
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid conversion of highly porous borate glass microspheres into hydroxyapatite†
Md Towhidul
Islam
 ab,
Laura
Macri-Pellizzeri
c,
Virginie
Sottile
cd and
Ifty
Ahmed
*a
ab,
Laura
Macri-Pellizzeri
c,
Virginie
Sottile
cd and
Ifty
Ahmed
*a
aAdvanced Materials Research Group, Faculty of Engineering, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: Ifty.Ahmed@nottingham.ac.uk
bDepartment of Applied Chemistry and Chemical Engineering, Faculty of Engineering, Noakhali Science and Technology University, Noakhali-3814, Bangladesh
cSchool of Medicine, University of Nottingham, Nottingham, NG7 2UH, UK
dDepartment of Molecular Medicine, The University of Pavia, 27100 Pavia, Italy
First published on 13th January 2021
Abstract
This paper reports on the rapid development of porous hydroxyapatite (HA) microspheres with large external pores and fully interconnected porosity. These porous microspheres were produced by converting borates glasses (namely 45B5, B53P4 and 13-93B) into HA by immersing them in potassium phosphate media and simulated body fluid (SBF). Solid (SGMS) non-porous and highly porous (PGMS) microspheres were prepared from borate glasses via a novel flame spheroidisation process and their physicochemical properties including in vitro biological response were investigated. Morphological and physical characterisation of the PGMS showed interconnected porosity (up to 75 ± 5%) with average external pore sizes of 50 ± 5 μm. Mass loss, ion release, X-ray diffraction (XRD) and Scanning electron microscopy (SEM) analysis confirmed complete conversion to HA in 0.02 M K2HPO4 solution for the PGMS (with exception of 13-93B glass) and at significantly faster rates compared to their SGMS counterparts. However, 13-93B microspheres only converted to HA in Na2HPO4 solution. The in vitro SBF bioactivity studies for all the borate compositions showed HA formation and much earlier for PGMS compared to SGMS. Direct cell culture studies using hMSCs revealed that the converted porous HA microspheres showed enhanced pro-osteogenic properties compared to their unconverted counterparts and such are considered as highly promising candidate materials for bone repair (and orthobiological) applications.
1. Introduction
Hydroxyapatite, [Ca10(PO4)6(OH)2], is one of the most stable forms of biological apatite and the major inorganic constituent of bone.1,2 Around 60–70% of bone tissue and 90% of tooth enamel is made up of hydroxyapatite.3 It has been extensively used clinically as a bone graft substitute for bone augmentation, as an orthopaedic and dental implant coating4 and has even been explored for drug delivery applications.5–7Microspheres have some key advantages for use in biomedical applications over other particle geometries. For example, delivery of microspheres to specific target sites via simple injection procedures can be better facilitated due to their enhanced flow properties.8 Moreover, porous microspheres can be even more advantageous over solid (non-porous) microspheres as they provide higher surface areas. They also exhibit lower mass density, have shown superior cell attachment/proliferation and are well suited for drug adsorption/absorption as they can exhibit controlled drug release kinetics.9
To prepare porous hydroxyapatite (HA), several methods have been developed such as template-assisted processes which utilise hard templates (e.g. silica and carbon spheres10) and soft templates (e.g. emulsion droplets,11 micelles12 and gas bubbles13). Other methods have included hydrothermal synthesis,14 self-assembly via solvothermal methods15 and by spraying and freezing emulsions.16 For example, Zhang et al.14 fabricated HA microspheres (∼7–9 μm) and microflowers (∼10 μm) via the hydrothermal synthesis method using hexadecyltrimethylammonium bromide (CTAB) as a surfactant. Ma et al.17 prepared hollow HA microspheres (∼3.6 μm) consisting of nanorods using potassium sodium tartrate as a chelating agent in water/N,N-dimethylformamide mixed solvents. Li et al.15 also prepared flower-like HA microspheres which were self-assembled using nanosheets via solvothermal treatment of calcined eggshells in hydrogen peroxide/N,N-dimethylformamide. Whilst Xiao et al.16 prepared hollow and porous HA microspheres (∼20 μm in diameter with pore sizes ∼0.6 μm) using an oil in water (O/W) emulsion spray freezing method. Most of the methods stated above required 2–7 days to obtain porous HA microspheres.
Borate-based glasses have recently received much interest due to their successful application in wound healing.18 However, prior to this they had been shown to undergo conversion to hydroxyapatite.19–24 Prof. Day and his research group explored the HA formation on a borate glass termed 45S5B1, which had the same composition as 45S5 but with all of the SiO2 replaced with B2O3. They found that not only did HA form on the surface of borate glass after immersion in K2HPO4 solution at 37 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25,26 it also formed more rapidly when compared to silicate glass (45S5). Furthermore, in vivo studies reported that 45S5B1 glass particles (which had been partially converted into HA in K2HPO4 solution) formed bone tissue more rapidly in comparison to 45S5 glass particles when implanted within tibial defects in rats.25 Other in vivo studies also showed that borate-based glass (e.g. 13-93B) scaffolds were able to regrow bone in a rat subcutaneous implantation model with no toxicity.27–29
25,26 it also formed more rapidly when compared to silicate glass (45S5). Furthermore, in vivo studies reported that 45S5B1 glass particles (which had been partially converted into HA in K2HPO4 solution) formed bone tissue more rapidly in comparison to 45S5 glass particles when implanted within tibial defects in rats.25 Other in vivo studies also showed that borate-based glass (e.g. 13-93B) scaffolds were able to regrow bone in a rat subcutaneous implantation model with no toxicity.27–29
There are only a few studies on the development of porous microspheres from borate-based glass materials. For example, Fu et al.30 prepared hollow HA microspheres with pore size of ∼13 nm by reacting solid microspheres of Li2O–CaO–B2O3 glass (size range 106–150 μm) in K2HPO4 solution. Amorphous dysprosium lithium–borate glass microspheres (wt% composition 30% Dy2O3, 8.8% Li2O and 61.2% B2O3) developed pore sizes of ∼30 nm after reaction with 0.25 M K2HPO4 solution at 37 °C.31 Flower-like porous magnesium borate (Mg3B2O6) microspheres with diameters between 0.6–1.0 μm and pore sizes ranging between 2–100 nm were synthesised using polyvinyl pyrrolidone as a template.32
Most studies fabricating porous HA microspheres have either used template-supports, structure-directing reagents, and/or harmful organic solvents. Moreover, these processes resulted in only achieving mesopores (ranging from 4 nm to 0.6 μm) which would not be suitable for cell incorporation. Likewise, studies on borate-based glass microspheres have similar drawbacks as they have only been reported to achieve pore sizes ranging between 2 and 100 nm. Therefore, the development of a rapid, effective and template-free method to prepare highly porous HA microspheres with pore sizes capable of supporting cell incorporation is an ongoing and challenging process.
This paper reports for the first time a methodology to prepare both non-porous solid glass microspheres (SGMS) and highly porous glass microspheres (PGMS) from three borate glass formulations (i.e. 45B5, B53P4 and 13-93B, derived from their silicate glass equivalents namely 45S5, S53P4 and 13-93). The microsphere materials were produced via a novel flame spheroidisation process and a comparative analysis between borate SGMS and PGMS conversion to HA, their ion release and in vitro bioactivity are reported. Moreover, borate PGMS and converted form of porous HA microspheres were also tested in vitro, using human mesenchymal precursor stem-cells as a clinically relevant cell type, to evaluate their ability to support cell growth and osteogenic potential.
2. Materials and methodology
2.1 Preparation of borate glass formulations investigated
Three different borate glasses (i.e. 45B5, B53P4 and 13-93B), which were derived from the original 45S5, S53P4 and 13-93 silicate glass equivalents, by fully replacing the SiO2 with B2O3, were prepared using sodium carbonate (Na2CO3), potassium carbonate (K2CO3), calcium carbonate (CaCO3), magnesium carbonate (MgCO3), calcium hydrogen phosphate (CaHPO4), magnesium hydrogen phosphate trihydrate (MgHPO4·3H2O), phosphorous pentoxide (P2O5) and boron oxide (B2O3) as starting materials (Sigma Aldrich, UK). The composition of the borate glasses investigated is shown in Table 1. The required amounts of precursors were weighed, mixed and transferred to a platinum rhodium alloy crucible (Birmingham Metal Company, U.K.) which was then placed into a furnace and followed a controlled program using 10 °C min−1 ramp for melting (i.e. 350 °C for 0.5 hours to remove any residual moisture, then at 800 °C for 0.5 hours to remove CO2 and at 1150 °C for 1.5 hours to allow for melting). The resulting molten glass was quenched between two steel plates.| Glass formulation | B2O3 (mol%) | P2O5 (mol%) | CaO (mol%) | Na2O (mol%) | MgO (mol%) | K2O (mol%) |
|---|---|---|---|---|---|---|
| 45B5 | 46.1 | 2.6 | 26.9 | 24.4 | 0 | 0 |
| B53P4 | 53.85 | 1.72 | 21.77 | 22.66 | 0 | 0 |
| 13-93B | 54.6 | 1.7 | 22.1 | 6 | 7.7 | 7.9 |
2.2 Manufacturing solid and porous borate glass microspheres
The three borate glass formulations (45B5, B53P4 and 13-93B, see Table 1) were processed into solid glass microspheres (SGMS) and highly porous glass microspheres (PGMS) utilising a novel single-stage manufacturing process developed in our group.The glasses made were ground using a ball mill (Retsch PM 100) and sieved into the particle size range of 63–125 μm and 125–200 μm. Particles in the size range of 125–200 μm were then processed via a flame spheroidisation process to prepare SGMS, which utilises an oxy/acetylene flame spray gun (MK 74, Metallisation Ltd, UK).33,34
To manufacture highly porous microspheres (PGMS), glass particles in the size range of 63–125 μm were then mixed with porogen (CaCO3) at a 1:3 ratio and processed via flame spheroidisation similar to above, again via a single-stage process.33,34
PGMS of B53P4 and 13-93B were washed using only deionised water for 1 min and the 45B5 PGMS were washed using 0.1 M acetic acid for 1 min, followed by washing using deionised water and industrial methylated spirit (IMS) for quick drying and then dried at 50 °C overnight. The resulting PGMS were then sieved again (to gain a similar size range to the SGMS) and those between 125–200 μm were utilised for further studies.
2.3 Phosphate solution used for conversion reaction
Two phosphate solutions with 0.02 M and 0.2 M concentration were used for the experiments to explore conversion of borate glass microspheres into HA and were prepared by dissolving K2HPO4 or Na2HPO4 (for 13-93B) (Reagent grade; Fisher Scientific, UK) in deionised water. The pH of the starting solution was adjusted to 7.4, by adding a few drops of dilute HCl.3. Characterisation methods
3.1 Compositional analysis
Elemental analysis for borate glasses and glass microspheres (both solid and porous) were conducted via inductively coupled plasma mass spectroscopy (ICP-MS, Thermo-Fisher Scientific iCAP-Q equipped with collision cell technology with energy discrimination, UK) to verify their composition. For analysis, 0.1 g of borate glass particles/microspheres (before and after degradation in dil. K2HPO4 and SBF) were digested in 50 ml 37% HCl until a clear solution was obtained. The solution was then 50% diluted with Milli-Q water (1:1). The resultant solution was then diluted with 2% HNO3 with 1:10 ratio. The final solution was then filtered through a 0.2 μm syringe filter for ICP-MS analysis. Moreover, blank samples of pure Milli-Q water were mixed with the same proportion of HCl and 2% HNO3 using standard calibration solutions. Three replicates were analysed and their averages are reported.
3.2 Scanning electron microscopy (SEM)
Scanning electron microscopy (Philips XL 30 SEM, UK) with accelerating voltage of 15 kV and a working distance of 10 m, was used to examine the surface morphology of microspheres prepared (solid and porous microspheres), before and after immersion in degradation media (i.e. SBF and dil. K2HPO4/dil. Na2HPO4). The microspheres were fixed onto aluminium stubs with conductive carbon sticky tabs and sputter coated (Agar Sputter Coater) with platinum prior to examination and viewed with a Philips XL30 scanning electron microscope. Cross-sectional analyses of the microspheres were achieved by embedding the microspheres in a cold set epoxy resin and polished with SiC paper followed by a diamond cloth and then coated.3.3 X-ray diffraction
X-ray diffraction analysis was used to explore the amorphous nature of each glass formulation including the solid and porous glass microspheres produced using a Bruker D8 Advanced diffractometer (BRUKER AXS, Germany). The instrument was operated at room temperature and ambient atmosphere with Ni-filtered CuKα radiation (λ = 0.15418 nm), generated at 40 kV and 35 mA. Scans were performed with a step size of 0.04° and step time of 8 s over an angular range 2θ from 8° to 50°. XRD analysis was also used to examine any deposits on the surface of microspheres during the 21 day immersion period in SBF and dil. K2HPO4. Phases were identified using the EVA software (DIFFRACplus suite, Bruker-AXS) and the International Centre for Diffraction Data (ICDD) database (2005).3.4 Thermal analysis
Thermal properties of borate glasses and glass microspheres (solid and porous); glass transition (Tg) (measured at midpoint), onset of crystallisation (Tx), crystallisation peak (Tc), melting peak (Tm) temperatures and glass stability against crystallisation, were characterised using a simultaneous thermal analysis instrument (SDT, TA Instruments SDT Q600, USA). Approximately 20 mg of glass samples were placed into a platinum pan and heated from room temperature to 1100 °C at 20 °C min−1 heating rate. An empty pan was also run to determine the baseline, which was then subtracted from the thermal traces using TA Universal Analysis 2000 software.3.5 Density measurement
The density of the glass microspheres was determined using a MicromeriticsAccuPyc 1330 helium pycnometer (Norcross, GA, USA). The equipment was calibrated using a standard calibration ball (3.18551 cm3) with error of ±0.03%. Glass microsphere samples, with an average weight of approximately 1 g, were used for the density measurements, and the analysis was performed in triplicate. The bulk or tap density of the microspheres were also carried out using the following eqn (1). | (1) |
3.6 Porosity measurements
The porosity of the porous microspheres was evaluated via mercury intrusion porosimetry (Micromeritics Autopore IV 9500). A 5 cc powder penetrometer (Micromeritics) with 1 cc intrusion volume was used for all of the glass formulations investigated. Before running the samples, an empty penetrometer test was also carried out as a blank. The porosity of the microspheres was also calculated using the following eqn (2).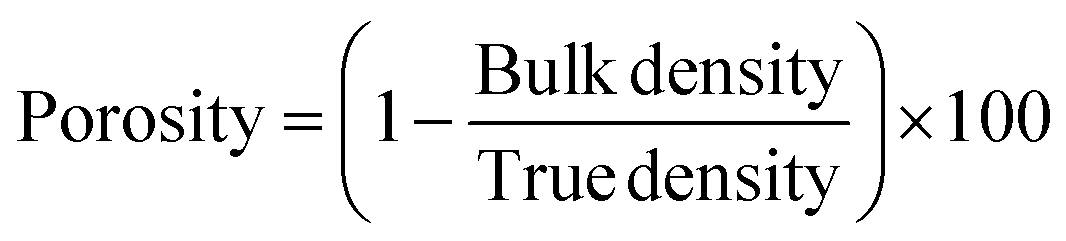 | (2) |
3.7 Mass loss, pH and ion release studies
The conversion of borate glass microspheres into HA in a phosphate solution was accompanied by a decrease in the mass of glass microspheres. Therefore, weight loss measurements provide a useful parameter for monitoring the kinetics of the conversion reaction. To evaluate the kinetics of material degradation rate, ion release and pH solution changes, 1% w/v of microspheres were immersed in dil. K2HPO4 or dil. Na2HPO4 and incubated at 37 °C. Assessments were performed on day 1, 3, 7, 10, 14 and 21. To determine the degradation rate, the microspheres were dried at 50 °C overnight and then weighted using a precision scale (Sartorius CP 225D). The percentage of mass loss was calculated according to the following eqn (3):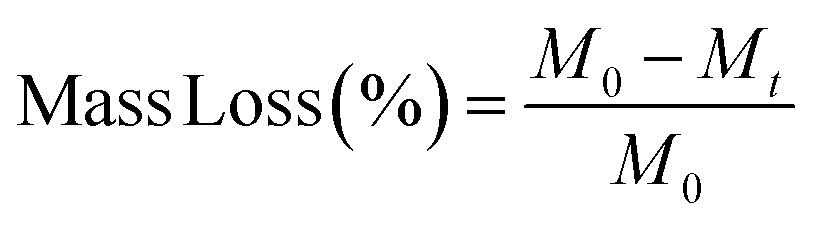 | (3) |
3.8 In vitro SBF bioactivity studies
In vitro bioactivity was tested in simulated body fluid (SBF) at day 1, 3, 7, 10, 14 and 21. The SBF solution was prepared following the standard procedure BS ISO 23317:2014. The SBF solution was kept at 5 °C for 48 h prior to use. 75 mg of microspheres (solid and porous) were immersed in 50 ml SBF solution at 37 °C in a polyethylene vial and agitated at 120 rpm. At each time point, the microspheres were filtered and washed with deionised water and then dried overnight at 50 °C in an oven. XRD, SEM and ICP-MS were utilised to explore the structural, morphological and compositional changes, respectively. The pH values of the SBF solution were measured at each time point. The concentration of boron, sodium, calcium, magnesium, phosphorous and potassium ions in SBF solution at each time point was also determined by ICP-MS (Thermo-Fisher iCAP-Q model, UK).3.9 Cell culture studies
Microspheres were sterilised throughout two washes of 15 minutes with 70% ethanol followed by complete evaporation at room temperature in sterile conditions as previously reported.33,35 For this study, GFP-labelled human mesenchymal stem cells (hMSCs) were seeded at a density of 10000 cells per cm2 on 10 mg of sterile material.33 Low-adherent 48-well plates were prepared using a coating with 200 μl of 1% w/v solution of poly(2-hydroxyethyl methacrylate) (poly-HEMA) (Sigma-Aldrich, UK) and 95% ethanol.36 Cells were cultured in standard medium (low glucose DMEM supplemented with 10% foetal calf serum, 1% penicillin and streptomycin, 1% L-glutamine, 1% of non-essential amino acid) at 37 °C and 5% CO2 for 12 days.
3.10 DNA quantitation assay
DNA amount was assayed at day 2 and 12 of culture using the Pico-Green® dsDNA quantitation kit according to the manufacturer's instructions. Briefly, medium was removed and 100 μl of sterile distilled water were added to each sample, after three cycles of freezing-thawing, 95 μl were transferred to a clear bottom 96-well plate for the measurement of fluorescence emission in the microplate reader (Infinite 200, Tecan, CH), setting 480 nm and 520 nm as excitation and emission wavelengths.3.11 Cell imaging
For bright-field and fluorescence images of living cells an EclipseT2 Nikon microscope coupled with a D3300 Nikon camera was used. Environmental Scanning Electron Microscopy was carried out in fixed cells using a FEI Quanta 650 (Thermo Fisher Scientific). Samples were washed twice with distilled water before imaging in order to remove any ion precipitation.3.12 Statistical analysis
For cell culture experiments, results of two independent experiments are presented as mean ± standard error of the mean (SEM). One-way ANOVA with Tukey's multiple comparison post hoc test was used. A 95% confidence level was considered significant. Statistical analysis was performed with the GraphPad PRISM 7.01 software package.4. Results
4.1 Characterisation of starting glass and microspheres
 | ||
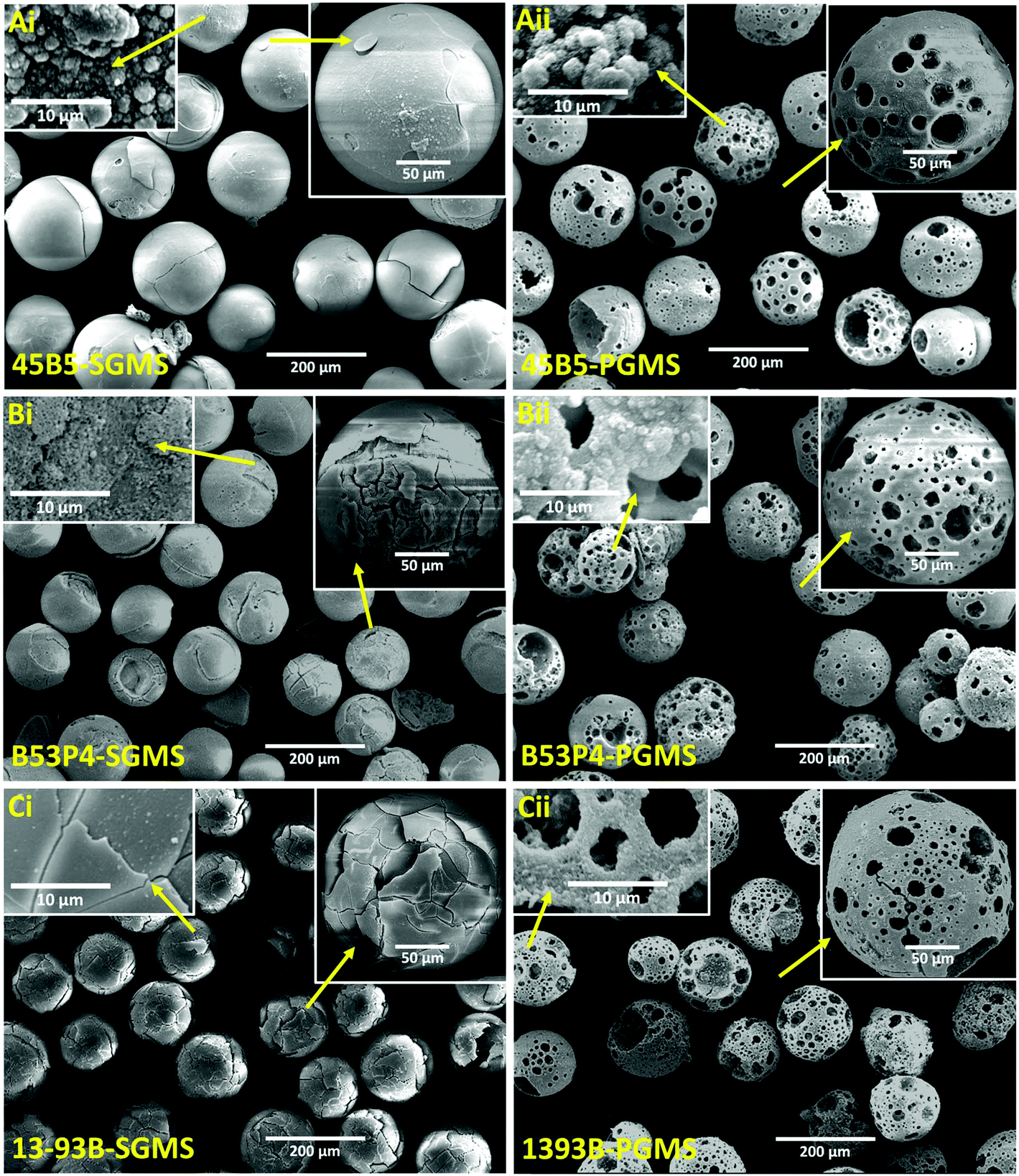
| Fig. 1 SEM images of Ai, Bi and Ci for SGMS and Aii, Bii and Cii for PGMS of 45B5, B53P4 and 13-93B glasses. Inset images Aiii, Biii and Ciii show porous microspheres at higher magnification and Aiv, Biv and Civ show the cross-sectional images of the porous microspheres of three borate glasses investigated. | ||
In order to examine the internal porous structure of the borate glass microspheres produced, they were embedded in a cold setting epoxy resin, ground and polished using SiC paper and diamond cloth, to a depth of a few microns to obtain microsphere cross-sections.
Fig. 1Aiii, Biii and Ciii represent higher magnification images of the porous microspheres produced. Whereas, Fig. 1Aiv, Biv and Civ show representative cross-sections of the porous 45B5, B53P4 and 13-93B glass microspheres produced, which not only revealed a large variation of internal pore sizes (ranging from meso to macropore scales), but also showed that the pores were fully interconnected throughout each microsphere produced.
The XRD profiles for the starting bulk glass (BG), SGMS and washed PGMS (i.e. 0.1 M acetic acid for 45B5 and water for B53P4 and 13-93B) are presented in Fig. 2A, B and C, respectively, where a single broad peak between 20° and 40° (2θ) was observed for BG and SGMS (with the exception of B53P4). The absence of any sharp crystalline peaks suggested that the glasses prepared were amorphous and that the SGMS retained their amorphous nature post processing. However, some peaks were observed for the SGMS from B53P4 which were matched to the sodium calcium phosphate (Na3Ca6(PO4)5) [JCPDS 11-0236] phase. Literature reported that the phase Na3Ca6(PO4)5 is highly water soluble and Na3Ca6(PO4)5 containing calcium phosphate cement could be used as bone substitute to enhance their bioresorbability.37,38 Peaks were also observed for the PGMS of each composition. The PGMS peaks were matched for CaCO3 which was used as the porogen material for manufacturing porous glass microspheres. This suggests that some residual porogen material remained in the pores of these microspheres, even after the wash-step.
 | ||
| Fig. 2 X-ray diffraction profiles for (A) starting bulk glass (BG), (B) SGMS and (C) washed PGMS of 45B5, B53P4 and 13-93B borate glasses investigated. ★ represents peaks matched to calcium carbonate (CaCO3) whilst ▼ represents peaks matched to sodium calcium phosphate (Na3Ca6(PO4)5). | ||
As EDX analysis can be unreliable for detecting Boron, the relative elemental concentrations (in mol%) for the BG, SGMS and PGMS produced from each formulation were analysed via ICP-MS instead and are summarised in Table 2. It can be seen that the respective oxide contents for the starting BG were within 1.5 mol% of their expected values with the exception of P2O5 and CaO content for B53P4 (which was 2.5 mol% higher and 2.3 mol% lower, respectively) as compared to their expected values. No significant variation in chemical composition between starting BG and SGMS were obtained for all three glass formulations. However, significantly higher CaO and lower B2O3 content was observed for the PGMS produced due to the addition of porogen in this process. For example, approximately 5 mol% lower B2O3 and 8 mol% higher CaO was observed for the PGMS produced as compared to the SGMS for 45B5 (see Table 2).
| Glass code | Different forms | Composition (mol%) | ||||||
|---|---|---|---|---|---|---|---|---|
| B2O3 | P2O5 | CaO | Na2O | MgO | K2O | |||
| 45B5 | BG | Expected | 46.1 | 2.6 | 26.9 | 24.4 | N/A | N/A |
| Actual | 47.08 ± 0.19 | 2.96 ± 0.01 | 26.33 ± 0.09 | 23.33 ± 0.05 | N/A | N/A | ||

|
47.70 ± 0.04 | 2.86 ± 0.02 | 26.42 ± 0.09 | 22.80 ± 0.02 | N/A | N/A | ||

|
42.90 ± 0.04 | 2.97 ± 0.01 | 34.12 ± 0.03 | 19.74 ± 0.02 | N/A | N/A | ||
| B53P4 | BG | Expected | 53.85 | 1.72 | 21.77 | 22.66 | N/A | N/A |
| Actual | 54.36 ± 0.08 | 4.22 ± 0.05 | 19.43 ± 0.02 | 21.81 ± 0.04 | N/A | N/A | ||

|
54.54 ± 0.01 | 4.22 ± 0.01 | 19.64 ± 0.01 | 21.42 ± 0.02 | N/A | N/A | ||
|
49.11 ± 0.26 | 3.85 ± 0.01 | 27.56 ± 0.21 | 19.26 ± 0.03 | N/A | N/A | ||
| 13-93B | BG | Expected | 54.6 | 1.7 | 22.1 | 6 | 7.7 | 7.9 |
| Actual | 55.43 ± 0.22 | 1.91 ± 0.01 | 21.61 ± 0.08 | 5.94 ± 0.12 | 7.35 ± 0.01 | 7.77 ± 0.01 | ||

|
55.40 ± 0.18 | 1.90 ± 0.01 | 21.88 ± 0.18 | 5.91 ± 0.01 | 7.33 ± 0.01 | 7.58 ± 0.01 | ||

|
49.68 ± 0.20 | 1.77 ± 0.01 | 29.77 ± 0.2 | 5.27 ± 0.03 | 6.68 ± 0.02 | 6.83 ± 0.02 | ||
| Formulation | Density | Tap density | % Porosity (calculated) | % Porosity (Hg porosimetry) | |
|---|---|---|---|---|---|
| 45B5 | SGMS | 2.496 ± 0.004 | 1.72 ± 0.03 | 31.10 | N/A |
| PGMS | 2.286 ± 0.004 | 0.82 ± 0.02 | 64.13 | 66 ± 4 | |
| B53P4 | SGMS | 2.432 ± 0.002 | 1.75 ± 0.02 | 28.05 | N/A |
| PGMS | 2.361 ± 0.005 | 0.65 ± 0.3 | 74.17 | 71 ± 5 | |
| 13-93B | SGMS | 2.425 ± 0.006 | 1.76 ± 0.02 | 27.82 | N/A |
| PGMS | 2.302 ± 0.006 | 0.50 ± 0.3 | 78.28 | 75 ± 4 | |
The pore diameters obtained (measured via mercury porosimetry versus the log differential pore volume) are presented in ESI Fig. 1.† It should be noted that mercury porosimetry determines the largest entrance to a space as a pore which is not always the actual “pore size” as it will also include inter-particulate gaps.39 The PGMS from all three glasses revealed multimodal pore size distribution with the first prominent peak showing a modal value range of ca. 43–52 μm, the second peak at ca. 20–28 μm and a third peak at ca. 2–5 μm. More interestingly, this analysis revealed pore size features at submicron scales ranging to nanoscale porosity levels down to 10 nm as shown in ESI Fig. 1.†
4.2 Immersion of solid and porous borate glass microspheres in phosphate solutions
 | ||
| Fig. 3 (A) Mass loss and (B) pH change as a function of immersion time (days) of SGMS and PGMS from three different borate glasses in 0.02 M K2HPO4 solution at 37 °C over 21 day duration. Solid symbols represent SGMS, whilst the open symbols represent the porous counterparts. | ||
In general, the pH values in phosphate solution increased from 7.4 to 10 by day 3, then gradually decreased to approximately neutral (i.e. 7.6) by day 10 and then remained relatively constant for the remainder of the study as seen in Fig. 3B.
 | ||
| Fig. 4 Cumulative ion release profile of (A) borate, (B) sodium, (C) phosphate, (D) potassium, (E) calcium and (F) magnesium ions as measured via ICP-MS for both SGMS and PGMS from the glasses investigated, over 21 days immersion in 0.02M K2HPO4. (Error bars are also included in the data above.) Black dashed lines represent for starting ion concentration of solution. | ||
The boron and sodium ion release profiles for both SGMS and PGMS from all three borate glasses increased with increasing immersion time to a plateau. The ion release rate was higher for PGMS compared to their SGMS counterpart. For example, cumulative boron release was 1311 ppm for PGMS and 836 ppm for SGMS of 45B5 at day 14. Similarly, higher K and Mg ion release were observed for PGMS of 13-93B compared to SGMS during the study. On the other hand, the opposite trend of Ca2+ ion release profile was observed for SGMS and PGMS, where the Ca2+ ion release for the SGMS was higher in comparison to their respective PGMS. However, the overall Ca2+ ion release was negligible in comparison to B or Na ion release.
Fig. 4C shows that the concentration of P ions in solution decreased for both SGMS and PGMS from the borate glasses with increasing immersion time. The reduction of P ion from the solution was higher for PGMS in comparison to their SGMS counterpart. For example, the concentration of P ion reduced from 623 ppm (starting concentration) to 68 ppm by day 10 for PGMS of B53P4. Whereas, P ion for SGMS of the same glass was found to be 466 ppm at the same time point. Interestingly, the P ion concentration reached a negative value for the PGMS of 45B5 at day 10 which then plateaued from Day 14 to 21. Similarly, depletion of K ions was observed for both SGMS and PGMS of 45B5 and B53P4 glasses (which did not have any K in their formulation) with the exception of 13-93B.
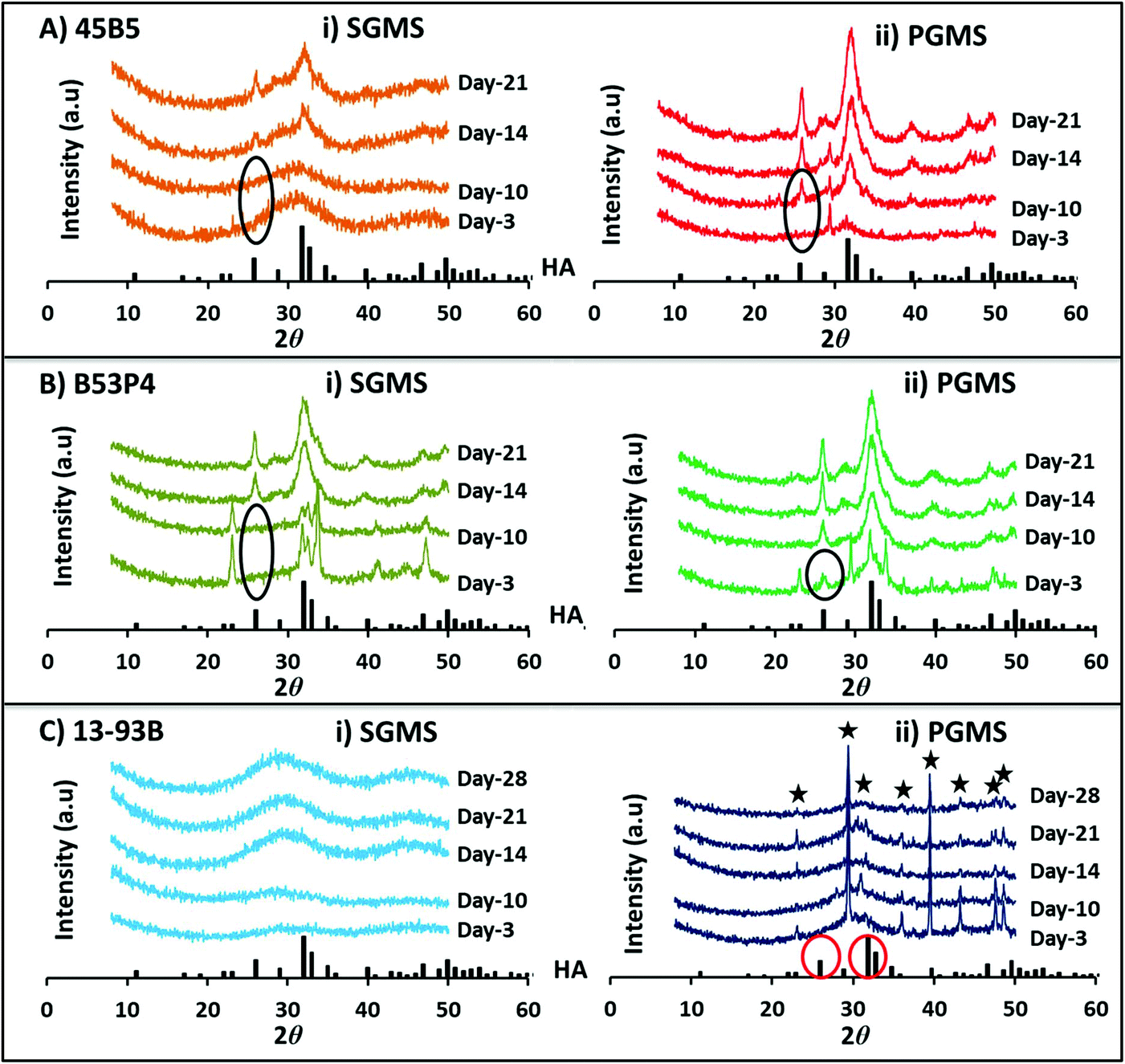 | ||
| Fig. 5 X-ray diffraction pattern for both (i) SGMS and (ii) PGMS of three borate glass formulations (A) 45B5, (B) B53P4 and (C) 13-93B during immersion of microspheres in 0.02 M K2HPO4 solution at 37 °C over time. XRD spectra for HA is shown at the base of each figure for comparison. ★ highlights peaks for calcium carbonate (CaCO3). | ||
For 45B5 a HA peak was observed at day-14 for the SGMS, whereas for the PGMS a HA peak was observed at a much earlier time point (i.e. formed between days 3 to 10) as seen in Fig. 5A. Moreover, the peak intensities were greater for all time points observed for the PGMS.
However, for the PGMS of B53P4 a HA peak was observed at day-3, which increased in intensity by day 21.
Surprisingly, no HA peak was observed for both the SGMS and PGMS of 13-93B even up to 28 days immersion in 0.02 M K2HPO4 solution (see Fig. 5C). The CaCO3 peaks observed were most likely due to the remaining presence of porogen residue in the PGMS even after washing.
 | ||
| Fig. 6 SEM images Ai, Bi and Ci for SGMS and Aii, Bii and Cii for PGMS of 45B5, B53P4 and 13-93B post immersion in 0.02 M K2HPO4 solution at 37 °C at day-21. Inset images show individual microsphere at higher magnification. | ||
In order to investigate if any changes had occurred to the internal morphology of both SGMS and PGMS post immersion in 0.02 M K2HPO4 solution at day 10 and 21, cross-sectional analysis of the microspheres was carried out and is presented in ESI Fig. 2.† The cross-sections at day-0 (i.e. starting microspheres) are also highlighted for comparison.
The SGMS behaved differently during the immersion in 0.02 M K2HPO4 solution. For example, the conversion reaction for the SGMS of 45B5 seemed to start from the outer surface and moved inward with increasing immersion time with multiple layers observed at day 21 (highlighted in dashed red circles, see ESI Fig. 2†). Surprisingly, hollow microspheres for SGMS of B53P4 were observed (as highlighted in dashed red circles with yellow arrow at day 10 and at day 21, see ESI Fig. 2†). On the other hand, a large solid core and small areas of degraded outer surfaces were observed for SGMS of 13-93B at day 21 (also highlighted in dashed red circles, see ESI Fig. 2†).
EDX analyses of the cores of the cross-sectioned microspheres (at day 0, 10 and 21) were also conducted to observe the presence (highlighted in red) and disappearance (highlighted in black) of boron and the representative EDX spectra are shown in Fig. 7. Point spectra at core from various microspheres were obtained (shown as ‘+’ symbol, see ESI Fig. 2†). The presence of boron was observed for SGMS of 45B5 at day 21 post immersion (see Fig. 7). Whereas, no boron was observed for PGMS at day 21, which disappeared at some point between day 10 and day 21. Interestingly, no boron was also observed for the SGMS of B53P4 at day 21. Furthermore, the boron peak also disappeared earlier than day 10 for the PGMS. However, the presence of boron was observed for both SGMS and PGMS of 13-93B even till day 21 (see Fig. 7).
 | ||
| Fig. 7 EDX analysis for the cross-sections of SGMS and PGMS of 45B5, B53P4 and 13-93B at day-0 (starting microspheres), day-10 and day-21 during the immersion of microspheres in 0.02 M K2HPO4 solution at 37 °C. EDX spectra shown for core of each cross-sections of microspheres, as a representative. Red circle highlights the presence of boron and black circle highlights the absence of boron. | ||
As mentioned earlier, it should be noted that quantitative analysis for boron containing compounds cannot be measured accurately via EDX analysis. Therefore, further compositional analysis of the microspheres was conducted via ICP-MS, post immersion in 0.02 M K2HPO4 solution for the day 21 samples.
| Components | 45B5 | B53P4 | 13-93B | |||
|---|---|---|---|---|---|---|
| SGMS (mol%) | PGMS (mol%) | SGMS (mol%) | PGMS (mol%) | SGMS (mol%) | PGMS (mol%) | |
| B2O3 | 35.96 ± 2.90 | 0.77 ± 0.09 | 0.84 ± 0.02 | 0.39 ± 0.04 | 50.74 ± 0.27 | 35.62 ± 1.23 |
| P2O5 | 9.68 ± 1.47 | 25.88 ± 0.09 | 26.01 ± 0.09 | 26.42 ± 0.06 | 4.11 ± 0.12 | 8.16 ± 0.31 |
| CaO | 47.86 ± 3.63 | 71.68 ± 0.22 | 71.41 ± 0.14 | 71.59 ± 0.14 | 26.81 ± 0.11 | 42.48 ± 1.17 |
| Na2O | 5.86 ± 2.22 | 0.30 ± 0.01 | 0.62 ± 0.01 | 0.34 ± 0.01 | 4.3 ± 0.01 | 2.05 ± 0.11 |
| MgO | — | — | — | — | 8.27 ± 0.08 | 8.78 ± 0.01 |
| K2O | 0.34 ± 0.01 | 0.92 ± 0.01 | 0.83 ± 0.09 | 0.83 ± 0.02 | 5.78 ± 0.03 | 2.92 ± 0.14 |
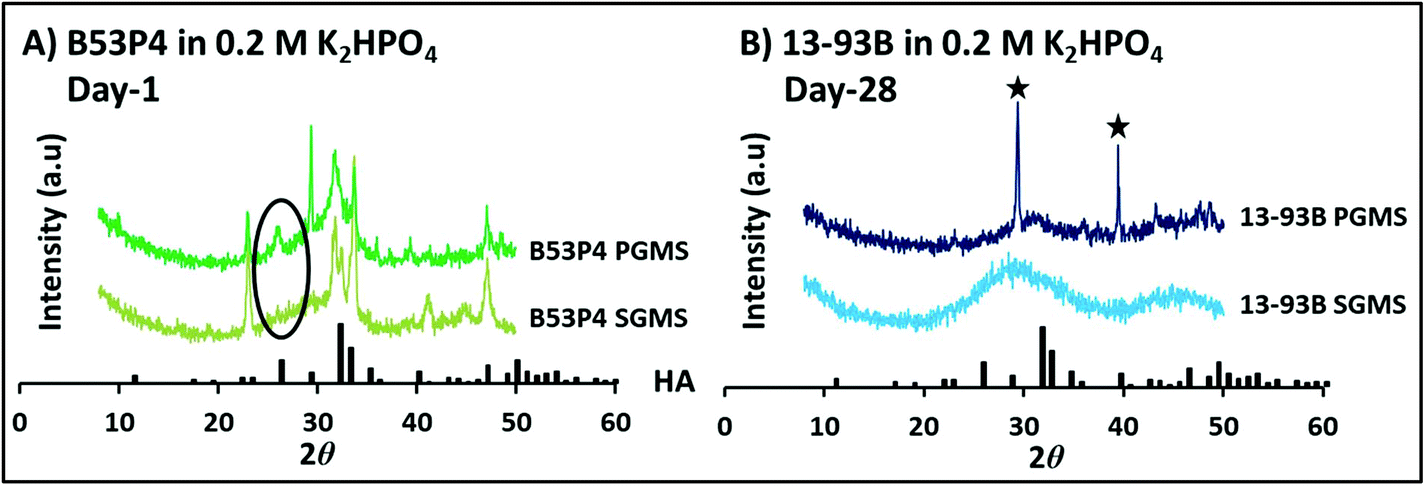 | ||
| Fig. 8 X-ray diffraction pattern for both SGMS and PGMS of (A) B53P4 at day 1 and (B) 13-93B at day 28 in higher 0.2 M K2HPO4 concentration solution at 37 °C. HA was identified at day 1 for the porous B53P4 microspheres in comparison to SGMS. Whereas, no HA was seen for 13-93B, even up to day 28. ★ highlights peaks for calcium carbonate (CaCO3). | ||
 | ||
| Fig. 9 (A) Highlights mass loss for PGMS of 13-93B in (0.02 M and 0.2 M) Na2HPO4versus immersion time (days), (B) shows the X-ray diffraction patterns at day 28 and SEM images for PGMS of 13-93B post immersion of microspheres in (C) 0.02 M Na2HPO4 and (D) 0.2 M Na2HPO4 solution at 37 °C at day-28. Inset images show microspheres at higher magnification. XRD spectra for HA shown at the bottom of B for comparison. ★ highlights peaks for calcium carbonate (CaCO3). | ||
Fig. 9B represents the XRD spectra for PGMS of 13-93B at day 28 post immersion in Na2HPO4 solution at the two different concentrations. Interestingly, a HA peak was observed for the microspheres in both concentrations (i.e. 0.02 M and 0.2 M). However, more intense HA crystalline peaks were seen for the microspheres in 0.2 M Na2HPO4 compared to 0.02 M Na2HPO4 solution. In addition, CaCO3 peaks were still present at day 28 post immersion in the lower 0.02 M Na2HPO4 solution.
Fig. 9C and D show the SEM images at different magnifications for PGMS of 13-93B at day 28 post immersion in 0.02 and 0.2 M Na2HPO4 solution at 37 °C, respectively. It can be seen from both figures at higher magnification that HA had formed on both the upper surface and within the inner porosity of the PGMS of 13-93B in both concentrations when Na2HPO4 solution was utilised (and did not form any HA when using K2HPO4).
4.3 In vitro bioactivity studies
 | ||
| Fig. 10 X-ray diffraction patterns for both (A) SGMS and (B) PGMS of 13-93B post immersion in SBF at 37 °C up to day 21. XRD spectra for HA is also shown at the bottom of each figure for comparison. SEM images (C) SGMS and (D) PGMS of 13-93B post immersion in SBF at day 21. Inset images shown at higher magnification. | ||
The boron, sodium and calcium ion release profiles for both SGMS and PGMS of all the borate glasses investigated increased with increasing immersion time before reaching a plateau. The ion release rate was higher for PGMS compared to their SGMS counterparts. For example, boron release was 178 ppm for PGMS of 45B5 at day 10 after that the value remained constant. Whereas, boron release was 134 ppm for the SGMS at day 10. Similarly, higher K and Mg ion release were observed for PGMS of 13-93B compared to SGMS before achieving a plateau at day 7.
ESI Fig. 5D† shows that the concentration of P ions decreased for both SGMS and PGMS from the borate glasses with increasing immersion time. The reduction of P ion from SBF was higher for PGMS in comparison to their SGMS counterpart. Interestingly, the P ion concentration achieved a negative value for both SGMS and PGMS at day-21. Similarly, depletion of Mg ions was observed for both SGMS and PGMS of 45B5 and B53P4 glasses over the bioactivity SBF study.
4.4 Cell culture study
A cell culture study was conducted to compare the influence of Boron containing microspheres versus the HA converted borate microspheres. Fluorescently-labelled human mesenchymal stem cells were seeded and cultured for 12 days on 45B5 and B53P4 PGMS sample as prepared and converted to HA (in 0.02 M K2HPO4). Live fluorescence imaging performed at day 2 showed the formation of macro-aggregates of cells and microspheres for the HA converted form of both formulations, while no signal was detected in cultures seeded on the boron containing unconverted counterparts (see Fig. 11A). Observation of the cultures at day 12 confirmed this trend, with significantly higher values for samples grown on the HA converted versus unconverted materials (see Fig. 11B). ESEM imaging performed at day 12, evaluated the cell–microsphere interactions and showed that cells appeared flat and adhered to the microsphere surface and pores of the HA converted forms (see Fig. 11C, arrowheads). Conversely, cells cultured on the boron containing unconverted microspheres appeared mainly rounded, with protrusions onto the material surface (see Fig. 11C, arrows). | ||
| Fig. 11 Evaluation of cell response to unconverted and converted (in 0.02 M K2HPO4) forms of PGMS of 45B5 and B53P4 borate glass at day 2 and day 12. (A) Representative live fluorescence images of cell-microspheres aggregates 48 hours after seeding. (B) Semi-quantitation of DNA amount at day 12 in the converted form normalised to the unconverted counterpart. Dotted line corresponds to the value of 1 of the normalisation. (C) Representative ESEM images obtained at day 12 of culture for unconverted and converted forms of both formulations. Yellow arrows point at round cells protruding from the material surface while yellow arrowheads points at flat cells attached to the microspheres. | ||
5. Discussion
It is not surprising that Hydroxyapatite (HA), the main inorganic constituent of bones and teeth, has been one of the most widely researched biomaterials, which exhibits natural bioactive behaviour and excellent biocompatibility.40,41 The performance of HA for bone regeneration greatly depends on its morphology, three-dimensional (3D) structure and chemical composition.42 As highlighted in the introduction, several studies report on developing porous HA microspheres which usually involve using template-based supports, harmful organic solvents, structure-directing reagents.10,11,13,17,43,44 Furthermore, these microspheres only possessed mesopores (ranging from ∼2 nm–0.6 μm), which would not be suitable for cell incorporation.31Studies also showed that borate-based glasses can undergo complete conversion to HA due to their higher solubility.26 However, the conversion rates can be influenced by their morphology and glass compositions. For example, Huang et al. (working with Professor Day and Professor Rahaman) investigated the conversion of 45B5 solid glass microspheres (i.e. a glass formulation which replaced all the SiO2 in the original 45S5 glass with B2O3) in dilute potassium phosphate solution.26 XRD and compositional analysis of the microspheres produced (which were in the size range of 800–1000 μm) revealed only partial conversion to HA after 310 h (i.e. 13 days).26 This paper set out to explore the hypothesis that producing highly porous glass microspheres (PGMS) from borate glasses would lead to complete conversion to HA and at a significantly faster rate. Three borate glass formulations were investigated (45B5, B53P4 and 13-93B) which were all derived based on 45S5, S53P4 and 13-93 biosilicate glasses, respectively by fully replacing the SiO2 with B2O3.
5.1 Manufacturing solid and porous borate glass microspheres
The results confirmed successful manufacture of both solid (SGMS) and highly porous glass microspheres (PGMS) from all three borate glass compositions. It has only recently become possible to produce such highly porous glass microspheres, due to a unique processing method established in our group. The cross-sectional SEM images of the PGMS for all three glass formulations investigated (see Fig. 1) revealed their internal porosity, which clearly showed that the pores were fully interconnected throughout each of the microspheres produced. The glass particles were transformed into microspheres due to surface tension forces during the cooling stage as the molten glass globules were ejected out of the flame as mentioned elsewhere.45 This novel and rapid method of manufacturing highly porous glass microspheres was first demonstrated for manufacturing highly porous microspheres from phosphate based glasses, which has now been transferred to borate glasses, for the first time.33 The yield in terms of porosity and pore sizes of the porous glass microspheres produced depends on various factors such as the size of the starting glass particles, glass powder to porogen ratio and the oxy-acetylene flame ratios used. Recent studies on phosphate glass microspheres from our group have reported on porosity levels ranging from 52 (±5) to 76 (±5) % by varying the glass particles to porogen ratios from 1:1 to 1:3.33 They also showed that the size of the starting microparticles effected pore size. For example, the porous microspheres between 63 and 125 μm revealed a significant number of larger surface pores (55 ± 8 μm) compared to the larger diameter size range porous glass microspheres (i.e. between 200 and 300 μm) which revealed a larger number of smaller surface pores (ranging from macro-porous sized features up to 30 μm).33
As borate glasses are highly degradable in acidic media, a very low concentration of acetic acid for 45B5 PGMS, whilst only water for B53P4 and 13-93B PGMS was utilised for the sample wash step to remove any residual porogen remnants post spheroidisation. However, the XRD results (see Fig. 2C) showed that porogen remnants were not completely removed, which was due to them being trapped deep inside the porous microspheres (confirmed via XRD; see Fig. 2C).
SGMS from 45B5 and 13-93B remained fully amorphous whereas the SGMS of B53P4 crystallised, revealing peaks for the phase sodium calcium phosphate [Na3Ca6(PO4)5] identified from XRD analysis (see Fig. 2B). This crystallisation was attributed to the lower glass stability (52 °C) of the B53P4 composition compared to 45B5 (104 °C) and 13-93B (198 °C), as highlighted in ESI Table 2 and ESI Fig. 7.†
In addition, the relatively higher (2.5 mol%) content of P2O5 in B53P4 compared to the expected value of P2O5 may also have aided the crystallisation process (see Table 2). A study on the influence of phosphorus speciation on the phase separation of Na2O–B2O3–SiO2 glasses by Muñoz et al. reported that the larger ionic field strength of P5+ cations (2.1) compared to those of Si4+ (1.57) and B3+ (1.63 and 1.34, for three and four-fold coordinated boron atoms, respectively) increased the tendency of phosphorus to separate from the borosilicate network, thus leading to crystalline phase separation of the phosphate species.46
5.2 Rapid conversion of borate glass microspheres to HA
The mechanism for HA formation on 45S5 silicate glass is well established where the first step is the formation of a SiO2-rich gel layer by ion exchange reactions, followed by dissolution of Ca2+ and PO43− ions and their diffusion through the SiO2-rich gel layer, leading to formation of an amorphous calcium phosphate (ACP) layer on the SiO2 gel which then converts to HA.47 The conversion of borate glasses to HA seems to follow a mechanism similar to that for 45S5 glass, however without formation of a SiO2-rich layer.20 Prof. Day and co-workers20,24,26,30,48 were the first to explore conversion of borate glasses to HA. They reported that borate glasses converted completely to HA at a faster rate compared to 45S5 glass and a wide range of HA structures, ranging from pseudomorphic nanoporous particles to pseudomorphic hollow microspheres, can be produced due to the structural changes occurring during the conversion reaction.Mass loss profiles for borate glass microspheres (both SGMS and PGMS) in this study correlated entirely to their ion release profiles, and especially to the borate and sodium release profiles (see Fig. 3A and 4A). The Na+ and BO43− ions were released into solution, whereas Ca2+ ions from the glass reacted with PO43− from the solution to form HA and consequently, depletion of phosphate ions from solution was observed (see Fig. 4C) which plateaued by day 14 for 45B5-PGMS and day 10 for B53P4-PGMS suggesting the reaction had completed.
Based on the results (i.e. mass loss, ion release, XRD and compositional analysis) obtained, comparisons for the conversion of SGMS and PGMS to HA post immersion in 0.02 M K2HPO4 solution are summarised in Table 5.
| Formulation | Between days 3–10 | Between days 10–14 | Between days 14–21 | Evidence | |
|---|---|---|---|---|---|
| 45B5 | SGMS | — | — | Partially converted (HA layers formed on periphery and not down to the inner core) | XRD + SEM + EDX + ICP-MS + cross-sections see Fig. 5, 6, and 7, Table 4 and, ESI Fig. 2† |
| PGMS | — | Completely converted | — | ||
| B53P4 | SGMS | — | — | Completely converted (formed hollow microspheres) | |
| PGMS | Completely converted | — | — | ||
| 13-93B | SGMS | — | — | Not converted | |
| PGMS | — | — | Not converted | ||
The conversion rates were much faster for the PGMS (of 45B5 and B53P4) in comparison to their SGMS counterparts, which was expected due to their increased surface area achieved from the starting materials.33 It was also apparent that conversion rate depended on glass composition, as the SGMS from three borate glasses revealed different conversion reaction profiles (e.g. 45B5-SGMS revealed formation of varying HA layers and remained unconverted at its core, whereas B53P4-SGMS revealed formation of hollow HA microspheres; see ESI Fig. 2†). The earlier complete conversion for SGMS of B53P4 compared to 45B5 could be attributed to the presence of highly water soluble Na3Ca6(PO4)5 phase in their structure which accelerated the conversion process.37,38 The compositional analysis at day 21 revealed that non-stoichiometric HA (Ca/P was approximately 1.4) formed for completely converted borate glass microspheres (i.e. PGMS of 45B5, and SGMS and PGMS of B53P4) in 0.02 M K2HPO4 solution due to the presence of trace amounts of Na, Mg and K in their HA crystal structure.49
The conversion of SGMS to HA started from the outer surface, moving inward with increasing immersion time. Whereas, the conversion reaction for PGMS occurred from both the upper surfaces and from within through the pores created during production. Based on the ion release profiles and compositional analysis (via ICP-MS), the mechanism for the conversion of borate SGMS vs. PGMS (i.e. 45B5) to HA is depicted in Fig. 12.
 | ||
| Fig. 12 (A) Mechanism for the conversion of borate glass microspheres into HA in phosphate solution and (B) outer surface and cross-sections of SGMS and PGMS at day 21 post immersion in 0.02 M K2HPO4 solution, shown as an evidence. | ||
5.3 Troublesome 13-93B
No conversion reaction was observed for both SGMS and PGMS of 13-93B in both the 0.02 M K2HPO4 and at the higher 0.2 M K2HPO4 concentration by day 28 (see Fig. 5C and 6C). Whereas, at the higher (0.2 M) concentration, HA peaks were observed much earlier (day 1) for PGMS of B53P4 as compared to 0.02 M K2HPO4 (at day 3). The lack of conversion of 13-93B glass microspheres to HA in K2HPO4 solution could be attributed to the presence of potassium in both the starting glass microsphere formulations and solution utilised, which may have inhibited the ion exchange reaction. However, PGMS of 13-93B did convert to HA in Na2HPO4 solution and a faster conversion rate was observed at higher concentration (i.e. 0.2 M) compared to 0.02 M Na2HPO4 solution.5.4 In vitro bioactivity in SBF
Both SGMS and PGMS from all three borate glasses showed bioactivity in SBF (see Fig. 10 and ESI Fig. 3, 4 and ESI Table 1†). However, the PGMS showed much earlier bioactivity as compared to their SGMS counterparts (as predicted in our hypothesis, due to their higher surface area). Compositional analysis at day 21 revealed that non-stoichiometric HA (Ca/P ratio 1.5) formed for both SGMS and PGMS of all borate glasses.49It was observed that borate leached out almost completely from SGMS of 45B5 and 13-93B (i.e. complete conversion) in SBF, which was not observed in 0.02 M K2HPO4 solution. This could be attributed to the presence of TRIS buffer in SBF which accelerated the dissolution of borate glass microspheres in comparison to the potassium phosphate solution.50 The effect of TRIS buffer on degradation rate of silicate 45S5 scaffolds has been investigated by Rohanová et al. who reported that the scaffold material dissolved twice as fast in TRIS buffer compared to dH2O.50 The significant reduction of material mass in the solutions containing TRIS buffer confirmed the accelerated dissolution of scaffolds, in comparison with solutions without TRIS buffer.50 Therefore, further rapid conversion rate for borate glass microspheres may well be achieved using TRIS buffer in K2HPO4 and/or Na2HPO4 solution.
5.5 Cell culture study
To evaluate biocompatibility of the HA PGMS developed, human mesenchymal cells were used as a clinically relevant cell model. PGMS of 45B5 and B53P4 which had been converted (in 0.02 M K2HPO4) to HA revealed significantly enhanced biological properties supporting cell adhesion and growth for up to 12 days in vitro, whilst no significant cell growth was detected on the unconverted forms. These observations are suggested to be due to a combined effect of glass surface stability and ion release rates. Approximately 25–50% mass loss was observed for the unconverted forms in 0.02 M K2HPO4 solution during the first 3 days, for both formulations, which could have inhibited the initial cell adhesion necessary to establish stable cell–material interaction.51 The second critical factor was the amount of ions released from the materials to the cell culture medium, particularly with regard to borate. In vitro studies using boron-doped bioglass materials have attributed a dual biological effect to this ion, with concentrations of borate in cell culture of up to 0.65–1.5 mmol associated with an increase of cell proliferation, while for higher values a decrease of cell growth has been reported.52,53 In the present study, the burst release of borate observed for the unconverted forms during the first 24 h resulted in a concentration of between 500 and 1100 ppm (46 mmol–102 mmol), which most likely contributed to inhibition of cell growth. Interestingly, a previous study reported a more favourable cell response using partially converted materials in comparison to fully converted ones, which may be due to a residual release of ions (calcium, borate and sodium) from the partially converted forms exerting a favourable effect on the cells.54The porous conformation of the materials presented in this study is a clear advantage for future applications in tissue engineering as it would enable cells to grow in 3D environments which more closely resemble the native tissue in vivo.55 In the context of porous materials, pore structure and porosity are both key factors of scaffolds in tissue engineering. However, the results available in the literature are still contrasting and the optimal values for successful outcomes are yet to be defined. For instance, studies comparing pore sizes in the range of 50–500 μm found more bone formation associated with larger pore values.56–58 Conversely, in another study scaffolds with 100 μm pore size seemed to perform better than the 250 and 400 μm-pore counterparts.59 It is worth noting that porous calcium phosphate microspheres with similar pore structure to that reported in this study promoted cell colonisation towards the core of the microsphere in vitro and induced tissue ingrowth and osteogenic onset in vivo.60
In summary, the present cell culture observations indicated much improved biological properties for PGMS post full conversion, which warrants a future comprehensive evaluation of their osteogenic potential in vitro and also of their regenerative potential in vivo. Indeed, formation of HA materials is still appealing for bone repair applications, as HA is known for its osteoinductive properties and HA-based devices are already in clinical use.61 The converted PGMS in this study combined the advantages of their spherical shape and porosity favouring tissue ingrowth, revealing enhanced pro-osteogenic properties provided by the HA conversion, and thus represent a promising novel material for bone repair applications.
To summarise overall, the novel rapid manufacturing process to produce highly porous glass materials, contributed towards rapid conversion of borate glasses to HA, which led to the development of unique porous HA microspheres from 45B5, B53P4 and 13-93B, with external pores large enough to support human stem cell incorporation.
6. Conclusions
Solid and porous glass microspheres from three different borate glasses (i.e. 45B5, B53P4 and 13-93B) were successfully manufactured into highly porous microspheres (porosity of 65–75% with pore size of 43–52 μm) via a flame spheroidisation process and their conversion to HA at different concentrations (i.e. 0.02 and 0.2 M) and media (i.e. K2HPO4, Na2HPO4 and SBF) were investigated. In addition, a cell culture study was conducted to compare the influence of Boron containing microspheres versus the HA converted microspheres.PGMS (with the exception of 13-93B glass) revealed complete conversion to HA and at much earlier timeframe in 0.02 M K2HPO4 solution compared to their SGMS counterparts, due to their higher surface area. The conversion rates also increased with increasing concentration of K2HPO4 solution. However, 13-93B microspheres only revealed conversion to HA in Na2HPO4 solution which was suggested to be due to the presence of K in both the glass formulation and immersion medium, inhibiting the ion exchange reaction. Borate glass microspheres (both SGMS and PGMS) of all compositions showed bioactivity in SBF and formation of HA was observed at much earlier for PGMS compared to SGMS. Interestingly, both SGMS and PGMS revealed full conversion to HA in SBF in comparison to 0.02 M K2HPO4 due to the presence of TRIS buffer which accelerated their conversion rate. Moreover, converted porous HA microspheres revealed enhanced pro-osteogenic properties compared to their unconverted counterparts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially funded through the Medical Technologies Innovation and Knowledge Centre (phase 2 – Regenerative Devices), funded by the EPSRC under grant number EP/N00941X/1 and supported by the University of Nottingham, Faculty of Engineering (the Dean of Engineering Research Scholarship for International Excellence). VS is funded by the Italian Ministry of Education, University and Research (MIUR) to the Department of Molecular Medicine of the University of Pavia under the ‘Dipartimenti di Eccellenza (2018–2022)’ initiative. The authors would also like to acknowledge and thank the Nanoscale and Microscale Research Centre (nmRC) and School of Life Science at the University of Nottingham for use of their instruments and help with analysis.References
- K. A. Hing, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 2821–2850 CrossRef CAS.
- A. Szczes, L. Holysz and E. Chibowski, Adv. Colloid Interface Sci., 2017, 249, 321–330 CrossRef CAS.
- A. Sáenz, E. Rivera, W. Brostow and V. M. Castaño, J. Mater. Educ., 1999, 21, 267–276 Search PubMed.
- E. Monroe, W. Votava, D. Bass and J. M. Mullen, J. Dent. Res., 1971, 50, 860–861 CrossRef CAS.
- D. Yu, C. Tsai, J. Wong and J. Fox, J. Formosan Med. Assoc., 1991, 90, 953–957 CAS.
- D. Loca, J. Locs, A. Dubnika, V. Zalite and L. Berzina-Cimdina, in Hydroxyapatite (HAp) for biomedical applications, Elsevier, 2015, pp. 189–209 Search PubMed.
- S. Jafari and K. Adibkia, J. Mol. Pharm. Org. Process Res., 2015, 3, 1–2 Search PubMed.
- A. Matamoros-Veloza, K. M. Z. Hossain, B. E. Scammell, I. Ahmed, R. Hall and N. Kapur, J. Mech. Behav. Biomed. Mater., 2020, 102, 103489 CrossRef CAS.
- Y. Cai, Y. Chen, X. Hong, Z. Liu and W. Yuan, Int. J. Nanomed., 2013, 8, 1111 Search PubMed.
- J. Moeller-Siegert, J. Parmentier, K. Anselme and C. Vix-Guterl, J. Mater. Sci., 2013, 48, 3722–3730 CrossRef CAS.
- H. C. Shum, A. Bandyopadhyay, S. Bose and D. A. Weitz, Chem. Mater., 2009, 21, 5548–5555 CrossRef CAS.
- G. Verma, K. Barick, N. Manoj, A. Sahu and P. Hassan, Ceram. Int., 2013, 39, 8995–9002 CrossRef CAS.
- X. Cheng, Z. Huang, J. Li, Y. Liu, C. Chen, R.-A. Chi and Y. Hu, Cryst. Growth Des., 2010, 10, 1180–1188 CrossRef CAS.
- C. Zhang, J. Yang, Z. Quan, P. Yang, C. Li, Z. Hou and J. Lin, Cryst. Growth Des., 2009, 9, 2725–2733 CrossRef CAS.
- S. Li, J. Wang, X. Jing, Q. Liu, J. Saba, T. Mann, M. Zhang, H. Wei, R. Chen and L. Liu, J. Am. Ceram. Soc., 2012, 95, 3377–3379 CrossRef CAS.
- Q. Xiao, K. Zhou, C. Chen, M. Jiang, Y. Zhang, H. Luo and D. Zhang, Mater. Sci. Eng., C, 2016, 69, 1068–1074 CrossRef CAS.
- M. G. Ma and J. F. Zhu, Eur. J. Inorg. Chem., 2009, 2009, 5522–5526 CrossRef.
- S. Naseri, W. C. Lepry and S. N. Nazhat, J. Mater. Chem. B, 2017, 5, 6167–6174 RSC.
- A. Yao, D. Wang, W. Huang, Q. Fu, M. N. Rahaman and D. E. Day, J. Am. Ceram. Soc., 2007, 90, 303–306 CrossRef CAS.
- W. Huang, D. E. Day, K. Kittiratanapiboon and M. N. Rahaman, J. Mater. Sci.: Mater. Med., 2006, 17, 583–596 CrossRef CAS.
- Q. Fu, M. N. Rahaman, H. Fu and X. Liu, J. Biomed. Mater. Res., Part A, 2010, 95, 164–171 CrossRef.
- A. Abdelghany, H. ElBatal and F. EzzElDin, Ceram. Int., 2012, 38, 1105–1113 CrossRef CAS.
- D. Day, J. White, R. Brown and K. McMenamin, Glass Technol., 2003, 44, 75–81 CAS.
- X. Han and D. E. Day, J. Mater. Sci.: Mater. Med., 2007, 18, 1837–1847 CrossRef CAS.
- M. Mizuno, D. Zhu and W. M. Kriven, Advances in Bioceramics and Biocomposites: A Collection of Papers Presented at the 29th International Conference on Advanced Ceramics and Composites, Jan 23–28, 2005, Cocoa Beach, FL, John Wiley & Sons, 2009 Search PubMed.
- W. Huang, M. N. Rahaman, D. E. Day and Y. Li, Phys. Chem. Glasses: Eur. J. Glass Sci. Technol., Part B, 2006, 47, 647–658 CAS.
- H. Wang, S. Zhao, J. Zhou, Y. Shen, W. Huang, C. Zhang, M. N. Rahaman and D. Wang, J. Mater. Chem. B, 2014, 2, 8547–8557 RSC.
- A. M. Deliormanlı, X. Liu and M. N. Rahaman, J. Biomater. Appl., 2014, 28, 643–653 CrossRef.
- L. Bi, M. N. Rahaman, D. E. Day, Z. Brown, C. Samujh, X. Liu, A. Mohammadkhah, V. Dusevich, J. D. Eick and L. F. Bonewald, Acta Biomater., 2013, 9, 8015–8026 CrossRef CAS.
- H. Fu, M. N. Rahaman, D. E. Day and R. F. Brown, J. Mater. Sci.: Mater. Med., 2011, 22, 579–591 CrossRef CAS.
- S. D. Conzone and D. E. Day, J. Biomed. Mater. Res., Part A, 2009, 88, 531–542 CrossRef.
- A.-M. Chen, P. Gu and Z.-M. Ni, Mater. Lett., 2012, 68, 187–189 CrossRef CAS.
- K. M. Z. Hossain, U. Patel, A. R. Kennedy, L. Macri-Pellizzeri, V. Sottile, D. M. Grant, B. E. Scammell and I. Ahmed, Acta Biomater., 2018, 72, 396–406 CrossRef CAS.
- M. T. Islam, L. Macri-Pellizzeri, K. M. Z. Hossain, V. Sottile and I. Ahmed, Mater. Sci. Eng., C, 2020, 111668 CrossRef.
- U. Patel, L. Macri-Pellizzeri, K. M. Zakir Hossain, B. E. Scammell, D. M. Grant, C. A. Scotchford, A. C. Hannon, A. R. Kennedy, E. R. Barney, I. Ahmed and V. Sottile, J. Tissue Eng. Regener. Med., 2019, 13, 396–405 CrossRef CAS.
- D. Gupta, D. M. Grant, K. M. Zakir Hossain, I. Ahmed and V. Sottile, J. Biomater. Appl., 2018, 32, 906–919 CrossRef.
- Z. Zyman, M. Epple, A. Goncharenko and D. Rokhmistrov, Materialwiss. Werkstofftech., 2013, 44, 259–263 CrossRef CAS.
- Y. Doi, Y. Shimizu, Y. Moriwaki, M. Aga, H. Iwanaga, T. Shibutani, K. Yamamoto and Y. Iwayama, Biomaterials, 2001, 22, 847–854 CrossRef CAS.
- H. Giesche, Part. Part. Syst. Charact., 2006, 23, 9–19 CrossRef.
- U. Heise, J. Osborn and F. Duwe, Int. Orthop., 1990, 14, 329–338 CrossRef CAS.
- K. Lin and J. Chang, in Hydroxyapatite (HAp) for biomedical applications, Elsevier, 2015, pp. 3–19 Search PubMed.
- K. Lin, Y. Zhou, Y. Zhou, H. Qu, F. Chen, Y. Zhu and J. Chang, J. Mater. Chem., 2011, 21, 16558–16565 RSC.
- C. Qi, Y. J. Zhu, B. Q. Lu, X. Y. Zhao, J. Zhao, F. Chen and J. Wu, Chem. – Eur. J., 2013, 19, 5332–5341 CrossRef CAS.
- C. Qi, Y.-J. Zhu, B.-Q. Lu, X.-Y. Zhao, J. Zhao and F. Chen, J. Mater. Chem., 2012, 22, 22642–22650 RSC.
- N. J. Lakhkar, J.-H. Park, N. J. Mordan, V. Salih, I. B. Wall, H.-W. Kim, S. P. King, J. V. Hanna, R. A. Martin and O. Addison, Acta Biomater., 2012, 8, 4181–4190 CrossRef CAS.
- F. Muñoz, L. Montagne and L. Delevoye, Phys. Chem. Glasses: Eur. J. Glass Sci. Technol., Part B, 2008, 49, 339–345 Search PubMed.
- M. T. Islam, R. M. Felfel, E. A. Abou Neel, D. M. Grant, I. Ahmed and K. M. Z. Hossain, J. Tissue Eng., 2017, 8, 2041731417719170 Search PubMed.
- W. Huang, M. N. Rahaman, D. E. Day and B. A. Miller, J. Mater. Sci.: Mater. Med., 2009, 20, 123 CrossRef CAS.
- E. M. Rivera-Muñoz, in Biomedical Engineering-Frontiers and Challenges, InTech, 2011 Search PubMed.
- D. Rohanová, A. R. Boccaccini, D. M. Yunos, D. Horkavcová, I. Březovská and A. Helebrant, Acta Biomater., 2011, 7, 2623–2630 CrossRef.
- K. L. Skelton, J. V. Glenn, S. A. Clarke, G. Georgiou, S. P. Valappil, J. C. Knowles, S. N. Nazhat and G. R. Jordan, Acta Biomater., 2007, 3, 563–572 CrossRef CAS.
- R. F. Brown, M. N. Rahaman, A. B. Dwilewicz, W. Huang, D. E. Day, Y. Li and B. S. Bal, J. Biomed. Mater. Res., Part A, 2009, 88, 392–400 CrossRef.
- H. Fu, F. Qiang, N. Zhou, W. Huang, M. N. Rahaman, D. Wang and X. Liu, Mater. Sci. Eng., C, 2009, 29, 2275–2281 CrossRef CAS.
- N. W. Marion, W. Liang, G. Reilly, D. E. Day, M. N. Rahaman and J. J. Mao, Mech. Adv. Mater. Struct., 2005, 12, 1–8 CrossRef.
- A. Di Luca, A. Longoni, G. Criscenti, C. Mota, C. van Blitterswijk and L. Moroni, Biofabrication, 2016, 8, 045007 CrossRef.
- V. Karageorgiou and D. Kaplan, Biomaterials, 2005, 26, 5474–5491 CrossRef CAS.
- B.-S. Chang, K.-S. Hong, H.-J. Youn, H.-S. Ryu, S.-S. Chung and K.-W. Park, Biomaterials, 2000, 21, 1291–1298 CrossRef CAS.
- O. Gauthier, J.-M. Bouler, E. Aguado, P. Pilet and G. Daculsi, Biomaterials, 1998, 19, 133–139 CrossRef CAS.
- J. Diao, J. OuYang, T. Deng, X. Liu, Y. Feng, N. Zhao, C. Mao and Y. Wang, Adv. Healthcare Mater., 2018, 7, 1800441 CrossRef.
- J. S. McLaren, L. Macri-Pellizzeri, K. M. Z. Hossain, U. Patel, D. M. Grant, B. E. Scammell, I. Ahmed and V. Sottile, ACS Appl. Mater. Interfaces, 2019, 11, 15436–15446 CrossRef CAS.
- V. S. Kattimani, S. Kondaka and K. P. Lingamaneni, Bone Tissue Regener. Insights, 2016, 7, 9–19 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Pore size distribution (ESI Fig. 1); cross-sectional analysis for SGMS and PGMS at day 0 (starting microspheres), day-10 and day-21 during the immersion of microspheres in 0.02 M K2HPO4 solution (ESI Fig. 2); X-ray diffraction patterns for SGMS and PGMS of (A) 45B5 and (B) B53P4 post immersion in SBF (ESI Fig. 3); SEM images for SGMS and PGMS of 45B5 and B53P4 glasses post immersion in SBF (ESI Fig. 4); ion release for SGMS and PGMS in SBF (ESI Fig. 5); pH change for SGMS and PGMS in SBF (ESI Fig. 6); DSC curves for bulk glass, solid and porous glass microspheres (ESI Fig. 7); compositional analysis for SGMS and PGMS post immersion in SBF at day 21 (ESI Table 1); thermal analysis data (ESI Table 2) (PDF). See DOI: 10.1039/d0bm01776k |
| This journal is © The Royal Society of Chemistry 2021 |