
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle chain variable fragment fused to maltose binding protein: a modular nanocarrier platform for the targeted delivery of antitumorals†
Francisco J.
Reche-Perez
 ab,
Simona
Plesselova
ab,
Eduardo
De los Reyes-Berbel
cb,
Mariano
Ortega-Muñoz
ab,
Francisco J.
Lopez-Jaramillo
cb,
Fernando
Hernandez-Mateo
cb,
Francisco
Santoyo-Gonzalez
*cb,
Rafael
Salto-Gonzalez
ab and
Maria D.
Giron-Gonzalez
*ab
ab,
Simona
Plesselova
ab,
Eduardo
De los Reyes-Berbel
cb,
Mariano
Ortega-Muñoz
ab,
Francisco J.
Lopez-Jaramillo
cb,
Fernando
Hernandez-Mateo
cb,
Francisco
Santoyo-Gonzalez
*cb,
Rafael
Salto-Gonzalez
ab and
Maria D.
Giron-Gonzalez
*ab
aDepartment of Biochemistry and Molecular Biology II, School of Pharmacy, University of Granada, E-18071 Granada, Spain. E-mail: mgiron@ugr.es
bUnit of Excellence in Chemistry Applied to Biomedicine and the Environment of the University of Granada, Spain
cDepartment of Organic Chemistry, School of Sciences, Biotechnology Institute, University of Granada, E-18071 Granada, Spain
First published on 4th January 2021
Abstract
The use of the specific binding properties of monoclonal antibody fragments such as single-chain variable fragments (ScFv) for the selective delivery of antitumor therapeutics for cancer cells is attractive due to their smaller size, low immunogenicity, and low-cost production. Although covalent strategies for the preparation of such ScFv-based therapeutic conjugates are prevalent, this approach is not straightforward, as it requires prior chemical activation and/or modification of both the ScFv and the therapeutics for the application of robust chemistries. A non-covalent alternative based on ScFv fused to maltose-binding protein (MBP) acting as a binding adapter is proposed for active targeted delivery. MBP-ScFv proves to be a valuable modular platform to synergistically bind maltose-derivatized therapeutic cargos through the MBP, while preserving the targeting competences provided by the ScFv. The methodology has been tested by using a mutated maltose-binding protein (MBP I334W) with an enhanced affinity toward maltose and an ScFv coding sequence toward the human epidermal growth factor receptor 2 (HER2). Non-covalent binding complexes of the resulting MBP-ScFv fusion protein with diverse maltosylated therapeutic cargos (a near-infrared dye, a maltosylated supramolecular β-cyclodextrin container for doxorubicin, and non-viral polyplex gene vector) were easily prepared and characterized. In vitro and in vivo assays using cell lines that express or not the HER2 epitope, and mice xenografts of HER2 expressing cells demonstrated the capability and versatility of MBP-ScFv for diagnosis, imaging, and drug and plasmid active targeted tumor delivery. Remarkably, the modularity of the MBP-ScFv platform allows the flexible interchange of both the cargos and the coding sequence for the ScFv, allowing ad hoc solutions in targeting delivery without any further optimization since the MBP acts as a pivotal element.
1. Introduction
Nanomedicine offers new tools for diagnosis, prevention, and treatment of diseases.1,2 In particular, cancer nanomedicine is rapidly becoming one of the leading areas of promise that rely mainly on the use of nanomaterials for targeted cancer therapy3 to overcome the limitations of conventional cancer chemotherapy.4–6 Active targeting requires the use of ligands that specifically recognize cancer cell receptors/biomarkers, triggering their docking and/or precise internalization.Targeted antitumor therapeutics (TATs) are constructed by either direct coupling of the therapeutic to the ligand targeting moiety or by conjugation to a soluble or insoluble nanoparticle carrier (NP) to load therapeutics.7,8 Currently, a wide variety of NPs are being investigated as platforms for cancer treatment, including lipid-based, polymer-based, inorganic, and viral conjugated nanoparticles.9
Amongst the different targeting ligands (antibodies, peptides, hormones, nucleic acids, and lipid derivatives), monoclonal antibodies (mAb) are the most frequently used to actively target tumor cells due to their high specificity and unique in vivo properties.10 Although the use of full-length antibodies is advantageous in terms of pharmacokinetics, certain inherent structural properties limit the applicability of mAb-based TATs for cancer therapy.11 The large size of mAb can hamper access to tumor cells, particularly in the treatment of solid tumors. To circumvent the shortcoming associated with full-sized mAb, antibody fragments have been introduced for the preparation of TATs.12,13 The most commonly used antibody fragments are single-chain variable fragments (ScFv) that consist of the antibody variable domains connected by a flexible linker. Although ScFv has a much lower binding affinity compared to whole antibody counterparts due to the use of a flexible link, and its blood half-life and stability are lower than the whole antibody, it has an improved therapeutic potential due to its smaller size, low immunogenicity, and low-cost production.14,15
Functionalizing NPs with mAb or ScFv is currently performed through a variety of covalent and non-covalent methodologies.16,17 Although mAb-NPs conjugates obtained through covalent strategies are prevalent due to their high stability, this is not a straightforward approach as it usually requires prior chemical activation of NPs and/or the chemical modification of the mAb or ScFv for the application of robust chemistries (carbodiimide, maleimide, and “click” chemistry). Furthermore, covalent functionalization depends on the nature of the protein and its amino acid composition and therefore it is difficult to standardize a functionalization method valid for any mAb or ScFv. Therefore, optimization in each case of the covalent functionalization is a time-consuming process.18 In contrast, less used non-covalent approaches rely on the use of physical adsorption, ionic bonding, or binding to an adapter molecule.19 The most relevant binding strategy based on an adapter molecule exploits the biotin–avidin interaction, being commonly implemented by biotinylation of the mAb/ScFv and functionalization of the NP with avidin or its analogs. Recently, a pre-targeting approach based on mAb-streptavidin fusion proteins has attracted considerable attention because of its modularity and improved cellular internalization.20,21
Recombinant ScFv can be produced in a variety of different systems ranging from bacteria to mammalian cells. However, in bacteria, several technical difficulties have to be addressed due to the reducing environment of the cytoplasm that causes misfolding of ScFv and the formation of inclusion bodies affecting ScFv stability. To solve this critical issue, different engineering approaches have been envisioned.22 Remarkably, the cytoplasmic expression of ScFvs as a C-terminal fusion to maltose-binding protein (MBP) provides a general solution, allowing a high-level production of stable, soluble, and functional MBP-ScFv fusion protein (MBP-ScFv).23,24 MBP is a part of the maltose/maltodextrin system of E. coli, which is responsible for the uptake and efficient catabolism of maltodextrins, and one of the most popular fusion partners used for producing recombinant proteins in bacterial cells. In this scenario, MBP functions as a molecular chaperone and a tag for maltose (Mal) based affinity purification of ScFv. Upon purification, the MBP tag is cleaved by a specific protease to obtain untagged ScFvs that are further used, including their applications in TATs.
To avoid the need for chemical conjugation of the targeting moiety in NPs, we proposed a novel strategy where the MBP-ScFv recombinant protein is not proteolytically processed but is directly used as a soluble NP. By using the whole fusion protein, the MBP moiety of an MBP-ScFv is used as a binding adapter molecule for the incorporation of Mal-containing therapeutic cargos while preserving the functionality of the ScFv as the targeting ligand for the specific recognition of antigen-bearing cells (Fig. 1A). We herein demonstrate the validity of our proposal in combination with the easy vinyl sulfone click-chemistry Mal glycosylation of the desired cargo, a methodology previously developed by us.25,26 This strategy prevents the need for covalent conjugation methodologies for the functionalization of NPs and can be used with any MBP-ScFv construct without the need for new chemical reagents. This is a modular and versatile approach, since by changing the maltosylated ligands, it is possible to generate NPs with the capability to visualize, transport or deliver genes. On the other hand, by just changing the coding sequence of the ScFv included in the expression plasmid, it is possible to target different surface receptors without any further optimization. As a proof of concept, a ScFv toward the human epidermal growth factor receptor 2 (HER2) was expressed as an MBP-ScFv containing a poly histidine affinity tag to allow purification. The platform was employed for the binding of a maltosylated NIR dye, a maltosylated supramolecular container of doxorubicin, and a maltosylated non-viral polyplex gene vector. The corresponding binding complexes were assayed in vitro and in vivo to test their capability for diagnosis and imaging, specific chemotherapeutic, and plasmid active targeted tumor delivery.
 | ||
| Fig. 1 MBP-ScFv recombinant protein as a platform for the delivery of therapeutic cargos. (A) Diverse maltosylated cargos bind to the MBP-ScFv through the MBP active center. The resulting complexes are directed by the ScFv to cells bearing complementary receptors. (B) HER2 expression in cell lines. HER2 was measured by western blot using either biotin-labeled transtuzumab or biotin-labeled MBP*-ScFv. A representative blot as well as the quantitation of the intensity of the detected bands are shown. Data are shown as means ± SEM (n = 4). *p < 0.05 vs. band intensity of MDA-MB-231 cells. (C) Fluorescence of unbound (IR783)Mal and binding complexes of (IR783)Mal to MBP based proteins. | ||
2. Materials and methods
2.1. General experimental procedures
Branched polyethylenimine 2 kDa and 25 kDa (2kPEI and 25kPEI), doxorubicin hydrochloride (DOX) and indocyanine green (ICG) were purchased from Merck (Madrid, Spain). Lipofectamine™ 2000 (LP) came from Invitrogen (Carlsbad, CA, USA). pEGFP-N3 plasmid (Genbank U57609), which encodes for an enhanced red-shifted variant of wild-type GFP (eGFP), was obtained from Clontech Laboratories (Palo Alto, CA). pGL3-control vector (Genbank U47296.2) expressing luciferase under the SV40 enhancer promoter was obtained from Promega (Mannheim, Germany). Endotoxin-free plasmids were purified from transformed bacteria using the EndoFree plasmid Maxi kit from Quiagen (Hilden, Germany). DNA concentration was measured by a fluorimetric method using the Hoechst 33258 dye. Trastuzumab (Herceptin) was offered by the Oncology Pharmacy at Cádiz General Hospital (Cádiz, Spain).2.2. Expression and purification of MBP-HER2 ScFv fusion proteins (MBP-ScFv)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g to remove cell debris. Protamine sulfate (0.1%) was added to the supernatant and maintained for 30 min at 4 °C while shaking. This suspension was centrifuged (30 min, 12000g), and the supernatant was filtered through a 0.45 μm filter. A HisTrap HP 1 ml column (GE Healthcare Life Sciences, Chicago, IL, USA) was equilibrated in 20 mM HEPES, 20 mM imidazole, 10% glycerol, 0.2% Triton X-100, pH 8 buffer, and the supernatant was then loaded onto the column. After washing, proteins were eluted using 20 mM HEPES, 500 mM imidazole, 10% glycerol, 0.2% Triton X-100, pH 8. Eluted samples were directly loaded onto a HiTrap Heparin HP 1 mL column (GE Healthcare Life Sciences) equilibrated with 20 mM HEPES, 500 mM NaCl 10% glycerol, 0.2% Triton X-100, pH 8. The column was washed with the same buffer and the protein of interest was eluted using a 20 mM HEPES, 1 M NaCl 10% glycerol, 0.2% Triton X-100, pH 8. The eluted MBPI334W-ScFv (MBP*-ScFv) and MBPI334W(MBP*) proteins were concentrated and buffer exchanged by centrifugation at 10000g using a PES (polyethersulfone) centrifugal filter (VWR International, Barcelona, Spain) with a 20 mM HEPES, 120 mM NaCl pH 7.4 buffer. Protein concentration was measured by BCA method.
000g to remove cell debris. Protamine sulfate (0.1%) was added to the supernatant and maintained for 30 min at 4 °C while shaking. This suspension was centrifuged (30 min, 12000g), and the supernatant was filtered through a 0.45 μm filter. A HisTrap HP 1 ml column (GE Healthcare Life Sciences, Chicago, IL, USA) was equilibrated in 20 mM HEPES, 20 mM imidazole, 10% glycerol, 0.2% Triton X-100, pH 8 buffer, and the supernatant was then loaded onto the column. After washing, proteins were eluted using 20 mM HEPES, 500 mM imidazole, 10% glycerol, 0.2% Triton X-100, pH 8. Eluted samples were directly loaded onto a HiTrap Heparin HP 1 mL column (GE Healthcare Life Sciences) equilibrated with 20 mM HEPES, 500 mM NaCl 10% glycerol, 0.2% Triton X-100, pH 8. The column was washed with the same buffer and the protein of interest was eluted using a 20 mM HEPES, 1 M NaCl 10% glycerol, 0.2% Triton X-100, pH 8. The eluted MBPI334W-ScFv (MBP*-ScFv) and MBPI334W(MBP*) proteins were concentrated and buffer exchanged by centrifugation at 10000g using a PES (polyethersulfone) centrifugal filter (VWR International, Barcelona, Spain) with a 20 mM HEPES, 120 mM NaCl pH 7.4 buffer. Protein concentration was measured by BCA method.
The fluorescence of unbound (IR783)Mal and binding complexes of (IR783)Mal to MBP based proteins was assayed by fluorimetry. Fluorescence spectra were recorded in a JASCO FP-8300 spectrofluorometer with 2.5 nm band windows using 50 μM (IR783)Mal (20 mM Na Hepes, 120 mM NaCl buffer pH 7,4) and 10 μM of either BSA, MBP, MBP* or MBP*-ScFv. Excitation was set at 550 nm.
2.3. Cell lines
Breast cancer cells HER2+ (SKBR3; ATCC HTB-30) and HER2− (MDA-MB-231; ATCC HTB-26) were grown at 37 °C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), 2 mM glutamine plus 100 U mL−1 penicillin, in an atmosphere of 5% CO2 and 95% air and maintained at sub-confluent densities in the growth media.MDA-MB-231 cells overexpressing the HER2 receptor (MDA-HER2+) were obtained by retroviral transduction using Phoenix Ampho cells (kindly offered by Dr J. L. Garcia-Perez, Centre for Genomics and Oncological Research: Pfizer, University of Granada, Granada, Spain), and the plasmid pBABEpuro-ERBB2 (a gift from Matthew Meyerson, Addgene plasmid # 40978).30 In brief, Phoenix-Ampho cells were seeded in 6 well plates at a density of 2.3 × 105 cells per well for 24 h to reach a cell confluence of 80–90%. Transfection was then performed using pBABEpuro-ERBB2 and LP following manufactured instructions. Retroviral supernatants were collected from sub-confluent cultures (48 h) and used to infect MDA-MB-231 cells. Then, MDA-HER2+ cells were selected by using 8 μg mL−1 puromycin. HER2 expression was confirmed by western blot (Fig. 1) using biotin labeled herceptin, and a high sensitivity streptavidin-HRP (Pierce, Rockford, IL, USA). Herceptin labeling was done using a vinyl sulfone derivatized bifunctional tag single-attachment-point reagent bearing biotin and a fluorescent tag31 (supplementary, vinyl sulfone based reagents). Alternatively, purified MBP*-ScFv was labeled with the same reagent.
2.4. MBP*-ScFv imaging assays
Imaging and detection of breast cancer cells by Mal-based ligands were evaluated by confocal microscopy using a Leica TCS-SP5 II multiphoton confocal microscope following previously reported procedures32 that prevent cross-talk of fluorophores by using distinct excitation laser lanes and non-overlapping detection channels. In all cases, cells were first seeded onto coverslips in 12-well plates (density equal to 9 × 105 cells per well) and incubated at 37 °C (24 h) to reach a cell confluence of 80–90%. For cell uptake assays, DOX- or ICG-based compounds were incubated for 2 h. Cells were fixed with paraformaldehyde (2% in PBS, 15 min at room temperature), washed and mounted for confocal microscopy. For detection of HER2 positive cells, HER2 expressing SKBR3 cells or HER2 negative MDA-MB-231 cells were seeded onto coverslips and 24 h later they were fixed and incubated with [(IR783)Mal]∼[MBP*-ScFv], washed and mounted.2.5. Targeted drug delivery assays
:nCD = 0:9) were incubated overnight at 4 °C to produce the corresponding inclusion complexes 2kPEI(DOX⊂CD)Mal or 2kPEI(ICG⊂CD)Mal. After freeze-drying, formation of the complexes was confirmed using UV-Vis and fluorescence spectroscopy. 2kPEI(DOX⊂CD)Mal or 2kPEI(ICG⊂CD)Mal were incubated with equimolecular amounts of MBP*-ScFv or MBP* (considering the number of Mal molecules in the complexes and Mal binding sites in the proteins) for 30 min at room temperature. Then, MDA-MB-231, MDA-HER2+ or SKBR3 cells were incubated, with concentrations corresponding to 1 μM DOX, with DOX, 2kPEI(DOX⊂CD)Mal, 2kPEI(DOX⊂CD)Mal∼[MBP*] or [2kPEI(DOX⊂CD)Mal]∼[MBP*-ScFv] for 2 h. Cells were lysed with 200 μL of 0.5% Triton X-100 in PBS and the fluorescence due to DOX was measured in a JASCO FP-8300 spectrofluorometer with 2.5 nm band windows at 499 nm excitation and 555 nm emission wavelengths.
2.6. Targeted gene delivery assays
2.7. In vivo studies
Female NSG immunodeficient mice (6–8 weeks of age, 25–30 g weight) were purchased from the Animal Facility at the University of Granada and maintained in accordance with guidelines established by Directive 2012/707/UE and the approval of the Committee on Animal Research at the University of Granada (15/11/2017/154). For xenografts models, SKBR3 cells were trypsinized and re-suspended in PBS (density equal to 2 × 107 cells per mL). Cells (1 × 106) were injected intradermally into the breast area of each female NSG mouse. When tumor sizes reached 1 to 6 mm in diameter, mice were injected in the tail vein with the different targeted antitumor therapeutics. The in vivo imaging over time assays were performed in an IVIS Spectrum (xCaliper Life Sciences, MA, USA). Isofluoran-anesthetized mice were placed in the dark chamber for luminiscence or fluorescence (excitation/emission, 675/720 nm) acquisition. Images were taken and analyzed with the Living Image 2.6 software package (Xenogen).2.8. Statistical analysis
Results are expressed as mean ± SEM. Statistical analysis was performed by two-way ANOVA followed by Tukey's test as appropriate. P < 0.05 was considered statistically significant.3. Results
3.1. Design, expression, purification and study of the functionalization of MBP-ScFv
The overexpression of the protein of interest as a fusion protein bearing a peptide/protein tag is a well-established strategy to facilitate its isolation.34 Different systems are commercially available and they rely on the reversible affinity of the tag for chromatographic matrix. We hypothesized that this affinity can be further exploited in the context of antibody–drug conjugates and we envisioned a two-tag platform bearing a His-tag for purification purposes and a maltose binding protein (MBP) tag as an attachment point of different elements that carry a maltose motif (Fig. 1A).However, for our purpose, the reversibility of the interaction between MBP tag and maltose was undesired and the wild type MBP tag was mutated to yield MBPI334W (MBP*) tag. The mutation is located at the hinge region that bends upon substrate binding to form the active site of the protein and MBP* exhibits enhanced ligand affinity (approximately 5 fold) and a decrease of the ligand Koff, without hampering its solubility and stability (unpublished data).
To test our hypothesis, the DNA encoding ScFv against HER2 was cloned between both tags to generate the plasmid pMALI334W-TEV-ScFv HER2-His. The overexpression in E. coli yielded the soluble recombinant protein MBP*-ScFv that was isolated by Ni2+-based IMAC chromatography and further purified by heparin chromatography to a final yield of 0.5 mg of protein per liter of culture (Fig. S1, ESI†).
The ability of MBP*-ScFv to recognize the HER2 epitope was assessed by western blot in HER2+ cell extracts such as SKBR3 and MDA-HER2+, using the HER2− cell line MDA-MB-231 as negative control, and Trastuzumab, the commercial antibody against HER2, as reference. As shown in Fig. 1B, both MBP*-ScFv and trastuzumab only recognized the HER2+ extracts and the intensity of the signal was in the same range for MDA-HER2+ and lower for SKBR3.
The functionality of the MPB* tag as an attachment point was studied using the NIR dye IR783 functionalized with maltose via a vinyl sulfone-based click strategy by the reaction of IR783 piperazine35 with vinyl sulfone maltose.25 As a cyanine-based fluorophore, in aqueous solution (IR783)Mal tends to self-assemble into aggregates, resulting in a quenching of the fluorescence that is restored upon disassembly.36 This feature was exploited to assess the interaction of MBP*-ScFv with the maltose-bearing element. As shown in Fig. 1C, the fluorescence of (IR783)Mal alone is quenched and neglectable. Both MBP and MBP* promoted the disaggregation, and the resulting fluorescence for MBP* was slightly blue-shifted and larger, as expected from the higher affinity resulting from the I334W mutation. As a tag, MBP*-ScFv was fully functional and the interaction with (IR783)Mal yielded a value of fluorescence similar to free MBP despite being fused to ScFv.
3.2. MBP*-ScFv complexes as specific imaging agents
A simultaneous evaluation of the capability of MBP*-ScFv to both recognize HER2 and form a complex with an element bearing the maltose moiety was carried out using (IR783)Mal as a probe. The combination of equimolecular amounts of MBP*-ScFv and (IR783)Mal yielded [(IR783)Mal]∼[MBP*-ScFv], which was assayed against the HER2+ SKBR3 cells and the HER2− MDA-MB-231 cells. As depicted in Fig. 2, SKBR3 cells showed a red fluoresce that was 5.7-fold more intense than that of MDA-MB-231, supporting the functionality of MBP*-ScFv and the feasibility of [(IR783)Mal]∼[MBP*-ScFv] as a diagnostic agent to detect HER2+ overexpressing cells. Further evaluation of mice bearing HER2+ xenografts showed that the fluorescence was located in both the xenograft and liver (Fig. 3A), with an intensity 16-fold and 3-fold higher respectively, when (IR783)Mal was attached to MBP*-ScFv (Fig. 3B). Analysis of the liver revealed the presence of metastasis that were labeled with [(IR783)Mal]∼[MBP*-ScFv] (Fig. 3C). These results demonstrate the functionality of MBP*-ScFv and the preferential concentration of [(IR783)Mal]∼[MBP*-ScFv] in tumors, supporting the feasibility of using complexes of suitable dyes with MBP*-ScFv as in vivo diagnostic agents to detect HER2+ overexpressing cells. | ||
| Fig. 2 In vitro imaging of HER2+ cells by [(IR783)Mal]∼[MBP*-ScFv] complex. HER2+ SKBR3 cells (A) or HER2− MDA-MB-231 cells (B) were seeded onto coverslips, fixed, incubated with [(IR783)Mal]∼[MBP*-ScFv] complex, washed, and mounted for confocal microscopy. Fluorescence (a), differential interference contrast (Nomarski) images (b), and merged images (c) are shown. (C) The average of the fluorescence/μ2 assigned to the cells is plotted. Data are shown as means ± SEM (n = 10). *p < 0.05 vs. fluorescence of MDA-MB-231 cells. | ||
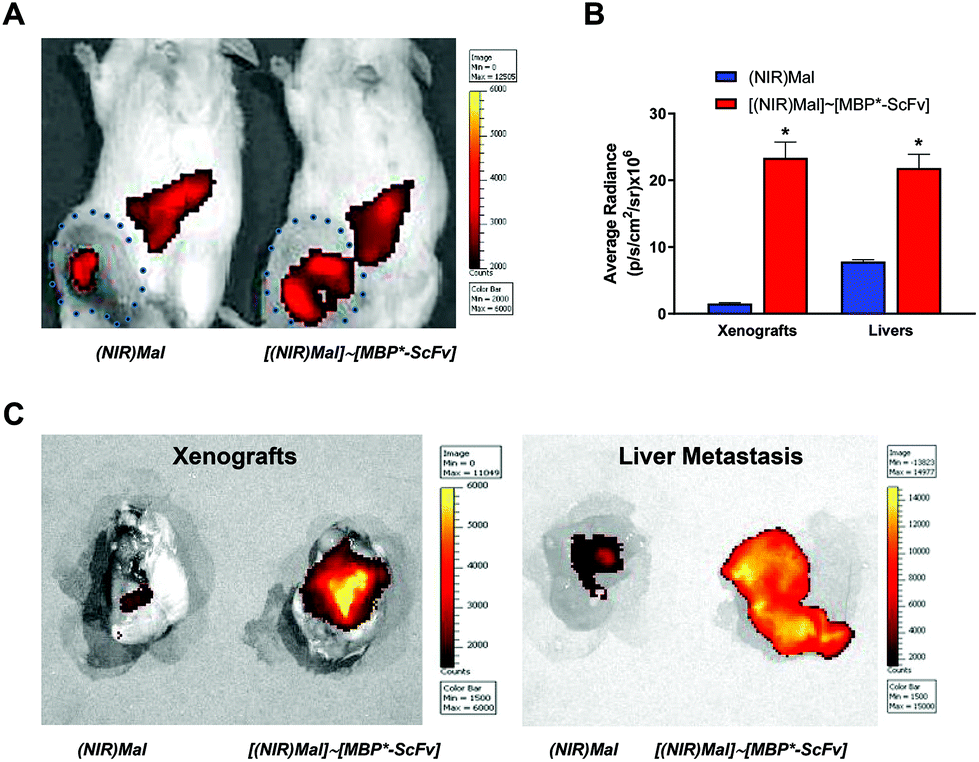 | ||
| Fig. 3 In vivo imaging of tumor HER2+ xenografts in mice by [(IR783)Mal]∼[MBP*-ScFv] complex. NSG mice bearing breast cancer HER2+ (SKBR3 cells) tumors were injected intravenously in the tail vein with (IR783)Mal or [(IR783)Mal]∼[MBP*-ScFv] and fluorescence was measured 24 h later. (A) NIR fluorescence images. The size of the xenografts is indicated by a dotted line. (B) Average radiance of the xenografts and liver metastases from the mice injected with (IR783)Mal or [(IR783)Mal]∼[MBP*-ScFv]. Data are shown as means ± SEM (n = 4). *p < 0.05 vs. (IR783)Mal xenografts or livers metastases. (C) Fluorescence imaging of dissected xenografts and livers metastases. | ||
3.3 MBP*-ScFv complexes for targeted chemotherapeutic delivery
Inspired from a previous paper describing a modular design of site-directed drug delivery systems that decouples the carrier function from the targeting function,37MBP*-ScFv was evaluated as the targeting module. A multivalent carrier module was designed through a combination of branched polyethylenimine 2 kDa (2kPEI) as a scaffold, β-cyclodextrin (βCD) as host, and maltose as the link to allow the coupling to MBP*-ScFv. 2kPEI and maltose vinyl sulfone were first reacted in a 1:1 ratio and then with 4 equivalents of βCD vinyl sulfone to obtain 2kPEI(CD)Mal. The incubation of 2kPEI(CD)Mal with doxorubicin (DOX) in a 1:3.6 ratio led to the formation of inclusion complexes termed 2kPEI(DOX⊂CD)Mal, as previously described.38 Then, the combination of 2kPEI(DOX⊂CD)Mal with equimolecular amounts of MBP*-ScFv yielded [2kPEI(DOX⊂CD)Mal]∼[MBP*-ScFv]. Despite the limitations and clinical relevance of DOX, its election as a model drug was based on the fact that its fluorescence allows the detection of [2kPEI(DOX⊂CD)Mal]∼[MBP*-ScFv]. The capability of [2kPEI(DOX⊂CD)Mal]∼[MBP*-ScFv] to deliver DOX was evaluated in vitro. Confocal microscopy (Fig. 4A and B) and fluorimetric quantitation of the DOX uptake (Fig. 4C) were carried out. HER2+ (MDA-HER2+ and SKBR3) and HER2− (MDA-MB-231) cells were selected for these assays. In HER2+ cells, DOX uptake was significantly higher when cells were incubated with the [2kPEI(DOX⊂CD)Mal]∼[MBP*-ScFv] complex compared to DOX alone or 2kPEI(DOX⊂CD)Mal. Furthermore, uptake was significantly decreased when DOX was occluded in 2kPEI(CD)Mal (i.e. the carrier module without the targeting module) or administrated as a [2kPEI(DOX⊂CD)Mal]∼[MBP*] complex (i.e. the carrier module with the attachment point but without the targeting function) compared to DOX. Similar results were obtained when the samples were assayed by flow cytometry (Fig. S2, ESI†). More interestingly, when the toxicity associated with the DOX administration was analyzed (Fig. 4D), the toxic effect of DOX as [2kPEI(DOX⊂CD)Mal]∼[MBP*-ScFv] was more pronounced than that of free DOX on HER2+ cells but less efficient in HER2− cells.
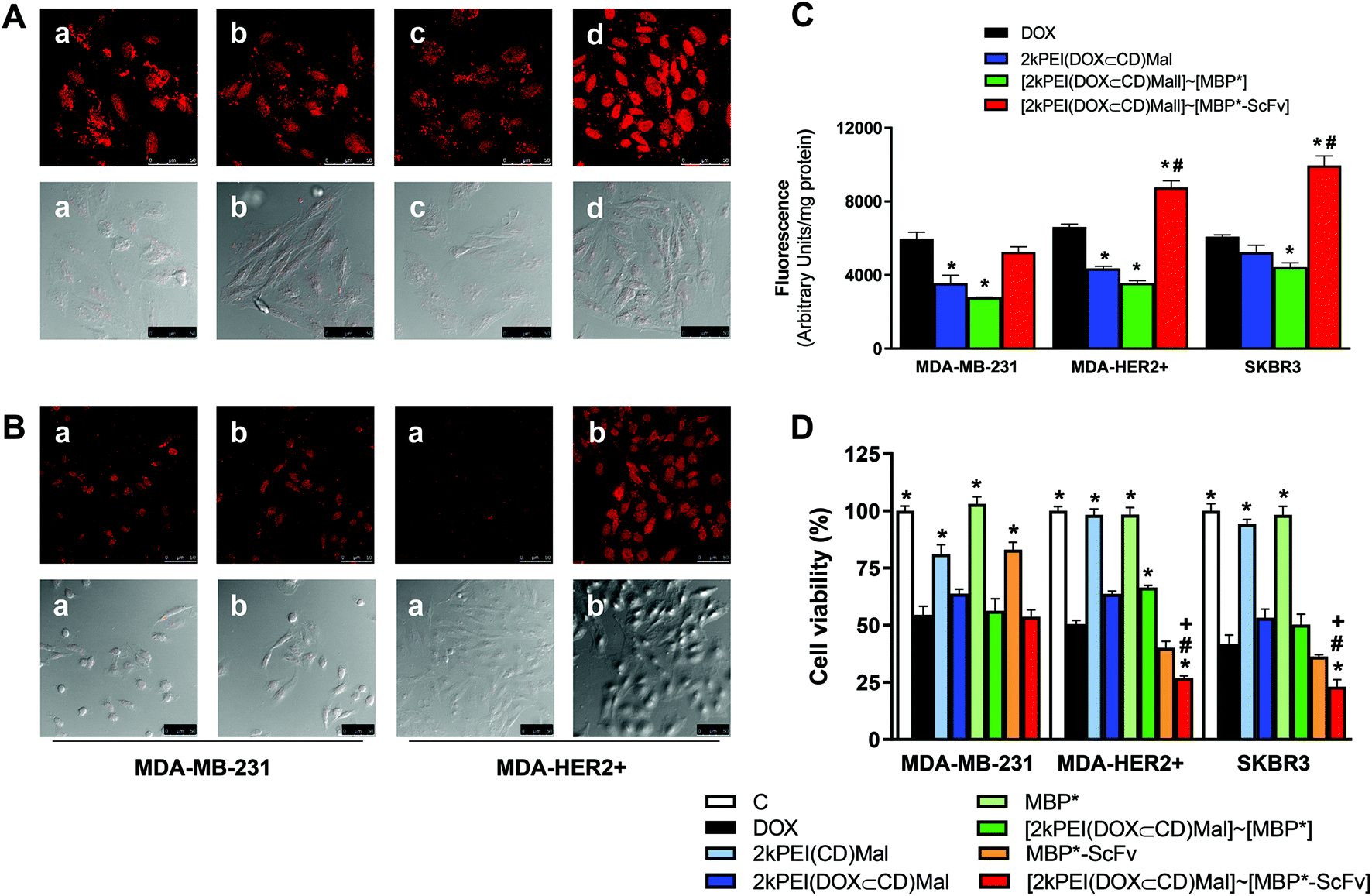 | ||
| Fig. 4 In vitro targeted DOX delivery and cytotoxicity mediated by a MBP*-ScFv binding complex. (A) Confocal microscopy for SKBR3 cells incubated for 2 h in the presence of DOX (a), 2kPEI(DOX⊂CD)Mal (b), [2kPEI(DOX⊂CD)Mal]∼[MBP*] (c), or [2kPEI(DOX⊂CD)Mal]∼[MBP*-ScFv] (d). Fluorescence and differential interference contrast (Nomarski) images are shown. (B) Confocal microscopy for MDA-MB-231 or MDA-HER2 cells incubated for 2 h with DOX (a) or [2kPEI(DOX⊂CD)Mal]∼[MBP*-ScFv] (b). (C) Fluorescence exhibit by MDA-MB-231, SKBR3 or MDA-HER2+ cells incubated with DOX and with Mal-based (DOX⊂CD) complexes: 2kPEI(DOX⊂CD)Mal, [2kPEI(DOX⊂CD)Mal]∼[MBP*] or [2kPEI(DOX⊂CD)Mal]∼[MBP*-ScFv]. Cells were lysed after 2 h and DOX fluorescence measured. Results are expressed as means ± S.E.M. (n = 8). *: p < 0.05 vs. DOX treated cells. #: p < 0.05 vs. [2kPEI(DOX⊂CD)Mal]∼[MBP*] treated cells. (D) In vitro cytotoxicity of DOX and Mal-based targeted (DOX⊂CD) inclusion complexes. Cell viability measured using the MTT method for MDA-MB-231, MDA-HER2+ or SKBR3 cells incubated for 48 h with DOX, 2kPEI(CD)Mal, [2kPEI(DOX⊂CD)Mal]∼[MBP*], MBP*-ScFv, [2kPEI(DOX⊂CD)Mal]∼[MBP*] or [2kPEI(DOX⊂CD)Mal]∼[MBP*-ScFv]. Results are expressed as means ± S.E.M. (n = 8). *: p < 0.05 vs. DOX treated cells; #: p < 0.05 vs.[2kPEI(DOX⊂CD)Mal]∼[MBP*] treated cells; +: p < 0.05 vs. MBP*-ScFv treated cells. | ||
To evaluate the potential of [2kPEI(DOX⊂CD)Mal]∼[MBP*-ScFv] as a site-directed drug delivery system, an in vivo analysis was carried out using immune-suppressed NSG mice bearing breast cancer HER2+ (SKBR3 cells) xenografts (Fig. 5). Since in vivo imaging systems are not optimal for detecting DOX due to the relatively low wavelength of its fluorescence emission, indocyanine green (ICG), a cyanine IR dye used in medical diagnostics,39 was assayed as a model drug. ICG was hosted in 2kPEI(CD)Mal and then complexed with either MBP* or MBP*-ScFv, to obtain [2kPEI(ICG⊂CD)Mal]∼[MBP*] or [2kPEI(ICG⊂CD)Mal]∼[MBP*-ScFv]. To test the validity of this approach, the (ICG⊂CD) inclusion complex distribution was first analyzed in vitro in SKBR3 cells by confocal microscopy (Fig. 5A). It was observed that ICG entered the cells and that the uptake was higher when cells were incubated with [2kPEI(ICG⊂CD)Mal]∼[MBP*-ScFv] in regard to [2kPEI(ICG⊂CD)Mal]∼[MBP*], supporting the results obtained with DOX. In animals, the analysis of fluorescence distribution 30 min after the injection in the tail vein revealed that only [2kPEI(ICG⊂CD)Mal]∼[MBP*-ScFv] was visible in the xenograft, although a significant amount of signal was located in the liver. Quantification of the fluorescence in the xenograft yielded a 7-fold higher fluorescence when ICG was targeted by MBP*-ScFv compared to MBP*. The above results with DOX and with ICG demonstrate the versatility of 2kPEI(CD)Mal as a carrier module and the feasibility of using MBP*-ScFv as a targeting module.
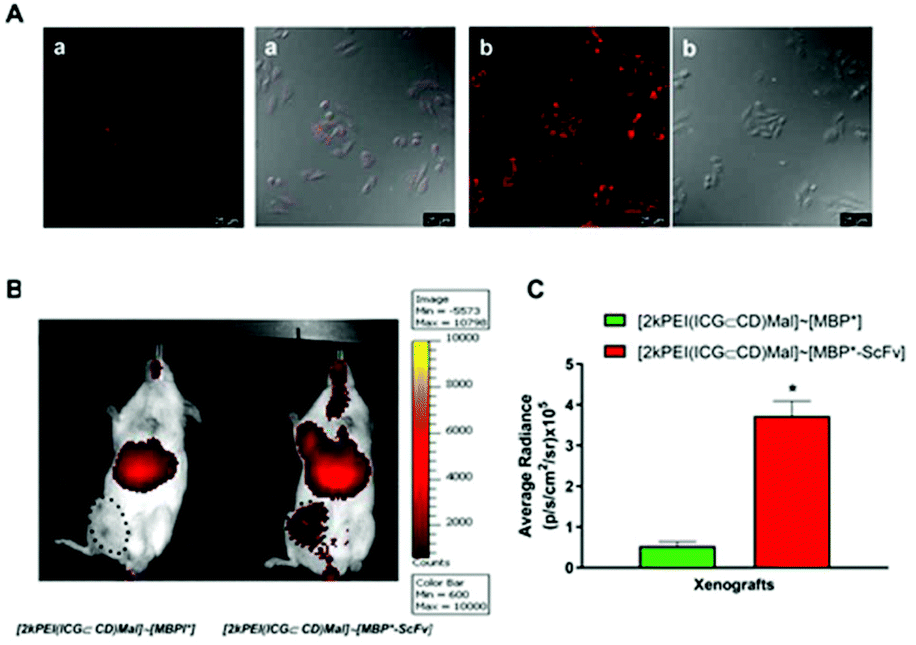 | ||
| Fig. 5 In vitro and vivo targeted ICG delivery mediated by Mal-based (ICG⊂CD) inclusion complexes. (A) Confocal microscopy images for SKBR3 cells incubated in the presence of [2kPEI(ICG⊂CD)Mal]∼[MBP*] (a) or [2kPEI(ICG⊂CD)Mal]∼[MBP*-ScFv] (b). Fluorescence and differential interference contrast (Nomarski) images are shown. (B) NSG mice bearing breast cancer HER2+ (SKBR3 cells) tumors injected intravenously in the tail vein with [2kPEI(ICG⊂CD)Mal]∼[MBP*] or [2kPEI(ICG⊂CD)Mal]∼[MBP*-ScFv]. Fluorescence distribution was measured 3 h later. Size of the xenografts is indicated by a dotted line. (C) Average radiance plotting of the xenografts from the mice injected with [2kPEI(ICG⊂CD)Mal]∼[MBP*] or [2kPEI(ICG⊂CD)Mal]∼[MBP*-ScFv]. Data are shown as means ± SEM (n = 4). *p < 0.05 vs. [2kPEI(ICG⊂CD)Mal]∼[MBP*] xenografts. | ||
3.4. MBP-ScFv complexes for targeted gene delivery
The use of PEI for gene transfection is a well-established methodology that works in vitro but the lack of specificity compromises the implementation in vivo.40 Additionally, the size of PEI is a critical factor to consider, since low molecular weight PEIs are ineffective while highly branched PEIs show significant toxicity. In this context, receptor-mediated site-directed delivery is an attractive approach and we hypothesized that the maltosylation of PEI is a feasible approach to link to the targeting module MBP*-ScFv while preserving the transfection activity. Branched polyethylenimine 25kPEI (25kPEI) was reacted with vinyl sulfone maltose using a 1:5 25kPEI/Mal stoichiometry ratio to get a multi-maltosylated PEI derivative, named 25kPEI(Mal), to maximize its binding capabilities to MBP*-ScFv. First, 25kPEI(Mal) was incubated with pDNA, followed by binding the resulting 25kPEI(Mal)/pDNA polyplex with equimolecular amounts of either MBP* or MBP*-ScFv. The analysis of the DNA binding affinity and protection from DNaseI showed that neither the maltosylation nor the link to either MBP* or MBP*-ScFv had any negative effect on these two critical features of 25kPEI (Fig. S3, ESI†). Transfection was assayed on the HER2+ cell lines (SKBR3 and MDA-HER2+) and MDA-MB-231 as a HER2− control, using the pEGFP-N3 plasmid encoding an enhanced red-shifted variant of wild-type GFP. The higher transfection efficiency was attained by using an N/P ratio of 7, as observed in optimization assays performed in the three types of cells (data non-shown). The transfection efficiency of native 25kPEI varied with the cell line (Fig. 6A). While PEI maltosylation improved the performance on MDA-HER2+, coupling to MBP* decreased the transfection efficiency. On the contrary, as expected, the coupling to MBP*-ScFv yielded an improvement in the transfection efficiency on the HER2+ cell lines whereas the values of fluorescence for MDA-MB-231 remained at the same level as those for 25kPEI.
 | ||
| Fig. 6 Targeted gene delivery to HER2 bearing cells mediated by MBP*-ScFv based polyplexes. (A) In vitro transfection efficiency of [25kPEI(Mal)/pDNA]∼[MBP*] and [25kPEI(Mal)/pDNA]∼[MBP*-ScFv] polyplex in HER2+ (SKBR3 and MDA-HER) and HER2− (MDA-MB-231) breast cancer cells. N = 8. * p < 0.5 compared to 25kPEI; # p < 0.5 compared to 25kPEI(Mal)∼[MBP*]. (B) Fluorescence values for SKBR3 cells pre-incubated with increasing concentrations of herceptin (30 min) and transfected with [25kPEI(Mal/pEGFP-N3)]∼[MBP*-ScFv] polyplex. Fluorescence values in the absence of herceptin have been normalized to 100%. * p < 0.5 compared with control transfection. (C) In vivo transfection for NSG mice bearing SKBR3 tumor-xenografts. Images were taken 24 h after LP/pGL3 or [25kPEI(Mal)/pGL3]∼[MBP*-ScFv] polyplexes intravenous injection in the tail vein. The size of the xenografts is indicated by a dotted line. (D) The average radiance of the xenografts from the mice injected with LP/pGL3 or [25kPEI(Mal)/pGL3]∼[MBP*-ScFv] polyplexes is plotted. Data are shown as means ± SEM (n = 4). *p < 0.05 vs.LP/pGL3 xenographs. | ||
To confirm the role of the MBP*-ScFv in the improvement of the transfection, SKBR3 cells were pre-treated with trastuzumab to block the HER2 receptor and then with [25kPEI(Mal)/pEGFP-N3]∼[MBP*-ScFv] (Fig. 6B). The transfection efficiency diminished as the concentration of trastuzumab in the pre-treatment was increased, demonstrating that the improvement in the efficiency of the transfection when the DNA is carried by [25kPEI(Mal)]∼[MBP*-ScFv] is a consequence of the targeting module MBP*-ScFv. In vivo experiments on NSG mice bearing SKBR3 tumor xenografts further confirmed the potential of [25kPEI(Mal)]∼[MBP*-ScFv] in gene delivery, the fluorescence signal being localized in the xenograft (Fig. 6C) and the intensity being 3-fold higher than that of LP (Fig. 6D).
4. Discussion
Antibody–drug conjugates represent a growing market, several of them approved by the FDA and more than 70 candidates currently in clinical trials.41 The technology seems simple and the majority of antibody–drug conjugates on the market use conjugation approaches that target naturally occurring amino acids such as lysine (i.e. gemtuzumab ozogamicin, trastuzumab emtansine, and inotuzumab ozogamicin) or interchain cysteines (i.e. brentuximab vedotin). However, this straightforward approach results in a random linkage of the drug to the antibody and yields heterogeneous mixtures with different drug-to-antibodies-ratios that impact the physicochemical properties, pharmacokinetics, and lot-to-lot reproducibility.42 At the cost of increasing the complexity, the next generation of antibody–drug conjugates rely on site-specific bioconjugation strategies such as THIOMABs technology, transglutaminase-mediated conjugations, click chemistry conjugation, bridge cysteine conjugations, glycosyl remodeling, to reduce the problems associated with heterogeneity and to enable antibody-like fragments to improve tumor penetration.Inspired by the biotin–(strept)avidin technology43 and the long history of the MBP as a tag that enhances the solubility29,30 and overexpression of recombinant proteins in both E. coli and mammalian cells,44 we envisaged a strategy of bioconjugation based on the overexpression of antibody-like fragments as MBP fusion proteins which, besides the aforementioned benefits,33,34,45,46 act as an attachment point for elements conveniently functionalized with a maltose moiety (Fig. 1A). As a proof of concept, we designed a construct containing an ScFv as a targeting element, a His-tag (6xHis-tag) as an affinity tag for purification, and the enhanced maltose affinity MBP I334W tag as the attachment point. An ScFv directed toward the human epidermal growth factor receptor 2 (HER2) was selected based on its relevance, on the fact that HER2 overexpression is associated with poor outcomes in breast and gastric cancer and on its key role in activating the downstream signaling pathways that control cell proliferation, survival, and apoptosis in breast cancer.27,47–49 Trastuzumab, a mAb that inhibits HER2 and is currently being exploited for targeting, was the first mAb approved for human use. It is the cornerstone of HER2+ breast cancer treatment despite the fact that other anti-HER2 Ab targeting the extracellular domain have been described.50–52
As expected from the MBP tag, the overexpression of the construct in E. coli rendered a soluble MBP*-ScFv which, after purification by IMAC chromatography, yields 0.5 mg L−1 of culture media. MBP*-ScFv bears a double functionality: ScFv being as selective towards cell lines that express HER2 as trastuzumab (Fig. 1B), and MBP* as a feasible attachment point of maltosylated elements. Our strategy was validated using [(IR783)Mal]∼[MBP*-ScFv], a complex resulting from the maltosylation of the NIR dye IR783 and later in combination with MBP*-ScFv. Furthermore, [(IR783)Mal]∼[MBP*-ScFv] could be clinically useful since the determination of Her2 expression levels is a prognostic biomarker for anti-HER2 treatment in a variety of tumor types including breast, gastric and gynecological cancers. In this context, [(IR783)Mal]∼[MBP*-ScFv] combines the selectivity to target HER2+ cells with the NIR emission of IR783, features that suggest a medical application in diagnosis.53 Nevertheless, although the selectivity toward cancer cells overexpressing HER2 could be compromised by the well-established affinity of IR783 for tumor cells or the interaction between (IR783)Mal and MBP*-ScFv could be disrupted in a biological environment, our in vitro experiments demonstrated that interaction is stable and that [(IR783)Mal]∼[MBP*-ScFv] shows a 5.7-fold-higher affinity for HER+ cells (Fig. 2), supporting a further evaluation in vivo on a xenograft model of HER2+ adenocarcinoma obtained by injection of SKBR3 cells in mice. In agreement with the general affinity of IR783 for tumor compound (IR783), Mal is found in the xenograft although the signal is more intense for [(IR783)Mal]∼[MBP*-ScFv], as expected from the targeting toward HER2 receptor (Fig. 3). It is important to recall that the potential of [(IR783)Mal]∼[MBP*-ScFv] may exceed the diagnosis application evaluated in this work since the reported therapeutic action of IR783 as an inhibitor of cancer cell proliferation54 and in phototherapy55 supports the use of [(IR783)Mal]∼[MBP*-ScFv] in theragnosis.
A key point in cancer therapy is dosage. Unfortunately, large doses, while ensuring therapeutic action, are associated with pronounced side effects. An interesting alternative is the design of methods for site-directed drug delivery where the amount of drug delivered to the site of action by antibody–drug conjugates is proportional to the number of receptors targeted. The increase in the drug–antibody ratio has been a classical approach not exempt from drawbacks, since it often affects pharmacokinetics and the increase of hydrophobicity is frequently one of the key factors to be considered.42,56 To approach this challenge and inspired by a previous work that demonstrated the feasibility of using a nanobody-cyclodextrin conjugate as a site-directed drug delivery system,37 we envisioned a modular design where the targeting module bears the MBP and the carrier module is maltosylated. As a proof of concept, PEI was functionalized with maltose and with β-cyclodextrin to obtain a multivalent platform that exploits the ability of cyclodextrin to form inclusion complexes with hydrophobic molecules such as chemotherapeutic anticancer drugs57 and the interaction of the maltose with MBP*-ScFv to yield the complex [2kPEI(CD)Mal]∼[MBP*-ScFv]. DOX was selected as a model drug for the evaluation of the complex because it shows a wide range of antitumor activity, being broadly used in cancer treatment. However, its potential is limited by cardiotoxicity, and the development of other ways to manage DOX is a challenge. In vitro experiments demonstrated that the uptake of DOX by HER2+ cells is higher when DOX is delivered as [2kPEI(DOX⊂CD)Mal]∼[MBP*-ScFv] and this effect measured as cytotoxicity was dependent on the expression HER2+ (Fig. 4). The In vivo evaluation using the diagnostic dye indocyanine green further confirmed the site-directed drug delivery capability of [2kPEI(ICG⊂CD)Mal]∼[MBP*-ScFv] (Fig. 5). In vivo assays indicate that the MBP*-ScFv platform is able to specifically target these xenographs, although some leaking of the ICG compound to liver takes place. This finding points to a partial dissociation of the complex that could be prevented in future developments by incorporating additional mutations into the MBP to increase its binding affinity for the maltosylated ligands.58 At this point, it is important to highlight the flexibility of [2kPEI(CD)Mal]∼[MBP*-ScFv] as a carrier of a wide range of molecules since the payload is only limited by the affinity towards CD to yield an inclusion complex.
Currently, cancer treatment relies on chemotherapy, radiotherapy and surgery as a multimodal therapy that often does not suffice to reverse the progress of cancer. The increasing advances in the knowledge of the characteristics of tumor cells and their microenvironment have led to novel therapeutic approaches based on gene therapy.59 However, the development of delivery systems that efficiently deliver genes to specific target tissues with minimal toxicity and immunogenicity remains a challenge. Nonviral vectors are an alternative to the immune and toxic side effects of viral vectors and among them branched 25kPEI acts as an efficient transfection agent. 25kPEI has been conjugated to trastuzumab for gene delivery60 but we hypothesized that our simpler and more versatile approach of bioconjugation could be used to transfect HER2+ cells. Our results demonstrate that the maltosylation of PEI 25k has no negative effect either on the affinity by DNA or the protection against DNaseI and it is a straightforward strategy to extend the application of MBP*-ScFv to deliver genes to HER2+ cells in vitro (Fig. 5) and in vivo (Fig. 6).
5. Conclusions
The interest in the widely used strategy of cloning the protein of interest into a plasmid encoding the MBP tag can be extended beyond its overexpression and purification. We have demonstrated that the MBP tag is an attachment point for maltosylated elements and that the use of the maltose-MBP interaction is a feasible bioconjugation strategy. In cancer therapy, antibody–drug conjugates represent a new generation of drugs and in this context, new strategies of bioconjugation are needed to overcome the limitations of those currently used. Our results demonstrate that the overexpression of a ScFv as a fusion protein with MBP transforms it into a versatile targeting element that can be combined with different carrier modules for in vivo diagnosis, site-directed drug delivery, and receptor-mediated gene therapy. This modular approach simplifies the design of antibody–drug conjugates and its implementation is feasible in standard molecular biology laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants CTQ2014-55474-C2-1-R, CTQ2014-55474-C2-2-R and CTQ2017-86125-P from the Ministerio Economia, Industria y Competitividad (co-financed by FEDER funds). SP is supported by a FPU fellowship (FPU17/04749). We acknowledge the University of Granada (Spain) cell culture, animal and microscopy central facilities (CIC-UGR).Notes and references
- A. Wicki, D. Witzigmann, V. Balasubramanian and J. Huwyler, J. Controlled Release, 2015, 200, 138–157 CrossRef CAS.
- V. K. Chaturvedi, A. Singh, V. K. Singh and M. P. Singh, Curr. Drug Metab., 2019, 20, 416–429 CrossRef CAS.
- S. Tran, P. J. DeGiovanni, B. Piel and P. Rai, Clin. Transl. Med., 2017, 6, 44 Search PubMed.
- V. J. Yao, S. D'Angelo, K. S. Butler, C. Theron, T. L. Smith, S. Marchio, J. G. Gelovani, R. L. Sidman, A. S. Dobroff, C. J. Brinker, A. R. M. Bradbury, W. Arap and R. Pasqualini, J. Controlled Release, 2016, 240, 267–286 CrossRef CAS.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS.
- L. Lu, W. Chen and Z. Sun, Angew. Chem., Int. Ed., 2020, 59, 2–20 CrossRef.
- A. Carvalho, A. R. Fernandes and P. V. Baptista, in Applications of Targeted Nano Drugs and Delivery Systems, 2019, pp. 257–295, DOI:10.1016/b978-0-12-814029-1.00010-7.
- M. Sharma, C. Pandey, N. Sharma, M. A. Kamal, U. Sayeed and S. Akhtar, Anticancer Agents Med. Chem., 2018, 18, 2078–2092 CrossRef CAS.
- P. Y. Liyanage, S. D. Hettiarachchi, Y. Zhou, A. Ouhtit, E. S. Seven, C. Y. Oztan, E. Celik and R. M. Leblanc, Biochim. Biophys. Acta, Rev. Cancer, 2019, 1871, 419–433 CrossRef CAS.
- J. M. Lambert and A. Berkenblit, Annu. Rev. Med., 2018, 69, 191–207 CrossRef CAS.
- M. R. Gordon, M. Canakci, L. Li, J. Zhuang, B. Osborne and S. Thayumanavan, Bioconjugate Chem., 2015, 26, 2198–2215 CrossRef CAS.
- A. Alibakhshi, K. F. Abarghooi, S. Ahangarzadeh, H. Yaghoobi, F. Yarian, R. Arezumand, J. Ranjbari, A. Mokhtarzadeh and M. de la Guardia, J. Controlled Release, 2017, 268, 323–334 CrossRef CAS.
- V. Crivianu-Gaita and M. Thompson, Biosens. Bioelectron., 2016, 85, 32–45 CrossRef CAS.
- Z. A. Ahmad, S. K. Yeap, A. M. Ali, W. Y. Ho, N. B. M. Alitheen and M. Hamid, Clin. Dev. Immunol., 2012, 2012, 980250 Search PubMed.
- P. Thanindratarn, D. C. Dean, S. D. Nelson, F. J. Hornicek and Z. Duan, Cancer Treat. Rev., 2020, 82, 101934 CrossRef CAS.
- A. C. Marques, P. J. Costa, S. Velho and M. H. Amaral, J. Controlled Release, 2020, 320, 180–200 CrossRef CAS.
- O. M. Kutova, E. L. Guryev, E. A. Sokolova, R. Alzeibak and I. V. Balalaeva, Cancers, 2019, 11, 68 CrossRef CAS.
- P. Zhang, H. Chen, J. Liu and G. Liu, Methods Mol. Biol., 2019, 2054, 283–294 CrossRef CAS.
- C. Q. Bie, D. H. Yang, X. J. Liang and S. H. Tang, World J. Hepatol., 2010, 2, 185–191 CrossRef.
- C. L. Parker, Q. Yang, B. Yang, J. D. McCallen, S. I. Park and S. K. Lai, Acta Biomater., 2017, 63, 181–189 CrossRef CAS.
- Q. Yang, C. L. Parker, Y. Lin, O. W. Press, S. I. Park and S. K. Lai, J. Controlled Release, 2017, 255, 73–80 CrossRef CAS.
- A. Sarker, A. S. Rathore and R. D. Gupta, Microb. Cell Fact., 2019, 18, 5 CrossRef.
- H. Bach, Y. Mazor, S. Shaky, A. Shoham-Lev, Y. Berdichevsky, D. L. Gutnick and I. Benhar, J. Mol. Biol., 2001, 312, 79–93 CrossRef CAS.
- R. Birnboim-Perach, Y. Grinberg, L. Vaks, L. Nahary and I. Benhar, Methods Mol. Biol., 2019, 1904, 455–480 CrossRef CAS.
- F. J. Lopez-Jaramillo, M. Ortega-Muñoz, A. Megia-Fernandez, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Bioconjugate Chem., 2012, 23, 846–855 CrossRef CAS.
- J. Morales-Sanfrutos, J. Lopez-Jaramillo, M. Ortega-Munoz, A. Megia-Fernandez, F. Perez-Balderas, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Org. Biomol. Chem., 2010, 8, 667–675 RSC.
- E. Song, P. Zhu, S. K. Lee, D. Chowdhury, S. Kussman, D. M. Dykxhoorn, Y. Feng, D. Palliser, D. B. Weiner, P. Shankar, W. A. Marasco and J. Lieberman, Nat. Biotechnol., 2005, 23, 709–717 CrossRef CAS.
- M. Torres, S. Uroz, R. Salto, L. Fauchery, E. Quesada and I. Llamas, Sci. Rep., 2017, 7, 943 CrossRef.
- M. Kucinska, M. D. Giron, H. Piotrowska, N. Lisiak, W. H. Granig, F. J. Lopez-Jaramillo, R. Salto, M. Murias and T. Erker, PLoS One, 2016, 11, e0145615 CrossRef.
- H. Greulich, B. Kaplan, P. Mertins, T. H. Chen, K. E. Tanaka, C. H. Yun, X. Zhang, S. H. Lee, J. Cho, L. Ambrogio, R. Liao, M. Imielinski, S. Banerji, A. H. Berger, M. S. Lawrence, J. Zhang, N. H. Pho, S. R. Walker, W. Winckler, G. Getz, D. Frank, W. C. Hahn, M. J. Eck, D. R. Mani, J. D. Jaffe, S. A. Carr, K. K. Wong and M. Meyerson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14476–14481 CrossRef CAS.
- J. Morales-Sanfrutos, F. J. Lopez-Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, J. Org. Chem., 2010, 75, 4039–4047 CrossRef CAS.
- E. De Los Reyes-Berbel, R. Salto-Gonzalez, M. Ortega-Munoz, F. J. Reche-Perez, A. B. Jodar-Reyes, F. Hernandez-Mateo, M. D. Giron-Gonzalez and F. Santoyo-Gonzalez, Bioconjugate Chem., 2018, 29, 2561–2575 CrossRef CAS.
- M. D. Giron-Gonzalez, R. Salto-Gonzalez, F. J. Lopez-Jaramillo, A. Salinas-Castillo, A. B. Jodar-Reyes, M. Ortega-Munoz, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Bioconjugate Chem., 2016, 27, 549–561 CrossRef CAS.
- M. E. Kimple, A. L. Brill and R. L. Pasker, Curr. Protoc. Protein Sci., 2013, 73, 9.9.1–9.9.23 Search PubMed.
- E. De Los Reyes-Berbel, R. Salto-Gonzalez, M. Ortega-Munoz, F. J. Reche-Perez, A. B. Jodar-Reyes, F. Hernandez-Mateo, M. D. Giron-Gonzalez and F. Santoyo-Gonzalez, Bioconjugate Chem., 2018, 29, 2561–2575 CrossRef CAS.
- T. C. Hou, Y. Y. Wu, P. Y. Chiang and K. T. Tan, Chem. Sci., 2015, 6, 4643–4649 RSC.
- T. del Castillo, J. Marales-Sanfrutos, F. Santoyo-Gonzalez, S. Magez, F. J. Lopez-Jaramillo and J. A. Garcia-Salcedo, ChemMedChem, 2014, 9, 383–389 CrossRef CAS.
- T. Yousef and N. Hassan, J. Inclusion Phenom. Macrocyclic Chem., 2017, 87, 105–115 CrossRef CAS.
- M. V. Marshall, J. C. Rasmussen, I. C. Tan, M. B. Aldrich, K. E. Adams, X. Wang, C. E. Fife, E. A. Maus, L. A. Smith and E. M. Sevick-Muraca, Open Surg. Oncol. J., 2010, 2, 12–25 Search PubMed.
- X. Wang, D. Niu, C. Hu and P. Li, Curr. Pharm. Des., 2015, 21, 6140–6156 CrossRef CAS.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaia, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS.
- J. W. Buecheler, M. Winzer, C. Weber and H. Gieseler, J. Pharm. Sci., 2020, 109, 161–168 CrossRef CAS.
- C. M. Dundas, D. Demonte and S. Park, Appl. Microbiol. Biotechnol., 2013, 97, 9343–9353 CrossRef CAS.
- R. Reuten, D. Nikodemus, M. B. Oliveira, T. R. Patel, B. Brachvogel, I. Breloy, J. Stetefeld and M. Koch, PLoS One, 2016, 11, e0152386 CrossRef.
- D. S. Waugh, Postepy Biochem., 2016, 62, 377–382 CAS.
- P. Sun, J. E. Tropea and D. S. Waugh, Methods Mol. Biol., 2011, 705, 259–274 CrossRef CAS.
- R. Schier, A. McCall, G. P. Adams, K. W. Marshall, H. Merritt, M. Yim, R. S. Crawford, L. M. Weiner, C. Marks and J. D. Marks, J. Mol. Biol., 1996, 263, 551–567 CrossRef CAS.
- M. M. Moasser, Oncogene, 2007, 26, 6469–6487 CrossRef CAS.
- S. Parakh, H. K. Gan, A. C. Parslow, I. J. G. Burvenich, A. W. Burgess and A. M. Scott, Cancer Treat. Rev., 2017, 59, 1–21 CrossRef CAS.
- J. Albanell and J. Baselga, Drugs Today, 1999, 35, 931–946 CAS.
- M. Mahdavi, M. Keyhanfar, A. Jafarian, H. Mohabatkar and M. Rabbani, Monoclonal Antibodies Immunodiagn. Immunother., 2015, 34, 213–221 CrossRef CAS.
- C. Ceran, M. Cokol, S. Cingoz, I. Tasan, M. Ozturk and T. Yagci, BMC Cancer, 2012, 12, 450 CrossRef CAS.
- X. Yang, C. Shi, R. Tong, W. Qian, H. E. Zhau, R. Wang, G. Zhu, J. Cheng, V. W. Yang, T. Cheng, M. Henary, L. Strekowski and L. W. Chung, Clin. Cancer Res., 2010, 16, 2833–2844 CrossRef CAS.
- P. Li, Y. Liu, W. Liu, G. Li, Q. Tang, Q. Zhang, F. Leng, F. Sheng, C. Hu, W. Lai, Y. Liu, M. Zhou, J. Huang, H. Zhou, R. Zhang and Y. Zhao, Int. J. Oncol., 2019, 55, 415–424 CAS.
- T. Duong, X. Li, B. Yang, C. Schumann, H. A. Albarqi, O. Taratula and O. Taratula, Nanomedicine, 2017, 13, 955–963 CrossRef CAS.
- K. J. Hamblett, P. D. Senter, D. F. Chace, M. M. Sun, J. Lenox, C. G. Cerveny, K. M. Kissler, S. X. Bernhardt, A. K. Kopcha, R. F. Zabinski, D. L. Meyer and J. A. Francisco, Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS.
- B. Tian, S. Hua and J. Liu, Carbohydr. Polym., 2020, 232, 115805 CrossRef CAS.
- J. S. Marvin and H. W. Hellinga, Nat. Struct. Biol., 2001, 8, 795–798 CrossRef CAS.
- C. Roma-Rodrigues, L. Rivas-Garcia, P. V. Baptista and A. R. Fernandes, Pharmaceutics, 2020, 12(3), 233 CrossRef CAS.
- O. Germershaus, T. Merdan, U. Bakowsky, M. Behe and T. Kissel, Bioconjugate Chem., 2006, 17, 1190–1199 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Vinyl sulfone based reagents, chemical synthesis of maltosylated ligands and additional table and figures including Table S1. List of oligonucleotides, Fig. S1. Expression and purification of MBP*-ScFv and Fig. S2. Gene delivery capabilities of MBP* based/pDNA polyplexes (PDF). See DOI: 10.1039/d0bm01903h |
| This journal is © The Royal Society of Chemistry 2021 |