
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomaterial screening of protein coatings and peptide additives: towards a simple synthetic mimic of a complex natural coating for a bio-artificial kidney†
Ronald C.
van Gaal
 ab,
Annika F.
Vrehen
ab,
Johnick F.
van Sprang
abc,
Peter-Paul K. H.
Fransen
bc,
Mark C.
van Turnhout
a and
Patricia Y. W.
Dankers
*abc
ab,
Annika F.
Vrehen
ab,
Johnick F.
van Sprang
abc,
Peter-Paul K. H.
Fransen
bc,
Mark C.
van Turnhout
a and
Patricia Y. W.
Dankers
*abc
aLaboratory for Cell and Tissue Engineering, Eindhoven University of Technology, PO Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: p.y.w.dankers@tue.nl
bInstitute for Complex Molecular Systems, Eindhoven University of Technology, PO Box 513, 5600 MB, Eindhoven, The Netherlands
cLaboratory of Chemical Biology, Eindhoven University of Technology, PO Box 513, 5600 MB, Eindhoven, The Netherlands
First published on 18th January 2021
Abstract
Bio-artificial kidneys require conveniently synthesized membranes providing signals that regulate renal epithelial cell function. Therefore, we aimed to find synthetic analogues for natural extracellular matrix (ECM) protein coatings traditionally used for epithelial cell culturing. Two biomaterial libraries, based on natural ECM-coatings and on synthetic supramolecular small molecule additives, were developed. The base material consisted of a bisurea (BU) containing polymer, providing supramolecular BU-additives to be incorporated via specific hydrogen bonding interactions. This system allows for a modular approach and therefore easy fractional factorial based screening. A natural coating on the BU-polymer material with basement membrane proteins, laminin and collagen IV, combined with catechols was shown to induce renal epithelial monolayer formation. Modification of the BU-polymer material with synthetic BU-modified ECM peptide additives did not result in monolayer formation. Unexpectedly, simple BU-catechol additives induced monolayer formation and presented similar levels of epithelial markers and apical transporter function as on the laminin, collagen IV and catechol natural coating. Importantly, when this BU-polymer material was processed into fibrous e-spun membranes the natural coating and the BU-catechol additive were shown to perfectly function. This study clearly indicates that complex natural ECM-coatings can be replaced by simple synthetic additives, and displays the potency of material libraries based on design of experiments in combination with modular, supramolecular chemistry.
Introduction
Biomaterials are at the fundament of many regenerative medicine therapies, providing a protective environment or supportive mechanical properties.1,2 Tailor-made bio-instructive materials are important to guide a desired cell response for a specific application.2 Functions have been engineered into biomaterials to allow cells to remodel their environment,3,4 or have cell specific adhesion sites.5,6The development of a bio-artificial kidney requires a bioactive membrane on which renal epithelial cells (RECs) form a tight polarized monolayer with efficient transporter activity to fine tune both pre-urine and blood composition.7,8 Several research groups have evaluated the effect of extracellular matrix (ECM) protein coatings on anti-fouling hemodialysis membranes to achieve a tight functional monolayer.9,10 Basement membrane components collagen type IV (Col IV) and laminin (Lam) were found to maintain epithelial monolayers for a prolonged period of time. Moreover, the addition of adhesive polymerized L-3,4-dihydroxyphenylalanine (L-DOPA) further improved monolayer formation.10 Albeit successful, the coating of hemodialysis membranes requires laborious protocols. Therefore, “off-the-shelf” bioactive membranes would be more desirable.
Alternatively, biomaterials have been functionalized with ECM protein mimicking peptides substituting ECM protein coatings.6,11,12 Short peptides do not suffer from batch to batch differences and are synthetically more accessible than proteins. Sharma et al. recently screened the adhesion of different cell types on a range of peptides, demonstrating cell specific responses to specific peptides.6 Fibronectin derived peptide sequence RGD, however showed to be a common anchoring point for cell types.6 The response of RECs to ECM mimicking peptides has been barely explored. Research by Enemchukwu et al. revealed that tubular genesis and polarity establishment is dependent on RGD density in artificial matrices.4
The incorporation of ECM derived peptides in biomaterials often relies on laborious covalent modification of the surface.13,14 Supramolecular chemistry can provide bioactive materials functionalized through a simple mix-and-match approach.5,15 Carefully designed supramolecular motifs interact through non-covalent interactions, thereby acting as modular building blocks.15 The conjugation of such supramolecular motifs to bioactive peptides or polymers allows for engineering complex materials that stably integrate the different properties brought by the individual components (e.g. mechanical properties, cell adhesion, or anti-fouling).5,16–18 A clear example is the work on ureido-pyrimidinone (UPy) modified polycaprolactone based (PCL) membranes. A mixture of UPy-modified ECM mimicking peptides was successfully integrated into the membranes, resulting in stable monolayer formation of RECs.17,19 However, the exact contribution and optimal concentration of each peptide were not investigated. Moreover it is unknown if peptides can completely replace complex protein coatings for phenotype induction.
Supramolecular bis-urea (BU) motifs are exceedingly suited as modular building blocks. BU-moieties assemble into ribbon structures through bifurcated hydrogen bonds, and three to six of these ribbons assemble into nanoscale fibers (Fig. 1A).20–24 Several (peptide) additives have been modified with BU-moieties, which enabled the integration of the additives into BU based polymers (Fig. 1B).25–29 BU-moieties allow for more efficient surface functionalization compared to UPy-units, resulting in a more pronounced cellular response.29 In combination with the aforementioned features, the intrinsic modular nature of supramolecular BU-moieties makes them eminently suitable for the creation of complex screening libraries.
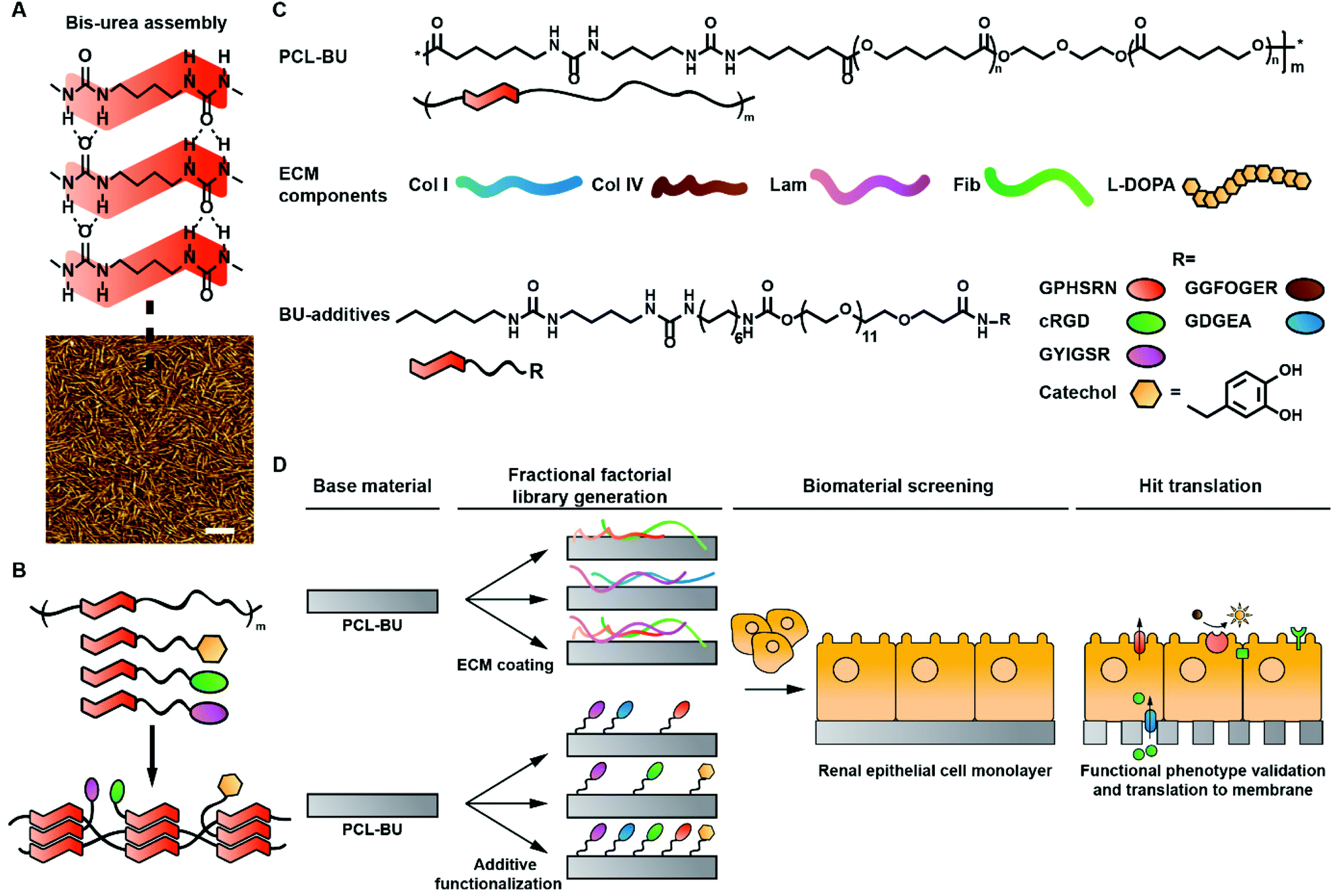 | ||
| Fig. 1 Schematic representation of library construction and chemical structures. A. Single stack assembly of bis-urea based hard phase motifs (top). Atomic force phase micrograph of a BU-modified polycaprolactone (PCL-BU) film depicting hard phase bis-urea fibers in a soft phase polymeric matrix. B. Schematic representation of BU-additive and -polymer assembly. C. Chemical structures of the base material PCL-BU and BU-modified additives. D. Schematic representation of library construction and evaluation. | ||
Until now the power of supramolecular biomaterials libraries has not been harnessed for screening elastomeric biomaterial compositions.30 Over the years a variety of covalent biomaterials libraries has been constructed, which mainly focused on basic cell adhesion or influencing stem cell behavior.6,31–34 In early work by Anderson et al., an impressive array was developed in which the polymer composition was varied using 24 different monomers to evaluate stem cell response.34 Other groups have evaluated biomaterial libraries based on protein coatings,32,33 peptide functionalizations,6,31 topography,35 and hydrophobicity.36 Protein and peptide functionalizations are seldom directly compared for a desired cell response, even though peptides are often selected to mimic proteins.37
In this study, a natural ECM protein coating library and a synthetic supramolecular additive library were investigated for their capacity to induce REC monolayers. Established statistical fractional factorial design of experiments (DoE) enables for creation and evaluation of both small libraries.38 The DoE approach is barely employed in synthetic polymer biomaterial screenings for surface modifications, due to difficulties faced when independently altering constituents in polymer systems.30 The natural ECM protein library consisted out of collagen type I (Col I), Col IV, Lam, fibronectin (Fib), and polymeric L-DOPA (Fig. 1B). L-DOPA is applied as an adhesive to improve protein anchoring to a material.10 The synthetic supramolecular additive library was comprised of several ECM derived peptides; (i) cyclic cRGD (cRGD) derived from laminin, collagens and fibronectin,14 (ii) PHSRN found in fibronectin,39 (iii) GFOGER and (iv) DGEA both derived from Col IV and Col I,13,40 and (v) YIGSR from laminin.12 The peptides were functionalized with BU-motifs to allow for modular integration into the base material (Fig. 1C, Scheme S1†). Additionally, a BU-functionalized monomeric catechol (BU-catechol) was included to potentially mimic a polymeric catechol L-DOPA coating (Fig. 1C). As a base material PCL-BU was selected, which has been shown to effectively present BU-peptides at the surface.29 Initial screening was performed with two different REC cell lines, i.e. renal proximal tubule epithelial cells (RPTEC) and human kidney 2 cells (HK-2). Their capacity to form a monolayer was assessed to elucidate cell specific responses to biomaterial functionalizations (Fig. 1D). Monolayer quality was characterized by quantifying epithelial cell–cell contact marker Zona Occludens 1 (ZO-1), as a measure for monolayer formation.41 Monolayers with matured ZO-1 indicate tight cell–cell contacts which are crucial for a cell based selective membrane in a bio-artificial kidney. Hits from both natural and synthetic libraries were directly compared in terms of monolayer coverage, presence of REC markers, apical transporters function, and matrix deposition. Finally, library hits were employed to create living electrospun membranes to move towards off-the-shelf bioactive membranes for a bio-artificial kidney.
Results & discussion
Library set-up
Biomaterial library combinations were generated by established fractional factorial design to statically leverage the ensuing data (Tables S1 and 2†). The five component ECM based library employed 16 initial screening conditions, while the six component synthetic library required 32 combinations. This resulted in low level aliasing of a primary effector with quaternary or pentanary interaction effects, for the 16 and 32 combination library, respectively. To quantify ZO-1 expression by REC monolayers a custom analysis script was developed inspired by previous work of Schophuizen et al.10 A grid was superimposed on images and the amount of intersections between the grid and ZO-1 positive cell–cell contacts was quantified (Fig. S1†).Natural ECM protein library screened for monolayer formation
The fractional factorial protein coating library revealed that pristine PCL-BU was incapable of maintaining a HK-2 monolayer with full coverage for a three week culture period (Fig. 2A, B and S2†). None of the protein coatings prevented defect and multilayer formation in HK-2 cell layers (Fig. 2A and B). The presence of laminin in a coating drastically improved the quality of the HK-2 monolayer in terms of confluency (Fig. 2A and B). The effect size, the size difference between groups, of laminin was significant compared to the base line (Fig. 2C). Indicating that laminin did not require other factors to exert this improvement.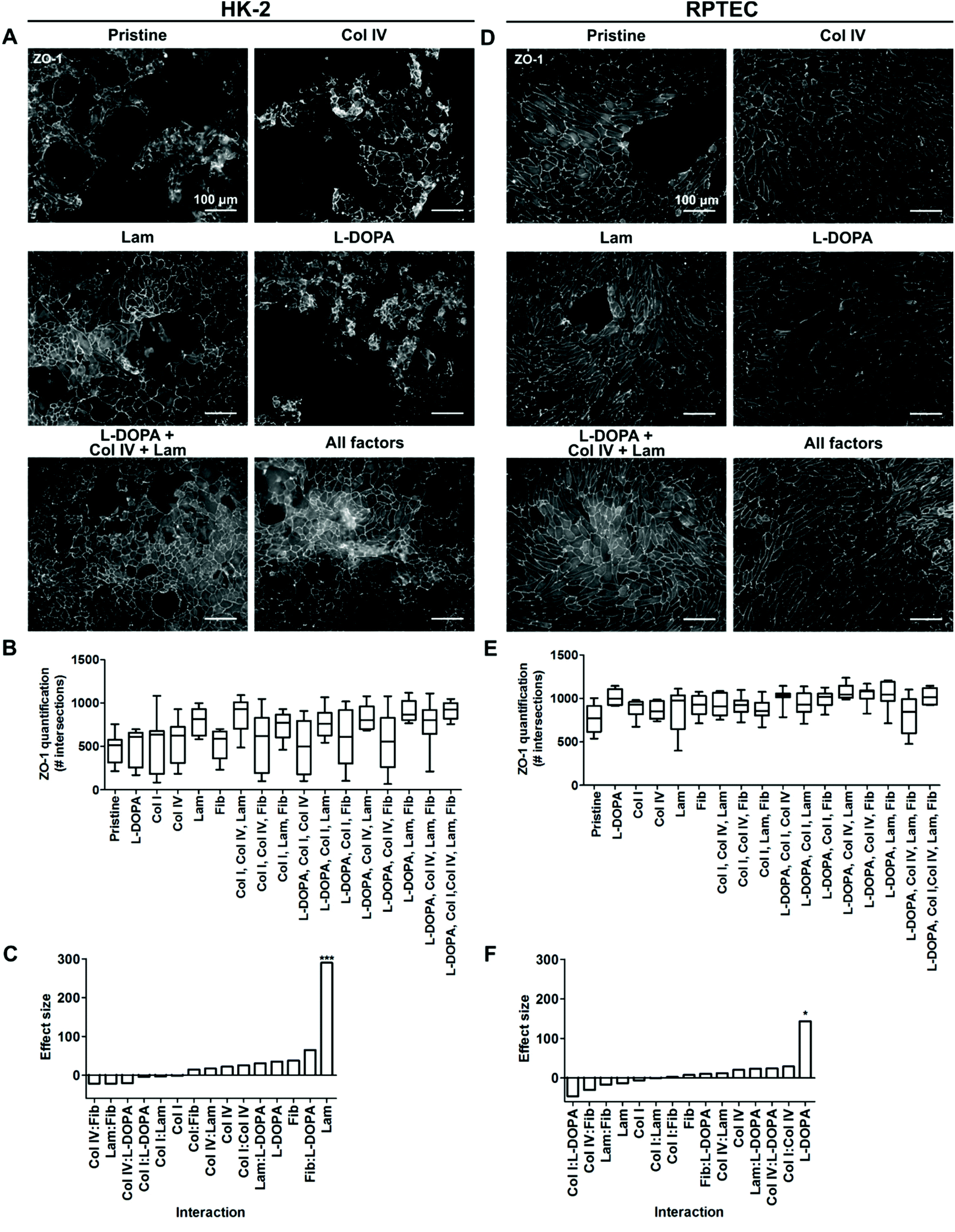 | ||
| Fig. 2 Renal epithelial cell monolayer response to a protein coating library. Selection of protein conditions depicting A HK-2 or D RPTEC monolayers stained for Zona Occludens-1 (ZO-1), scale bars are 100 μm. Pristine is uncoated PCL-BU, collagen type I (Col I) and type IV (Col IV), laminin (Lam), fibronectin (Fib). B, E Quantification of ZO-1 expression by HK-2 and RPTEC, respectively. C, F Effect size of single components and interactions by two coating components. The effect size is determined as the size difference between the groups including and excluding a component. n = 9, *p ≤ 0.05, ***p ≤ 0.001. | ||
Full confluency of RPTEC monolayers was not achieved on pristine PCL-BU. The addition of protein coatings did partly improve cell coverage (Fig. 2D, E and S3†). RPTEC monolayers with full to near full coverage were observed in most combinations of L-DOPA with proteins (Fig. 2D, E and S3†). The promiscuous synergistic behavior of L-DOPA with Col IV, Lam or Fib resulted in a large effect size of L-DOPA as a single factor, albeit the modest effect observed as single coating (Fig. 2D and F). To further investigate synergistic effects between L-DOPA and various ECM proteins, a full factorial screening was performed where L-DOPA was set as a constant and Col IV, Lam and Fib as variables. The presence of Col IV was shown to be the most important synergistic partner of L-DOPA with a significant effect size (Fig. S4†). The combination of L-DOPA with Col IV and Lam produced confluent monolayers with the highest intersection score, while large to small defects can be observed in the monolayers of the other combinations, i.e. including L-DOPA with only Col IV (Fig. 2D and S4†).
Taken together these results indicated cell specific differences between the two REC cell lines in response to ECM protein coatings. HK-2 monolayer formation benefitted from a Lam coating. RPTECs required a complex coating consisting of L-DOPA, Col IV and Lam to reach full confluency. The data are in line with previous reports by Zhang et al. who observed cell specific responses to ECM coatings between different types of REC.9 Secondly, our data confirms previous reports that L-DOPA, Col IV and Lam are of positive influence on REC monolayer formation.9,10 Catechols in the polymeric L-DOPA coating likely effectively tether proteins to the surface through both covalent and supramolecular interactions.42 The improved protein adhesion to the surface is postulated to improve cellular adhesion to the protein coating. The native basement membrane of REC is abundant in Col IV and Lam, thereby indicating that the coatings mimic parts of native micro-environmental cues.43
Synthetic supramolecular biomaterial library screening for monolayer formation
The employed BU-additive design entailing C6-urea-C4-urea-C12-(OEG)12-R, where R can be a variety of functional groups, was shown to be successful in presenting functional peptides at the biomaterial surface.29 Screening of the synthetic additive library indicated that all BU-additives were unable to exert a meaningful positive effect on monolayer quality of HK-2, with overgrowth and defects observed in the cell layer (Fig. 3A–C and S6†). The quality of the monolayer coverage was equal to that of pristine PCL-BU. The addition of BU-peptides to PCL-BU, used individually or in combinations, resulted in a patchy RPTEC layer on the materials after three weeks of culture (Fig. 3D, E and S7†). According to previous studies, BU-peptides additives were estimated to be effective at a concentration of 1 mol%.17,19,29 BU-peptide ineffectiveness could be caused by insufficient peptide presentation. Nevertheless, a concentration increase from 1 mol% to ultimately 5 mol% did not improve monolayer confluency for RPTEC cells (Fig. S8†).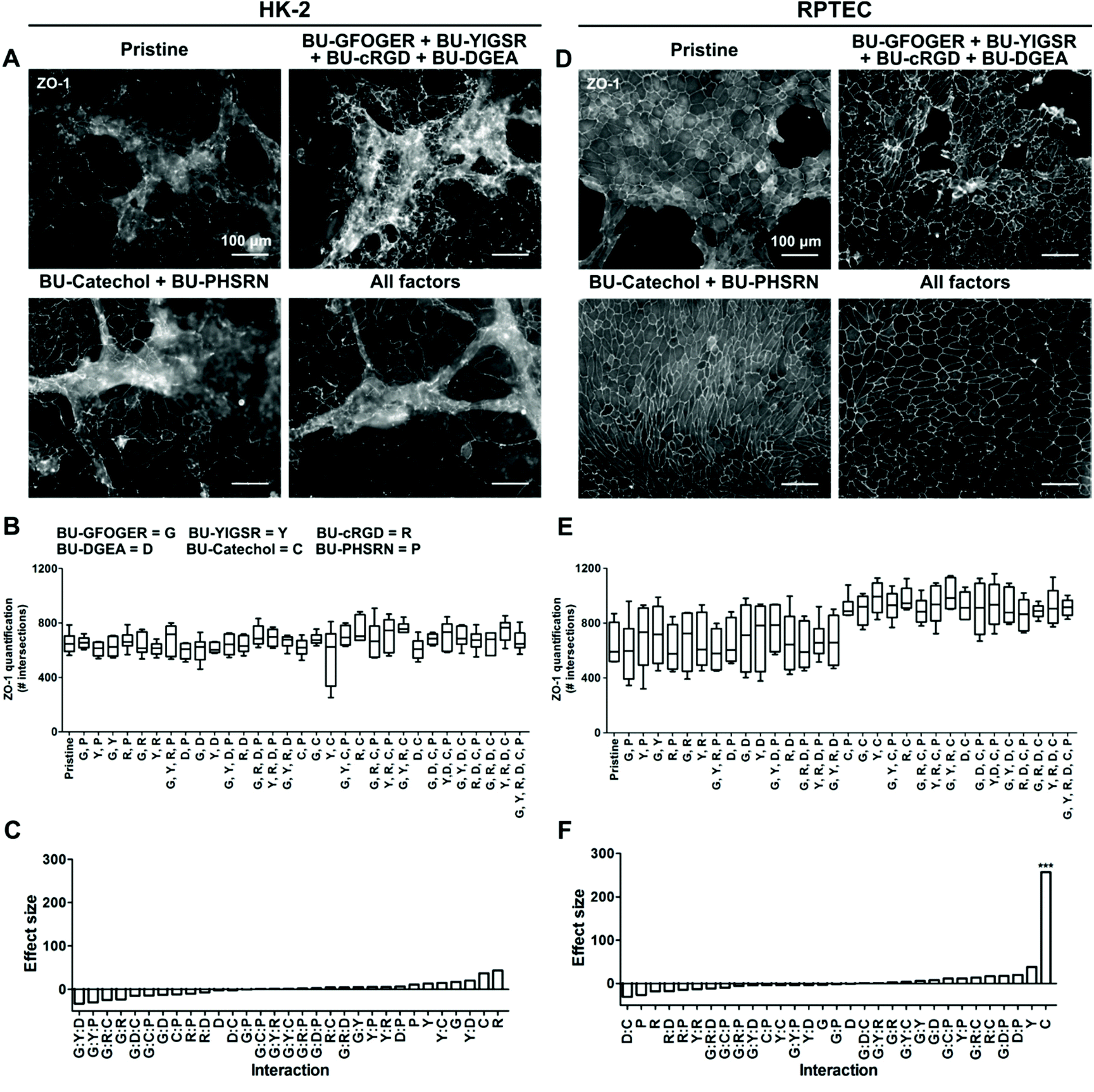 | ||
| Fig. 3 Renal epithelial cell monolayer response to an additive library. Selection of BU-additive conditions depicting A HK-2 or D RPTEC monolayers stained for Zona Occludens-1 (ZO-1), scale bar = 100 μm. Pristine is uncoated PCL-BU. B, E Quantification of ZO-1 expression by HK-2 and RPTEC, respectively. Pristine is unmodified PCL-BU, BU-GFOGER (G), BU-YIGSR, (Y), BU-cyclic RGD (R), BU-DGEA (D), BU-catechol (C), and BU-PHSRN (P) C, F Effect size of single components and interactions by two coating components. The effect size is determined as the size difference between the groups including and excluding a component. n = 6, ***p ≤ 0.001. | ||
The screening approach enabled us to identify a positive hit for monolayer formation. The addition of BU-catechol to PCL-BU did result in perfect to near perfect RPTEC monolayer formation in all combinations (Fig. 3D and E). The BU-catechol additive presented a significant effect size on monolayer coverage (Fig. 3F). Interestingly, subsequent investigation revealed BU-catechol to be active as a single component which retained its activity when its concentration was decreased from 5 mol% to 2.5 mol% (Fig. S8†). More frequent defect formation was observed in the RPTEC monolayers when the addition of BU-catechol was below 2.5 mol%. However, BU-catechol induced monolayers displayed better coverage quality than those found on peptide functionalized surfaces, further demonstrating its beneficial effect (Fig. S8†). Hereby, it is shown that the strong effect size produced by BU-catechol is independent of other factors (Fig. 3F).
In conclusion, screening of the synthetic library revealed the selected peptides were incapable of recapitulating the beneficial effect on monolayer formation found by protein coatings for both cell types under these experimental conditions. Remarkably, the simple reactive BU-catechol additive outperformed all additives and was able to induce RPTEC monolayer formation.
Reports in literature describe variable abilities of ECM mimicking peptides to induce a REC monolayer.17,19 In a previous studies, primary human REC formed confluent monolayers on peptide functionalized membranes, in the absence of flow.17 In contrast, this was not achieved by HK-2s, indicating cell specific responses.19 The presently employed BU-system has superior effective peptide presenting properties compared to the previously employed material system.29 This suggests that any initial cues provided by the BU-peptides are unable to steer the currently selected cell types to form full coverage monolayers. Monomeric catechol surface functionalization by BU-catechol addition resulted in fully confluent RPTEC monolayers, yet not for HK-2 cells. Interestingly, sole polymeric catechol L-DOPA coating was unable to promote RPTEC monolayer formation. This discrepancy can potentially be explained by work of Choi et al., where it was speculated that polymerized L-DOPA coatings can detach and exert detrimental effects on cells, acting as a double edge sword.44 Stable anchoring of monomeric catechols to the surface prevents detachment and retains the adhesive character of the surface.44 We speculate that a subsection of BU-catechol reacts to proteins bound on the cell membrane which initiates initial attachment, afterwards proteins deposited by the cells are bound to the surface. Unraveling the exact mechanism of catechol mediated cell adhesion is an interesting topic for future studies.
Comparison of natural and synthetic modifications on monolayer formation
Bio-artificial kidneys are based on the premise that a confluent monolayer of REC can act as a selective barrier for filtrate fine tuning. HK-2 were unable to achieve a confluent monolayer on both coatings and additive modifications. Moreover, HK-2 lack several key transporters required for filtrate fine-tuning compared to RPTECs.45,46 Initial quantification of ZO-1 positive cell–cell contacts revealed the degree of cell coverage and the basic premise of the cells epithelial nature to be similar on a complex protein coated surface and on a simple reactive surface functionalization, i.e.L-DOPA with Col IV and Lam, and BU-catechol, for RPTEC.41 However, BU-catechol cannot provide complex signaling in the same manner as the natural basement membrane proteins. Therefore we assume the mechanism behind the similar monolayer coverage are initially different. In a direct comparison it was assessed whether the monolayers were of equal quality in terms of coverage, epithelial marker expression and function.ZO-1 expression and quantification revealed similar levels of cell coverage between L-DOPA with Col IV and Lam coated samples on one hand, and BU-catechol functionalized materials on the other hand (Fig. 4A). In this experiment both conditions outperformed pristine PCL-BU in monolayer formation. Additional epithelial markers, i.e. primary cilia, megalin expression, and γ-glutamyltransferase activity, were found to be conserved over all material conditions (Fig. 4B–D), indicating that the RPTEC cell line stably preserves its epithelial nature over the applied culture period. Furthermore, the deposition of basement membrane proteins Col IV and Lam γ1 by the RPTECs was investigated. Under all material conditions Col IV and Lam is expressed, with Col IV more abundantly visible (Fig. 4E). RPTECs cultured on pristine PCL-BU presented Col IV in a punctuated form and which was mainly situated intracellular. In response to the L-DOPA with Col IV and Lam coating, RPTECs deposited small Col IV fibrils homogenously over the surface under the cells. Cells cultured on BU-catechol functionalized surfaces deposited Col IV in similar fibrils, but in lesser extent and more heterogeneously in comparison to cells cultured L-DOPA with Col IV and Lam coating. Additionally, larger Col IV aggregates were observed on BU-catechol surfaces (Fig. 4E). These deposition pattern are unlike the initial pattern provided by the L-DOPA with Col IV and Lam coating pre-cell culture, which presented small globular clusters of co-localized Col IV and Lam γ1 (Fig. S9†).
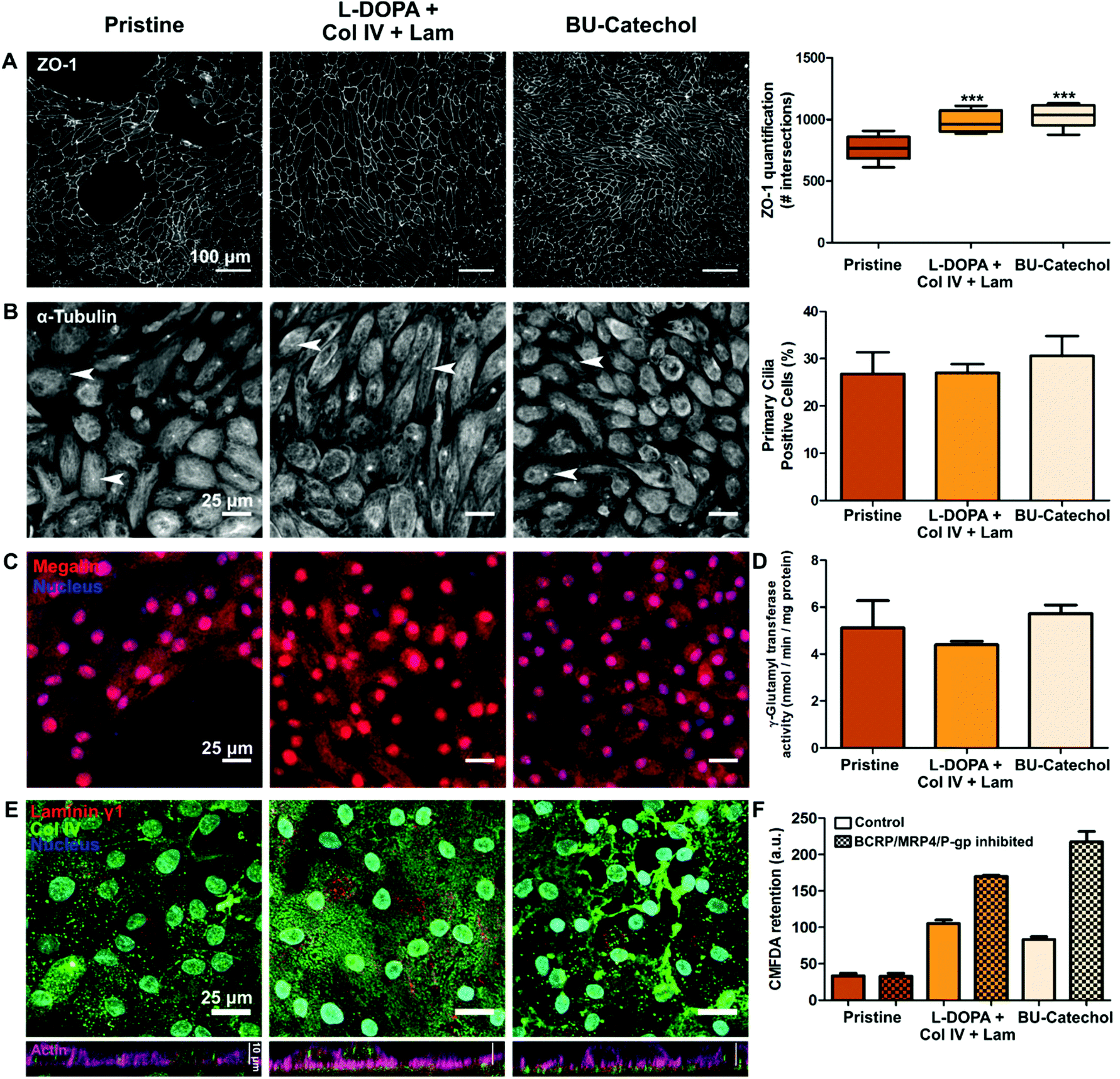 | ||
| Fig. 4 Epithelial phenotype of monolayers on natural vs. synthetic modified materials. Left column pristine PCL-BU, middle column PCL-BU coated with L-DOPA, collagen type IV (Col IV) and laminin (Lam), right column PCL-BU functionalized with 5 mol% BU-catechol. A Monolayer formation of RPTECs, cells stained for Zona Occludens-1 (ZO-1; left), and quantification of ZO-1 (right). Scale bar is 100 μm. B Cells stained for primary cilia (α-tubulin; left), arrows indicate examples of primary cilia. Scale bar = 25 μm. Amount of primary cilia positive cells (right). C Megalin (red) expression by cells, nucleus (blue), scale bar = 25 μm. D γ-Glutamyltransferase activity of cells corrected for cell numbers. E Extra cellular matrix deposition by cells, top row depicting top view with laminin γ1 (red), Col IV (green), and nucleus (blue), scale bar is 25 μm. Bottom row side view laminin γ1 (red), Col IV (green) and actin (pink), scale bar is 10 μm. F Ratio of fluorescent model substrate presence in cells where apical efflux transporters are inhibited and uninhibited. Mean ± SEM depicted from n = 3, ***p ≤ 0.001. | ||
The collective apical efflux transporter function of breast cancer resistance protein (BCRP), multidrug resistance protein 4 (MRP4) and P-glycoprotein (P-gp) was assessed by the clearance efficiency of 5-chloromethylfluorescein diacetate (CMFDA). Cells cultured on pristine PCL-BU showed no effective transporter activity, however cells on L-DOPA with Col IV/Lam, or BU-catechol functionalized PCL-BU surfaces showed clearance of CMFDA in an inhibited state (Fig. 4F). Cells on BU-catechol functionalized PCL-BU exhibited the strongest collective efflux transport function of all material conditions. Transporter function was shown to improve over time as a REC monolayer matures.46,47 RPTECs cultured on pristine PCL-BU were incapable of forming a monolayer with full coverage, hindering monolayer maturation, and thereby potentially limiting efflux transporter expression.
Overall the data indicated that both the complex L-DOPA with Col IV and Lam protein coating, and a simple synthetic BU-catechol functionalization resulted in a monolayers of similar quality, in terms of cell coverage, REC marker expression and transporter activity. Moreover, both functionalizations showed improved matrix deposition by cells compared to pristine PCL-BU. Taken together the data implies that a simple synthetic modification can replace a complex protein coating.
Both natural and synthetic modifications enable living membrane formation
Membranes which permit efficient diffusion and potentially mimic topographical ECM features are desired for a bio-artificial kidney. Both features are provided by the highly interconnected fibrous electrospun membranes. Here, we assessed whether hits from both libraries could be translated from flat polymer films towards living membranes. PCL-BU and PCL-BU with 5 mol% BU-DOPA were successfully electrospun in membranes with similar fiber morphology, respective fiber diameters of 0.79 ± 0.20 μm and 0.65 ± 0.19 μm (Fig. 5A). An Arnow's staining revealed accessible catechols in BU-catechol functionalized materials, while pristine samples remained negative (Fig. 5A). Fully confluent monolayers were observed on L-DOPA with Col IV and Lam, or BU-catechol modified membranes, in line with results found on flat polymer films (Fig. 5B). These monolayers effectively limited passive diffusion of FITC-inulin from the basolateral to the apical compartment (Fig. 5C). RPTECs showed effective basolateral uptake transporter organic cation transporter 2 (OCT2) activity under all material conditions, with the highest activity found in cells cultured on L-DOPA with Col IV and Lam coated membranes (Fig. 5D).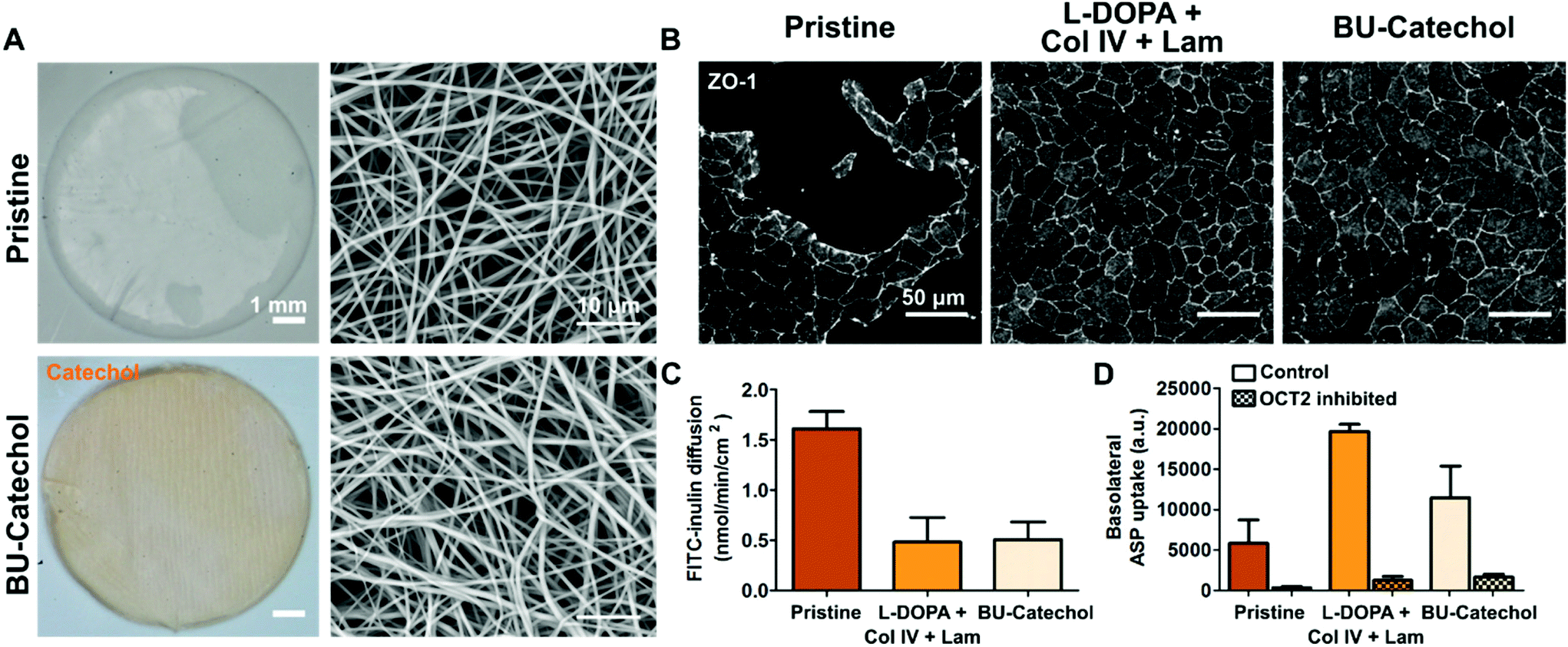 | ||
| Fig. 5 Achieving functional living membranes through different bio-activation routes. A Left panels show macroscopic images of PCL-BU membranes, either pristine or with 5 mol% BU-catechol incorporation subjected to an Arnow's staining for catechols (orange). The right panels show scanning electron micrographs of the membranes. B RPTEC cultured on pristine PCL-BU (left column), PCL-BU coated with L-DOPA, collagen type IV (Col IV) and laminin (Lam; middle column), PCL-BU functionalized with 5 mol% BU-catechol (right column) for 3 weeks, cells stained for Zona Occludens-1 (ZO-1; grey). Scale bar is 50 μm. C Membrane permeability defined by the level of passive FITC-inulin transport between the basolateral and apical compartment. D Basolateral transporter OCT2 activity assessment by fluorescent model substrate (4-(4-(dimethylamino)styryl)-N-methylpyridinium iodide, ASP) uptake presence in cells where OCT2 is inhibited and uninhibited. Mean ± SEM depicted from n = 3. | ||
Conclusion
We reported a powerful screening approach, based on parallel screening of two, natural or synthetic, biomaterial libraries to identify the ideal biomaterial composition for renal monolayer formation. The complete set of tested biomaterials comprised of a library of commonly used natural ECM-proteins coatings and a synthetic supramolecular additive biomaterial library (based on bioactive peptide sequences). Natural basement membrane components Col IV and Lam with polymerized L-DOPA achieved monolayer formation. In contrast, small ECM mimicking peptides were unable to induce stable monolayer formation, whereas a simple monomeric catechol additive did. In addition, the developed approach was suitable to distinguish cell-specific responses between employed REC cell lines. Importantly, the simple catechol modification yielded cell monolayers of equal quality in terms of epithelial phenotype and transporter function compared to the complex coating mixture of L-DOPA with Col IV and Lam. These findings can be translated to produce bioactive membranes for applications such as the bio-artificial kidney or nephrotoxicity assays as demonstrated by the formation of bioactive spun membranes. Moreover, this study displayed the strength of the mix-and-match principle of supramolecular BU based biomaterials.Outlook
Currently, only one base polymer and a few selected ECM peptide mimics were selected to screen for a response. The library has the potential to be expanded with different base polymers and to include more ECM protein mimicking peptides such as IKVAV, full length GFOGER or PHSRNKRGDS.12,48 In addition, other cell environment cues could be incorporated such as cell–cell contact mimics (e.g. HAVS), and glycosaminoglycan mimics (e.g. sulfated peptides).49,50 Overall the supramolecular approach is eminently suited for the fast, easy construction and screening of versatile biomaterial libraries aimed at identifying the ideal biomaterial for a biomedical applications.Materials & methods
Synthesis of BU functionalized peptides and catechol
Peptide synthesis of GDGEA, GPHSRN, GYIGSR and GGFOGER and subsequent conjugation to BU-synthon (SyMO-Chem) are extensively described in the ESI (Schemes S2–5†). The synthesis of BU-catechol is additionally reported in the ESI (Scheme S6, Fig. S5†). BU-cRGD was synthesized as reported before.29Biomaterial library preparation
A solution of 15 mg mL−1 PCL-BU (Mn = 2.7 kg mol−1; per segmented unit, SyMO-Chem) in hexafluoroisopropanol (HFIP; Fluorochem) was prepared and left to dissolve overnight. A glass bottom 96-well plate (Greiner Bio-one) was coated with PCL-BU by pipetting 40 μL of solution per well. Through solvent evaporation phase separation a thin polymer film was formed on the glass under a maximum relative humidity of 40%. The well plate was placed in vacuo overnight to ensure evaporation of the organic solvent. Samples were UV-sterilized for 10 min and subsequently pre-wetted with PBS (Sigma-Aldrich) for 10 min. ECM protein screening conditions were applied according to Table S1.† 2 mg mL−1L-DOPA (Sigma-Aldrich) in 10 mM TRIS-buffer (Sigma-Aldrich) pH 8.5 was left to dissolve and polymerize for 1 h at 37 °C. PBS was aspirated and sterile filtered L-DOPA solution was applied where applicable and incubated for 4 min at 37 °C. Subsequently, samples were washed with PBS and left to dry for 5 min at RT. Four times concentrated stocks of ECM protein were prepared in PBS, Rat tail Col I (1200 μg mL−1, Corning), bovine Fib (120 μg mL−1, Biomedical Technologies Inc.), Col IV from human placenta (100 μg mL−1, Sigma-Aldrich), and Lam from Engelbreth-Holm-Swarm murine sarcoma basement membrane (120 μg mL−1, Sigma-Aldrich). According to Table S1,† 25 μL of protein coating solution and PBS were added in each well to achieve an end volume of 100 μL. The ECM protein solutions were applied for 30 minutes at 37 °C and were subsequently aspirated and washed with PBS. The films were left to dry for 5 minutes after which samples were immediately used for cell culture purposes. The conditions were performed in triplo, and replicated three times. The screening combinations for the BU-peptides and BU-catechol were prepared as according to Table S2.† Per condition a 15 mg mL−1 PCL-BU solution in HFIP was complemented with additives to achieve 1 mol% of each of the applicable BU-peptides and 5 mol% of BU-catechol in the final casting solution. Thin polymer films were produced in triplo per condition as described before for pristine PCL-BU. Additionally, samples were prepared with 1.25 mol%, 2.5 mol%, and 5 mol% of single BU-peptide functionalized surfaces, or PCL-BU was functionalized with 0.3 mol%, 0.6 mol%, 1.25 mol%, 2.5 mol% or 5 mol% BU-catechol. Samples were UV-sterilized for 10 min before usage for cell culture.Membrane fabrication and characterization
Electrospun membranes were produced from a 200 mg mL−1 PCL-BU in HFIP solution with or without 5 mol% BU-catechol. Electrospinning was performed on an EC-CLI (IME Technologies), polymer fibers were collected on a cylindrical target (∅ = 28 mm) wrapped in aluminum foil while rotating at 500 rpm. The polymer solution was fed through a nozzle (∅ = 1.0–0.8 mm) with a rate of 20 μL min−1, the nozzle tip was 21.5 cm removed from the collector with a scanning distance of 100 mm. A voltage difference of 25 kV was applied to enable spinning of around 800 μL of solution. The chamber temperature was set to 23 °C and a relative humidity of 30%. Scanning electron microscopy and an Arnows staining (i.e. staining for catechols) were performed as described in literature.51 The experiments were performed in triplicate. Circular membranes were punched with a diameter of 8 mm and mounted in custom modified 24-well transwell inserts.Culture of renal epithelial cell lines
HK-2 cells (ATCC) were cultured in complete medium consisting of Dulbecco's modified Eagle medium (DMEM; Gibco) supplemented with 10% v/v fetal bovine serum (FBS; Greiner Bio-one) and 1% v/v penicillin–streptomycin solution (Invitrogen) in a humidified atmosphere at 37 °C and 5% CO2. RPTEC cells (RPTECs-TERT1; ATCC) were cultured in complete medium consisting of DMEM:F-12 nutrient mixture (Gibco), L-glutamine and 15 mM HEPES. Furthermore, the medium was supplemented with 1% v/v penicillin–streptomycin solution, RPTEC growth kit of ATCC (PCS-999-058, PCS-999-059) and 0.1 mg mL−1 G418 (Sigma-Aldrich). HK-2 and RPTECs cells were seeded respectively with a density of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 160000 cells per cm2 on samples to form a monolayer 3 to 4 days. Monolayers are defined as a (confluent) single cell layer. Cells were kept in culture for 3 weeks, with medium changes every 2 to 3 days, to evaluate screening conditions over a long-term.
000 and 160000 cells per cm2 on samples to form a monolayer 3 to 4 days. Monolayers are defined as a (confluent) single cell layer. Cells were kept in culture for 3 weeks, with medium changes every 2 to 3 days, to evaluate screening conditions over a long-term.
Immunostaining and imaging of cells
Cells were washed with PBS and fixated with 3.7% formaldehyde in PBS for 10 minutes. This was followed by a second wash with PBS and a permeabilization step with 0.5% v/v Triton X-100 in PBS for 10 minutes. The solution was aspirated and the cells were washed twice with PBS. To prevent non-specific binding of antibodies, samples were incubated with 5% BSA (Roche) in washing buffer (0.05% v/v Triton X-100 in PBS). The blocking solution was aspirated and samples were incubated with primary antibodies against selected biomarkers (Table S3†) in 2% BSA in washing buffer for 1 h at RT. Cells were subsequently washed with washing buffer and incubated with secondary antibodies and phallodin-488 (Sigma-Aldrich; Table S3†) for 45 minutes. During the final 10 min DAPI nuclei staining (1:500 dilution, D9542, Sigma) was added. Samples were washed three times with PBS. Cells were imaged using an Axiovert 200M microscope (Zeiss) or TCS SP5X confocal microscope (Leica). Amount of cell nuclei were quantified with ImageJ (v1.48, NIH), cilia manually counted in a blinded fashion.
ZO-1 quantification model
Per well, three images were acquired of the ZO-1 localization in the sample. A custom automatic MatLab script was written to quantify ZO-1 expression in cells. Briefly, the following steps were executed. Firstly images were subjected to a sharpening filter and contrast stretching followed by subtraction of grey values of the DAPI staining to reduce the signal of non-specific ZO-1 staining around the nuclei of the cells. With an automatically calculated global threshold images converted into binary images. Remaining noise was removed through the application of a Wiener filter. An identical twenty by twenty grid with a 3 pixel line width was imposed on the images. For each grid line the intensity was averaged over these 3 pixels to create a 1D intensity profile. The profiles per image were analyzed for peaks with Matlab® findpeaks function. Peak properties for detection (width, minimum area and minimum peak height) were established by visual inspection of pilot data in order to minimize the amount of false positives and false negatives. For each image the total number of detected peaks on the grid lines are presented and the model calculates the total amount of peaks per image. The term “intersections” is used to refer to these measured intensity peaks. The script has been made publicly available on gitlab.tue.nl/stem/ZOlab.γ-Glutamyltransferase activity
γ-Glutamyltransferase (GGT) is an enzyme secreted by renal epithelial cells. Cells were incubated with assay mixture containing 1 mM L-glutamic acid γ-(4-nitroanilide) and 20 mM glycylglycine in DMEM:F12 medium for 1 h at 37 °C. Cells were incubated with 20 mM glycylglycine in DMEM:F12 as control. A serial diluted p-nitroaniline standard curve was prepared with a range of 5 mM to 2.5 μM in DMEM:F12 medium. Absorbance was measured at 405 nm (SynergyHT, BioTEK). Cells were lysed in 20 mM TRIS-buffer pH 8 supplemented with 150 mM NaCl, 1 mM EDTA and 1% v/v Triton X-100. Protein content was determined through a BCA assay (Thermo Scientific), which was performed according to manufacturer's protocol. The experiment was performed in triplicate.Transporter activity
Efflux transporter functionality was assessed through inhibition of BCRP, MRP4, and P-gp by an inhibitor cocktail adapted from Caetano-Pinto et al.52 In short, cell were washed with PBS and exposed either to an inhibitor cocktail containing 5 μM KO143 (K2144, Sigma), 5 μM MK571 (M7571, Sigma), and 2 μM PSC833 (4042, Tocris) in completed growth medium, or to completed growth medium for 30 min at 37 °C. After pre-inhibition, the cells were incubated with 1.25 μM CMFDA (C7025, Molecular Probes) in completed growth medium correspondingly with or without inhibitors. Cells were incubated for 40 min at 37 °C and subsequently washed with ice cold completed medium. Cells were lysed with 0.5% v/v Triton X-100 in PBS for 30 min at 37 °C. The lysate was collected and fluorescence was measured (Ex: 492 nm, Em: 517 nm, SynergyHT). The experiment was performed in triplicate.Basolateral uptake receptor OCT-2 activity was assessed through adaptation of previously reported protocols.46,53 In short, cells were washed with PBS and pre-exposed either in the presence or absence of 100 μM OCT-2 inhibitor tetrapentyl ammonium chloride (Sigma-Aldrich) in completed growth medium without antibiotics for 30 min at 37 °C. Subsequently, fluorescent substrate 4-(4-(dimethylamino)styryl)-N-methylpyridinium iodide (ASP) was added in the basolateral compartment to achieve a final concentration of 12.5 μM. Cells were lysed with 1% v/v Triton X-100 in PBS for 30 min after substrate incubation for 24 h at 37 °C. The lysate was collected and fluorescence was measured (Ex: 485 nm, Em: 590 nm, SynergyHT). The experiment was performed in triplicate.
Monolayer permeability
A FITC-inulin diffusion assay was performed to assess monolayer permeability, as described in literature.54Statistics
An established standard statistical fractional factorial design was employed to reduce the number of ECM protein and peptide combinations in the initial screening library. Design and analysis of the experiments was done using R (v3.4.4, R Foundation) with Rcmdr.DoE plugin (v0.12-3, Ulrike Groemping). A 2 level fractional factorial design with 5 factors for ECM coatings and 6 factors for ECM peptides was selected with 16 or 32 unique conditions (Tables S1 and 2†). This resulted in respectively a design resolution of V and IV. Level III ≥ design resolution is found appropriate for screening purposes. Main effects, two- and three-factor interactions along with statistical significance were calculated according to standard fractional factorial analysis by R and the Rcmdr.DoE plugin. Significance was assumed α ≤ 0.05. Cell phenotype and function data was subjected to a Kruskal–Wallis test with a Dunns post-test in which all conditions were compared in Prism (GraphPad Software Inc.).Author contributions
RG, AV and PD conceptualized the experiments. RG and AV preformed the screening of the protein coating library. RG preformed screening of the BU-additive library, and subsequent studies on epithelial phenotype and membrane production. MT and AV developed quantification script for ZO-1. JS and PF synthesized all BU-additives. Manuscript was written by RG and PD, and all other authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the European Research Council (FP7/2007–2013) ERC Grant Agreement 308045, and the Ministry of Education, Culture and Science (Gravity programs 024.001.035 and 024.003.013).References
- A. J. Keung, S. Kumar and D. V. Schaffer, Annu. Rev. Cell Dev. Biol., 2010, 26, 533–556 CrossRef CAS PubMed.
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Nature, 2009, 462, 433–441 CrossRef CAS PubMed.
- N. Gjorevski, N. Sachs, A. Manfrin, S. Giger, M. E. Bragina, P. Ordóñez-Morán, H. Clevers and M. P. Lutolf, Nature, 2016, 539, 560–564 CrossRef CAS PubMed.
- N. O. Enemchukwu, R. Cruz-Acuña, T. Bongiorno, C. T. Johnson, J. R. García, T. Sulchek and A. J. García, J. Cell Biol., 2016, 212, 113–124 CrossRef CAS PubMed.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed.
- S. Sharma, M. Floren, Y. Ding, K. R. Stenmark, W. Tan and S. J. Bryant, Biomaterials, 2017, 143, 17–28 CrossRef CAS.
- P. Aebischer, T. K. Ip, G. Panol and P. M. Galletti, Life Support Syst., 1987, 5, 159–168 CAS.
- J. Jansen, M. Fedecostante, M. J. Wilmer, J. G. Peters, U. M. Kreuser, P. H. van den Broek, R. A. Mensink, T. J. Boltje, D. Stamatialis, J. F. Wetzels, L. P. van den Heuvel, J. G. Hoenderop and R. Masereeuw, Sci. Rep., 2016, 6, 26715 CrossRef CAS PubMed.
- H. Zhang, F. Tasnim, J. Y. Ying and D. Zink, Biomaterials, 2009, 30, 2899–2911 CrossRef CAS PubMed.
- C. M. S. Schophuizen, I. E. De Napoli, J. Jansen, S. Teixeira, M. J. Wilmer, J. G. J. Hoenderop, L. P. W. Van den Heuvel, R. Masereeuw and D. Stamatialis, Acta Biomater., 2015, 14, 22–32 CrossRef CAS PubMed.
- E. Battista, F. Causa, V. Lettera, V. Panzetta, D. Guarnieri, S. Fusco, F. Gentile and P. A. Netti, Biomaterials, 2015, 45, 72–80 CrossRef CAS PubMed.
- G. Delaittre, A. M. Greiner, T. Pauloehrl, M. Bastmeyer and C. Barner-Kowollik, Soft Matter, 2012, 8, 7323 RSC.
- I. W. Hamley, Chem. Rev., 2017, acs.chemrev.7b00522.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS.
- G. C. van Almen, H. Talacua, B. D. Ippel, B. B. Mollet, M. Ramaekers, M. Simonet, A. I. P. M. Smits, C. V. C. Bouten, J. Kluin and P. Y. W. Dankers, Macromol. Biosci., 2016, 16, 350–362 CrossRef CAS PubMed.
- P. Y. W. Dankers, J. M. Boomker, A. Huizinga-van der Vlag, E. Wisse, W. P. J. Appel, F. M. M. Smedts, M. C. Harmsen, A. W. Bosman, W. Meijer and M. J. A. van Luyn, Biomaterials, 2011, 32, 723–733 CrossRef CAS PubMed.
- B. D. Ippel, E. E. Van Haaften, C. V. C. Bouten and P. Y. W. Dankers, ACS Appl. Polym. Mater., 2020, 2, 3742–3748 CrossRef CAS PubMed.
- B. B. Mollet, I. L. J. Bogaerts, G. C. van Almen and P. Y. W. Dankers, J. Tissue Eng. Regener. Med., 2017, 11, 1820–1834 CrossRef CAS PubMed.
- N. E. Botterhuis, S. Karthikeyan, A. J. H. Spiering and R. P. Sijbesma, Macromolecules, 2010, 43, 745–751 CrossRef CAS.
- R. A. Koevoets, R. M. Versteegen, H. Kooijman, A. L. Spek, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 2999–3003 CrossRef CAS.
- M. C. Etter, Z. Urbanczyk-Lipkowska, M. Zia-Ebrahimi and T. W. Panunto, J. Am. Chem. Soc., 1990, 112, 8415–8426 CrossRef CAS.
- J. Van Esch, S. De Feyter, R. M. Kellogg, F. De Schryver and B. L. Feringa, Chem. – Eur. J., 1997, 3, 1238–1243 CrossRef CAS.
- R. M. Versteegen, R. Kleppinger, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 772–783 CrossRef CAS.
- E. Wisse, A. J. H. Spiering, E. N. M. van Leeuwen, R. A. E. Renken, P. Y. W. Dankers, L. A. Brouwer, M. J. A. van Luyn, M. C. Harmsen, N. A. J. M. Sommerdijk and E. W. Meijer, Biomacromolecules, 2006, 7, 3385–3395 CrossRef CAS.
- E. Ressouche, S. Pensec, B. Isare, J. Jestin and L. Bouteiller, Langmuir, 2016, 32, 11664–11671 CrossRef CAS.
- Y. Xiang, E. Moulin, E. Buhler, M. Maaloum, G. Fuks and N. Giuseppone, Langmuir, 2015, 31, 7738–7748 CrossRef CAS PubMed.
- B. D. Ippel, H. M. Keizer and P. Y. W. Dankers, Adv. Funct. Mater., 2019, 29, 1805375 CrossRef.
- R. C. van Gaal, A. B. C. Buskermolen, B. D. Ippel, P. P. K. H. Fransen, S. Zaccaria, C. V. C. Bouten and P. Y. W. Dankers, Biomaterials, 2019, 224, 119466 CrossRef CAS PubMed.
- J. P. Jung, J. V. Moyano and J. H. Collier, Integr. Biol., 2011, 3, 185 CrossRef CAS PubMed.
- D. Zhang, J. Lee, M. B. Sun, Y. Pei, J. Chu, M. U. Gillette, T. M. Fan and K. A. Kilian, ACS Cent. Sci., 2017, 3, 381–393 CrossRef CAS PubMed.
- C. J. Flaim, S. Chien and S. N. Bhatia, Nat. Methods, 2005, 2, 119–125 CrossRef CAS PubMed.
- S. Gobaa, S. Hoehnel, M. Roccio, A. Negro, S. Kobel and M. P. Lutolf, Nat. Methods, 2011, 8, 949–955 CrossRef CAS PubMed.
- D. G. Anderson, S. Levenberg and R. Langer, Nat. Biotechnol., 2004, 22, 863–866 CrossRef CAS PubMed.
- H. V. Unadkat, M. Hulsman, K. Cornelissen, B. J. Papenburg, R. K. Truckenmüller, A. E. Carpenter, M. Wessling, G. F. Post, M. Uetz, M. J. T. Reinders, D. Stamatialis, C. A. van Blitterswijk and J. de Boer, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16565–16570 CrossRef CAS PubMed.
- Q. Zhou, L. Ge, C. F. Guimarães, P. T. Kühn, L. Yang and P. van Rijn, Adv. Mater. Interfaces, 2018, 5, 1800504 CrossRef.
- E. H. Nguyen, W. T. Daly, N. N. T. Le, M. Farnoodian, D. G. Belair, M. P. Schwartz, C. S. Lebakken, G. E. Ananiev, M. A. Saghiri, T. B. Knudsen, N. Sheibani and W. L. Murphy, Nat. Biomed. Eng., 2017, 1, 1–14 CrossRef.
- J. Antony, Design of Experiments for Engineers and Scientists, Elsevier, 2014 Search PubMed.
- S. Aota, M. Nomizu and K. M. Yamada, J. Biol. Chem., 1994, 269, 24756–24761 CrossRef CAS.
- C. G. Knight, L. F. Morton, a. R. Peachey, D. S. Tuckwell, R. W. Farndale and M. J. Barnes, J. Biol. Chem., 2000, 275, 35–40 CrossRef CAS PubMed.
- M. P. Rastaldi, F. Ferrario, L. Giardino, G. Dell'Antonio, C. Grillo, P. Grillo, F. Strutz, G. A. Müller, G. Colasanti and G. D'Amico, Kidney Int., 2002, 62, 137–146 CrossRef PubMed.
- J. Yang, M. A. Cohen Stuart and M. M. G. Kamperman, Chem. Soc. Rev., 2014, 43, 8271–8298 RSC.
- J. H. Miner, Kidney Int., 1999, 56, 2016–2024 CrossRef CAS PubMed.
- J. S. Choi, P. B. Messersmith and H. S. Yoo, Macromol. Biosci., 2014, 14, 270–279 CrossRef CAS PubMed.
- S. E. Jenkinson, G. W. Chung, E. van Loon, N. S. Bakar, A. M. Dalzell and C. D. A. Brown, Pflügers Arch. Eur. J. Physiol., 2012, 464, 601–611 CrossRef CAS PubMed.
- L. Aschauer, G. Carta, N. Vogelsang, E. Schlatter and P. Jennings, Toxicol. In Vitro, 2015, 30, 95–105 CrossRef CAS PubMed.
- S. P. Hämmerle, B. Rothen-Rutishauser, S. D. Krämer, M. Günthert and H. Wunderli-Allenspach, Eur. J. Pharm. Sci., 2000, 12, 69–77 CrossRef.
- J. Valdez, C. D. Cook, C. C. Ahrens, A. J. Wang, A. Brown, M. Kumar, L. Stockdale, D. Rothenberg, K. Renggli, E. Gordon, D. Lauffenburger, F. White and L. Griffith, Biomaterials, 2017, 130, 90–103 CrossRef CAS PubMed.
- N. Kobayashi, J. Pharmacol. Exp. Ther., 2005, 317, 309–316 CrossRef PubMed.
- H. D. Maynard and J. A. Hubbell, Acta Biomater., 2005, 1, 451–459 CrossRef.
- L. E. Arnow, J. Biol. Chem., 1937, 118, 531–537 CrossRef CAS.
- P. Caetano-Pinto, M. J. Janssen, L. Gijzen, L. Verscheijden, M. J. G. Wilmer and R. Masereeuw, Mol. Pharm., 2016, 13, 933–944 CrossRef CAS.
- C. M. S. Schophuizen, M. J. Wilmer, J. Jansen, L. Gustavsson, C. Hilgendorf, J. G. J. Hoenderop, L. P. Van Den Heuvel and R. Masereeuw, Pflugers Arch. Eur. J. Physiol., 2013, 465, 1701–1714 CrossRef CAS PubMed.
- J. Jansen, C. M. Schophuizen, M. J. Wilmer, S. H. Lahham, H. A. Mutsaers, J. F. Wetzels, R. A. Bank, L. P. van den Heuvel, J. G. Hoenderop and R. Masereeuw, Exp. Cell Res., 2014, 323, 87–99 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01930e |
| This journal is © The Royal Society of Chemistry 2021 |