
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cues from human atrial extracellular matrix enrich the atrial differentiation of human induced pluripotent stem cell-derived cardiomyocytes†
Fernanda C. P.
Mesquita
a,
Jacquelynn
Morrissey
a,
Po-Feng
Lee
a,
Gustavo
Monnerat
b,
Yutao
Xi
a,
Helen
Andersson
a,
Fabio C. S.
Nogueira
 b,
Gilberto B.
Domont
b,
Luiz C.
Sampaio
ac,
Camila
Hochman-Mendez
*a and
Doris A.
Taylor
*ad
b,
Gilberto B.
Domont
b,
Luiz C.
Sampaio
ac,
Camila
Hochman-Mendez
*a and
Doris A.
Taylor
*ad
aTexas Heart Institute, 6770 Bertner Avenue, MC 1-135, Houston, TX 77030, USA. E-mail: cmendez@texasheart.org; taylordorisa2020@gmail.com; Fax: +1 832-355-9552; Fax: +1 334-263-7010; Tel: +1 832-355-8994 Tel: +1 713-882-9945
bInstitute of Chemistry, Federal University of Rio de Janeiro, Rio de Janeiro, RJ 21941–909, Brazil
cMichael E. DeBakey Department of Surgery, Baylor College of Medicine, Houston, TX 77030, USA
dRegenMedix Consulting LLC, Houston, TX 77030, USA
First published on 16th April 2021
Abstract
New robust and reproducible differentiation approaches are needed to generate induced pluripotent stem cell (iPSC)-derived cardiomyocytes of specific subtypes in predictable quantities for tissue-specific disease modeling, tissue engineering, and eventual clinical translation. Here, we assessed whether powdered decellularized extracellular matrix (dECM) particles contained chamber-specific cues that could direct the cardiac differentiation of human iPSCs toward an atrial phenotype. Human hearts were dissected and the left ventricle (LV) and left atria (LA) were isolated, minced, and decellularized using an adapted submersion decellularization technique to generate chamber-specific powdered dECM. Comparative proteomic analyses showed chamber-specific dECM segregation, with atrial- and ventricle-specific proteins uniquely present in powdered dECM-hA and dECM-hV, respectively. Cell populations differentiated in the presence of dECM-hA showed upregulated atrial molecular markers and a two-fold increase in the number of atrial-like cells as compared with cells differentiated with dECM-hV or no dECM (control). Finally, electrophysiological data showed an increase in action potentials characteristic of atrial-like cells in the dECM-hA group. These findings support the hypothesis that dECM powder derived from human atria retained endogenous cues to drive cardiac differentiation toward an atrial fate.
1. Introduction
Harnessing the inherent biological capabilities of the extracellular matrix (ECM) and the pluripotent capacity of human-induced pluripotent stem cells (hiPSCs) together is a promising platform for disease modeling, preclinical cardiovascular research, and cardiac tissue engineering. Advancements in stem cell technologies have yielded cell differentiation protocols that guide hiPSCs toward a cardiac fate, typically by using chemically defined media and growth factors that help recapitulate cardiac development.1 These protocols yield mixed cell populations of predominantly ventricular-like cells, with only a small percentage of nodal- and atrial-like cells.2–4To model ventricular and atrial-specific diseases and defects, investigators need access to large quantities of purified chamber-specific human cardiomyocytes free from unwanted cell subtypes. Atrial and ventricular myocytes are distinguishable at the molecular, electrophysiological, and protein levels.5,6 A wide range of genes is preferentially expressed in different regions of the heart and can be used as markers to identify the origin or type of cells.7,8 In the various anatomical regions of the heart, protein composition differs to reflect the unique function of the particular area. Novel protein targets that express chamber specificity have been identified.9 In terms of electrophysiology, atrial myocytes have a lower and shorter action potential with larger conductance and faster activation kinetics than do ventricular cells, whereas the latter express more inward rectifier currents and have a greater negative resting membrane potential.5,6,10
Recent efforts have focused on improving the ability to control lineage development in hiPSC differentiation cultures and to promote efficient atrial cardiomyocyte differentiation. Most techniques for generating purified atrial-like cell populations rely on the use of retinoic acid in culture. In the developing heart, retinoic acid signaling commits progenitor cells toward a cardiac fate11 and contributes to chamber specification and morphogenesis.12,13 Differentiation protocols that use retinoic acid yield cardiomyocytes with increased expression of atrial molecular markers and the characteristic contraction kinetics and drug responsiveness of endogenous atrial-like cardiomyocytes.8,14 However, despite its wide use in cell culture, retinoic acid is unstable in serum-free media,15 which can compromise experimental reproducibility.
The extracellular matrix (ECM) can directly modulate cell proliferation, migration, and differentiation by regulating various growth factor and signaling interactions; however, the details of these processes are not well defined.1,16,17 During development, ECM plays an essential role in modulating growth factor activity and signaling pathways to influence primitive streak formation and cellular differentiation, respectively.1 Furthermore, ECM has been shown to promote mesoderm formation. During cardiomyocyte specification, the ECM in the myoendocardial space (cardiac jelly) is a key contributor, and its composition and stiffness direct the cells toward an atrial or ventricular fate.18–20 Because of the ability of PSCs to differentiate into all three germ layers, we sought to determine the endogenous ability of region-specific ECM to drive chamber-specific cardiac cell differentiation. We reasoned that atrial ECM could serve as a driver to direct hiPSCs toward an atrial specification.
Here, we examined the ability of powdered decellularized ECM (dECM) to promote chamber-specific cardiac differentiation of hiPSCs. Particles (powder) of atrial and ventricular dECM derived from human myocardium were generated using an adapted submersion decellularization method.21,22 The decellularization and structure of atrial and ventricular powder were validated by fractal dimension analysis; quantification of sodium dodecyl sulfate (SDS), DNA, and glycosaminoglycan (GAG) content; and proteomic analysis. Atrial dECM (dECM-hA) powder could drive cardiac differentiation toward an atrial fate yielding a twofold increase in the number of cells with a functional atrial phenotype in a mixed cardiac cell population as compared with cells differentiated in the presence of ventricular dECM (dECM-hV) or with no dECM (control). These findings demonstrate a critical step toward generating human cardiomyocytes that exhibit regional specificity using decellularized chamber powders, without additional chemical reagents.
2. Materials and methods
2.1. Human heart samples
De-identified human hearts unsuitable for clinical transplantation were provided by a local organ procurement organization (LifeGift, Houston, TX, USA) after being exempted by the Texas Heart Institute's institutional review board (Project #3239 Exempt) because of the collection of tissue from deceased donors. Sex, age, ethnicity, cause of death, and risk factors are presented in Table S1.† To maintain organ viability during cold storage, hearts were flushed antegrade through the coronary arteries with preservation solution (Celsior, Sanofi, Bridgewater, NJ) after aortic cross clamping. The hearts were then immersed in the same solution, kept at 4 °C, and processed within 12 hours of procurement inside a workstation flow hood (AirClean 6000 Workstation, AirClean Systems, Creedmoor, NC, USA).2.2. TAD assembly and tissue decellularization
Human hearts were dissected into the left ventricle (LV) free wall, the right ventricle (RV) free wall (for ventricular tissue), and the atrial appendage (for atrial tissue). The coronary sinus and the posterior and anterior interventricular arteries were used to guide the dissection of the ventricle free walls. To avoid including vascular tissue, we obtained the atria by dissecting only the atrial appendage. Processing included removal of the pericardium. The regions were separately minced with scissors into ∼5 mm pieces under a sterile environment. LV and LA minced pieces were decellularized using a modified turbulent agitation device (TAD) (Fig. 1a). The device consists of a 1 L-jar with a lid containing inflow and outflow ports. A solution reservoir fills liquid into the jar at the desired speed via hose clamps. Placed in the middle of the jar is an octagonal-shaped pole with slit-inserted turbulence-inducing plates (TIPs). The TIP is perforated, promoting tissue contact with the system to enhance the decellularization process (Fig. 1a).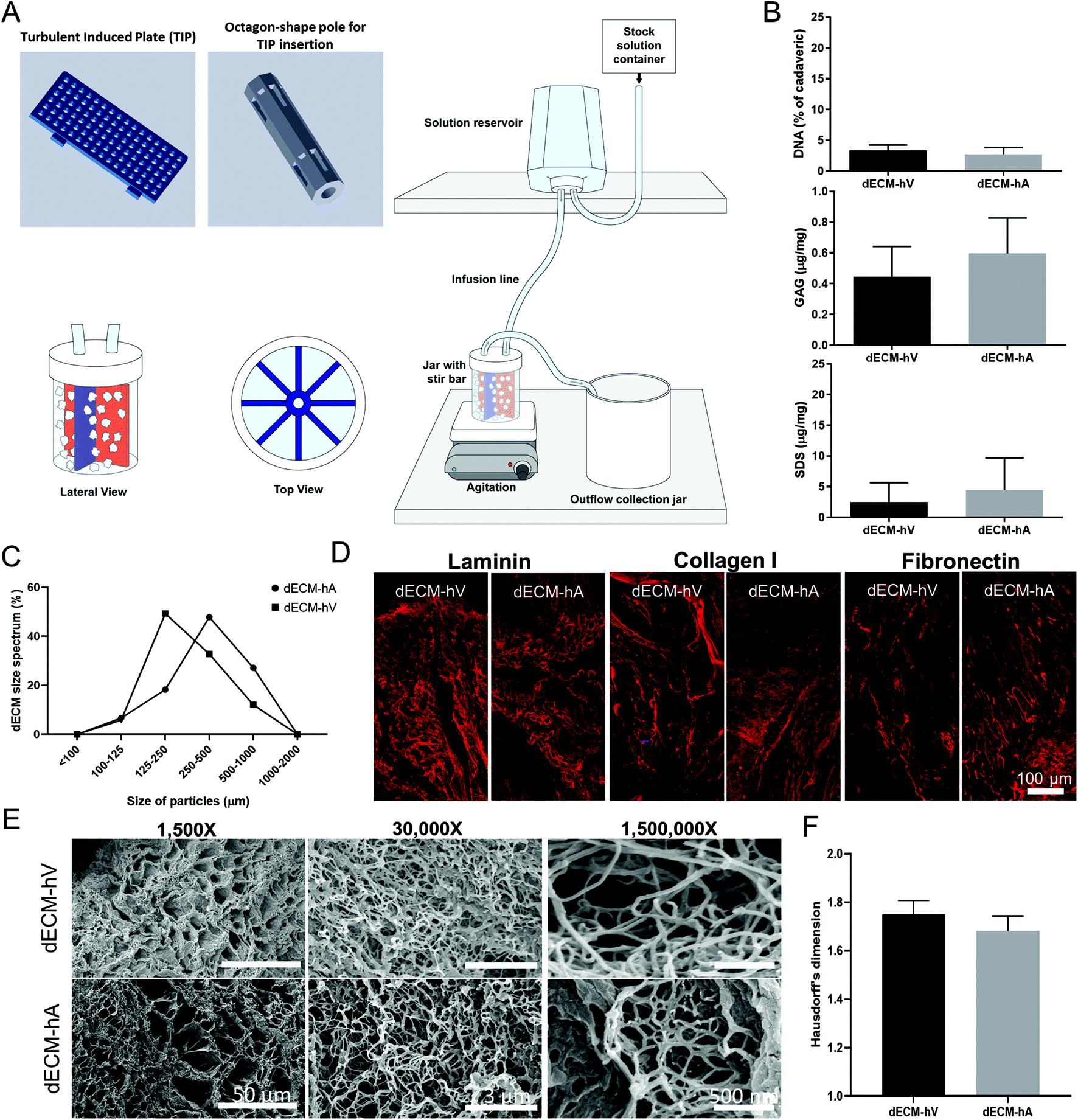 | ||
| Fig. 1 Decellularization using the turbulent agitation device. (a) Schematic of device assembly and components for decellularization. (b) Biochemical quantification of DNA (n = 3), GAG (n = 5), and SDS (n = 5) after decellularization of dECM-hV and dECM-hA. (c) Percentage of different sized powdered dECM particles (n = 3) from atria (circle) and ventricles (square). (d) Immunofluorescence for laminin, collagen I, and fibronectin of dECM-hV and dECM-hA. (e) Scanning electron microscopy of powdered dECM particles derived from ventricles (top) and atria (bottom). (f) Quantification of Hausdorff's dimension (n = 5) in powdered dECM-hV and dECM-hA. Error bars represent SD. Scale bars are indicated in each figure. | ||
We used the following succession of solutions combined with deionized water: hypertonic solution (500 mM NaCl, Sigma-Aldrich, Milwaukee, WI, USA) (24 hours); hypotonic solution (20 mM NaCl) (24 hours); 1% sodium dodecyl sulfate (SDS, v/v, Thermo Fisher Scientific, Fair Lawn, NJ, USA) (72 hours); and phosphate-buffered saline (PBS, Corning, Manassas, VA, USA) (24 hours).
2.3. Biochemical assays for dECM characterization
We quantified the DNA, sulfated glycosaminoglycan (GAG), and SDS content of each sample as previously described22 after powderization of the dECM samples. DNA content was quantified using the Quan-iT™ PicoGreen™ dsDNA assay kit (Invitrogen Corp., Carlsbad, CA, USA), according to the manufacturer's instructions. Briefly, we digested all samples with 1N NaOH in a heating block for 3 hours at 65 °C. Samples were neutralized with 10× Tris-EDTA buffer, and the pH was adjusted to 7.0. Samples were plated in duplicate against a calf thymus standard prepared according to kit instructions and were read using a fluorescence microplate reader (Infinite M200 Pro, TECAN, Switzerland) with the absorbances set to 480 nm excitation and 530 nm emission.Sulfated GAG content was determined using the Blyscan Sulfated Glycosaminoglycan Assay Kit (Biocolor Ltd, Carrickfergus, UK), per the manufacturer's instructions. Briefly, samples were digested using a papain extraction reagent and placed in a heating block for 3 hours at 65 °C. After completing the assay per kit instructions, the samples were plated in duplicate against a sulfated GAG standard and were read using a fluorescence microplate reader with the absorbance set to 656 nm.
SDS content in the samples was quantified by obtaining the dry weights of the tissues as previously described.21 Briefly, samples were rehydrated and phase-separated with a chloroform-methylene blue solution. The SDS that bound to methylene blue (in the organic phase of the sample) was measured by using a fluorescence microplate reader set to an absorbance of 655 nm, and the SDS concentration in the samples was calculated by using a known SDS concentration curve.
2.4. Powderization
After decellularization, dECM fragments were lyophilized (Labconoco 6L Console Freeze Dry System, Kansas City, MO, USA), according to the manufacturer's instructions with a pre-cooling step to guarantee minimal alteration of mechanical properties.23 Then, the dECM fragments were cryomilled (Retsch Cryomill, Haan, Germany), and sieved (Retsch AS 200, Haan, Germany) to obtain separate decellularized human atrial and ventricular extracellular matrix (dECM-hA and dECM-hV, respectively). The hearts were processed and used as individual samples for ECM characterization, cardiac differentiation, and accompanying experiments (ESI Fig. 1a†).Cryomilling was performed using the Retsch Cryomill under the grinding with cooling cycles, according to the manufacturer's instructions. We performed a total of 6 cryocycles (cooling/grinding cycles) for each sample, with a grinding frequency of 5 Hz. Grinding time settings were as follows: precooling–auto set, grinding time – 2 minutes, and intermediate cooling – 40 seconds.
2.5. ECM characterization
dECM-hA and dECM-hV samples were fixed in 4% paraformaldehyde for 20 minutes at room temperature. After washing, the samples were blocked with 10% bovine serum albumin (BSA, Sigma-Aldrich, St Louis, MO, USA) in PBS for 60 minutes. Primary antibodies (anti-laminin, Abcam, Cambridge, MA, cat# ab11575; anti-fibronectin, Abcam, cat# ab2413; and anti-collagen I, Abcam, cat# ab34710) were diluted in 1% BSA in PBS and incubated overnight at 4 °C. After washing, samples were incubated for 2 hours at room temperature with the secondary antibodies (Alexa Fluor 594 goat anti-rabbit, Thermo Fisher Scientific Inc., Waltham, MA, USA, cat# A11012). The slides were mounted in Vectashield® Mounting Medium (Vector Laboratories, Burlingame, CA, USA), and the images were acquired with a A1 MP+ multiphoton confocal microscope (Nikon, Melville, NY, USA). For negative controls, the primary antibodies were omitted.2.6. Scanning electron microscopy
dECM-hA and dECM-hV samples were fixed in 2.5% glutaraldehyde for 20 minutes, post-fixed 1 hour in 1% osmium tetroxide, and then dehydrated through increasing concentrations of ethanol. Supercritical dried samples were sputter-coated with a thin layer of carbon and imaged on a S-5000 scanning electron microscope (Hitachi High-Tech, Inc., Tokyo, Japan).2.7. Fractal dimension
Fractal dimension was calculated as previously described.24,25 Briefly, scanning electron microscope images of both dECM-hA and dECM-hV were subjected to binarization, and each binarized image was superimposed with grids of variable sizes (ESI Fig. 2†). The box-counting algorithm was applied using a software developed in MATLAB (The MathWorks, Natick, MA, USA).2.8. Proteomics
Proteomics were performed as previously described.26–28 Protein samples from five different dECM-hA and dECM-hV powders were macerated in the presence of liquid nitrogen, precipitated with cold 10% (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 v/v) trichloroacetic acid (Sigma-Aldrich) in acetone (Sigma-Aldrich), and centrifuged for 15 minutes at 4 °C and 15000 rpm. The samples were washed three times with cold acetone and air-dried. Proteins were suspended in 15 μL of 7 M urea/2 M thiourea (Sigma-Aldrich) and were quantified using a Qubit Protein Assay Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). Proteins were reduced with 10 mM dithiothreitol (Sigma-Aldrich, St Louis, MO, USA) by incubation for 1 hour at 30 °C followed by alkylation with 55 mM iodoacetamide (Sigma-Aldrich) for 30 minutes in the dark at room temperature. After alkylation, mass spectrometry-grade trypsin (Promega Corp., Madison, WI, USA) dissolved in 50 mM NH4HCO3 (10:1 v/v; Sigma-Aldrich) was added to the protein samples at a ratio of 50:1 (protein: trypsin), which were incubated overnight at 35 °C. After digestion, the samples were acidified until the final concentration of 0.1% trifluoroacetic acid (TFA; Sigma-Aldrich). The peptides were cleaned with an in-house prepared reverse-phase POROS® R2 (Thermo Fisher Scientific Inc.) stage-tip column and eluted in 50 μL of a 50% acetonitrile (ACN)/0.1% TFA solution followed by 50 μL of 70% ACN/0.1% TFA, dried in a SpeedVac concentrator (Thermo Fisher Scientific Inc.), and resuspended in 15 μL of 0.1% formic acid (Sigma-Aldrich). Peptides were quantified using a Qubit Protein Assay Kit and suspended to a final concentration of 0.25 μg μL−1 in 0.1% formic acid.
:4 v/v) trichloroacetic acid (Sigma-Aldrich) in acetone (Sigma-Aldrich), and centrifuged for 15 minutes at 4 °C and 15000 rpm. The samples were washed three times with cold acetone and air-dried. Proteins were suspended in 15 μL of 7 M urea/2 M thiourea (Sigma-Aldrich) and were quantified using a Qubit Protein Assay Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). Proteins were reduced with 10 mM dithiothreitol (Sigma-Aldrich, St Louis, MO, USA) by incubation for 1 hour at 30 °C followed by alkylation with 55 mM iodoacetamide (Sigma-Aldrich) for 30 minutes in the dark at room temperature. After alkylation, mass spectrometry-grade trypsin (Promega Corp., Madison, WI, USA) dissolved in 50 mM NH4HCO3 (10:1 v/v; Sigma-Aldrich) was added to the protein samples at a ratio of 50:1 (protein: trypsin), which were incubated overnight at 35 °C. After digestion, the samples were acidified until the final concentration of 0.1% trifluoroacetic acid (TFA; Sigma-Aldrich). The peptides were cleaned with an in-house prepared reverse-phase POROS® R2 (Thermo Fisher Scientific Inc.) stage-tip column and eluted in 50 μL of a 50% acetonitrile (ACN)/0.1% TFA solution followed by 50 μL of 70% ACN/0.1% TFA, dried in a SpeedVac concentrator (Thermo Fisher Scientific Inc.), and resuspended in 15 μL of 0.1% formic acid (Sigma-Aldrich). Peptides were quantified using a Qubit Protein Assay Kit and suspended to a final concentration of 0.25 μg μL−1 in 0.1% formic acid.
Samples were analyzed in two technical replicates by nano-liquid chromatography-tandem mass spectrometry (nLC-MS/MS).27 Briefly, 4 μL of the diluted samples was applied to an EASY-nLC 1000 system (Thermo Fisher Scientific Inc.) coupled online to an nESI-Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific Inc.). The peptides were loaded into a trap column (EASY-ColumnTM, 2 cm, ID100 μm, 5 μm, 120 A, C18-A1, Thermo Fisher Scientific Inc.) and eluted in an analytical column (75 μm × 25 cm) packed in-house with ReproSil-Pur 120 C18-AQ, 3 μm (Dr Maisch, Ammerbuch, Germany). Peptide separations were performed using a gradient from 95% solution A (0.1% formic acid, 5% acetonitrile) to 5–20% solution B (0.1% formic acid, 95% acetonitrile; Sigma-Aldrich) over 60 minutes followed by 20–40% solution B over 20 minutes and then 40–95% solution B over 3.5 minutes. They were maintained in 95% solution B for 6.5 minutes. MS1 spectra were acquired in a positive mode using the data-dependent acquisition (DDA) method. Each mass spectra (MS) acquired in the DDA consisted of a survey scan in the m/z range of 350–2000 and a resolution of 70000 (at m/z 200) with automatic gain control (AGC) target value of 1 × 10−6 ions. The 20 most intense ions were subjected to fragmentation to acquire the MS2 using higher-energy Collisional Dissociation of previously selected ions, a resolution of 17500, and an AGC of 1 × 10−6 ions.
The MS data were analyzed with Proteome Discoverer 2.1.0.81 (Thermo Fisher Scientific Inc.) using the UniProt Homo sapiens database downloaded in June 2019 and SequestHT algorithm. Search parameters were semi-tryptic hydrolysis, two missed cleavages, oxidation of methionine, and n-terminal protein acetylation as variable and carbamidomethylation as fixed modifications, and a peptide and fragment tolerance of 10 ppm and 0.05 Da, respectively. For the processing workflow, we used the percolator node for peptide-spectrum matches validation and for setting up the false discovery rate (FDR). A cutoff score was established to accept an FDR < 1%. Proteins were grouped according to the maximum parsimony approach, and the protein to be considered identified should be identified in at least 30% of the analytical runs of each group. Protein quantification was based on the node Precursor Ion Area Detection using the average of the peptide peak areas.
2.9. Human iPSC expansion and cardiac differentiation
The hiPSCs (SCVI273) used in this study were derived from blood mononuclear cells using the non-integrative Sendai virus system. The cells were kindly donated by the Joseph Wu Lab (Stanford Medicine, Department of Medicine and Radiology, Stanford Cardiovascular Institute Biobank).Cells were cultured and maintained in a feeder-free system of hESC-qualified Matrigel™ (Corning) and TeSR1™ E8™ (StemCell Technologies Inc., Cambridge, MA, USA) under standard culture conditions (37 °C at 5% CO2). Briefly, we coated 100 mm Petri dishes with Matrigel for at least 1 hour at 37 °C and plated 1 × 105 cells in TeSR E8™ media supplemented with ROCK Inhibitor Y-27632 (10 μM, ATCC, Manassas, VA, USA) for 24 hours. The medium was changed daily, and the cells were passaged using the cell dissociation recombinant enzymatic solution TrypLE™ Express (Gibco, Waltham, MA, USA). Cell viability was determined by staining with Trypan blue and counting cells in a hemocytometer.29
The cardiomyocytes generated in this study were obtained by differentiating the hiPSCs using the STEMdiff™ Cardiomyocyte Differentiation Kit (StemCell Technologies Inc., Vancouver, Canada) (ESI Fig. 1b†). Briefly, we coated 24-well tissue-culture plates with hESC-qualified Matrigel™ for at least 1 hour at 37 °C and plated 0.25–0.4 × 106 cells in 1 mL of TeSR™ E8™ media supplemented with ROCK Inhibitor Y-27632 (10 μM, ATCC) per well for 24 hours. After this period, the cells reached over 95% confluency, and the medium was replaced with 1 mL of TeSR™ E8™ media. Cardiac differentiation started on day 0, when we added 1 mL of STEMdiff™ Cardiomyocyte Differentiation Medium A, supplemented with Matrigel™ Matrix Basement Membrane, Growth Factor Reduced diluted 1 in 100. Before use in cell culture experiments, dECM powder particles were pretreated for 24 hours with 1% penicillin–streptomycin (10000 U mL−1, Gibco, Grand Island, NY, USA) in PBS. After sterilization, the particles were washed 3 times with PBS. For the dECM-hV and dECM-hA groups, we resuspended the powder particles in STEMdiff™ Cardiomyocyte Differentiation Medium A, with a final concentration of 10 μg ml−1.
On day 2, the powder particles were attached to the cells, and the medium was removed and replaced with STEMdiff™ Cardiomyocyte Differentiation Medium B, and on days 4 and 6, with STEMdiff™ Cardiomyocyte Differentiation Medium C. On days 8–15, the cells were cultured using STEMdiff™ Cardiomyocyte Maintenance Medium, and the medium was changed every other day.
Cells were harvested using the STEMdiff™ Cardiomyocyte Dissociation Kit (StemCell Technologies). The cells were washed two times with PBS, and 500 μL of Cardiomyocyte Dissociation Medium (37 °C) was added per well. Culture plates were incubated for 15 minutes at 37 °C and 5% CO2. Afterwards, the cells were dislodged by adding Cardiomyocyte Support Medium and pipetting up and down 5–10 times. The cells were centrifugated at 300g for 5 minutes, the pellet was resuspended, and the cardiomyocytes were replated or used for our experiments (ESI Fig. 1b†).
2.10. Immunophenotype characterization
After differentiation, the cardiomyocytes were fixed and permeabilized using the BD Cytofix/Cytoperm™ kit (BD Biosciences, San Diego, CA, USA), per the manufacturer's instructions. Briefly, cells were resuspended and incubated in 250 μL of BD Cytofix/Cytoperm solution for 20 minutes at 4 °C and then centrifuged (300g for 5 minutes). The cells were resuspended in BD Perm/Wash buffer for 30 minutes at 4 °C and then were stained with Alexa Fluor® 647 mouse anti-cardiac troponin T (BD Biosciences, cat# 565744). The isotype Alexa Fluor® 647 mouse IgG1 (BD Biosciences, cat# 557714) was used as a negative control.For non-conjugated antibodies, the cardiomyocytes were fixed with 4% paraformaldehyde for 20 minutes at 37 °C, permeabilized using 0.5% Triton X-100 in PBS for 5 minutes at room temperature, and blocked with 10% BSA for 30 minutes at room temperature. Primary antibodies MLC2a (BD Biosciences, cat# 565496), MLC2v (Abcam, cat# ab79935), and cTNT (Thermo Fisher Scientific Inc., cat# MA5-12960 and Abcam, cat# ab45932) were diluted in a solution containing 5% BSA and 0.1% Triton X-100 in PBS and incubated for 1 hour at room temperature. Next, the cells were washed and incubated for 1 hour at room temperature with the secondary antibodies: donkey anti-mouse IgG (H + L) highly cross-adsorbed secondary antibody, Alexa Fluor Plus 488 (Thermo Fisher Scientific Inc., cat# A-32766) or goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody, Alexa Fluor 555 (Thermo Fisher Scientific Inc., cat# A-21428). Samples were analyzed using BD LSRFortessa and FlowJo v10 software.
2.11. Gene expression
RNA was extracted using the RNeasy Mini kit (Qiagen, Hilden, Germany), and total RNA was quantified with a NanoDrop spectrophotometer. Reverse-transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems™, Waltham, MA, USA) according to the manufacturer's instructions. The gene expression profiles were determined by using SYBR™ Select Master Mix (Applied Biosystems, Carlsbad, CA, USA). Gene expression was analyzed using the ΔCt method in which the gene was compared to the housekeeping gene GAPDH and TNNT2. The list of primers used is presented in Table S2.†2.12. Cytotoxicity assay
The cell toxicity assay was performed using the Cell Counting Kit – 8 (CCK-8, Sigma-Aldrich), per the manufacturer's instructions. Cells were plated into 48-well plates (0.75 × 106 cells per well). Cell viability was evaluated at days −1, 0, 2, 4, 6, 8, and 10 of cardiac differentiation in the presence of dECM-hA and dECM-hV. Briefly, cells were incubated with 0.01 μl ml−1 of CCK-8 solution under standard culture conditions (37 °C at 5% CO2) for 1 hour. Cell supernatant was collected, and the absorbance was measured at 450 nm using a microplate reader (Infinite M200 Pro, TECAN).2.13. Electrophysiology
Recordings were performed on cardiomyocytes 18–25 days after starting the cardiac differentiation protocol. The cardiomyocytes were transferred to acrylic plates that were pre-treated with Matrigel. The cells were maintained in culture for 3 days before microelectrode AP recording.Action potentials were recorded as described previously.30 Cardiomyocyte preparations were perfused with Tyrode's solution containing (in mM) 140 NaCl, 5 KCl, 1.8 CaCl2, 1.0 MgCl2, 11 D-glucose, and 5 HEPES (pH 7.4 adjusted with NaOH) at 37 ± 1 °C using a temperature controller (TC-344C, Warner Instruments LLC., Hamden, CT, USA) saturated with oxygen at a perfusion flow rate of 0.5 ml min−1 (Master Flex C L−1, Cole-Parmer, Vernon Hills, IL, USA). The transmembrane potential was recorded using glass microelectrodes (40–80 MΩ DC resistance) filled with 3 M KCl connected to a microelectrode amplifier (MultiClamp 700A, Molecular Devices, San Jose, CA, USA). Amplified signals were digitized (1440 digidata A/D interface, Molecular Devices) and analyzed using LabChart 7.3 software (ADInstruments, Sydney, Australia). The following parameters were analyzed: APD10 repolarization, APD20 repolarization, APD40 repolarization, and APD90 repolarization from at least 10 consecutive APs for each cell.
2.14. Statistical analysis
Data are shown as the mean ± standard deviation. dECM-hA, dECM-hV, and control samples were compared by using a Student t-test or two-way ANOVA depending on the necessity, and p < 0.05 was considered significant. The GraphPad Prism® software, version 8.0 (GraphPad Software Inc., La Jolla, CA, USA), was used for statistical analyses.For statistical analysis of the proteomic data, a non-parametric test (Wilcoxon rank-sum test) using adjusted P-value (FDR) cutoff of 0.1 was performed with the use of the MetaboAnalyst online platform.
3. Results
3.1. Powdered particles from dECM show fractal dimension and preserved morphology and composition
To produce the chamber-specific dECM powders used in these experiments, we obtained 5 human hearts from male donors (average age, 35 ± 13.47 years) that were considered unsuitable for clinical transplantation. The human atria (hA) and ventricles (hV) were separated, minced, and decellularized by using TAD, which is a modified submersion decellularization technique (Fig. 1a and ESI Fig. 1a†). After decellularization, individual dECM-hA and dECM-hV were lyophilized, subjected to cryomilling, and strained through a sieve to sort powder particles by size (100–1000 μm). We have previously described the chemical components and experimental steps.22 Decellularized ECM-hA and dECM-hV, respectively, had 3.37 ± 0.5052% and 2.77 ± 0.62% of DNA content relative to cadaveric samples, 0.44 ± 0.08 μg mg−1 and 0.59 ± 0.13 μg mg−1 of GAG, and 2.46 ± 1.20 μg mg−1 and 4.46 ± 1.98 μg mg−1 of SDS (Fig. 1b). As anticipated, no difference in decellularization efficiency was noted between dECM-hA and dECM-hV. Powder particles ranging in size from 125–500 μm were the most abundant (66.14% and 82.13% of total dECM-hA and dECM-hV, respectively, Fig. 1c). Particles outside of this size range were excluded from use because of their small percentage among the total particles generated, limiting the number of experimental replicates. Immunostaining showed that the ECM components—laminin, collagen I, and fibronectin—were preserved in powder particles (Fig. 1d). Interestingly, scanning electron microscopy images at increased magnification (up to 1000-fold) showed that both dECM-hV and dECM-hA had similar architectural morphology (Fig. 1e). Using the box-counting algorithm and power spectrum analysis,24,25 we estimated that the fractal (Hausdorff's) dimensions for our particles were 1.75 ± 0.057 (dECM-hV) and 1.63 ± 0.061 (dECM-hA) (Fig. 1f and ESI Fig. 2†).3.2. ECM molecules are selectively expressed in the atria and ventricles
To chemically characterize the cardiac dECM and identify chamber-specific molecules, we performed nanoflow liquid chromatography combined with high-resolution mass spectrometry (nLC-MS/MS)-based proteomics on 5 dECM-hA and 5 dECM-hV powder samples in a technical duplicate analysis. We identified a total of 683 proteins in both atrial and ventricle-derived powders (Table S3†), of which 91 were uniquely present in dECM-hA (Table S4†) and 135 in dECM-hV (Table S5†). A principal component analysis (PCA) with two components was applied as a multivariate statistical model for our proteomic data and showed a clear distinction between dECM-hA and dECM-hV samples (Fig. 2a). The clustering of the top 25 proteins identified in our data also clearly indicates a different protein expression pattern in the two cardiac-specific chamber samples (Fig. 2b).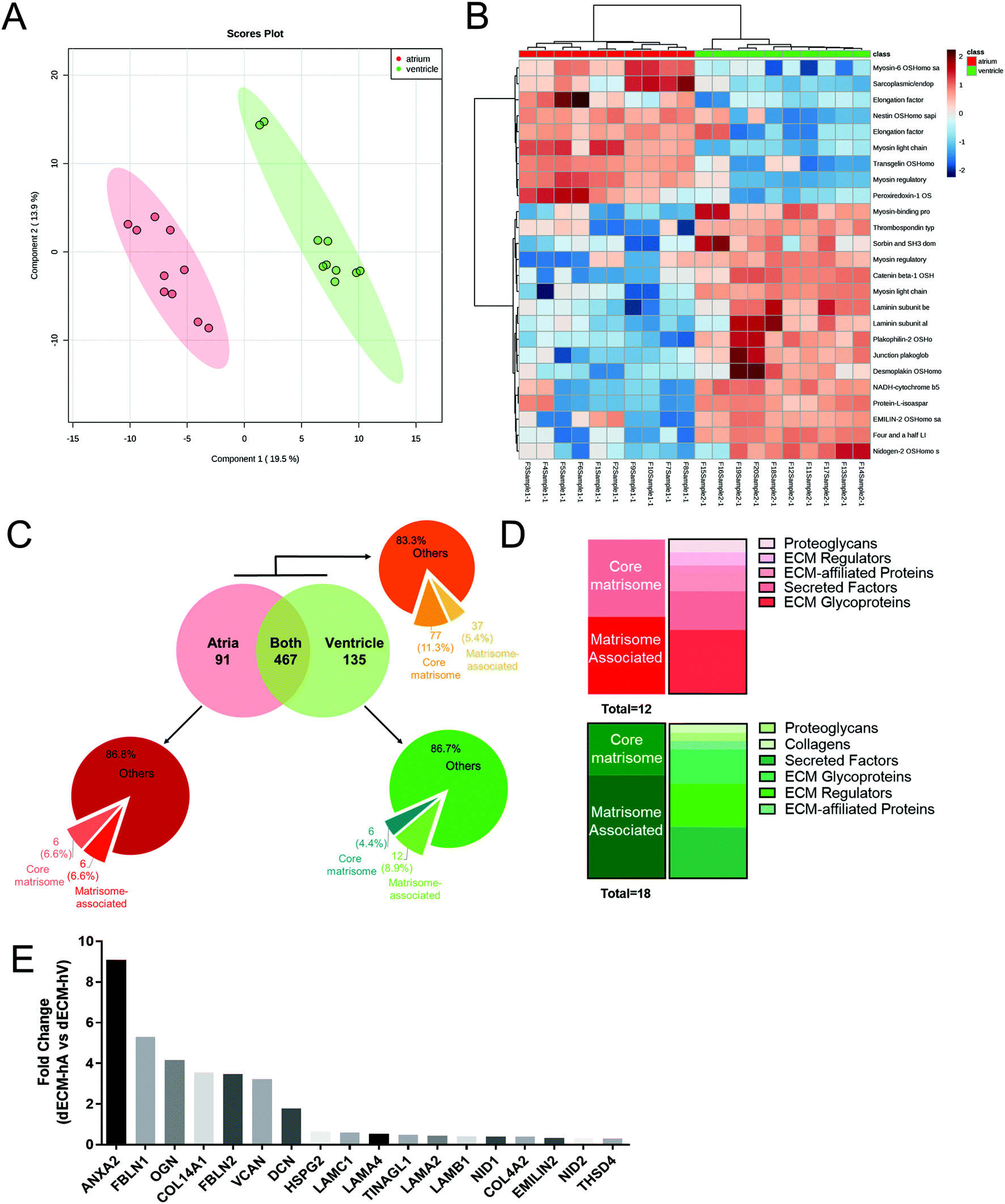 | ||
| Fig. 2 Proteomic analysis of human dECM-hA and dECM-hV. (a), principal component analyses of powdered dECM obtained from atrial (red) and ventricular (green) regions (n = 5). (b) Heat map with the top 25 proteins presented in dECM-hA (red) and dECM-hV (green). (c) Matrisome characterization of all detected proteins (orange) and those uniquely present in dECM-hA (red) or dECM-hV (green). (d) Stacked bar charts illustrating the major components of core matrisome and matrisome-associated proteins of dECM-hA (top, red) and dECM-hV (bottom, green). (e) Statistically significant matrisome proteins presented in dECM-hA compared to dECM-hV organized by abundance. | ||
Using the UniProt Homo sapiens database, we also demonstrated that 77 (11.3%) proteins were assigned to the core matrisome, including different types of collagen, ECM glycoproteins, and proteoglycans as illustrated in Fig. 2c (orange pie chart) and summarized in Tables S3–S5.† Furthermore, 37 (5.4%) matrisome-associated proteins were detected, including ECM-secreted factors, ECM-affiliated proteins, and ECM regulators. For the atrial-specific samples, we identified 6 (6.6%) core matrisome proteins and an additional 6 (6.6%) matrisome-associated proteins (Fig. 2c, red pie chart; Fig. 2d, top panel). For the ventricular-specific samples, we identified 6 (4.4%) core matrisome proteins and 12 (8.9%) matrisome-associated proteins (Fig. 2c, green pie chart; Fig. 2d, bottom panel). We then examined the quantitative differences between the core matrisome and matrisome-related proteins identified in dECM-hA versus dECM-hV. We found statistically significant overexpression among a total of 18 proteins in dECM-hA (ANXA2, HSPG2, COL14A1, COL4A2, DCN, EMILIN2, FBLN1, FBLN2, LAMA2, LAMA4, LAMB1, LAMC1, OGN, NID1, NID2, THSD4, TINAGL1, and VCAN) compared to dECM-hV (Fig. 2e).
3.3. dECM drives chamber-specific cardiomyocyte phenotypes
To evaluate the ability of the ECM components to selectively drive differentiation of hiPSC toward atrial-like or ventricular-like cardiomyocyte phenotypes in cell culture, we added powder particles (125–500 μm) of dECM-hA or dECM-hV to the cell culture medium on day 0 of the differentiation protocol as described in the Methods (ESI Fig. 1b†). After 48 hours, no cell death was observed (ESI Fig. 1c†). On day 10, beating areas were visualized in both dECM groups (dECM-hA and dECM-hV) and in the no powder (control) group as expected (Videos S1–S3†). The differentiation protocol was completed at day 15 (ESI Fig. 1d†), and cells were maintained in culture up to day 25 for characterization experiments (ESI Fig. 1†). Flow cytometry analysis on day 15 revealed no difference in cardiomyocyte differentiation efficiency, with 66.61 ± 10.21% and 65.97 ± 11.19% expression of a cardiac-specific marker (cardiac troponin T [cTNT]) in dECM-hA and dECM-hV, respectively, as compared to cells differentiated without dECM (control group; 65.45 ± 12.73%) (Fig. 3a). However, the phenotype of cells differentiated in the presence of dECM differed from that in the control group (Fig. 3b and c).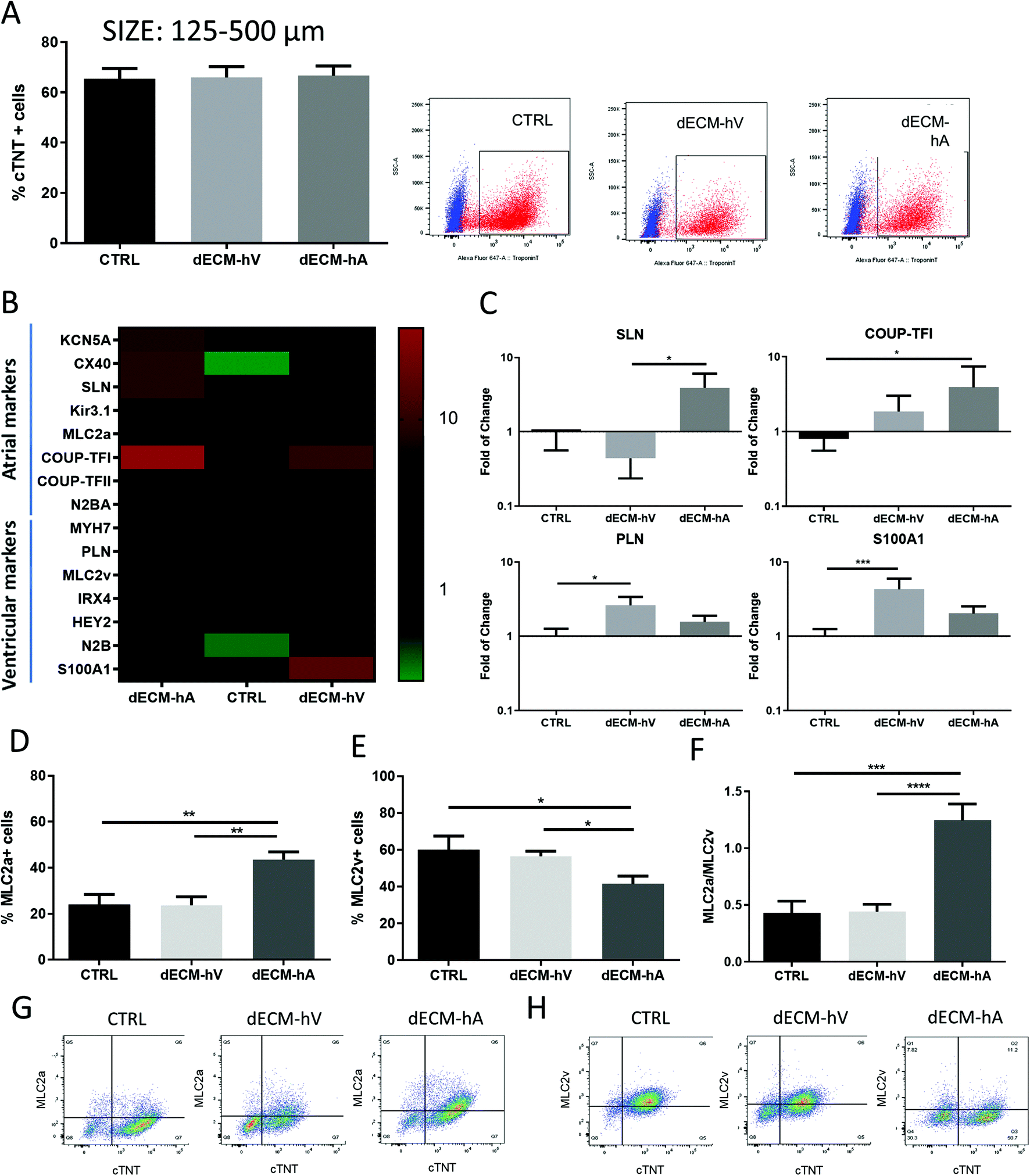 | ||
| Fig. 3 Evaluation of the effect of dECM on cardiac differentiation. (a) Percentage of cTNT+ cells differentiated with dECM-hA, dECM-hV, and no dECM (CTRL) (n = 7), with their representative dot plot [cTNT (red) and isotype (blue)]. (b) Heat map of atrial and ventricular markers (n = 10) in all groups. (c) Gene expression of sarcolipin (SLN), COUP-TFI, phospholamban (PLN) and S100A1 in cells differentiated without dECM (CTRL) and with dECM-hA and dECM-hV. (d–h) Expression of MLC2a (d, n = 13), MLC2v (e, n = 10) and MLC2a:MLC2v ratio (f, n = 6–11) after 15 days of differentiation in the absence of dECM (CTRL) and the presence of dECM-hA or dECM-hV. Representative dot plot of cardiomyocytes (cTNT+) co-expressing MLC2a (g) or MLC2v (h) differentiated without dECM (CTRL) and in the presence of dECM-hA or dECM-hV. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. | ||
Cells differentiated with dECM-hA showed an upregulation of atrial-related markers (Fig. 3b); sarcolipin expression was significantly higher in dECM-hA than in dECM-hV (p < 0.05), and COUP transcription factor 1 expression was significantly higher in dECM-hA than in control samples (p < 0.05) (Fig. 3c). Cells differentiated with dECM-hV demonstrated a clustered expression of ventricular-related markers (Fig. 3b), with a statistical increase in phospholamban (p < 0.05) and S100A1 (p < 0.001) expression compared to control levels (Fig. 3c). Flow cytometric analysis of atrial (MLC2a) and ventricular (MLC2v) markers (Fig. 3d–h) showed that 43.53 ± 12.17% of the cardiomyocytes in the dECM-hA group expressed MLC2a, which was significantly more than the percentage of cardiomyocytes in the dECM-hV (23.71 ± 12.09, p < 0.01) and control (23.99 ± 10.85, p < 0.01) groups (Fig. 3d and g). MLC2v was expressed similarly between the dECM-hV group (56.57 ± 8.45) and controls (60.07 ± 18.09) but was significantly decreased in cells differentiated with dECM-hA (41.45 ± 12.10, p < 0.05) (Fig. 3e and h). We found an increase in the MLC2a:MLC2v ratio of 1.24 ± 0.40 in cells differentiated with dECM-hA as compared to 0.42 ± 0.23 (p < 0.001) in the control group and 0.44 ± 0.21 (p < 0.0001) in the dECM-hV group (Fig. 3f).
To evaluate the impact of the dECM on cardiomyocyte electrophysiological activity, we recorded and classified action potentials (APs) according to the criteria defined by Ma et al.10 APs characteristic of atrial-like cells were seen more often in cells differentiated with dECM-hA (37.5%) than in those differentiated with dECM-hV (5.4%) or in the absence of dECM (control group) (13.04%) (Fig. 4a). Representative APs of atrial-like and ventricular-like cardiomyocytes derived from each of the three groups (controls, dECM-hV, and dECM-hA) are shown in Fig. 4b. Although each cell type could be found in each group, the chamber-specific AP characteristics of the cells differed among the groups, suggesting a balance between ionic currents across the cell membrane. The corrected AP duration (cAPD) at 10% and 20% of repolarization (cAPD10 and cAPD20, respectively) was significantly shorter in cardiomyocytes differentiated with dECM-hA (57.15 ± 20.28 ms and 76.20 ± 22.03 ms, respectively) than in those differentiated with dECM-hV (117.8 ± 16.15 ms and 139 ± 17.85 ms, p < 0.05) and in the control group (104.4 ± 48.71 ms and 126 ± 47.41 ms, p < 0.05) (Fig. 4c and d); these results suggest faster electrical kinetics in dECM-hA cardiomyocytes. In examining cAPD10, 20, and cAPD40, we observed no difference in AP duration in ventricular-like cells among all groups (Fig. 4c–e). cAPD90 was significantly prolonged in ventricular-like cells in cardiomyocytes differentiated with dECM-hV (468.8 ± 108.3 ms) when compared to those differentiated with dECM-hA (367.3 ± 112.5 ms, p < 0.01) or no dECM group (367.8 ± 97.44 ms, p < 0.001) (Fig. 4f). The triangulation, approximated as the duration of phase 3 repolarization (duration between cAPD90 and cAPD40), was higher in the dECM-hV group than in the control group (p < 0.05), implying a more prolonged phase 3 of the AP (Fig. 4g).
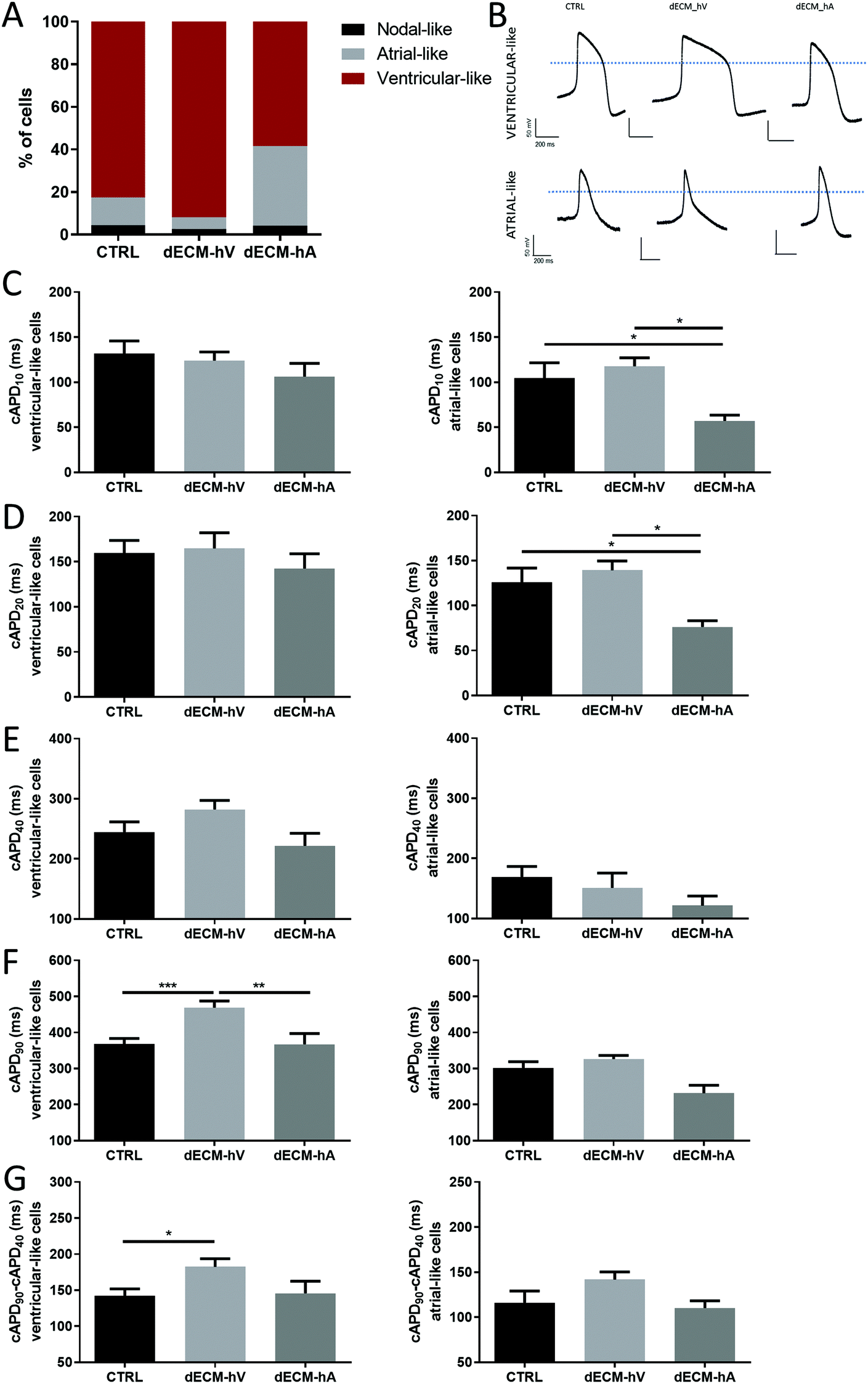 | ||
| Fig. 4 Electrophysiological characterization of dECM-hA and dECM-hV cardiomyocytes. (a) Percentage of atrial-, nodal- and ventricular-like action potentials (APs) in cardiomyocytes differentiated in the presence of dECM-hA, dECM-hV, and no dECM (CTRL). (b) Representative AP recordings of spontaneously contracting cardiomyocytes. (c–f) AP duration of ventricular- (left) and atrial-like (right) cardiomyocytes in CTRL, dECM-hA, dECM-hV groups. AP duration with 10% (c), 20% (d), 40% (e) and 90% (f) of repolarization. (g) Triangulation of APs in all groups. Ventricular-like cells: CTRL (n = 40), dECM-hV (n = 34) and dECM-hA (n = 14). Atrial-like cells: CTRL (n = 8), dECM-hV (n = 3) and dECM-hA (n = 11). *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
4. Discussion
Established hiPSC-CM differentiation protocols typically yield mixed ventricular cell-dominant populations. Human iPSC-derived ventricular cardiomyocytes have been used to recapitulate several cardiac disease phenotypes.30–32 In contrast, little progress has been made toward atrial cardiomyocyte disease modeling.33 Thus, there is a need to improve differentiation processes to yield a predictable amount of cell subtypes, particularly atrial-like cardiomyocytes. Here, we report the successful use of chamber-specific dECM to drive atrial differentiation, resulting in a greater quantity of high-quality atrial cells within a mixed population.Since our group first described the whole organ decellularization process,34 we have made strides toward better preserving native ECM characteristics.21,35,36 Decellularized samples are traditionally obtained from the whole organ, and most protocols do not distinguish the individual regions of the organs.37 Emerging research emphasizes the need for chamber-specific samples because different regions of the heart have distinct gene expression and protein profiles.7,38 In this study, we isolated the atria and ventricles by dissecting human heart chambers to produce chamber-specific dECM. Atria and ventricles were separately minced into small pieces and decellularized by using optimal modifications to our standard protocol.21
The preservation of essential matrix components in our dECM powder particles led us to believe that region-specific ECM may be able to induce differentiation of chamber-specific cell phenotypes. As previously described, ECM molecules have auto symmetry.24,25,39 This standard characteristic of the ECM can be used to calculate ECM deposition and organization in normal and failing hearts.40,41 Using a similar approach, we report a quantitative description of auto symmetry by calculating the Hausdorff's (fractal) dimension, demonstrating that our dECM powder particles are in the range of cadaveric young-to-middle age hearts,42 preserving their native architectural structure.
Classically, single purified ECM proteins have been used in in vitro studies43,44 to describe their ability to induce cellular responses. However, reproducing the molecular complexity and heterogeneity of cardiac ECM may increase the relevance of in vitro studies. In recent years, ECM from decellularized tissues (dECM) has been used in in vitro models to better mimic the in vivo microenvironment and to examine tissue-specific effects on cellular behavior.45 Here, we sought to determine if atrial dECM could drive an increase in the differentiation of atrial-like cells. In comparison, another strategy currently uses chemical mediators during differentiation to drive cardiomyocyte fate toward a specific subtype. Adding retinoic acid at days 4–7 of differentiation has been shown to increase the atrial subtype percentage of heterogeneous cultures.2,4,8,11,14 However, after differentiation with high doses (1 μmol l−1) of retinoic acid, the increase of atrial-like cells is accompanied by a decrease in the differentiation efficiency of other cell subtypes.11,14 Thus, the drawback of using this exogenous chemical is its instability and unintended effects on the whole population. In the current study, cells differentiated using dECM-hA demonstrated an increase in the expression level of MLC2a and in the AP of atrial-like cells versus control cells. Importantly, although the distinct phenotypic subtypes of the cell populations were altered by the presence of chamber-specific powder, the differentiation efficiency was not compromised by the presence of atrial or ventricular dECM. These data suggest that enriching cell phenotypes is possible without relying on exogenous chemical mediators that can diminish the quality and quantity of the differentiated population.
Molecular and protein analyses revealed a cluster of chamber-specific cardiac markers from cells differentiated with atrial or ventricular dECM. We noted a statistically significant increase in sarcolipin in dECM-hA as compared to dECM-hV and a statistically significant increase in COUP-TFI as compared to control. Sarcolipin inhibits the sarcoplasmic reticulum Ca2+-ATPase (SERCA) and is restricted to the atrial lineage.46,47 COUP-TFI belongs to the steroid receptor superfamily of genes, displays a distinct pattern of expression in all the three germ layers, and is detected only in the atria of the human fetal heart.14,48 By using flow cytometry, we also observed a corresponding increase in MLC2a and a decrease in MLC2v expression in cardiomyocytes differentiated using dECM-hA compared to dECM-hV and control cells. Moreover, a marked clustering pattern in transcription factors was observed in cardiomyocytes differentiated using dECM-hV, with an increased expression of phospholamban and S100A1 compared to control. Phospholamban is robustly expressed in adult cardiomyocytes and has a crucial role in β-adrenergic signaling and Ca2+ handling during cardiac maturation.49 S100A1 is a Ca2+ binding protein that is expressed in high concentrations in human ventricular myocardium and in low concentrations in human atria.50
Our electrophysiological data suggested a robust increase of atrial-like cells in the dECM-hA group as determined by AP parameters.10,51 Atrial-like cells differentiated using dECM-hA fulfilled one of the criteria for classifying atrial cells described by Ma et al. (APD30–40/APD70–80 < 1.5) with a value of 0.69 ± 0.28. A further indication of electrical activity characteristic of atrial cells was that cells differentiated with dECM-hA exhibited a decreased plateau phase and a shorter APD10 and APD20 compared with cells differentiated using dECM-hV and control cells. Although similar AP results were observed when hiPSCs were differentiated using retinoic acid,2,4,14 our data compares favorably in contrast to cells differentiated using this chemically mediated differentiation strategy to promote specification of cardiomyocyte subtypes. Additionally, in our study, when cells were differentiated using dECM-hV, we observed a prominent ventricular morphology, consisting of a longer phase 2 of the AP and a significant increase in APD90 in ventricular-like cells compared to the control group.52,53 This suggests that dECM-hV may promote electrical maturation of the ventricular-like cardiomyocytes.
The heart's chemical composition has been elucidated by proteomic experiments,37,38,54 resulting in chamber-specific mapping of the human heart by cell and protein type.38,55 Doll et al.9 performed proteomic analyses of 16 segregated, distinct areas of the human heart, helping to elucidate region-specific functional differences at the subcellular level. However, these studies analyzed cadaveric hearts, and the cellular constituents of these samples dominated over ECM molecules, hindering sensitive identification of ECM molecules by proteomics. In contrast, Guyette et al.37 used decellularized hearts in their study, which allowed them to identify a large number of ECM proteins, similar to our results. However, they used a perfusion decellularization technique and analyzed the whole organ dECM, which did not allow them to evaluate region-specific differences. Using TAD, we were able to demonstrate a high association of dECM-hA with atrial-specific proteins, whereas dECM-hV presented ventricle-specific proteins. Based on reports that region-specific proteins can affect cardiac fate during in vitro differentiation,45,56 we believe our data support the hypothesis that atrial-specific dECM proteins influence the differentiation of iPSCs toward an atrial phenotype.
4.1 Limitations
Our study had limitations. Although our findings are an early step toward specifying lineage differentiation of cardiomyocytes in cardiac bioengineering, we did not identify the mechanisms underlying this process, which will require further study. Minor technical limitations in tissue preparation included the use of scissors for mincing tissue, which is not an ideal approach; however, our mincing technique resulted in less tissue damage than other methods we also pursued. In addition, other limitations included the lack of homogeneous distribution of dECM particles in culture wells and the unknown individual impact of factors secreted by the dECM as drivers of specification during the cardiac differentiation protocol. Nevertheless, all preparations were identical between experimental conditions tested, and our data were consistent in all groups, suggesting minimal effects of these limitations on our results.5. Conclusion
Taken together, these results demonstrate that adding decellularized atrial ECM in powdered form during hiPSC-CM differentiation drives cardiomyocyte subtype specification toward an atrial-like phenotype. These findings suggest the potential for using dECM-enriched differentiation media as a robust and high-throughput model for atrial-selective pharmacology. The implications of these data extend to other research applications using decellularized organ powders or particles.Conflicts of interest
DAT holds a financial interest in Miromatrix Medical, Inc. and is entitled to sales royalty through the University of Minnesota for products related to the research described in this paper. This relationship has been reviewed and managed by the University of Minnesota and the Texas Heart Institute in accordance with its conflict of interest policies.Acknowledgements
The authors would like to acknowledge Kimberly Macellaro, PhD, and Rebecca Bartow, PhD, of Texas Heart Institute for editorial assistance. The authors would like to thank Abdelmotagaly Elgalad, MD, PhD, for recovering the human hearts used in this project. FCSN is supported by FAPERJ (grant E-26/202.650/2018). GBD has financial support from grants 88887.130697 (CAPES) and 440613/2016-7 (CNPq). DAT received funding from the Houston Endowment and the Texas Emerging Technology Fund.References
- J. Zhang, M. Klos, G. F. Wilson, A. M. Herman, X. Lian, K. K. Raval, M. R. Barron, L. Hou, A. G. Soerens, J. Yu, S. P. Palecek, G. E. Lyons, J. A. Thomson, T. J. Herron, J. Jalife and T. J. Kamp, Circ. Res., 2012, 111, 1125–1136 CrossRef CAS PubMed.
- V. Schwach, A. O. Verkerk, M. Mol, J. J. Monshouwer-Kloots, H. D. Devalla, V. V. Orlova, K. Anastassiadis, C. L. Mummery, R. P. Davis and R. Passier, Stem Cell Rep., 2017, 9, 1765–1779 CrossRef CAS PubMed.
- Y. Yoshida and S. Yamanaka, Circ. Res., 2017, 120, 1958–1968 CrossRef CAS PubMed.
- M. Lemme, B. M. Ulmer, M. D. Lemoine, A. T. L. Zech, F. Flenner, U. Ravens, H. Reichenspurner, M. Rol-Garcia, G. Smith, A. Hansen, T. Christ and T. Eschenhagen, Stem Cell Rep., 2018, 11, 1378–1390 CrossRef CAS PubMed.
- S. Y. Ng, C. K. Wong and S. Y. Tsang, Am. J. Physiol.: Cell Physiol., 2010, 299, C1234–C1249 CrossRef CAS PubMed.
- C. Kane and C. M. N. Terracciano, Stem Cells, 2017, 35, 1881–1897 CrossRef CAS PubMed.
- J. Asp, J. Synnergren, M. Jonsson, G. Dellgren and A. Jeppsson, Physiol. Genomics, 2012, 44, 89–98 CrossRef CAS PubMed.
- J. H. Lee, S. I. Protze, Z. Laksman, P. H. Backx and G. M. Keller, Cell Stem Cell, 2017, 21, 179–194 CrossRef CAS PubMed . e174.
- S. Doll, M. Dressen, P. E. Geyer, D. N. Itzhak, C. Braun, S. A. Doppler, F. Meier, M. A. Deutsch, H. Lahm, R. Lange, M. Krane and M. Mann, Nat. Commun., 2017, 8, 1469 CrossRef PubMed.
- J. Ma, L. Guo, S. J. Fiene, B. D. Anson, J. A. Thomson, T. J. Kamp, K. L. Kolaja, B. J. Swanson and C. T. January, Am. J. Physiol.: Heart Circ. Physiol., 2011, 301, H2006–H2017 CrossRef CAS PubMed.
- Q. Zhang, J. Jiang, P. Han, Q. Yuan, J. Zhang, X. Zhang, Y. Xu, H. Cao, Q. Meng, L. Chen, T. Tian, X. Wang, P. Li, J. Hescheler, G. Ji and Y. Ma, Cell Res., 2011, 21, 579–587 CrossRef CAS PubMed.
- J. Xavier-Neto, N. Rosenthal, F. A. Silva, T. G. Matos, T. Hochgreb and V. L. Linhares, Genesis, 2001, 31, 97–104 CrossRef CAS PubMed.
- A. M. Merks, M. Swinarski, A. M. Meyer, N. V. Muller, I. Ozcan, S. Donat, A. Burger, S. Gilbert, C. Mosimann, S. Abdelilah-Seyfried and D. Panakova, Nat. Commun., 2018, 9, 2161 CrossRef PubMed.
- H. D. Devalla, V. Schwach, J. W. Ford, J. T. Milnes, S. El-Haou, C. Jackson, K. Gkatzis, D. A. Elliott, S. M. Chuva de Sousa Lopes, C. L. Mummery, A. O. Verkerk and R. Passier, EMBO Mol. Med., 2015, 7, 394–410 CrossRef CAS PubMed.
- K. A. Sharow, B. Temkin and M. A. Asson-Batres, Int. J. Dev. Biol., 2012, 56, 273–278 CrossRef CAS PubMed.
- T. Y. Lu, B. Lin, J. Kim, M. Sullivan, K. Tobita, G. Salama and L. Yang, Nat. Commun., 2013, 4, 2307 CrossRef PubMed.
- A. C. Silva, S. C. Rodrigues, J. Caldeira, A. M. Nunes, V. Sampaio-Pinto, T. P. Resende, M. J. Oliveira, M. A. Barbosa, S. Thorsteinsdottir, D. S. Nascimento and O. P. Pinto-do, Biomaterials, 2016, 104, 52–64 CrossRef CAS PubMed.
- M. Lockhart, E. Wirrig, A. Phelps and A. Wessels, Birth Defects Res., Part A, 2011, 91, 535–550 CrossRef CAS PubMed.
- S. Zaffran, R. G. Kelly, S. M. Meilhac, M. E. Buckingham and N. A. Brown, Circ. Res., 2004, 95, 261–268 CrossRef CAS PubMed.
- J. Manner and T. M. Yelbuz, J. Cardiovasc. Dev. Dis., 2019, 6(1), 12 CrossRef PubMed.
- D. A. Taylor, L. C. Sampaio, R. Cabello, A. Elgalad, R. Parikh, R. P. Wood, K. A. Myer, A. T. Yeh and P. F. Lee, J. Visualized Exp., 2018, 141 DOI:10.3791/58123.
- P. F. Lee, E. Chau, R. Cabello, A. T. Yeh, L. C. Sampaio, A. S. Gobin and D. A. Taylor, Acta Biomater., 2017, 49, 181–191 CrossRef PubMed.
- W. S. Sheridan, G. P. Duffy and B. P. Murphy, Tissue Eng., Part C, 2013, 19, 981–990 CrossRef CAS PubMed.
- C. Hochman-Mendez, M. Cantini, D. Moratal, M. Salmeron-Sanchez and T. Coelho-Sampaio, PLoS One, 2014, 9, e109388 CrossRef PubMed.
- A. J. Kent, N. Mayer, J. L. Inman, C. Hochman-Mendez, M. J. Bissell and C. Robertson, Biomaterials, 2019, 218, 119337 CrossRef CAS PubMed.
- C. Hochman-Mendez, D. B. Pereira de Campos, R. S. Pinto, B. Mendes, G. M. Rocha, G. Monnerat, G. Weissmuller, L. C. Sampaio, A. B. Carvalho, D. A. Taylor and A. C. C. de Carvalho, J. Tissue Eng., 2020, 11, 2041731420921482 Search PubMed.
- J. R. Murillo, L. Goto-Silva, A. Sanchez, F. C. S. Nogueira, G. B. Domont and M. Junqueira, EuPa Open Proteomics, 2017, 16, 1–11 CrossRef CAS PubMed.
- E. Velasquez, D. Martins-de-Souza, I. Velasquez, G. R. A. Carneiro, A. Schmitt, P. Falkai, G. B. Domont and F. C. S. Nogueira, J. Proteome Res., 2019, 18, 4240–4253 CrossRef PubMed.
- F. C. Mesquita, T. H. Kasai-Brunswick, F. Gubert, T. Borgonovo, D. Silva-dos-Santos, D. S. de Araujo, A. C. Campos-de-Carvalho and A. B. Carvalho, Stem Cell Res., 2015, 15, 445–448 CrossRef CAS PubMed.
- F. C. P. Mesquita, P. C. Arantes, T. H. Kasai-Brunswick, D. S. Araujo, F. Gubert, G. Monnerat, D. Silva Dos Santos, G. Neiman, I. C. Leitão, R. A. Q. Barbosa, J. L. Coutinho, I. M. Vaz, M. N. Dos Santos, T. Borgonovo, F. E. S. Cruz, S. Miriuka, E. H. Medei, A. C. Campos de Carvalho and A. B. Carvalho, Sci. Rep., 2019, 9, 19203 CrossRef CAS PubMed.
- J. M. Inacio, M. Almeida, F. Cristo and J. A. Belo, Stem Cell Res., 2020, 42, 101677 CrossRef CAS PubMed.
- A. S. Smith, J. Macadangdang, W. Leung, M. A. Laflamme and D. H. Kim, Biotechnol. Adv., 2017, 35, 77–94 CrossRef CAS PubMed.
- M. Marczenke, I. Piccini, I. Mengarelli, J. Fell, A. Ropke, G. Seebohm, A. O. Verkerk and B. Greber, Front. Physiol., 2017, 8, 469 CrossRef PubMed.
- H. C. Ott, T. S. Matthiesen, S. K. Goh, L. D. Black, S. M. Kren, T. I. Netoff and D. A. Taylor, Nat. Med., 2008, 14, 213–221 CrossRef CAS PubMed.
- H. Lyu, M. John, D. Burkland, B. Greet, Y. Xi, L. C. Sampaio, D. A. Taylor, A. Babakhani and M. Razavi, J. Cardiovasc. Electrophysiol., 2018, 29, 1588–1593 CrossRef PubMed.
- A. Porzionato, E. Stocco, S. Barbon, F. Grandi, V. Macchi and R. De Caro, Int. J. Mol. Sci., 2019, 20(7), 1701 CrossRef PubMed.
- J. P. Guyette, J. M. Charest, R. W. Mills, B. J. Jank, P. T. Moser, S. E. Gilpin, J. R. Gershlak, T. Okamoto, G. Gonzalez, D. J. Milan, G. R. Gaudette and H. C. Ott, Circ. Res., 2016, 118, 56–72 CrossRef CAS PubMed.
- Z. Q. Lu, A. Sinha, P. Sharma, T. Kislinger and A. O. Gramolini, J. Proteome Res., 2014, 13, 5869–5878 CrossRef CAS PubMed.
- F. F. Franchi, M. P. Hernandes, A. L. Coalho Ferreira, V. A. Vieira de Lima, L. de Oliveira Mendes, A. Musa de Aquino, W. R. Scarano and A. Cesar de Souza Castilho, Biochem. Biophys. Res. Commun., 2020, 523, 823–828 CrossRef CAS PubMed.
- P. F. Favero, V. A. Vieira de Lima, P. Helena Dos Santos, A. P. Marques Andrade, L. O. Mendes, F. L. Pacagnelli and A. Cesar de Souza Castilho, Biochem. Biophys. Res. Commun., 2019, 516, 888–893 CrossRef CAS PubMed.
- F. A. Zouein, M. Kurdi, G. W. Booz and J. W. Fuseler, Microsc. Microanal., 2014, 20, 1134–1144 CrossRef CAS PubMed.
- L. B. Mostaco-Guidolin, M. S. D. Smith, M. Hewko, B. Schattka, M. G. Sowa, A. Major and A. C. Ko, J. Appl. Physiol., 2019, 126, 638–646 CrossRef PubMed.
- D. Wang, Y. Wang, H. Liu, C. Tong, Q. Ying, A. Sachinidis, L. Li and L. Peng, J. Cell. Mol. Med., 2019, 23, 3629–3640 CrossRef CAS PubMed.
- Y. Yang, Z. Ren, F. Xu, Y. Meng, Y. Zhang, N. Ai, Y. Long, H. I. Fok, C. Deng, X. Zhao, L. Huang, Q. Zhao, J. Wang, W. Liu, W. Ge and G. Chen, Cell Rep., 2019, 29, 3374–3384 CrossRef CAS PubMed . e3375.
- I. Ullah, J. F. Busch, A. Rabien, B. Ergün, C. Stamm, C. Knosalla, S. Hippenstiel, P. Reinke and A. Kurtz, Adv. Sci., 2020, 7, 1901198 CrossRef CAS PubMed.
- R. Josowitz, J. Lu, C. Falce, S. L. D'Souza, M. Wu, N. Cohen, N. C. Dubois, Y. Zhao, E. A. Sobie, G. I. Fishman and B. D. Gelb, PLoS One, 2014, 9, e101316 CrossRef PubMed.
- S. A. Shaikh, S. K. Sahoo and M. Periasamy, J. Mol. Cell. Cardiol., 2016, 91, 81–91 CrossRef CAS PubMed.
- F. A. Pereira, M. J. Tsai and S. Y. Tsai, Cell. Mol. Life Sci., 2000, 57, 1388–1398 CrossRef CAS PubMed.
- G. Chen, S. Li, I. Karakikes, L. Ren, M. Z. Chow, A. Chopra, W. Keung, B. Yan, C. W. Chan, K. D. Costa, C. W. Kong, R. J. Hajjar, C. S. Chen and R. A. Li, Circ.: Arrhythmia Electrophysiol., 2015, 8, 193–202 CAS.
- D. Rohde, J. Ritterhoff, M. Voelkers, H. A. Katus, T. G. Parker and P. Most, J. Cardiovasc. Transl. Res., 2010, 3, 525–537 CrossRef PubMed.
- R. Zhu, M. A. Millrod, E. T. Zambidis and L. Tung, Sci. Rep., 2016, 6, 18544 CrossRef CAS PubMed.
- J. C. Wu, P. Garg, Y. Yoshida, S. Yamanaka, L. Gepstein, J. S. Hulot, B. C. Knollmann and P. J. Schwartz, Circ. Res., 2019, 125, 653–658 CrossRef CAS PubMed.
- A. Barbuti, P. Benzoni, G. Campostrini and P. Dell'Era, Dev. Dyn., 2016, 245, 1145–1158 CrossRef CAS PubMed.
- H. Suryawanshi, R. Clancy, P. Morozov, M. K. Halushka, J. P. Buyon and T. Tuschl, Cardiovasc. Res., 2020, 116(8), 1446–1457 CrossRef CAS PubMed.
- A. Naba, K. R. Clauser, H. Ding, C. A. Whittaker, S. A. Carr and R. O. Hynes, Matrix Biol., 2016, 49, 10–24 CrossRef CAS PubMed.
- S. C. den Hartogh, K. Wolstencroft, C. L. Mummery and R. Passier, Sci. Rep., 2016, 6, 19386 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01686a |
| This journal is © The Royal Society of Chemistry 2021 |