
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green synthesis of methoxy-poly(ethylene glycol)-block-poly(L-lactide-co-glycolide) copolymer using zinc proline as a biocompatible initiator for irinotecan delivery to colon cancer in vivo†
Prabhanjan S.
Giram
abc,
Julie Tzu-Wen
Wang
 *c,
Adam A.
Walters
c,
Priyanka P.
Rade
ab,
Muhammad
Akhtar
cd,
Shunping
Han
c,
Farid N.
Faruqu
c,
Hend M.
Abdel-Bar
ce,
Baijayantimala
Garnaik
*ab and
Khuloud T.
Al-Jamal
*c
*c,
Adam A.
Walters
c,
Priyanka P.
Rade
ab,
Muhammad
Akhtar
cd,
Shunping
Han
c,
Farid N.
Faruqu
c,
Hend M.
Abdel-Bar
ce,
Baijayantimala
Garnaik
*ab and
Khuloud T.
Al-Jamal
*c
aPolymer Science and Engineering Division, CSIR-National Chemical Laboratory, Pune-411008, India. E-mail: b.garnaik@ncl.res; Tel: +91 20 25902304
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
cSchool of Cancer and Pharmaceutical Sciences, Faculty of Life Sciences & Medicine, King's College London, London, SE1 9NH, UK. E-mail: khuloud.al-jamal@kcl.ac.uk; tzu-wen.wang@kcl.ac.uk; Tel: +44 (0)2078484525
dDepartment of Pharmacy, Faculty of Pharmacy and Alternative Medicine, The Islamia University of Bahawalpur, 63100, Pakistan
eDepartment of Pharmaceutics, Faculty of Pharmacy, University of Sadat City, Egypt
First published on 11th November 2020
Abstract
Poly(lactic-co-glycolic acid) (PLGA) is the most commonly described biocompatible copolymer used in biomedical applications. In this work, a green synthetic approach based on the biocompatible zinc proline complex, as an initiator for PLGA synthesis, is reported for the first time for the synthesis of methoxy-poly(ethylene glycol)-block-poly(L-lactic-co-glycolic acid) (mPEG–PLGA). mPEG–PLGA with controlled molecular weight and narrow polydispersity was synthesised. Its potential for delivery of irinotecan (Ir), a poorly water-soluble chemotherapeutic drug used for the treatment of colon and pancreatic cancer, was studied. Nanoparticles of controlled size (140–160 nm), surface charge (∼−10 mV), release properties and cytotoxicity against CT-26 (colon) and BxPC-3 (pancreatic) cancer cells, were prepared. Tumor accumulation was confirmed by optical imaging of fluorescently labelled nanoparticles. Unlike Tween® 80 coated NP-Ir, the Pluronic® F-127 coated NP-Ir exhibits significant tumor growth delay compared to untreated and blank formulation treated groups in the CT-26 subcutaneous tumor model, after 4 treatments of 30 mg irinotecan per kg dose. Overall, this proof-of-concept study demonstrates that the newly synthesized copolymer, via a green route, is proven to be nontoxic, requires fewer purification steps and has potential applications in drug delivery.
1. Introduction
Polyesters such as poly(L-lactic acid) (PLA), poly(lactic-co-glycolic acid) (PLGA), poly-ε-caprolactone (PCL) and their copolymers synthesized from ring-opening polymerization (ROP) of cyclic lactone monomers have shown great promise in biomedical applications including controlled drug-delivery (microparticles, nanoparticles, or micelles)1–5 due to their in vivo degradability and biocompatibility.6,7 PLGA copolymers and poly (ethylene glycol) methyl ether-block-poly (lactic-co-glycolic acid) (mPEG–PLGA) copolymers constitute an important class of biologically relevant copolymers.8,9 They have the propensity to self-assemble into micelles when dispersed in an aqueous medium, depending on composition, making them ideal drug delivery carriers to solubilise hydrophobic drugs.10,11In polymer synthesis, stannous octoate is used worldwide as an inexpensive commercial catalyst. However, it suffers from toxicity and removal of trace metal impurities is tedious and costly. The synthesis involves many side reactions which often results in a broad dispersity. For instance, Zhang et al. investigated ring opening polymerization of L-lactide using stannous octoate and found linear or branched polymer chains with various molecular weights.12 The reaction between stannous octoate and alcohol produces stannous alkoxide, which initiates the polymerization by co-ordinately inserting into polymer chains, known as the alkoxide initiation mechanism. A few of zinc complexes, served as catalysts, have been reported to be non-toxic in the literature.13,14 We have previously synthesised PLAs using a green approach in the presence of the biocompatible zinc proline complex in bulk polymerization.15 In the present study, we attempted to synthesize mPEG–PLGA from L-lactide, glycolide monomers and m-PEG–OH in the presence of zinc proline complex using solvent-free bulk ROP.
Irinotecan, a semi-synthetic derivative of camptothecin, is a chemotherapeutic drug used alone or in combination with fluorouracil to treat advanced colon cancer or other types of cancers, respectively.16 Only one camptothecin nanoformulation, the PEGylated liposome Onivyde®, received FDA approval for treatment of metastatic pancreatic cancer in 2015.17 NKTR102 (etirinotecan pegol), an irinotecan conjugated, via a proprietary biodegradable ester-based linker, to polyethylene glycol (PEG), with extended release characteristic has reached phase III clinical trials to treat metastatic breast cancer.18,19
In this study, mPEG–PLGA was synthesized using green chemistry requiring minimal purification steps. mPEG–PLGA polymeric nanoparticles were formulated using nanoprecipitation method and two types of hydrophilic surfactants namely Tween® 80 (T-NP-Ir) and Pluronic® F-127 (P-NP-Ir), for irinotecan (NP-Ir) delivery to colon cancer. NP were evaluated and characterized for their physicochemical properties, drug loading efficiency and release profiles. The in vitro cytotoxicity profiles were tested in murine CT-26 colon and human BxPC-3 pancreatic cancer cells. Tumor uptake and organ biodistribution profiles of the two types of irinotecan-loaded NPs after intraveneous administration were studied in CT-26 tumor-bearing mice following intravenous administration by optical imaging. The therapeutic efficacy and overall immunological assessments were assessed in CT-26 tumor-bearing mice.
2. Experimental
2.1. Material and reagents
Irinotecan was a generous gift from Emcure, Pune (India). 1,1′-Dioctadecyl-3,3,3′,3′Tetramethylindotricarbocyanine Iodide (DiR), dialysis tubing (MWCO 2000 Da), Tween® 80, Pluronic® F-127, methylene chloride, acetone, absolute ethanol, dimethyl sulfoxide (DMSO) and diethyl ether (ultra-pure grades) were purchased from Sigma-Aldrich (UK). Methoxy poly(ethylene glycol) (MW ∼ 2000) was purchased from Fluka Chemical (USA). SnakeSkin® dialysis tubing (MWCO 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) was purchased from Thermo-fisher (USA). Soybean lecithin (Epikuron™) was a kind gift from Cargill Pharmaceuticals. PD-10 desalting column was obtained from GE Healthcare Life Sciences (UK). RPMI-1640 media, fetal bovine serum (FBS), penicillin/streptomycin, trypsin/EDTA, and phosphate buffered saline (PBS) were obtained from Gibco, Thermo Fisher Scientific Inc. (UK). Isoflurane (IsoFlo®) for anesthesia was purchased from Abbott Laboratories Ltd (UK). CT-26 and BxPC-3 cells were obtained from ATCC (USA). All reagents were used without further purification. Antibodies for immunological studies (anti CD8-PE, anti Ly-6G-APC, anti CD45-FITC, anti FOXP3-APC), RBC fix/lyse solution, Precision count beads and True-Nuclear Transcription Factor Buffer Set were purchased from Biolegend (UK). Cell strainers at 70 μm were purchased from BD Biosciences (UK).
000 Da) was purchased from Thermo-fisher (USA). Soybean lecithin (Epikuron™) was a kind gift from Cargill Pharmaceuticals. PD-10 desalting column was obtained from GE Healthcare Life Sciences (UK). RPMI-1640 media, fetal bovine serum (FBS), penicillin/streptomycin, trypsin/EDTA, and phosphate buffered saline (PBS) were obtained from Gibco, Thermo Fisher Scientific Inc. (UK). Isoflurane (IsoFlo®) for anesthesia was purchased from Abbott Laboratories Ltd (UK). CT-26 and BxPC-3 cells were obtained from ATCC (USA). All reagents were used without further purification. Antibodies for immunological studies (anti CD8-PE, anti Ly-6G-APC, anti CD45-FITC, anti FOXP3-APC), RBC fix/lyse solution, Precision count beads and True-Nuclear Transcription Factor Buffer Set were purchased from Biolegend (UK). Cell strainers at 70 μm were purchased from BD Biosciences (UK).
2.2. Synthesis of mPEG–PLGA by ring-opening polymerization
mPEG–PLGA copolymer was synthesized according to the method developed by Parwe et al. employed for PLA synthesis with some modifications (Scheme 1).15 Briefly, polymerization was carried out in sealed glass ampoules (inner diameter of 2 cm and 10 cm in height) in absence of solvents. Glass ampoules were passivated using 20% trimethylsilyl chloride (Me3SiCl) in acetone before polymerization. This is to convert all active hydroxyl groups (–OH) on the inner surface of glass ampoules to OSiMe3 groups which are inert and do not interfere the polymerization reaction. Addition of monomers and the initiator was carried out in a MBRAUN UNilab™ glove box to maintain dry conditions. Ampoules were subjected to 3–4 freeze pump–thaw cycles in vacuo. Ampoules containing L-lactide (5.208 mmole, 750 mg), glycolide (2.192 mmole, 250 mg), mPEG-2000 (0.025 mmole, 10 mg) and zinc proline (0.032 mmole, 5 mg), used as a catalyst/coinitiator, were sealed under vacuum. Ampoules were then immersed in Techne SBL-2D™ Fluidized sand bath previously set at 180 °C temperature for 1 h. L-Lactide to glycolide molar feed ratio used was 70:30. Ampoules were cooled down and broken at different time intervals to monitor the reaction. At the end of the reaction, solid residue containing m-PEG–PLGA copolymer and the unreacted m-PEG-2000, zinc proline and unreacted L-lactide, glycolide were dissolved in a small amount of dichloromethane and precipitated in excess ice-cold methanol to obtain the copolymer. Copolymer samples were dried at 40 °C under vacuum for 48 h.
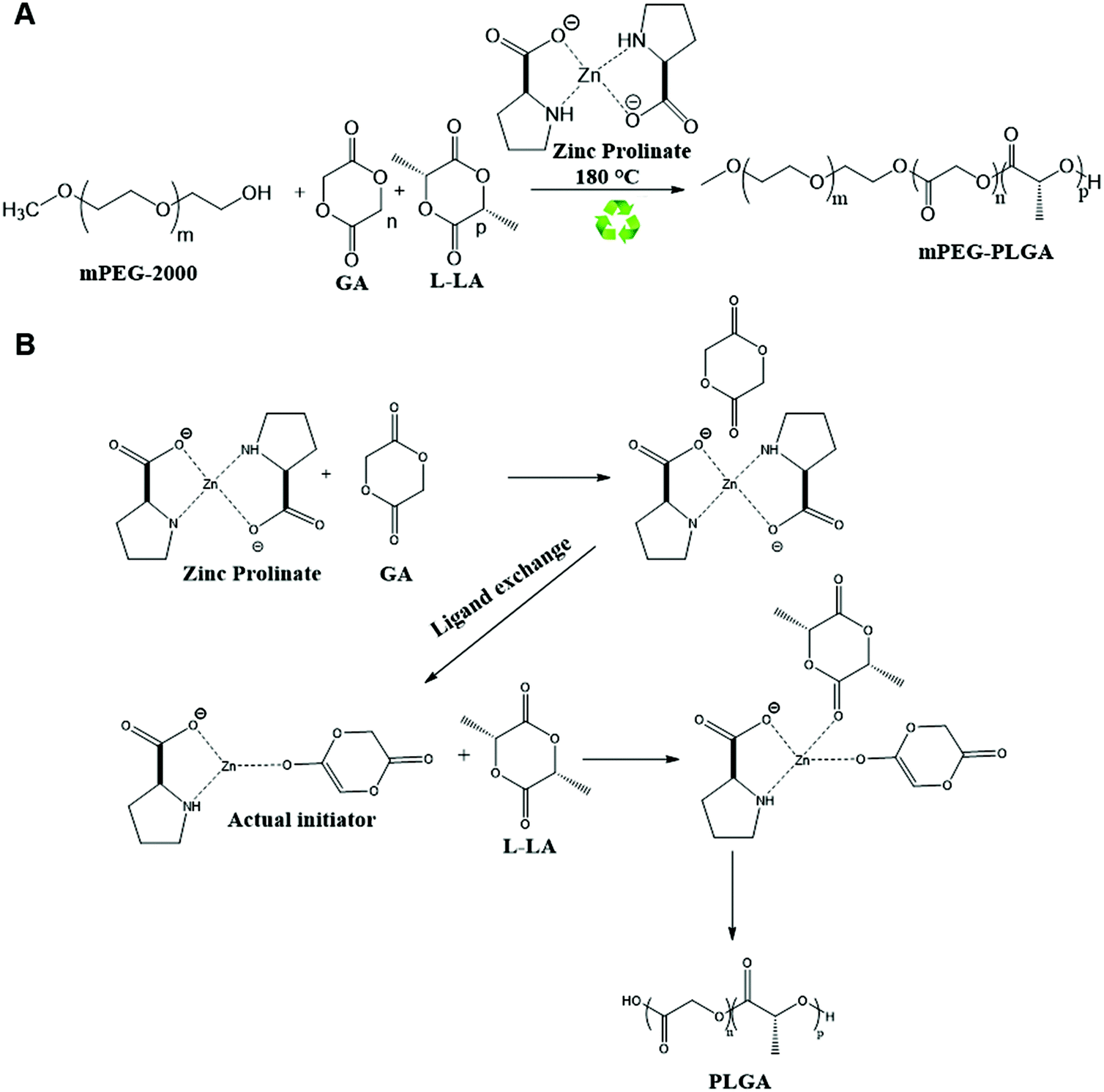 | ||
| Scheme 1 The reaction scheme and proposed mechanism. (A) Synthesis of mPEG–PLGA copolymer. (B) Proposed reaction mechanisms for ring-opening polymerization of L-lactide and glycolide in presence of zinc proline. | ||
2.3. Characterization of mPEG–PLGA polymer
The copolymers were characterized by attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR), gel permeation chromatography (GPC), 1H NMR, 13C NMR and matrix assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) as detailed in ESI.†2.4. Formulation of the NPs
Irinotecan loaded polymeric NPs were prepared using the nanoprecipitation technique as described previously20 and presented in Scheme 2. Briefly, m-PEG–PLGA copolymer (12.5 mg), irinotecan (1, 3 or 5 mg) and soybean lecithin (12.5 mg) were dissolved in 2.5 mL of acetone/ethanol (60:40 v/v) mixture. This organic phase was added drop-wise into the aqueous phase (5 mL) containing Tween® 80 (0.2%, 5 mg) or Pluronic® F-127 (0.5%, 25 mg) as a hydrophilic surfactant. The mixture was maintained under magnetic stirring in the chemical hood for 30 min to allow the solvent to diffuse as a result of the difference in surface tension of two phases and form NPs. Organic solvents were then eliminated by evaporation under reduced pressure using a rotavapor. The final volume of the colloidal suspension was adjusted to 5 mL using deionized water.
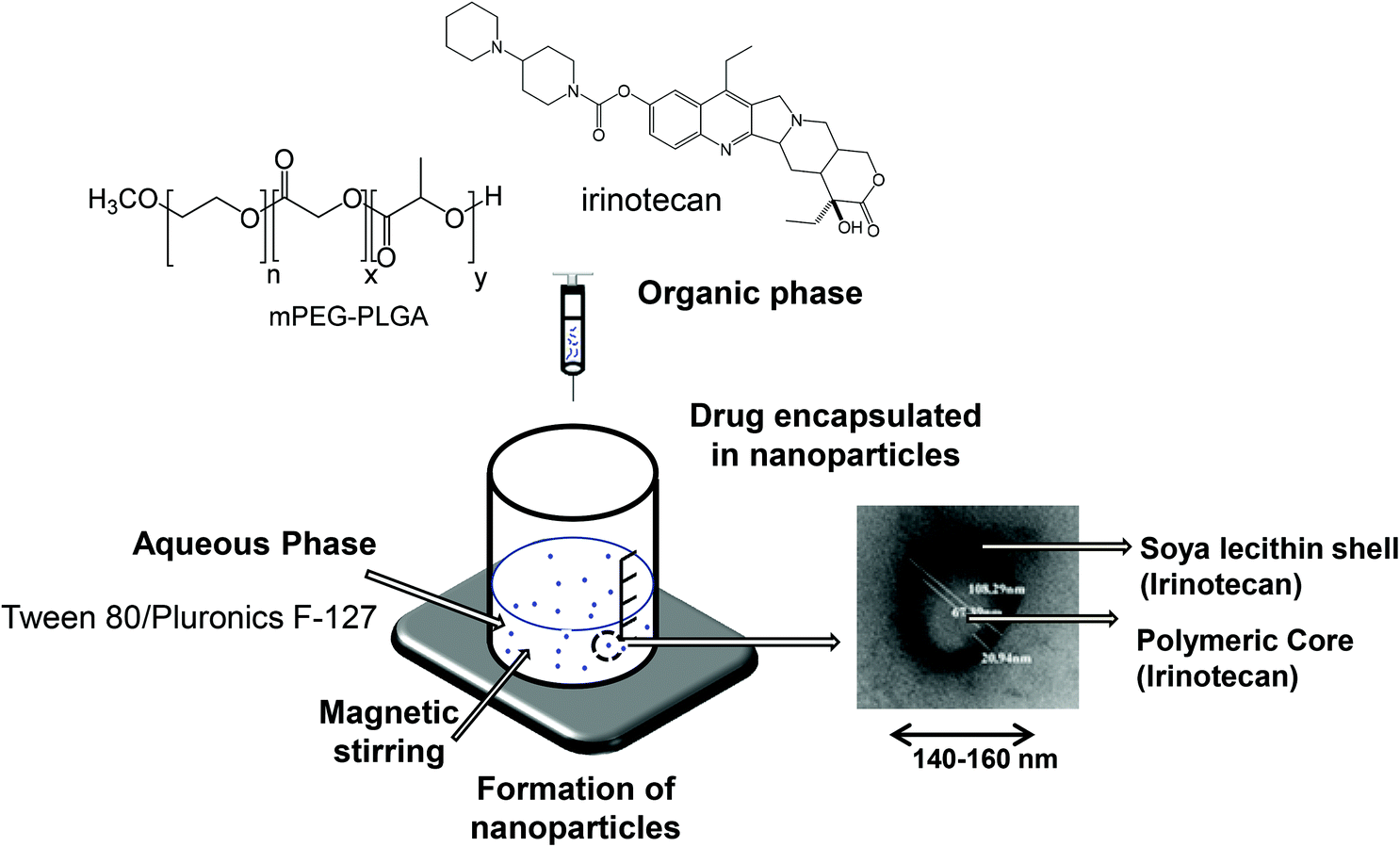 | ||
| Scheme 2 Preparation of irinotecan loaded NPs was carried out by the nanoprecipitation method. Schematic representation of the formulation design of Tween® 80 coated NP (T-NP) and Pluronic® F-127 coated NP (P-NP). | ||
2.5. Characterization of the formulated NPs
:3 v/v) and the amount of irinotecan was determined by measuring the absorbance at 360 nm using UV-vis spectrophotometer (Shimadzu, Japan). The amount of irinotecan in NPs was calculated from a standard curve established from known irinotecan concentrations prepared using identical organic phase conditions. The EE (%) was then calculated using the following equation: | EE (%) = (([drug] total − [drug] free)/[drug] total) × 100. |
The percentage of drug loading efficiency (% LE) was calculated using the following equation:
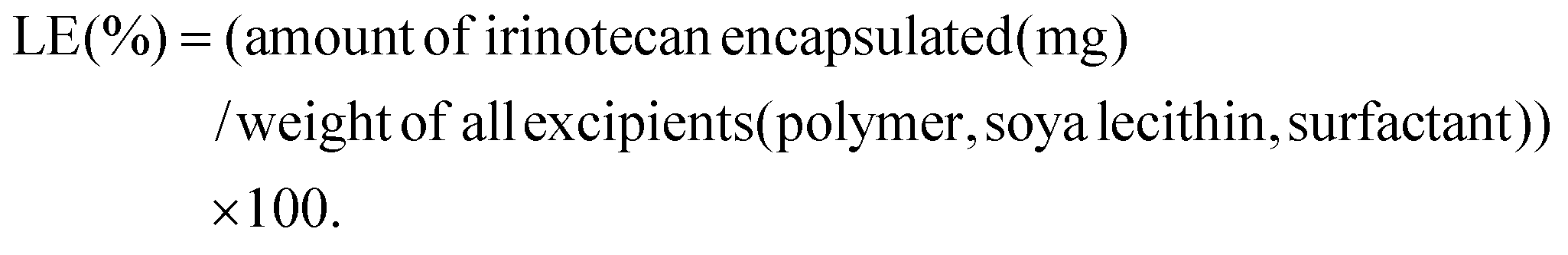
2.6. Shelf life stability
NP suspensions were sealed in 7 mL glass vials and stored at 4 °C. The stability of NP dispersions was tested after 0, 7, 14 and 28 days of preparation by visual inspection of the physical properties (color and opacity) and also by size and zeta-potential measurements. The measurements were carried out in triplicate and presented as an average ± SD.2.7. Release profiles in vitro
Five milliliters of NP formulations containing 5 mg irinotecan were transferred into a 10 kDa dialysis bag, with or without 50% FBS. This MWCO was selected so that serum was confined inside the dialysis bag not to interfere with drug quantification in the dialysate. The samples were dialyzed against 40 mL of 1% (w/v) Tween® 80 in PBS, pH 7.4. The release was studied at 37 °C at 250 strokes per min. At predetermined time points, 1.5 mL aliquots were taken and replaced by the addition of an equal volume of the dialysate to maintain sink conditions. Drug concentration in the dialysate was assessed by spectroscopy as described above. A control experiment was set up alongside in which the same amount of irinotecan was dissolved in DMSO and dialyzed for comparison. This control was set up to eliminate nonspecific adsorption of the drug to the dialysis membrane.2.8. Cytotoxicity studies in vitro
The CT-26 murine colon carcinoma (CT-26; ATCC, CRL-2638) and BxPC3 human pancreatic carcinoma (BxPC3, ATCC® CRL1687™) were cultured in RPMI-1640 media supplemented with 10% FBS, 50 U mL−1 penicillin, 50 μg mL−1 streptomycin, 1% L-glutamine, at 37 °C in 5% CO2. Cells were routinely grown in 75 cm2 canted-neck tissue culture flasks and passaged twice a week using trypsin/EDTA at 80% confluence.CT-26 or BxPC3 cells were seeded in 96-well plates at 4000 cells per well and incubated with different concentrations of irinotecan or its NP (0.1–100 μM irinotecan concentration) in complete media for 72 h. Cells were also treated with blank NP formulations (P-NP and T-NP) at the polymer concentrations equivalent to drug NP formulations (P-NP-Ir AND T-NP-Ir). Cytotoxicity was examined by MTT assay. Briefly, at the end of the incubation period, the media was removed and replaced with 120 μL of MTT solution at a final concentration of 0.5 mg mL−1. Cells were incubated for 3 h at 37 °C and 5% CO2. At the end of the incubation, formazan was dissolved in 200 μL of DMSO and the plate was read at 570 nm in an FLUO star OPTIMA plate reader (BMG Labtech) and the results were expressed as the percentage cell survival (mean ± SD) and calculated using the following equation: % cell survival = (A570 nm of treated cells/A570 nm of untreated control cells) × 100.
2.9. Organ biodistribution studies in vivo by optical imaging
All animal experiments were performed in compliance with the UK Animals (Scientific Procedures) Act 1986 and UK Home Office Code of Practice for the Housing and Care of Animals Used in Scientific Procedures (Home Office 1989). In vivo experimentation was adhered to the project licence approved by the King's College London animal welfare and ethical review body (AWERB) and UK Home Office. Female Balb/c mice (∼20 g) aged 4–6 weeks (Envigo) were inoculated subcutaneously with CT-26 cells (1 × 106 cells in 0.1 mL PBS) at both lower flanks. DiR was incorporated in the formulation at 2.5 wt% of m-PEG–PLGA copolymer and DiR-labeled P-NP (P-NP-DiR) was prepared as described previously.10 Mice (n = 3) were intravenously injected with P-NP-DiR in PBS solution and scanned at 1, 4, and 24 h post injection using an IVIS Lumina Series III In Vivo Imaging System (PerkinElmer, UK). Untreated animals were included as controls. Animals were anesthetized with 1.5% isoflurane/98.5% oxygen to maintain sedation during the imaging procedure. Ex vivo imaging was carried out for excised major organs (heart, lung, liver, spleen, and kidney) including tumors. Fluorescence images were obtained using DiR filter (740/790 nm for excitation/emission wavelengths) and analysed using Living Image® 4.3.1 Service Pack 2 software (PerkinElmer, UK).2.10. Tumor growth delay in vivo studies
To determine the therapeutic effect of NPs containing irinotecan, tumor-bearing Balb/c mice were randomly divided into five groups (n = 8), anesthetized using isoflurane and injected via a tail vein with (i) PBS (ii) irinotecan at 30 mg kg−1 in 200 μL (positive control) (iii) P-NP Blank (iv) P-NP-Ir at 30 mg irinotecan per kg in 200 μL (v) T-NP-Ir at 30 mg irinotecan per kg in 200 μl. All solutions were prepared in PBS and injected on days 6, 11, 14 and 17 post tumor inoculation for a total of 4 doses. Mice were sacrificed by cervical dislocation when tumors reached 1500 mm3. Data are given as mean value ± SEM (standard error of the mean).2.11. Histological examination of major organs
For histological examination, major organs (heart, lung liver, spleen, and kidney) and tumors from all treatments and the control group were excised at the end of therapy studies. Organs were immediately fixed in 10% neutral buffered formalin as 5 mm2 pieces, paraffin-embedded, and sectioned for hematoxylin and eosin staining (H&E) according to standard histological protocols at the Royal Veterinary College (UK). All stained sections were analyzed using a Leica DM 1000 LED Microscope (Leica Microsystems, UK) coupled with CCD digital camera (QImaging, UK).2.12. Enumeration of the blood leukocyte population
Whole heparinized blood was obtained at the terminal time point by cardiac puncture. Blood (50 μl) was incubated with relevant fluorophore conjugated monoclonal antibody (anti CD8-PE, anti Ly-6G-APC, anti CD45-FITC) at previously optimized concentration for 15 min in the dark. Following incubation, red blood cells were lysed and cells fixed with 1× RBC fix/lyse solution (450 μL) for 30 min. An exact quantity of precision count beads (25 μL) was added to each sample before the sample being acquired by a FACSCalibur flow cytometer. The analysis was performed using Flowjo V10 software (Tree Star). Cells were enumerated by first gating on an appropriate forward/side scatter profile then on the fluorophore positive population. Cell counts in each population were then normalized to bead number then to blood volume following the manufacturer's datasheet.2.13. Isolation and phenotyping of cells from tumor-draining lymph node
The inguinal lymph node of tumor-bearing mice was extracted following termination of in vivo experiment. Lymph nodes were macerated through a 70 μm cell strainer with a syringe to obtain a uniform single-cell suspension. Cells were collected and washed three times in PBS (300 μL). Cells were stained with fluorophore-conjugated monoclonal antibody against cell surface marker (anti CD4-FITC, anti CD8-PE) for 15 min. Cells were washed three times in PBS, fixed and permeabilized using 1× fixation and 1× permeabilization buffer from True-Nuclear Transcription Factor Buffer Set following the manufacturer's protocol. FOXP3 transcription factor was stained by the addition of anti FOXP3-APC diluted in permeabilization buffer and incubated for 1 hour. The following staining, cells were washed three times in permeabilization buffer before being resuspended in PBS and acquired on a FACSCalibur flow cytometer. Data were analyzed using Flowjo V10 (Tree Star) initially gating on an appropriate forward/side scatter profile before individual fluorophores were analyzed.2.14. Statistical analysis
For all experiments, data were presented as mean ± SD except for the therapy experiments; data were presented as mean ± SEM, where n denotes the number of repeats. Significant differences were examined using one-way ANOVA. p < 0.05 was considered statistically significant in all studies. In the case of therapy experiment, significant differences were examined using Mann Whitney for the tumor growth delay study.3. Results
3.1. Synthesis and characterization of mPEG–PLGA copolymer
mPEG–PLGA copolymer was synthesized using biocompatible, non-toxic zinc proline as an alternative initiator to avoid the huge cost and efforts associated with tin removal after stannous octoate catalyzed polymerization reactions (Scheme 1). In our previous work synthesizing PLA,15 computational modelling suggested monomer lactide first coordinated with zinc proline and replaced one of the ligands to form a complex which acted as the initiator and such ligand exchange initiated the ROP. In the present mPEG–PLGA synthesis, since glycolide is more reactive than L-lactide, it is likely that one ligand of zinc proline was replaced with glycolide to form zinc proline glycolide complex which acted as the actual initiator. The zinc proline glycolide complex underwent propagation with L-lactide. m-PEG acted as a co-initiator and assisted the ROP with chain end mechanism (Scheme 1B).1H NMR spectrum of mPEG–PLGA copolymer is shown in Fig. 1A. The peaks in the 1H NMR copolymer spectra at 1.55 (d, 3H; –CO–CH (CH3)–O–) and 5.18 (q, 1H; –CO–CH (CH3)–O–) were assigned to the lactic backbone protons, the peak at 4.81 (m, 2H; –CO–CH2–O––) was assigned to the glycolic protons and the peaks at 3.67 (m for mPEG2K) and 3.38 (s, 3H; CH3O–) corresponded to the mPEG protons. 13C NMR spectrum of mPEG–PLGA is shown in Fig. 1B. L-Lactyl carbonyl and glycolyl carbonyl appear at 169 and 166 ppm, respectively.
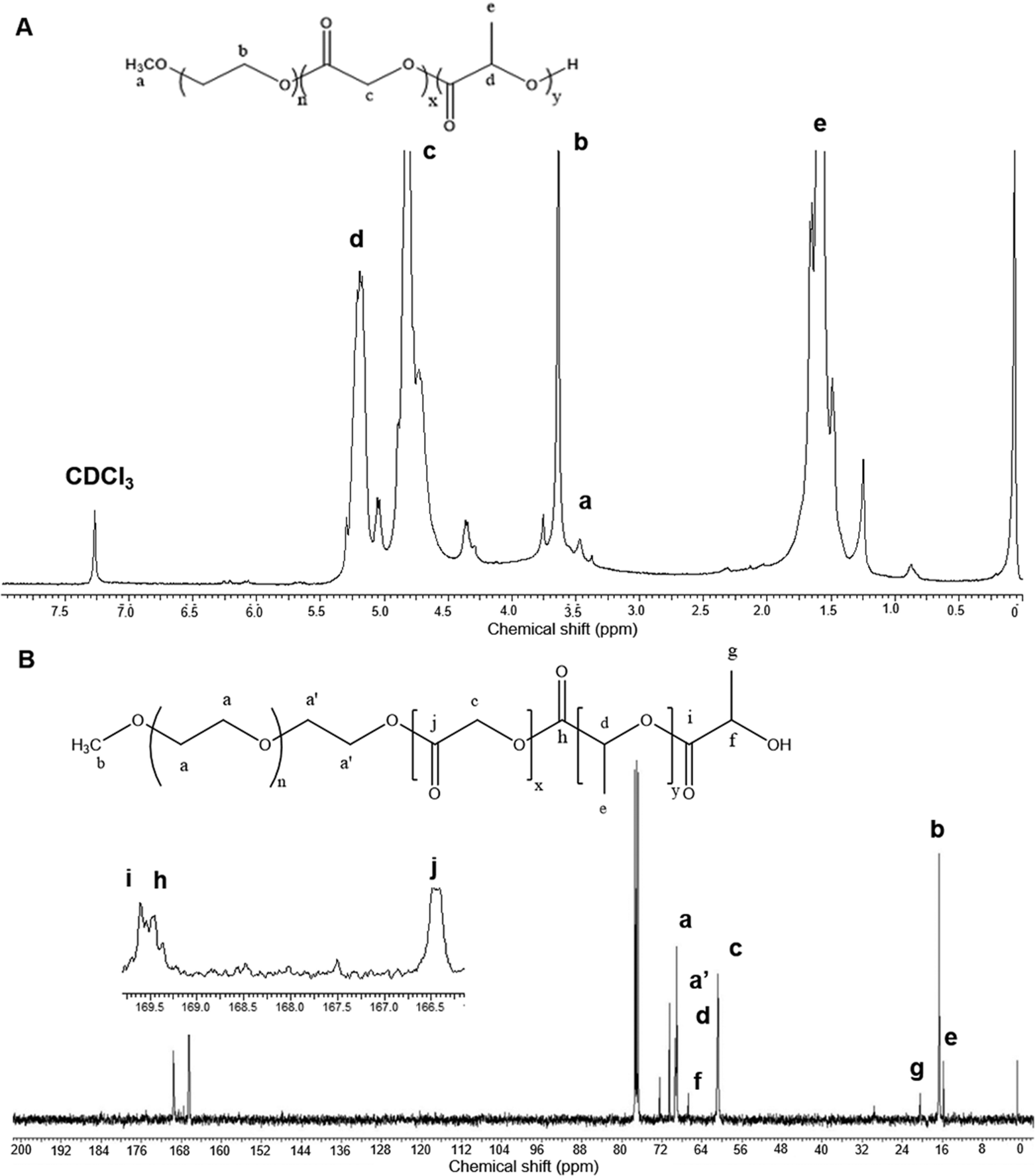 | ||
| Fig. 1 NMR spectrum analyses of the synthesized mPEG–PLGA copolymer in CDCl3. (A) 1H NMR spectrum (B) 13C NMR spectrum. | ||
GPC curve for mPEG–PLGA corresponded to an average molecular weight (Mw), number average molecular weight (Mn) of 6700 and 5171 Da, respectively (Fig. S1A†). The polydispersity index (PDI) of 1.2 was obtained. The FTIR spectrum of mPEG–PLGA (Fig. S1B†) showed major peaks at 1065 cm−1 (O–CH2 stretching) and 1740 cm−1 (ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching). The peaks appear at 3500 cm−1 (O–H stretching) belong to the terminal hydroxyl groups in the copolymer. The absorptions at the range 2900–3000 cm−1 represent –CH– stretching of –CH– and –CH2 groups, respectively. These assigned peaks provided structural confirmation of mPEG–PLGA. MALDI-TOF spectra of mPEG–PLGA copolymers is shown in Fig. S2.†
O stretching). The peaks appear at 3500 cm−1 (O–H stretching) belong to the terminal hydroxyl groups in the copolymer. The absorptions at the range 2900–3000 cm−1 represent –CH– stretching of –CH– and –CH2 groups, respectively. These assigned peaks provided structural confirmation of mPEG–PLGA. MALDI-TOF spectra of mPEG–PLGA copolymers is shown in Fig. S2.†
3.2. Formulation, physico-chemical, morphological and thermal characterization of NPs
mPEG–PLGA copolymer was used for the formulation of irinotecan containing NPs using two types of surfactants, Tween® 80 (T-NP-Ir) or Pluronic® F-127 (P-NP-Ir), by the nanoprecipitation method (Scheme 2). The non-encapsulated drug was removed by the PD-10 column using distilled water. Characterization of nanoparticle formation including hydrodynamic diameter and zeta potential is shown in Table 1, Tables S1 and S2.† A slightly larger hydrodynamic diameter was reported for T-NP-Ir (163.8 ± 2.9 nm) than P-NP-Ir (145.3 ± 7.3 nm). The optimized EE % and LE % obtained for T-NP-Ir were ∼47.0% and 4.7%, respectively. The corresponding values for P-NP-Ir were ∼57.7% and 5.7% respectively. No change in size was observed up to 7 days of storage 4 °C. The NP appeared spherical in shape and in the size range ∼35–95 nm according to TEM images (Fig. S3†).| Formulation | Day | Hydrodynamic diametera,e (nm) | PDIa | Zeta-potentialb (mV) | Encapsulation efficiencyc,e (EE%) | Loading efficiencyd,e (LE%) |
|---|---|---|---|---|---|---|
| a Measured in deionized water by dynamic light scattering. b Surface charge was measured in deionized water. c Calculated as the percentage of the drug incorporated over total added drug determined by spectrophotometry. d Calculated as the percentage of the incorporated drug divided by the weight of excipients (polymer, soya lecithin, surfactant), determined by spectrophotometry. e Expressed as mean ± SD (n = 3). | ||||||
| P-NP-Ir | 0 | 145.3 ± 7.3 | 0.261 | −9.3 | 57.7 ± 0.5 | 5.7 ± 0.02 |
| 7 | 148.0 ± 3.5 | 0.267 | −9.2 | |||
| 28 | 166.0 ± 8.9 | 0.321 | −9.0 | |||
| T-NP-Ir | 0 | 163.8 ± 2.9 | 0.391 | −10.3 | 47.0 ± 5.6 | 4.7 ± 0.02 |
| 7 | 165.0 ± 6.1 | 0.399 | −10.0 | |||
| 28 | 205.1 ± 8.6 | 0.412 | −9.8 | |||
3.3. Drug release study
The release pattern of irinotecan from T-NP-Ir and P-NP-Ir was studied up to 96 h in the presence or absence of 50% serum in PBS (Fig. 2). The solution in DMSO was used as a 100% release control. The presence of serum did not affect the release profiles for all solutions/formulations. In the case of irinotecan/DMSO control, a 100% release was achieved within 24 h with or without serum suggesting lack of interactions between the drug and serum or dialysis membrane. In contrast, both NPs showed sustained drug release profiles over the study period. The release profile plateaued after 48 h reaching ∼50–60% release.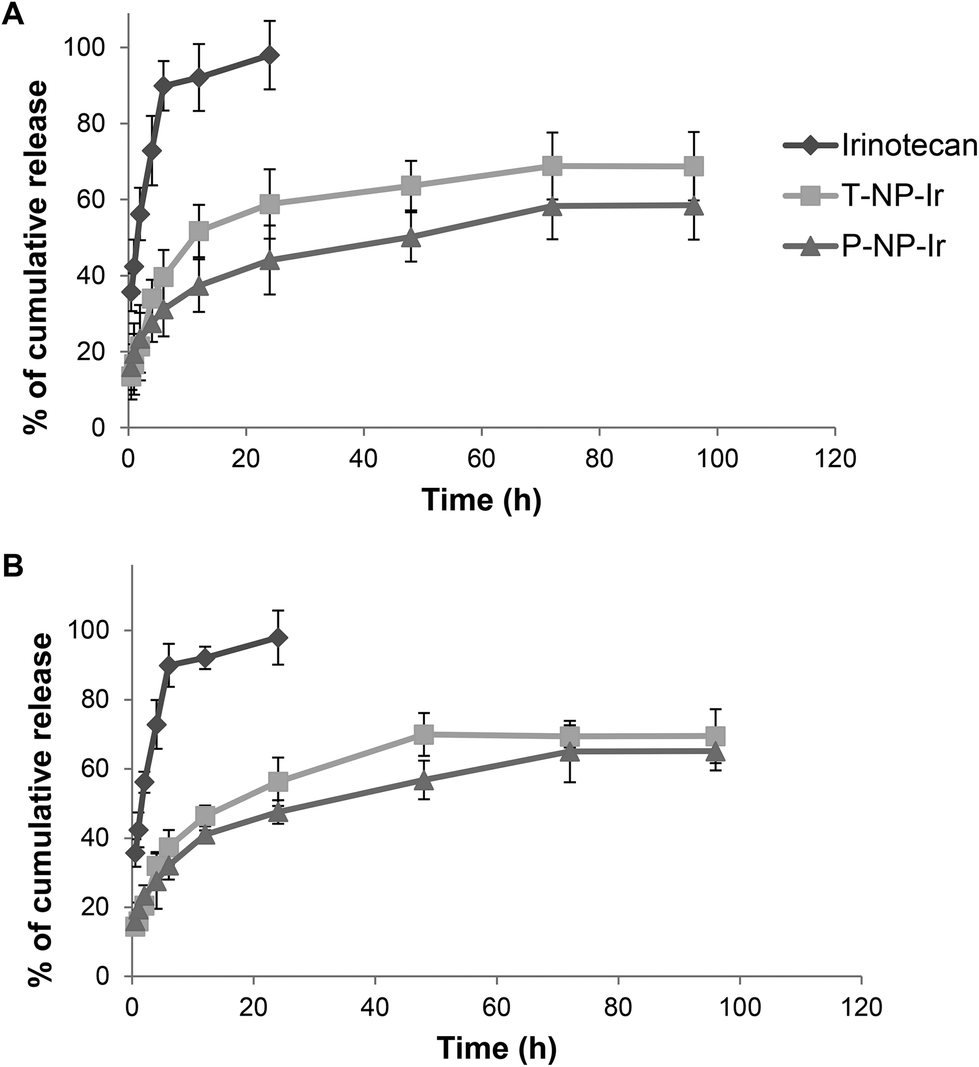 | ||
| Fig. 2 The release profiles of irinotecan from the NPs. NPs were dialyzed against 1% w/v Tween® 80 in phosphate buffered PBS (PBS), pH 7.4 in the presence (A) or absence (B) of 50% FBS. Drug concentration in the dialyzate was assessed by measuring the absorbance at 360 nm. A control experiment was set up concurrently in which the same amount of irinotecan was dissolved in DMSO and dialyzed for comparison. NPs exhibited a sustained release pattern as compared to the free drug. | ||
3.4. In vitro cytotoxicity studies in CT-26 and BxPC-3 cells
The cytotoxicity of NPs was assessed in CT-26 and BxPC-3 cells in vitro. Cells were exposed to 0.1–100 μM free irinotecan or the NPs at equivalent drug concentrations for 72 h (Fig. 3). Blank NPs exhibited no significant toxicity to cells. A dose-dependent reduction in cell viability was observed in all drug treatments (Irinotecan, P-NP-Ir, and T-NP-Ir) with no significant differences between them at the studied concentrations for both cell lines. All drug treatments induced significant cytotoxicity compared to blank NPs (P-NP and T-NP) at the concentration above 10 μM (p < 0.001). BxPC-3 cells were more susceptible to drug treatments than CT-26 cells as less than 50% cell viability was measured at the concentrations >10 μM.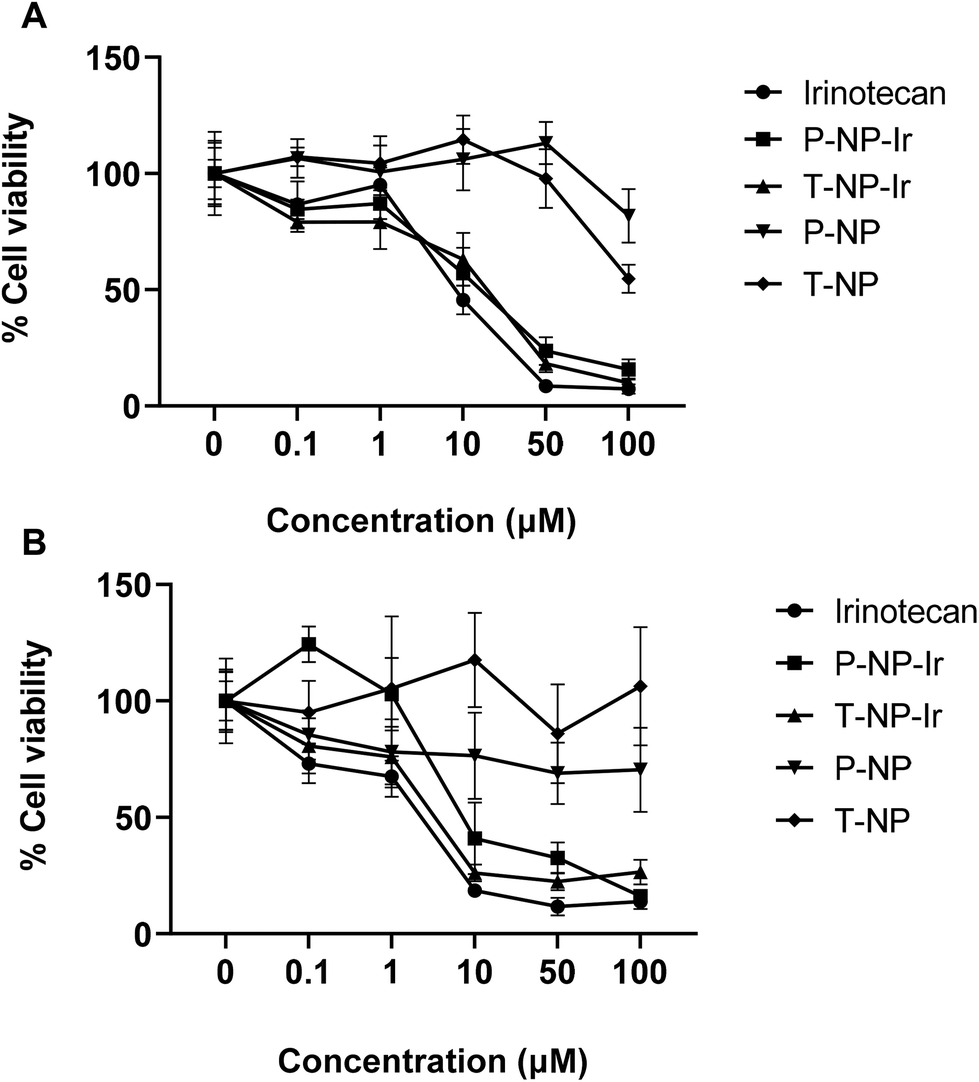 | ||
| Fig. 3 Cytotoxicity of irinotecan-loaded NPs in cancer cells in vitro. (A) Murine colon cancer CT-26 cells and (B) human primary pancreatic adenocarcinoma BxPC-3 cells were incubated with the free irinotecan (Ir), Ir-NPs (T-NP-Ir and P-NP-Ir) or the blank formulations (T-NP and P-NP) at increasing drug concentrations (0.1–100 μM) for 72 h. The viability was assessed by MTT assay and expressed as percentage cell viability from untreated cells. Values are expressed as mean ± SD (n = 5). | ||
3.5. In vivo tumor uptake and organ biodistribution studies after intravenous administration
Whole body imaging of DiR-labelled & Pluronic F-127 coated NPs (P-NP-DiR) in CT-26 tumor confirmed tumor uptake with increasing signals detected over 24 h following intravenous administration (Fig. 4). Highest uptake was observed in liver and spleen.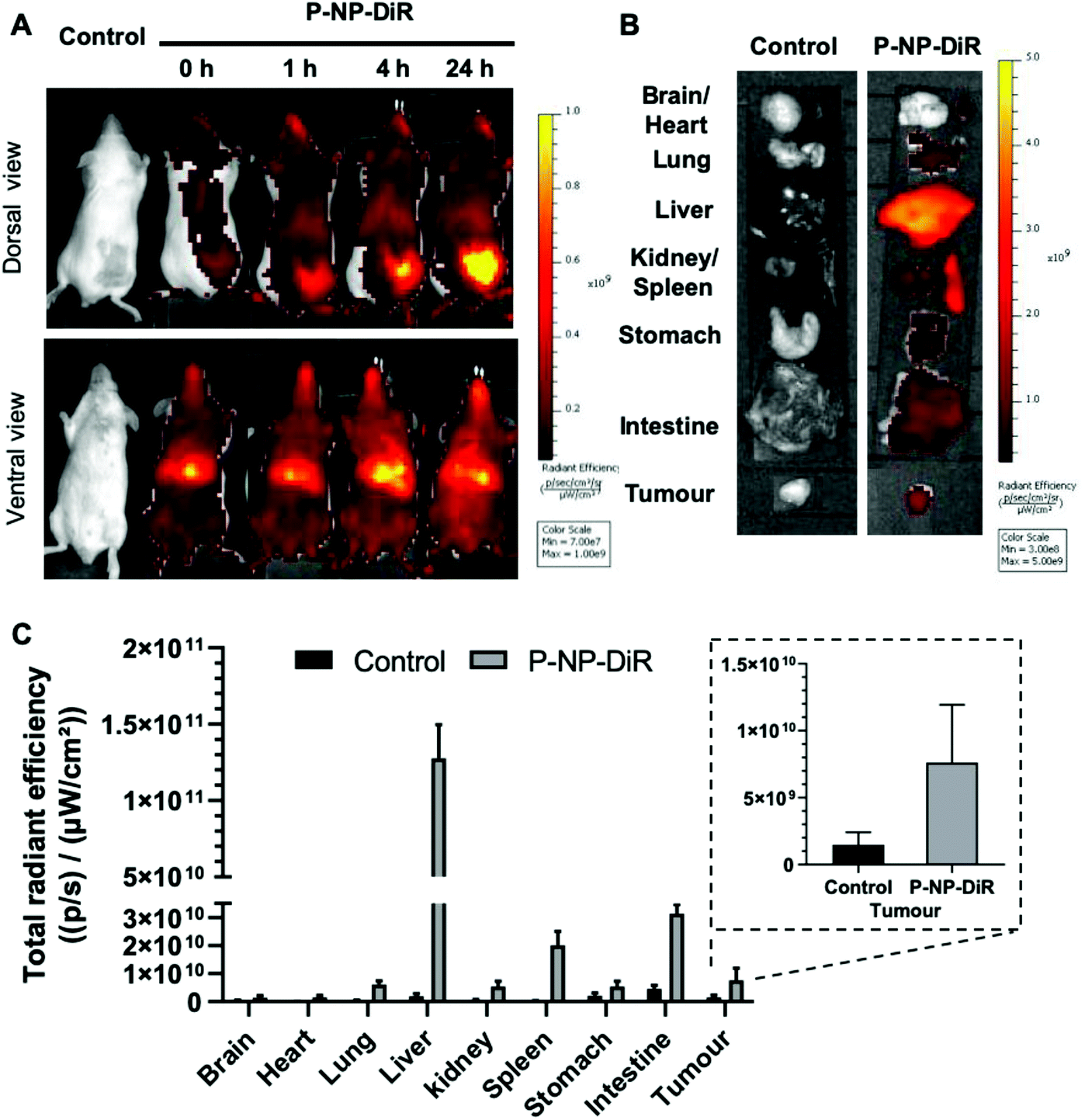 | ||
| Fig. 4 In vivo and ex vivo organ biodistribution of DiR-labelled P-NP (P-NP-DiR) in CT-26 tumor-bearing Balb/c mice after intravenous administration. Mice were transplanted subcutaneously with CT-26 tumors at the right flank. When tumors reached ∼70–80 mm3, mice were i.v. injected with P-NP-DiR in 200 ul PBS. (A) Representative whole-body in vivo images were obtained at 0, 1, 4, and 24 h post-injection. (B) Representative ex vivo images of excised organs at 24 h post-injection. (C) Ex vivo quantification of fluorescence signals per organ at 24 h. Zoom-in of tumor uptake is shown in the insert. All images were obtained by IVIS Lumina III (λex: 740; λem: 790 nm). Data were analyzed by Living Image® 4.3.1 Service Pack 2 software. | ||
3.6. Tumor growth delay studies in CT-26 tumor-bearing Balb/c mice
The anti-cancer potential of NPs loaded with irinotecan was determined in the CT-26 tumor model. Mice were given 4 doses of irinotecan, irinotecan loaded NPs (T-NP-Ir or P-NP-Ir) at 30 mg irinotecan per kg or the P-NP (matched NP amount), on day 7, 11, 14 and 17 post tumor inoculation. The dose of irinotecan at 30 mg kg−1 is used in the clinic for cancer patients. As shown in Fig. 5A, P-NP-Ir therapy resulted in significant tumor growth delay compared to PBS, on day 22 (p < 0.01). No significant differences between the other therapy groups (irinotecan, P-NP, and T-NP-Ir) and the PBS group were observed throughout the studied period. No obvious toxicity was observed in all mice. The whole-body weight changes during the therapy study and the weight of vital organs measured at sacrifice between different treatments groups were insignificant (Fig. 5B and C). In line with further histological analysis, no obvious histological changes were observed in the lung, kidney, liver, heart, and spleen in treatment groups compared to the PBS control (Fig. S4†). The overall results indicated the biocompatibility of the NPs in vivo under the studied conditions.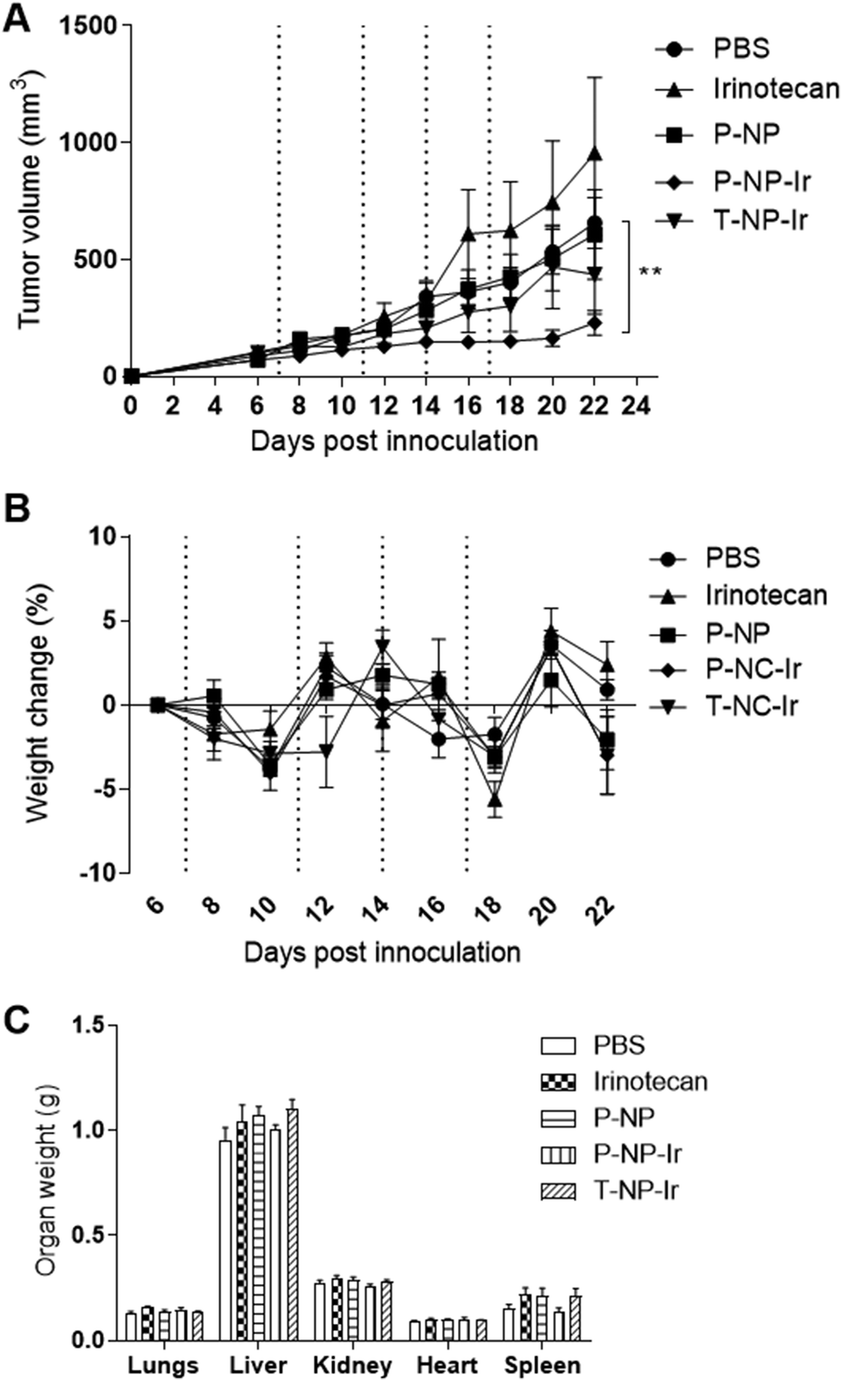 | ||
| Fig. 5 Evaluation of in vivo anti-tumor effect of irinotecan NPs in CT-26 tumor-bearing Balb/c mice. (A) Tumor growth delay curves (B) percentage weight change and (C) organ weight at the sacrifice. Mice were transplanted subcutaneously with 1 × 106 CT-26 cells at the right flanks. When tumors reached ∼70–80 mm3, mice were randomly divided into five groups: PBS (control), 30 mg kg−1 Ir (free drug), P-NP (empty NP) matching excipients weight in P-NP-Ir formulation, P-NP-Ir or T-NP-Ir at Ir dose of 30 mg kg−1. Mice were intravenously administered with a total number of 4 injections of the corresponding treatment on days 6, 11, 14 and 17 post-tumor inoculation (vertical dashed lines). Mice were weighed and tumors were measured three times a week. Data are presented as mean value ± SEM (n = 8). Tumors volume analysis was performed using Mann Whitney test. **p < 0.01. | ||
3.7. Immunological modulation study
The effect of irinotecan-loaded NPs on the immune system was studied. Clinically, irinotecan has been associated with neutropenia.21 Hematological studies evaluated the total numbers of leukocytes, CD8+ cells and granulocytes. The results showed that the irinotecan dose studied has no detectable effect on total blood leukocyte counts nor analyzed subpopulations (Fig. 6A–C). It has previously been reported that irinotecan can selectively deplete regulatory T cells.22 We observed a slight, but significant, decrease in the frequency on FOXP3+ regulatory T cells in the tumor-draining lymph node in the P-NP-Ir group. This trend was maintained in the T-NP-Ir groups, albeit non significantly. The CD4+ to CD8+ ratio was maintained across all groups suggesting the relative levels of CD4+ cells are comparable and the FOXP3+ cell depletion is selective (Fig. 6D and E).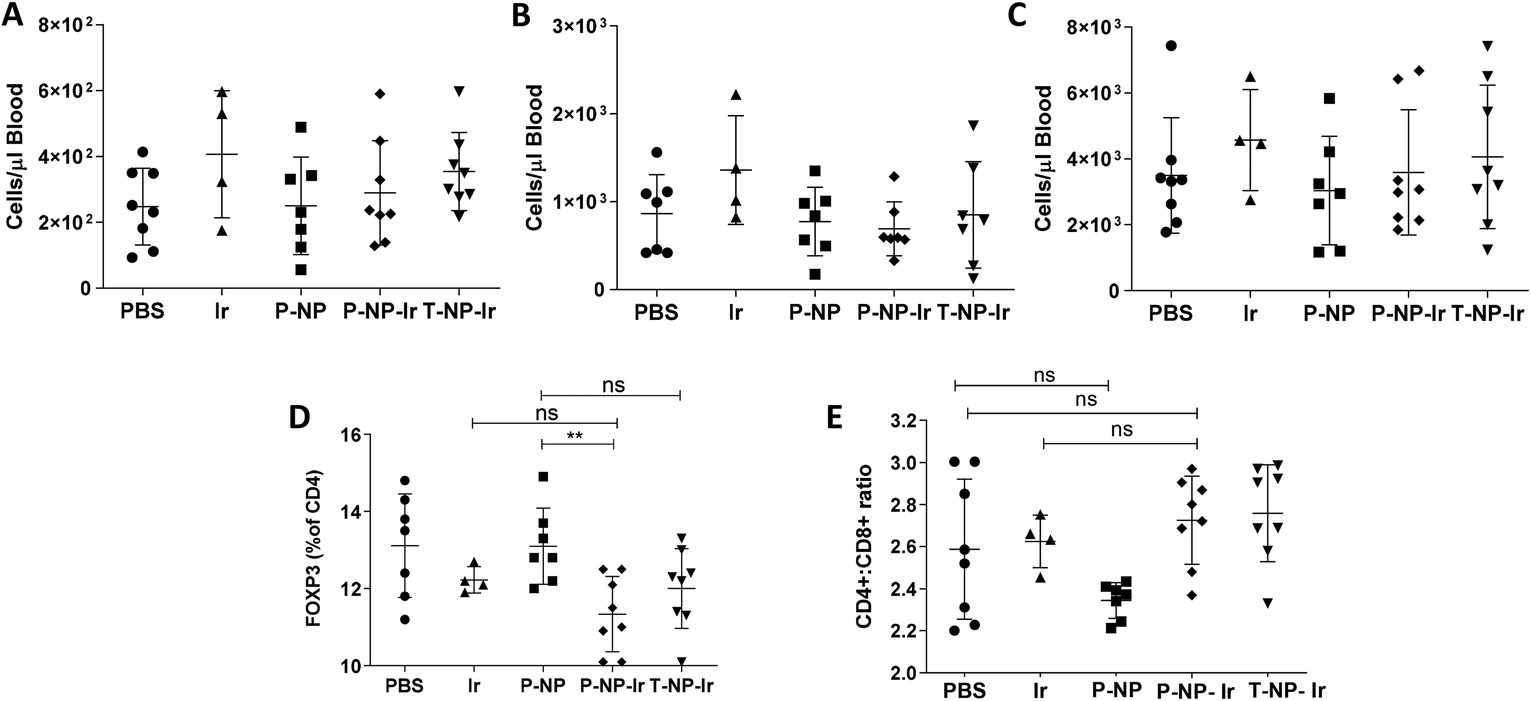 | ||
| Fig. 6 Profiling leukocytes population in whole blood and tumor-draining lymph nodes after therapy studies. Whole blood acquired at the final time point by cardiac puncture was stained using anti CD8-PE for CD8+ T cells (A), anti Ly-6G-FITC for granulocytes (B), or anti CD45-APC for leukocytes (C). Before acquisition on FACs Calibur, a fixed quantity of Precision count counting beads was added to the sample. Absolute cell numbers were calculated from the relative numbers of cells staining positive for the aforementioned marker as a proportion of total sample volume acquired before being back calculated to blood volume. In addition to cardiac blood tumor, cells isolated from draining lymph nodes were stained with anti-CD4 FITC and anti-CD8 PE before being intracellularly stained with anti-FOXP3APC for regulatory T cells. The ratio CD4: CD8+ are shown in (E) while the FOXP3+ cells in lymph node as a percentage of total CD4+ is shown in (D). Data was analysed with Graph pad prism 5 using the Students T-test. **p < 0.01. ns, non-significant. | ||
4. Discussion
mPEG–PLGA based polyester copolymers have been widely used in the clinic as drug carriers. There are approximately 15 FDA-approved PLA/PLGA based drug products, available in the US market used for applications in cancer,23 alcohol withdrawal,24 opioid dependence,25 dental disorder,26 and hormonal replacement therapy27,28 with advantages including reduced dosing frequency, improved patient compliance, enhanced therapeutic actions and reduced drug toxicities.PLGA copolymers have been synthesized using ROP of lactide and glycolide monomers using commercial catalysts predominantly the FDA approved stannous octoate, which raised toxicity concerns especially when used in biomedical applications. Many zinc based complexes such as zinc stearate,29 zinc acetate,30 and zinc acetylacetonate,31 have been utilized for ROP of lactides and lactones as being stereoselective and active catalysts,32–34 non-toxic in nature and of low cost. In this study, mPEG–PLGA copolymers were synthesized using zinc proline, proposed here as a safe biocompatible initiator. The polymerization followed the insertion-coordination mechanism and resulted in linear copolymers substituted with methoxy and hydroxyl end groups. The synthesised copolymers are cheap to make and of desirable solubility profiles, molecular weight, PDI, and in vitro and in vivo biocompatibility profiles.
Low tumor uptake of chemotherapeutics drugs in tumors constitutes one of the obstacles in cancer chemotherapy. One reason is the poor physicochemical properties of drugs such as poor solubility, permeability, rapid degradation causing poor tumor bioavailability. Irinotecan in particular has poor solubility in water, associated with the unpredictable side effects observed in patients.35 Several strategies have been adapted to improve the water solubility of irinotecan and the consequent pharmacokinetics. Zhang et al.36 have reported tuneable self-assembly and polarity of irinotecan by esterification of its hydroxyl group with short and long chain fatty acid. Meng et al. reported irinotecan delivery to pancreatic cancer using lipid-coated mesoporous silica nanoparticles.37 Zhang et al. developed liposomes co-loaded with oxaliplatin and irinotecan for combinational chemotherapy against colorectal cancer.38 Giarra et al. have observed the spontaneous arrangement of a tumor-targeting hyaluronic acid shell on irinotecan loaded PLGA nanoparticles. This led to improved CD44-expressing breast carcinoma cells targeting and cytotoxicity.39 Wang et al. developed a nanoparticle system consisting of PLGA, Pluronic F127, chitosan and hyaluronic acid to encapsulate both irinotecan and doxorubicin to target cancer stem cells.40 The formulation allowed pH and thermal responsive drug release, resulting in enhanced anti-tumor effect on an orthotopic triple-negative breast cancer model. Co-encapsulation of irinotecan and metformin in PLGA NPs has been shown to delay glioma growth.41 Tseng et al. used an electronic spinning technique to formulate PLGA nanofibers containing three chemotherapeutic agents: irinotecan, cisplatin and carmustine for treatment of glioblastoma multiforme. PLGA nanofibers were implanted into the brain surface after craniectomy in rats.42,43 Sustained in vivo release and high concentration of drugs were detected in cerebral parenchyma of rats for more than 8 weeks with no obvious inflammation observed histopathologically. In situ drug release has also been utilized by Ci et al. that a PLGA–PEG–PLGA thermogel containing irinotecan was developed to achieve tumor regression in subcutaneously implanted human colon tumors.44
Encouraged by positive studies in the literature, we have in this study synthesized biologically safe mPEG–PLGA copolymer and investigated its applicability in drug delivery. Irinotecan-loaded mPEG–PLGA nanoparticles were formulated using either Tween® 80 or Pluronic® F-127 as hydrophilic surfactants. Sizes and zeta potential of NP obtained are in the range reported in our previous studies using commercially sourced PLGA copolymers.10,11 Similarly, in vitro release studies demonstrated an initial burst release in the first 4 h due to the discharge of surface-bound irinotecan followed by slow and sustained release by diffusion/erosion of polymeric matrix. This release behaviour is in agreement with release pattern observed for hydrophobic drugs such as curcumin and quercetin previously reported.10,11
Optical imaging demonstrated increasing uptake of the fluorescently labelled P-NP-DiR in CT-26 tumors over time after intravenous injection. The results suggest the proposed nanoformulation of irinotecan has altered the organ biodistribution compared to free drug, in which most of irinotecan (>90% injected dose) is excreted in urine and faeces following intravenous injection.35 Interestingly, we observed that P-NP-Ir was more efficient than T-NP-Ir in delaying tumor growth. Previous studies have reported that Pluronic® F-127 inhibited P-gp efflux pumps and improved the therapeutic efficacy of several chemo drugs.40,45–47 Additional studies are however required to understand the molecular function of Pluronic® F-127 in the formulation. Intestinal toxicity is one of the major adverse effects induced by irinotecan and its metabolites including SN-38 which is 100–1000-fold more toxic than the parent drug. Lower luminal SN-38 concentration was found to be correlated with lower incidence of diarrhea.48 In this study, no diarrhea and weight loss was observed in mice treated with irinotecan, P-NP-Ir or T-NP-Ir. No obvious toxicity was observed in major organs post-mortem.
Although the role of irinotecan in tumor immunity is not fully understood, it has been reported that irinotecan can deplete immunosuppressive regulatory T cells.22 In this study, neither the free drug nor the NP-Ir caused alterations in blood leukocyte populations. Reasons could be that irinotecan dose was not sufficient to deplete the cells populations or the cell numbers had rebounded by the time of sampling (5 days between the final dose and blood sampling). In either case the study proves that the anti-tumor activity was achieved without the neutropenia associated with irinotecan. It is worth to note that a slight depletion of regulatory T cells in tumor-draining lymph nodes was observed in our study in the syngeneic CT-26 tumor model only for P-NP-Ir not the free drug (Fig. 6). This may have provided some beneficial effects by relieving the immune suppression.49 However, it should be noted, while statistically different, the differences in values were slight and may not be biologically relevant. This phenomenon would make for interesting future study, especially when combined with immunotherapy.
5. Conclusion
This is the first report on the synthesis of mPEG–PLGA using solvent-free and stannous octoate catalyst-free bulk ROP in the presence of biocompatible and safe zinc proline complex as a catalyst. The molecular weight and well-controlled PDI of mPEG–PLGA were confirmed by GPC. Polymeric formulations prepared by the nanoprecipitation method, using Tween® 80 or Pluronic®-127 as surfactants, achieved ∼50% encapsulation efficiency of irinotecan and exhibited sustained drug release profiles in the presence or absence of 50% serum. Effective cell killing was achieved in colon and pancreatic cancer cells in vitro. P-NP-Ir showed good tumor accumulation associated with a significant delay in tumor growth delay after multiple intravenous injections in CT26 tumor-bearing mice. No signs of toxicity or irinotecan-associated neutropenia was reported in the study. Overall, this proof-of-concept study demonstrated that the newly synthesised copolymer exhibits good in vivo biocompatibility profiles and that the developed irinotecan polymeric nanoparticles have a great potential as a new formulation for treatment of colon cancer.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
P. S. G. acknowledges the University Grant Commission and Department of Biotechnology New Delhi, and British Council, UK for Newton Bhabha Fellowship (345719944), India for his doctoral research fellowship. P. P. R. acknowledges the Department of Science and Technology, New Delhi, India for the INSPIRE fellowship for her doctoral research. This work was financially supported by the Council of Scientific and Industrial Research, Government of India, New Delhi (CSC-0302 and CSC-0134). F. N. F. is funded by the Malaysian government agency Majlis Amanah Rakyat (MARA). M. A. is a recipient of Foreign Post-Doctoral Fellowships Program FY 2016–2017 funded by Punjab Higher Education Commission, Government of Punjab, Pakistan. H. A. is a recipient of Newton Musharafa Fellowship. S. H. is a recipient of the King's-China Scholarship Council Scholarship. This project has received funding from the Brain Tumour Charity (GN-000398), British Council Newton Fund Institutional Link (337313).References
- C. E. Astete and C. M. Sabliov, J. Biomater. Sci., Polym. Ed., 2006, 17, 247–289 CrossRef CAS.
- B. Xie, Y. Liu, Y. Guo, E. Zhang, C. Pu, H. He, T. Yin and X. Tang, Pharm. Res., 2018, 35, 62 CrossRef.
- D. Luo, K. A. Carter and J. F. Lovell, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 169–188 CAS.
- B. Pelaz, C. Alexiou, R. A. Alvarez-Puebla, F. Alves, A. M. Andrews, S. Ashraf, L. P. Balogh, L. Ballerini, A. Bestetti, C. Brendel, S. Bosi, M. Carril, W. C. W. Chan, C. Chen, X. Chen, X. Chen, Z. Cheng, D. Cui, J. Du, C. Dullin, A. Escudero, N. Feliu, M. Gao, M. George, Y. Gogotsi, A. Grünweller, Z. Gu, N. J. Halas, N. Hampp, R. K. Hartmann, M. C. Hersam, P. Hunziker, J. Jian, X. Jiang, P. Jungebluth, P. Kadhiresan, K. Kataoka, A. Khademhosseini, J. Kopeček, N. A. Kotov, H. F. Krug, D. S. Lee, C.-M. Lehr, K. W. Leong, X.-J. Liang, M. Ling Lim, L. M. Liz-Marzán, X. Ma, P. Macchiarini, H. Meng, H. Möhwald, P. Mulvaney, A. E. Nel, S. Nie, P. Nordlander, T. Okano, J. Oliveira, T. H. Park, R. M. Penner, M. Prato, V. Puntes, V. M. Rotello, A. Samarakoon, R. E. Schaak, Y. Shen, S. Sjöqvist, A. G. Skirtach, M. G. Soliman, M. M. Stevens, H.-W. Sung, B. Z. Tang, R. Tietze, B. N. Udugama, J. S. VanEpps, T. Weil, P. S. Weiss, I. Willner, Y. Wu, L. Yang, Z. Yue, Q. Zhang, Q. Zhang, X.-E. Zhang, Y. Zhao, X. Zhou and W. J. Parak, ACS Nano, 2017, 11, 2313–2381 CrossRef CAS.
- A. Z. Wang, R. Langer and O. C. Farokhzad, Annu. Rev. Med., 2012, 63, 185–198 CrossRef CAS.
- C. E. Soma, C. Dubernet, D. Bentolila, S. Benita and P. Couvreur, Biomaterials, 2000, 21, 1–7 CrossRef CAS.
- R. A. Jain, Biomaterials, 2000, 21, 2475–2490 CrossRef CAS.
- Y. Su, J. Hu, Z. Huang, Y. Huang, B. Peng, N. Xie and H. Liu, Drug Des., Dev. Ther., 2017, 11, 659 CrossRef CAS.
- Q. Guan, S. Sun, X. Li, S. Lv, T. Xu, J. Sun, W. Feng, L. Zhang and Y. Li, J. Mater. Sci.: Mater. Med., 2016, 27, 24 CrossRef.
- R. I. El-Gogary, N. Rubio, J. T.-W. Wang, W. T. Al-Jamal, M. Bourgognon, H. Kafa, M. Naeem, R. Klippstein, V. Abbate and F. Leroux, ACS Nano, 2014, 8, 1384–1401 CrossRef CAS.
- R. Klippstein, J. T. W. Wang, R. I. El-Gogary, J. Bai, F. Mustafa, N. Rubio, S. Bansal, W. T. Al-Jamal and K. T. Al-Jamal, Small, 2015, 11, 4704–4722 CrossRef CAS.
- X. Zhang, D. A. MacDonald, M. F. A. Goosen and K. B. McAuley, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2965–2970 CrossRef CAS.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- H. R. Kricheldorf and D. O. Damrau, Macromol. Chem. Phys., 1997, 198, 1753–1766 CrossRef CAS.
- S. P. Parwe, S. D. Warkad, M. V. Mane, P. S. Shedage and B. Garnaik, Polymer, 2017, 111, 244–251 CrossRef CAS.
- C. Bailly, Pharmacol. Res., 2019, 148, 104398 CrossRef CAS.
- Y. N. Lamb and L. J. Scott, Drugs, 2017, 77, 785–792 CrossRef CAS.
- E. A. Perez, A. Awada, J. O'Shaughnessy, H. S. Rugo, C. Twelves, S.-A. Im, P. Gómez-Pardo, L. S. Schwartzberg, V. Diéras, D. A. Yardley, D. A. Potter, A. Mailliez, A. Moreno-Aspitia, J.-S. Ahn, C. Zhao, U. Hoch, M. Tagliaferri, A. L. Hannah and J. Cortes, Lancet Oncol., 2015, 16, 1556–1568 CrossRef CAS.
- D. Tripathy, S. M. Tolaney, A. D. Seidman, C. K. Anders, N. Ibrahim, H. S. Rugo, C. Twelves, V. Dieras, V. Müller, M. Tagliaferri, A. L. Hannah and J. Cortés, Future Oncol., 2019, 15, 2211–2225 CrossRef CAS.
- Y.-P. Li, Y.-Y. Pei, X.-Y. Zhang, Z.-H. Gu, Z.-H. Zhou, W.-F. Yuan, J.-J. Zhou, J.-H. Zhu and X.-J. Gao, J. Controlled Release, 2001, 71, 203–211 CrossRef CAS.
- H. Hamano, M. Mitsui, Y. Zamami, K. Takechi, T. Nimura, N. Okada, K. Fukushima, M. Imanishi, M. Chuma, Y. Horinouchi, Y. Izawa-Ishizawa, Y. Kirino, T. Nakamura, K. Teraoka, Y. Ikeda, H. Fujino, H. Yanagawa, T. Tamaki and K. Ishizawa, Supportive Care Cancer, 2019, 27, 849–856 CrossRef.
- C. Fernandes, C. W. S. Wanderley, C. M. S. Silva, H. A. Muniz, M. A. Teixeira, N. R. P. Souza, A. G. F. Cândido, R. B. Falcão, M. H. L. P. Souza, P. R. Almeida, L. M. C. Câmara and R. C. P. Lima-Júnior, Eur. J. Pharm. Sci., 2018, 115, 158–166 CrossRef CAS.
- S. Acharya and S. K. Sahoo, Adv. Drug Delivery Rev., 2011, 63, 170–183 CrossRef CAS.
- E. Khodaverdi, F. S. M. Tekie, S. A. Mohajeri, F. Ganji, G. Zohuri and F. Hadizadeh, AAPS PharmSciTech, 2012, 13, 590–600 CrossRef CAS.
- M. Kovaliov, S. Li, E. Korkmaz, D. Cohen-Karni, N. Tomycz, O. B. Ozdoganlar and S. Averick, RSC Adv., 2017, 7, 47904–47912 RSC.
- B. M. Priyadarshini, K. Mitali, T. B. Lu, H. K. Handral, N. Dubey and A. S. Fawzy, Dent. Mater., 2017, 33, 830–846 CrossRef CAS.
- S. Malathi, P. Nandhakumar, V. Pandiyan, T. J. Webster and S. Balasubramanian, Int. J. Nanomed., 2015, 10, 2207–2218 CAS.
- Y. Wang, Q. Wen and S. Choi, Am. Pharm. Rev., 2016, 19, 5–9 Search PubMed.
- A. Kowalski, J. Libiszowski, T. Biela, M. Cypryk, A. Duda and S. Penczek, Macromolecules, 2005, 38, 8170–8176 CrossRef CAS.
- R. R. Gowda and D. Chakraborty, J. Mol. Catal. A: Chem., 2010, 333, 167–172 CrossRef CAS.
- M. Pastusiak, P. Dobrzynski, B. Kaczmarczyk, J. Kasperczyk and A. Smola, Polymer, 2011, 52, 5255–5261 CrossRef CAS.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS.
- C. Kan, J. Hu, Y. Huang, H. Wang and H. Ma, Macromolecules, 2017, 50, 7911–7919 CrossRef CAS.
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS.
- R. H. Mathijssen, R. J. Van Alphen, J. Verweij, W. J. Loos, K. Nooter, G. Stoter and A. Sparreboom, Clin. Cancer Res., 2001, 7, 2182–2194 CAS.
- C. Zhang, S. Jin, X. Xue, T. Zhang, Y. Jiang, P. C. Wang and X.-J. Liang, J. Mater. Chem. B, 2016, 4, 3286–3291 RSC.
- X. Liu, A. Situ, Y. Kang, K. R. Villabroza, Y. Liao, C. H. Chang, T. Donahue, A. E. Nel and H. Meng, ACS Nano, 2016, 10, 2702–2715 CrossRef CAS.
- B. Zhang, Y. Song, T. Wang, S. Yang, J. Zhang, Y. Liu, N. Zhang and S. Garg, Int. J. Nanomed., 2017, 12, 2871 CrossRef CAS.
- S. Giarra, C. Serri, L. Russo, S. Zeppetelli, G. De Rosa, A. Borzacchiello, M. Biondi, L. Ambrosio and L. Mayol, Carbohydr. Polym., 2016, 140, 400–407 CrossRef CAS.
- H. Wang, P. Agarwal, S. Zhao, R. X. Xu, J. Yu, X. Lu and X. He, Biomaterials, 2015, 72, 74–89 CrossRef CAS.
- A. Taghizadehghalehjoughi, A. Hacimuftuoglu, M. Cetin, A. B. Ugur, B. Galateanu, Y. Mezhuev, U. Okkay, N. Taspinar, M. Taspinar, A. Uyanik, B. Gundogdu, M. Mohammadzadeh, K. A. Nalci, P. Stivaktakis, A. Tsatsakis, T. W. Jung, J. H. Jeong and A. A. El-Aty, Nanomedicine, 2018, 13, 1595–1606 CrossRef.
- Y.-Y. Tseng, Y.-C. Wang, C.-H. Su, T.-C. Yang, T.-M. Chang, Y.-C. Kau and S.-J. Liu, Colloids Surf., B, 2015, 134, 254–261 CrossRef CAS.
- Y. Y. Tseng, T. C. Yang, Y. C. Wang, W. H. Lee, T. M. Chang, Y. C. Kau and S. J. Liu, Int. J. Nanomed., 2017, 12, 1265–1276 CrossRef CAS.
- T. Ci, L. Chen, L. Yu and J. Ding, Sci. Rep., 2014, 4, 5473 CrossRef CAS.
- Z. Wei, S. Yuan, J. Hao and X. Fang, Eur. J. Pharm. Biopharm., 2013, 83, 266–274 CrossRef CAS.
- N. Shaik, G. Pan and W. F. Elmquist, J. Pharm. Sci., 2008, 97, 5421–5433 CrossRef CAS.
- X. Cheng, X. Lv, J. Xu, Y. Zheng, X. Wang and R. Tang, Eur. J. Pharm. Sci., 2020, 146, 105275 CrossRef CAS.
- R. Sun, L. Zhu, L. Li, W. Song, X. Gong, X. Qi, Y. Wang, R. Ghose, S. Gao, M. Hu and Z. Liu, Toxicol. Appl. Pharmacol., 2020, 398, 115032 CrossRef CAS.
- Y. Togashi, K. Shitara and H. Nishikawa, Nat. Rev. Clin. Oncol., 2019, 16, 356–371 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01421d |
| This journal is © The Royal Society of Chemistry 2021 |