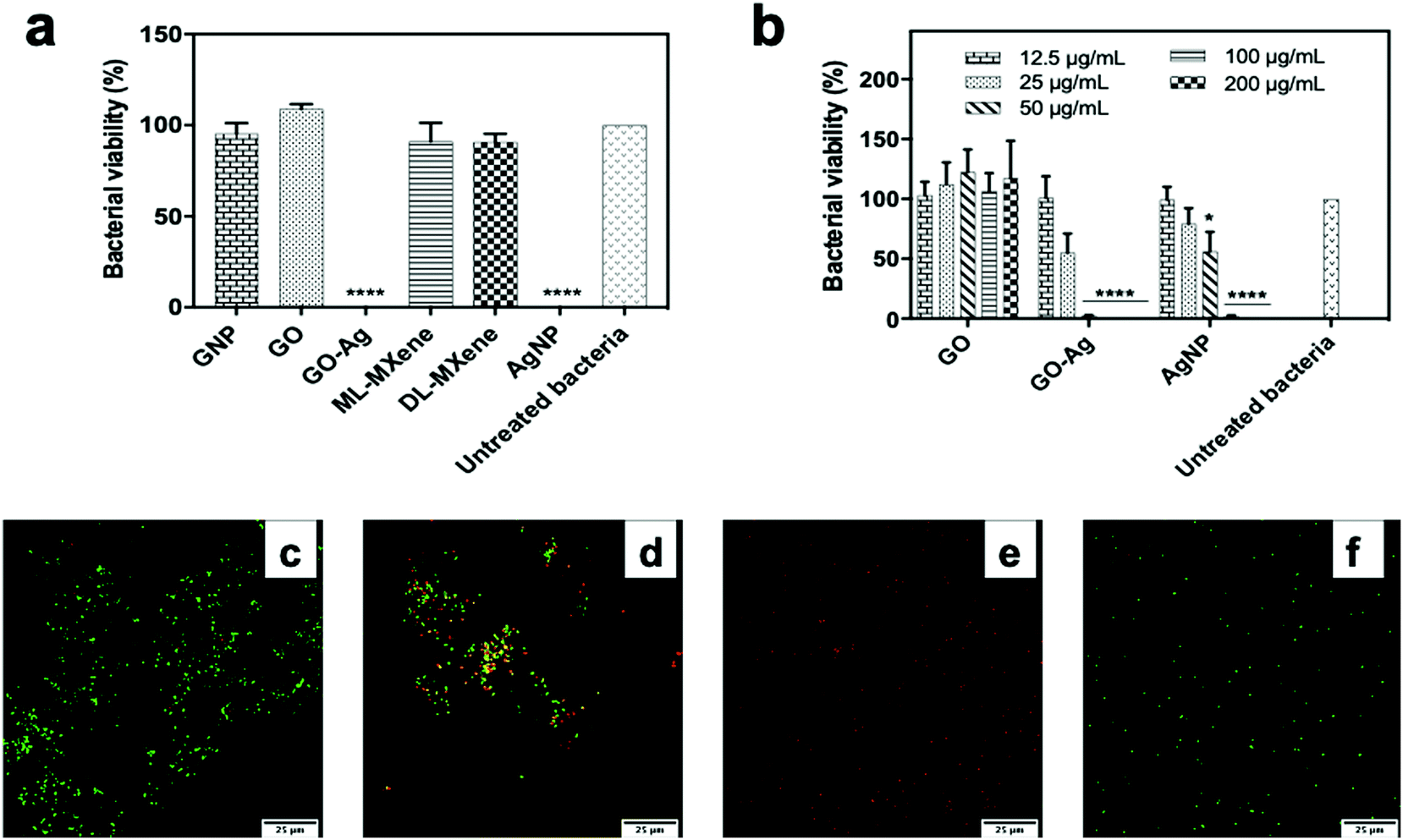 | ||
| Fig. 7 Antibacterial activity of the nanomaterials following 4-hour incubation with E. coli measured using colony count at concentrations of 200 μg mL−1 (a) and 12.5–200 μg mL−1 (b) (n = 3, mean ± SEM). Data were analysed using one-way ANOVA and a Dunnett post hoc test by comparing treated bacteria to untreated bacteria (*p < 0.05, ****p < 0.0001). Confocal microscopy images showing E. coli viability after 4-hour incubation with GO (c), GO-Ag (d), AgNP (e) at 200 μg mL−1 and untreated E. coli (f). Bacteria were stained with SYTO 9 and propidium iodide – the green spots represent live bacteria, red spots represent dead bacteria and yellow spots, a combination of both dyes. | ||
While there are multiple reports on the bactericidal properties of 2D nanomaterials such as GO in the literature, other studies have also reported non-toxicity of these materials towards bacteria.64,65,73–77 These differences may have resulted from variations in nanomaterial synthesis routes, experimental conditions and bacterial strains used. For example, Barbolina et al.64 demonstrated that residual acidic impurities from graphite oxidation imparted a ‘false’ antibacterial activity to GO sheets against Escherichia coli and Staphylococcus aureus. This antibacterial effect was eliminated upon extensive washing of the GO product and neutralisation of the acidic pH. It is difficult to directly compare the findings in this study with other published studies due to differences in synthesis and experimental protocols which could have impacted bacterial viability.
2.4 Suppression of inflammatory stimulus
GNP and GO induced significant removal of IL-8 (Fig. 8a), IL-6 (Fig. 8b) and TNF-α (Fig. 8c) from plasma at all the masses tested compared to the negative no nanomaterial control (p < 0.01). Compared to the graphenes, removal of the largest cytokine TNF-α was reduced for DL-MXene and in the case of ML-MXene, adsorption of IL-6 and TNF-α was reduced. This could be attributed to the contribution of hydrophobic bonds and π–π stacking interactions which enhanced binding to aromatic amino acid residues in the cytokines.78 Ultimately, the carbon surface and hydrophobic interactions, not surface area, seem to be most important for cytokine adsorption as GO exhibited a similar adsorptive performance to GNP despite having a lower surface area.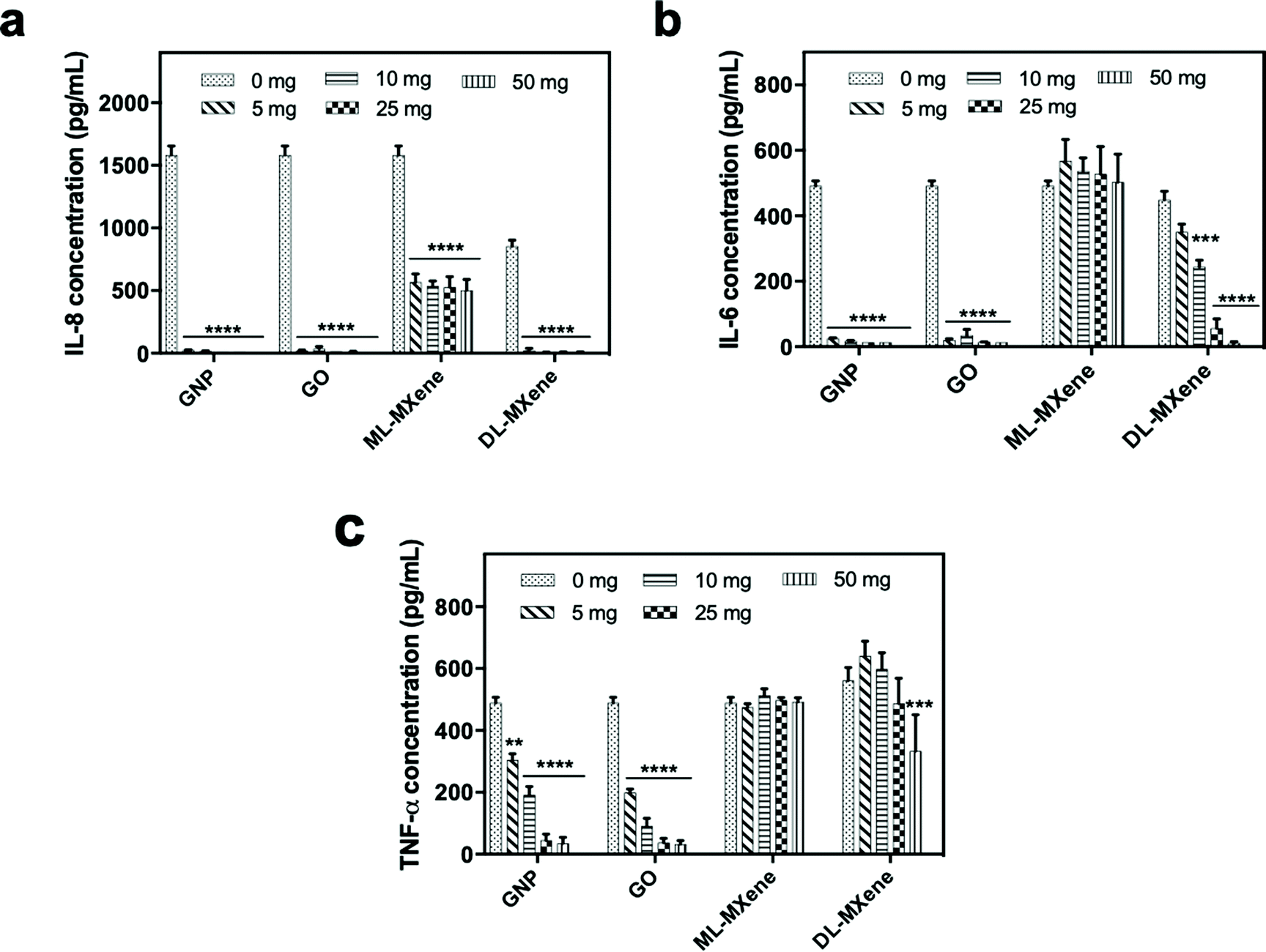 | ||
| Fig. 8 Residual IL-8 (a), IL-6 (b) and TNF-α (c) concentrations after 4-hour incubation of cytokine spiked plasma (∼1000 pg mL−1) with GNP, GO, ML-MXene and DL-MXene at different mass loadings (5–50 mg) compared to the control, spiked plasma without adsorbent (0 mg). Cytokine concentrations were measured using ELISA (n = 3, mean ± SEM). Data were analysed using two-way ANOVA and a Dunnett post hoc test by comparing treated groups to the control (**p < 0.01, ***p < 0.001, ****p < 0.0001). | ||
GNP, GO, ML-MXene and DL-MXene significantly reduced IL-8 (Fig. 9a), IL-6 (Fig. 9b) and TNF-α (Fig. 9c) levels in LPS stimulated THP-1 monocytes at a concentration of 12.5 mg compared to the no nanomaterial LPS-stimulated cell controls. However, the nanomaterials did not significantly adsorb LPS at the concentrations used (p < 0.05) (Fig. 9d) which suggested that the reduction of cytokine levels in stimulated THP-1 cells was predominantly driven by direct cytokine adsorption rather than LPS removal or inactivation. Apart from adsorption of pro-inflammatory cytokines secreted by the stimulated THP-1 monocytes, the nanomaterials may have impacted cellular cytokine levels via other mechanisms such as disrupting LPS attachment to the LPS-binding protein.79
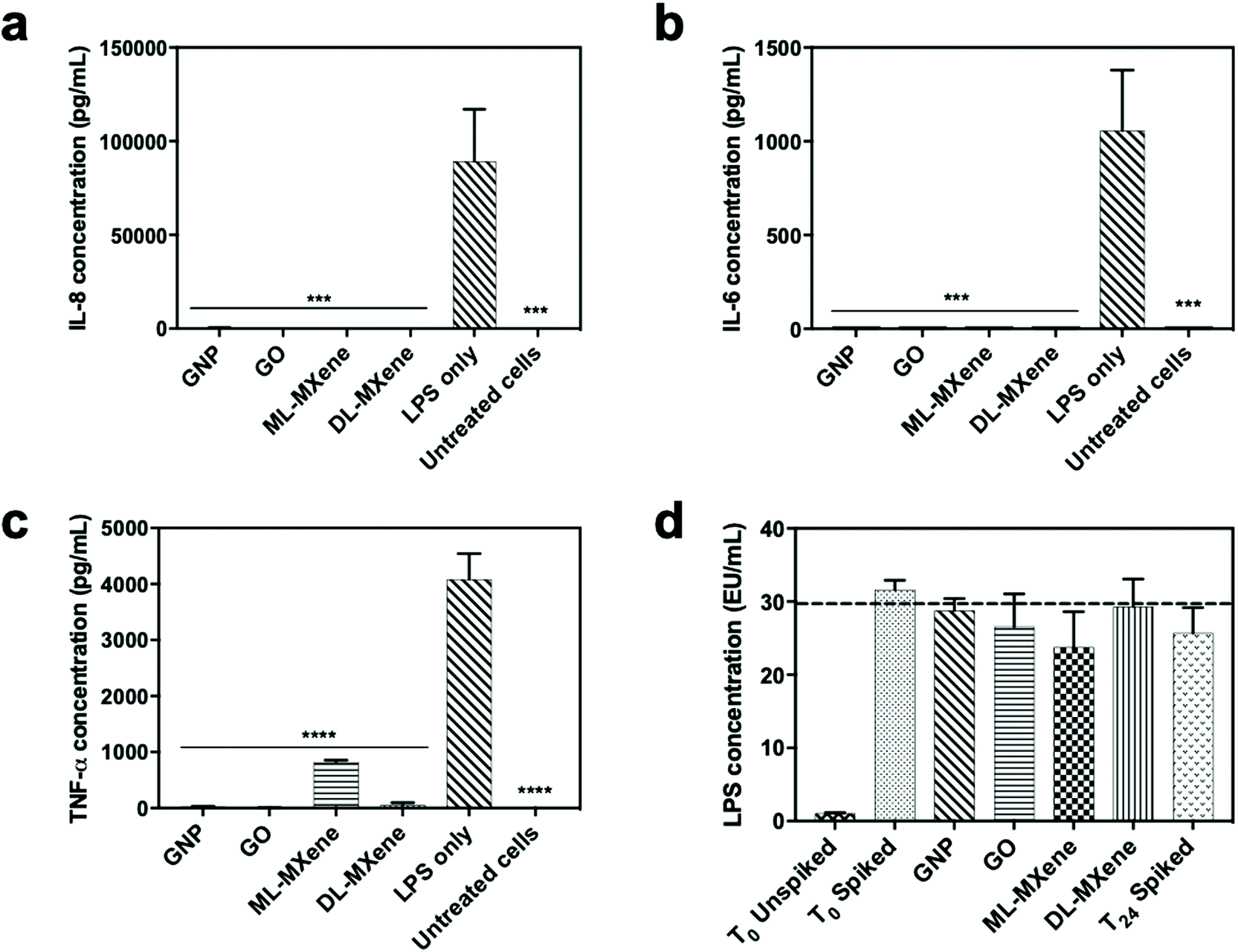 | ||
| Fig. 9 Residual IL-8 (a), IL-6 (b) and TNF-α (c) levels in LPS-stimulated THP-1 cells after incubation with GNP, GO, ML-MXene and DL-MXene (12.5 mg) for 24, 24 and 8 hours respectively (n = 3, mean ± SEM). Cytokine concentrations were measured using ELISA. Data were analysed using one-way ANOVA and a Dunnett post hoc test by comparing stimulated cells with nanomaterials to stimulated cells without nanomaterial (LPS only) (***p < 0.001, ****p < 0.0001). Residual LPS levels in spiked RPMI 1640 media (2000 ng mL−1) after 24 hours-incubation with GNP, GO, ML-MXene and DL-MXene (12.5 mg) (d) (n = 3, mean ± SEM). LPS concentration was measured using the LAL assay. Data were analysed using one-way ANOVA by comparing treated groups to spiked media without adsorbent (T24 spiked). | ||
3 Conclusions
This study is the first to directly compare the biological interactions of graphene and titanium carbide MXene nanomaterials using cell based models. The results indicate no cytotoxicity and the feasible use of these materials in medical devices as adsorbents for the suppression of cytokine driven inflammatory stimulus highlighting the complex interplay of numerous factors including material surface chemistry and external surface area which combine to influence biological interactions. No significant induction of haemolysis or platelet activation occurred on incubation of blood with GNP, ML-MXene and DL-MXene. Whilst the carbon based graphene adsorbents and delaminated Ti3C2Tx removed significantly more of the high molecular weight cytokines in direct studies per mass compared to the multilayered Ti3C2Tx, all materials repressed bacterial LPS stimulation of cytokines through a mechanism that was not direct LPS adsorption at the concentrations used. It is hypothesised that the nanomaterials may have adsorbed the monocyte secreted cytokines. Whilst antibacterial properties were only observed in this study in the Ag modified GO variants, further work should investigate impact of flake size on cellular uptake as well as the tendency of these materials to undergo oxidation and participate in redox reactions which could potentially alter cellular oxidative state.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by a University of Brighton PhD studentship award to Tochukwu Ozulumba. This work is based on the NOMAD project which was supported by the British Council and the UK Department for Business, Innovation and Skills through the Global Innovation Initiative. The authors acknowledge the Electron Microscopy Imaging centre at the University of Sussex, funded by the School of Life Sciences, the Wellcome Trust (095605/Z/11/A, 208348/Z/17/Z) and the RM Phillips Trust. The authors thank Dr Pascale Schellenberger from the Electron microscopy imaging centre at the University of Sussex for her help with TEM imaging and support in this work. The authors also thank Kanit Hantanasirisakul (Drexel University) for assistance with XPS analysis of MXene.References
- D. M. Silverstein, Pediatr. Nephrol., 2009, 24, 1445–1452 CrossRef.
- V. Chiurchiù, A. Leuti and M. Maccarrone, Front. Immunol., 2018, 9, 38 CrossRef.
- C. A. Howell, S. R. Sandeman, G. J. Phillips, A. W. Lloyd, J. G. Davies, S. V. Mikhalovsky, S. R. Tennison, A. P. Rawlinson, O. P. Kozynchenko, H. L. H. Owen, J. D. S. Gaylor, J. J. Rouse and J. M. Courtney, Biomaterials, 2006, 27, 5286–5291 CrossRef CAS.
- J. A. Del Campo, P. Gallego and L. Grande, World J. Hepatol., 2018, 10, 1–7 CrossRef.
- T. S. Blackwell and J. W. Christman, Br. J. Anaesth., 1996, 77, 110–117 CrossRef CAS.
- N. J. Trengove, H. Bielefeldt-Ohmann and M. C. Stacey, Wound Repair Regen., 2000, 8, 13–25 CrossRef CAS.
- C. Huang, Y. Wang, X. Li, L. Ren, J. Zhao, Y. Hu, L. Zhang, G. Fan, J. Xu, X. Gu, Z. Cheng, T. Yu, J. Xia, Y. Wei, W. Wu, X. Xie, W. Yin, H. Li, M. Liu, Y. Xiao, H. Gao, L. Guo, J. Xie, G. Wang, R. Jiang, Z. Gao, Q. Jin, J. Wang and B. Cao, Lancet, 2020, 395, 497–506 CrossRef CAS.
- J. F. Winchester, J. A. Kellum, C. Ronco, J. A. Brady, P. J. Quartararo, J. A. Salsberg and N. W. Levin, Blood Purif., 2003, 21, 79–84 CrossRef CAS.
- T. Rimmelé and J. A. Kellum, Crit. Care, 2011, 15, 205 CrossRef.
- G. Yushin, E. N. Hoffman, M. W. Barsoum, Y. Gogotsi, C. A. Howell, S. R. Sandeman, G. J. Phillips, A. W. Lloyd and S. V. Mikhalovsky, Biomaterials, 2006, 27, 5755–5762 CrossRef CAS.
- B. Malard, C. Lambert and J. A. Kellum, Intensive Care Med. Exp., 2018, 6, 12 CrossRef.
- I. K. Herrmann, M. Urner, F. M. Koehler, M. Hasler, B. Roth-Z'Graggen, R. N. Grass, U. Ziegler, B. Beck-Schimmer and W. J. Stark, Small, 2010, 6, 1388–1392 CrossRef CAS.
- I. K. Herrmann, A. Schlegel, R. Graf, C. M. Schumacher, N. Senn, M. Hasler, S. Gschwind, A. M. Hirt, D. Gunther, P. A. Clavien, W. J. Stark and B. Beck-Schimmer, Nanoscale, 2013, 5, 8718–8723 RSC.
- V. Stadlbauer, P. Krisper, R. Aigner, B. Haditsch, A. Jung, C. Lackner and R. E. Stauber, Crit. Care, 2006, 10, R169 CrossRef.
- S. Frimmel, M. Hinz, J. Schipper, S. Bogdanow, S. Mitzner and S. Koball, Int. J. Artif. Organs, 2019, 42, 658–664 CrossRef.
- Y. Zheng, N. Pescatore, Y. Gogotsi, B. Dyatkin, G. Ingavle, V. Mochalin, T. Ozulumba, S. Mikhalovsky and S. Sandeman, J. Nanomater., 2018, 2018, 6274072 Search PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- A. Peigney, C. Laurent, E. Flahaut, R. R. Bacsa and A. Rousset, Carbon, 2001, 39, 507–514 CrossRef CAS.
- Kenry, A. Geldert, Y. Liu, K. P. Loh and C. T. Lim, NPG Asia Mater., 2017, 9, e422 CrossRef CAS.
- K. P. Loh, Q. Bao, P. K. Ang and J. Yang, J. Mater. Chem., 2010, 20, 2277–2289 RSC.
- S. Vijay Kumar, N. M. Huang, H. N. Lim, A. R. Marlinda, I. Harrison and C. H. Chia, Chem. Eng. J., 2013, 219, 217–224 CrossRef CAS.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS.
- G. Deysher, C. E. Shuck, K. Hantanasirisakul, N. C. Frey, A. C. Foucher, K. Maleski, A. Sarycheva, V. B. Shenoy, E. A. Stach, B. Anasori and Y. Gogotsi, ACS Nano, 2020, 14, 204–217 CrossRef CAS.
- N. K. Chaudhari, H. Jin, B. Kim, D. S. Baek, S. H. Joo and K. Lee, J. Mater. Chem. A, 2017, 5, 24564–24579 RSC.
- Z. Ling, C. E. Ren, M.-Q. Zhao, J. Yang, J. M. Giammarco, J. Qiu, M. W. Barsoum and Y. Gogotsi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16676–16681 CrossRef CAS.
- F. Meng, M. Seredych, C. Chen, V. Gura, S. Mikhalovsky, S. Sandeman, G. Ingavle, T. Ozulumba, L. Miao, B. Anasori and Y. Gogotsi, ACS Nano, 2018, 12, 10518–10528 CrossRef CAS.
- Z. Liu, Q. Liu, Y. Huang, Y. Ma, S. Yin, X. Zhang, W. Sun and Y. Chen, Adv. Mater., 2008, 20, 3924–3930 CrossRef CAS.
- G. W. Zhang, F. Z. Wang, J. Dai and Z. X. Huang, Materials, 2016, 9, 92 CrossRef.
- M. A. Hope, A. C. Forse, K. J. Griffith, M. R. Lukatskaya, M. Ghidiu, Y. Gogotsi and C. P. Grey, Phys. Chem. Chem. Phys., 2016, 18, 5099–5102 RSC.
- F. Perreault, A. F. de Faria, S. Nejati and M. Elimelech, ACS Nano, 2015, 9, 7226–7236 CrossRef CAS.
- W. Shao, X. Liu, H. Min, G. Dong, Q. Feng and S. Zuo, ACS Appl. Mater. Interfaces, 2015, 7, 6966–6973 CrossRef CAS.
- E. Satheeshkumar, T. Makaryan, A. Melikyan, H. Minassian, Y. Gogotsi and M. Yoshimura, Sci. Rep., 2016, 6, 32049 CrossRef CAS.
- N. M. Dat, V. N. P. Linh, L. A. Huy, N. T. Huong, T. H. Tu, N. T. L. Phuong, H. M. Nam, M. T. Phong and N. H. Hieu, Mater. Technol., 2019, 34, 369–375 CrossRef.
- S. Wang, F. Tristan, D. Minami, T. Fujimori, R. Cruz-Silva, M. Terrones, K. Takeuchi, K. Teshima, F. Rodríguez-Reinoso, M. Endo and K. Kaneko, Carbon, 2014, 76, 220–231 CrossRef CAS.
- M. Megawati, C. K. Chua, Z. Sofer, K. Klimova and M. Pumera, Phys. Chem. Chem. Phys., 2017, 19, 15914–15923 RSC.
- L. T. Drzal and H. Fukushima, United States Pat, US8501858B2, 2006 Search PubMed.
- K. Suttiponparnit, J. Jiang, M. Sahu, S. Suvachittanont, T. Charinpanitkul and P. Biswas, Nanoscale Res. Lett., 2010, 6, 27 Search PubMed.
- K. W. Powers, M. Palazuelos, B. M. Moudgil and S. M. Roberts, Nanotoxicology, 2007, 1, 42–51 CrossRef CAS.
- G. Liu, J. Zou, Q. Tang, X. Yang, Y. Zhang, Q. Zhang, W. Huang, P. Chen, J. Shao and X. Dong, ACS Appl. Mater. Interfaces, 2017, 9, 40077–40086 CrossRef CAS.
- J. Ma, J. Zhang, Z. Xiong, Y. Yong and X. S. Zhao, J. Mater. Chem., 2011, 21, 3350–3352 RSC.
- J. Lu, I. Do, H. Fukushima, I. Lee and L. T. Drzal, J. Nanomater., 2010, 2010, 186486 Search PubMed.
- M. Lotya, A. Rakovich, J. F. Donegan and J. N. Coleman, Nanotechnology, 2013, 24, 265703 CrossRef.
- K. H. Liao, Y. S. Lin, C. W. MacOsko and C. L. Haynes, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CrossRef CAS.
- S. Gurunathan, J. W. Han, A. A. Dayem, V. Eppakayala and J. H. Kim, Int. J. Nanomed., 2012, 7, 5901–5914 CrossRef CAS.
- K. Maleski, C. E. Ren, M.-Q. Zhao, B. Anasori and Y. Gogotsi, ACS Appl. Mater. Interfaces, 2018, 10, 24491–24498 CrossRef CAS.
- S. Liu, T. H. Zeng, M. Hofmann, E. Burcombe, J. Wei, R. Jiang, J. Kong and Y. Chen, ACS Nano, 2011, 5, 6971–6980 CrossRef CAS.
- A. Casey, E. Herzog, M. Davoren, F. M. Lyng, H. J. Byrne and G. Chambers, Carbon, 2007, 45, 1425–1432 CrossRef CAS.
- S. J. Oh, H. Kim, Y. Liu, H. K. Han, K. Kwon, K. H. Chang, K. Park, Y. Kim, K. Shim, S. S. A. An and M. Y. Lee, Toxicol. Lett., 2014, 225, 422–432 CrossRef CAS.
- X. Han, R. Gelein, N. Corson, P. Wade-Mercer, J. Jiang, P. Biswas, J. N. Finkelstein, A. Elder and G. Oberdörster, Toxicology, 2011, 287, 99–104 CrossRef CAS.
- L. Liang, M. H. Cui, M. Zhang, P. W. Zheng, Z. Y. Deng, S. S. Gao, X. S. Wang, X. Y. Zhang, C. Wang, Y. Liu and L. M. Xie, RSC Adv., 2015, 5, 67327–67334 RSC.
- A. R. Gliga, S. Skoglund, I. O. Wallinder, B. Fadeel and H. L. Karlsson, Part. Fibre Toxicol., 2014, 11, 11 CrossRef.
- S.-R. Ryoo, Y.-K. Kim, M.-H. Kim and D.-H. Min, ACS Nano, 2010, 4, 6587–6598 CrossRef CAS.
- L. Q. Chen, L. Fang, J. Ling, C. Z. Ding, B. Kang and C. Z. Huang, Chem. Res. Toxicol., 2015, 28, 501–509 Search PubMed.
- S. Gurunathan, J. W. Han, V. Eppakayala, M. Jeyaraj and J.-H. Kim, BioMed Res. Int., 2013, 2013, 535796 Search PubMed.
- D. Ali, S. Alarifi, S. Alkahtani and R. S. Almeer, Int. J. Nanomed., 2018, 13, 5685–5699 Search PubMed.
- S. Gurunathan, M.-H. Kang, M. Jeyaraj and J.-H. Kim, Int. J. Mol. Sci., 2019, 20, 247 Search PubMed.
- A. M. Jastrzebska, A. Szuplewska, T. Wojciechowski, M. Chudy, W. Ziemkowska, L. Chlubny, A. Rozmyslowska and A. Olszyna, J. Hazard. Mater., 2017, 339, 1–8 CrossRef CAS.
- X. Guo and N. Mei, J. Food Drug Anal., 2014, 22, 105–115 CrossRef CAS.
- L. Ou, B. Song, H. Liang, J. Liu, X. Feng, B. Deng, T. Sun and L. Shao, Part. Fibre Toxicol., 2016, 13, 57 CrossRef.
- T. Lammel, P. Boisseaux, M.-L. Fernández-Cruz and J. M. Navas, Part. Fibre Toxicol., 2013, 10, 27 CrossRef CAS.
- J. Yuan, H. Gao, J. Sui, H. Duan, W. N. Chen and C. B. Ching, Toxicol. Sci., 2011, 126, 149–161 CrossRef.
- K. Srikanth, L. S. Sundar, E. Pereira and A. C. Duarte, J. Appl. Toxicol., 2018, 38, 504–513 CrossRef CAS.
- Z. B. Tang, L. Zhao, Z. X. Yang, Z. H. Liu, J. Gu, B. Bai, J. L. Liu, J. Y. Xu and H. L. Yang, Int. J. Nanomed., 2018, 13, 2907–2919 CrossRef CAS.
- I. Barbolina, C. R. Woods, N. Lozano, K. Kostarelos, K. S. Novoselov and I. S. Roberts, 2D Mater., 2016, 3, 025025 CrossRef.
- O. N. Ruiz, K. A. S. Fernando, B. Wang, N. A. Brown, P. G. Luo, N. D. McNamara, M. Vangsness, Y. P. Sun and C. E. Bunker, ACS Nano, 2011, 5, 8100–8107 CrossRef CAS.
- R. Feng, Y. Yu, C. Shen, Y. Jiao and C. Zhou, J. Biomed. Mater. Res., Part A, 2015, 103, 2006–2014 CrossRef CAS.
- ASTM International, ASTM F756-17, Standard Practice for Assessment of Haemolytic Properties of Materials, ASTM International, Philadelphia, USA, 2017 Search PubMed.
- J. K. Kim, J. H. Shin, J. S. Lee, J. H. Hwang, J. H. Lee, J. E. Baek, T. G. Kim, B. W. Kim, J. S. Kim, G. H. Lee, K. Ahn, S. G. Han, D. Bello and I. J. Yu, Nanotoxicology, 2016, 10, 891–901 CrossRef CAS.
- C. J. Pan, L. Q. Pang, F. Gao, Y. N. Wang, T. Liu, W. Ye and Y. H. Hou, Mater. Sci. Eng., C, 2016, 63, 333–340 CrossRef CAS.
- R. Feng, Y. Yu, C. Shen, Y. Jiao and C. Zhou, J. Biomed. Mater. Res., Part A, 2015, 103, 2006–2014 CrossRef CAS.
- C. J. Pan, L. Q. Pang, F. Gao, Y. N. Wang, T. Liu, W. Ye and Y. H. Hou, Mater. Sci. Eng., C, 2016, 63, 333–340 CrossRef CAS.
- K. Rasool, M. Helal, A. Ali, C. E. Ren, Y. Gogotsi and K. A. Mahmoud, ACS Nano, 2016, 10, 3674–3684 CrossRef CAS.
- M. R. Das, R. K. Sarma, R. Saikia, V. S. Kale, M. V. Shelke and P. Sengupta, Colloids Surf., B, 2011, 83, 16–22 CrossRef CAS.
- Q. Bao, D. Zhang and P. Qi, J. Colloid Interface Sci., 2011, 360, 463–470 CrossRef CAS.
- J. Tang, Q. Chen, L. Xu, S. Zhang, L. Feng, L. Cheng, H. Xu and Z. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 3867–3874 CrossRef CAS.
- S. V. Kumar, N. M. Huang, H. N. Lim, A. R. Marlinda, I. Harrison and C. H. Chia, Chem. Eng. J., 2013, 219, 217–224 CrossRef.
- A. F. de Faria, D. S. T. Martinez, S. M. M. Meira, A. C. M. de Moraes, A. Brandelli, A. G. S. Filho and O. L. Alves, Colloids Surf., B, 2014, 113, 115–124 CrossRef.
- G. Shi and X. Wang, Phys. Chem. Chem. Phys., 2015, 17, 28484–28504 RSC.
- P. P. Wibroe, S. V. Petersen, N. Bovet, B. W. Laursen and S. M. Moghimi, Biomaterials, 2016, 78, 20–26 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01953d |
| This journal is © The Royal Society of Chemistry 2021 |