
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Smart nanomicelles with bacterial infection-responsive disassembly for selective antimicrobial applications†
Dicky
Pranantyo
 ab,
En-Tang
Kang
*b and
Mary B.
Chan-Park
*acd
ab,
En-Tang
Kang
*b and
Mary B.
Chan-Park
*acd
aCentre of Antimicrobial Bioengineering, School of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore 637459. E-mail: mbechan@ntu.edu.sg
bDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Kent Ridge, Singapore 117585. E-mail: cheket@nus.edu.sg
cLee Kong Chian School of Medicine, Nanyang Technological University, 59 Nanyang Drive, Singapore 636921
dSchool of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371
First published on 7th January 2021
Abstract
New generation antimicrobial agents are expected to exhibit non-metabolic killing mechanisms, high killing potency and biocompatibility. We synthesized a cationic chitosan derivative and an anionic chitosan derivative – specifically an α-poly(L)lysine side-grafted chitosan (CS-PLL) and an anionic citraconyl anhydride (CA) modified polylysine side graft for chitosan (CS-PLL-CA). The β-carboxylic amide of CS-PLL-CA is pH-labile and self-cleavable under pH 6 or below. When we mixed the cationic (CS-PLL) and anionic (CS-PLL-CA) peptidosaccharide copolymers, they self-assembled, due to electrostatic charge interactions, into nanomicelles (NMs) with the oppositely charged peptides in the core and the chitosan polysaccharide arms on the shell. The NMs exhibited high hemo- and cytocompatibility (nontoxic) at physiological pH of 7.4, due to the chitosan protection on the shell and charge neutralization on the core. Upon reaching the bacterial infection site, the chitosan shell interacted and accumulated around the bacteria. The bacterial infection sites in the body usually show localized acidity as a result of the combined actions of bacterial metabolism and host immune response, and the pH can decrease to as low as 5.5. At this low pH, the β-carboxylic amide bond of the anionic polypeptide gradually hydrolyzed to expose the initial cationic amine moieties, causing the NMs to ‘decompose’ into individual CS-PLL and ‘spill’ the cationic molecules which then disrupted and killed the bacteria. This ‘smart’ bacteria-recognizing chitosan-decorated nanosystem opens the pathway to explore other anionic and cationic and biocompatible polymers for ‘stealth’ delivery of antimicrobial polypeptide, and ‘on-demand’ recovery of the cationic parts to kill bacteria at infection sites.
1. Introduction
Pathogenic infections cause various life-threatening diseases and have become a leading cause of global morbidity and mortality rates in the past three decades.1 This threat is aggravated by the rapidly growing emergence of resistant bacterial clonal lineages, such as in the ESKAPE pathogens, which are recalcitrant to the current antibiotic treatment. In the United States, more than 2.8 million infections are caused by antibiotic-resistant bacteria every year, leading to the death of more than 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people.2 Excessive usage of antibiotics in human medicine and livestock farming over the past seven decades, and transmission by horizontal gene transfer, have contributed to the selective expansion and mutagenic development of multidrug-resistant bacteria.3–5 Worldwide extensive efforts to develop new antibiotics are lagging behind the rapid spread of resistant pathogens, as indicated by no new class of antibiotic being approved to treat Gram-negative bacteria in over five decades.6 In general, the mechanisms for resistance involve intrinsic or metabolic functions in bacteria, such as enzymatic scission or inactivation of antibiotic structure, modification of antibiotic target binding sites, efflux pumps, and biofilm formation. Therefore, there is motivation to develop alternative remedies which bypass the resistance mechanisms and are able to treat bacterial infections. Cationic antimicrobial agents, such as antimicrobial peptides (AMPs), are a superior treatment for bacterial infections because they can permeabilize the cytoplasmic membrane without triggering bacterial resistance reactions.7 Due to this direct killing mode, cationic molecules and AMPs are among the most widely studied anti-infective agents,8–11 and more than 3000 AMPs have been discovered and listed in the Antimicrobial Peptide Database.12 However, cationic molecules such as AMPs often show biocompatibility issues in terms of lack of selectivity, leading to acute toxicity and hemolytic activity.13 Therefore, it is of high interest to develop a system to render cationic AMPs nontoxic in the host body, which can automatically engage their killing mode in the presence of bacteria.
000 people.2 Excessive usage of antibiotics in human medicine and livestock farming over the past seven decades, and transmission by horizontal gene transfer, have contributed to the selective expansion and mutagenic development of multidrug-resistant bacteria.3–5 Worldwide extensive efforts to develop new antibiotics are lagging behind the rapid spread of resistant pathogens, as indicated by no new class of antibiotic being approved to treat Gram-negative bacteria in over five decades.6 In general, the mechanisms for resistance involve intrinsic or metabolic functions in bacteria, such as enzymatic scission or inactivation of antibiotic structure, modification of antibiotic target binding sites, efflux pumps, and biofilm formation. Therefore, there is motivation to develop alternative remedies which bypass the resistance mechanisms and are able to treat bacterial infections. Cationic antimicrobial agents, such as antimicrobial peptides (AMPs), are a superior treatment for bacterial infections because they can permeabilize the cytoplasmic membrane without triggering bacterial resistance reactions.7 Due to this direct killing mode, cationic molecules and AMPs are among the most widely studied anti-infective agents,8–11 and more than 3000 AMPs have been discovered and listed in the Antimicrobial Peptide Database.12 However, cationic molecules such as AMPs often show biocompatibility issues in terms of lack of selectivity, leading to acute toxicity and hemolytic activity.13 Therefore, it is of high interest to develop a system to render cationic AMPs nontoxic in the host body, which can automatically engage their killing mode in the presence of bacteria.
Such systems with auto-trigger mechanisms have been realized, largely by employing stimuli-responsive materials, and in particular, pH is the most commonly exploited stimulus in the biological environment. Due to the combined actions of microbial metabolism and host immune response, bacterial infection sites usually show localized acidity in which the pH can reach as low as 5.5,14 and in the case of S. aureus infection the bacteria colonies even grow at pH 4.2.15 This local acidosis is a characteristic feature of the inflammatory locus, and it is attributed to the local increase of lactic-acid production by the anaerobic, glycolytic activity of infiltrated neutrophils, and to the presence of short-chain, fatty acid by-products of bacterial metabolism.16 For example, subcutaneous abscesses are localized skin infections that are characterized by a build-up of acidic pus of pH 6.0–6.6, which contains pathogenic bacteria such as S. aureus.17,18 Another example is tooth cavities or dental caries caused by the build-up of oral biofilm on the enamel and gum surfaces, which produce acids as bacteria digest sugars found in the human diet. This local acidosis of the infection sites can be exploited by designing an antimicrobial drug delivery systems (DDS) with pH-responsive activation, in which the system can render the drug inactive during circulation in the body under physiological conditions, but activate the drug killing effect under the acidic conditions at the infection sites. With this approach, antimicrobial agents involving highly cytotoxic AMPs or cationic polymers can be administered to selectively target bacteria at infection sites such as subcutaneous abscesses and tooth cavities.
Extensive work has been carried out on the delivery of antibiotic drugs, quaternary ammonium groups, phosphonium groups, polycations, and AMPs via biodegradable polymeric nanosystems, which incorporate labile chemical linkages such as carbonate, ester, amide, and phosphoester bonds.19 On the other hand, many DDS with pH-activatable polymers have been developed for anticancer treatment,20–24 but they are seldom used in antimicrobial therapy. For example, polylysine protected with acid-labile β-carboxylic amides (PLL/amide) could undergo hydrolysis in cancer cells’ acidic lysosomes to expose primary amines, which could then traverse the anticancer drug camptothecin (CPT) into the nucleus.25 In order to achieve extracellular delivery, this PLL/amide-CPT was conjugated with folic acid to target the folate-receptor overexpressing cancer cells. For antimicrobial applications, Liu et al. designed a charge-convertible polymer with zwitterionic groups that can switch to cationic quaternary ammonium ions under low pH.26 In a more recent report, cationic AMP-coated gold nanoclusters (Au NCs) were protected with acid-labile β-carboxylic amides to form anionic NCs.27 In both cases, the zwitterionic polymer and anionic NCs showed low toxicity, and they could readily convert to cationic charge under low pH and kill bacteria. However, the zwitterionic polymer and anionic NCs are generally bacterial-repulsive and they do not target or accumulate on bacteria surfaces, so they might escape prematurely from the infection sites in their intact non-bactericidal mode, because the hydrolysis of β-carboxylic amides requires time to progress.
In this work, we report the application of a pH-switchable antimicrobial system to develop nontoxic nanomicelles (NMs) that can switch to bactericidal mode under local acidic conditions such as at the infection sites. The NMs are prepared via electrostatic interaction between cationic and anionic polymer conjugates. The cationic conjugate is α-poly(L)lysine side-grafted onto chitosan backbone (CS-PLL), while the anionic conjugate is CS-PLL with the amine moieties functionalized by citraconyl amide (CS-PLL-CA). Thus, the cationic and anionic segments form the NMs core, and the chitosan segments form the NMs corona. The NMs exhibit ultralow hemo- and cytotoxicity due to the charge neutralization in the core, and the protection of biocompatible chitosan on the corona. The chitosan corona is slightly protonated and therefore is expected to bind with the bacterial surface, which promote the NMs accumulation at the local infection sites. Further at pH 6 and below, the citraconyl amide functionalities can undergo self-hydrolysis and re-expose the amine moieties, causing the NMs to lose electrostatic stability and burst, spilling out their cationic segments core. The liberated cationic CS-PLL then interact and kill Gram-positive and Gram-negative bacteria via membrane disruption. The electrostatic NMs thus represent a ‘smart’ delivery system that can render the cationic polypeptide in a nontoxic mode at physiological pH, and auto-switch to bactericidal mode at the infection sites.
2. Experimental section
2.1. Materials
Methanesulfonic acid (MSA, 99%), 6-bromohexanoyl chloride (97%), sodium azide (99.5%), dibenzocyclooctyne-amine (DBCO-NH2), Nε-carbobenzoxy-L-lysine (CbzLys, 99%), triphosgene (98%), trifluoroacetic acid (99%), hydrobromic acid (33 wt% in acetic acid), citraconic anhydride (CA, 98%), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 98%), 3,3′-dipropylthiadicarbocyanine iodide (DiSC3(5), 98%), N-phenyl-1-naphthylamine (NPN, 98%), fluorescamine (98%), calcein, and Triton X-100 were purchased from Sigma-Aldrich Corp. (St Louis, MO). 1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC, 99%) was purchased from Avanti Polar Lipids Inc. (Alabaster, AL). LIVE/DEAD BacLight bacterial viability kit (containing SYTO 9 and propidium iodide) and CyQUANT LDH cytotoxicity assay kit were purchased from Thermo Fisher Sci. Inc. (Waltham, MA). Dichloromethane (DCM), dimethylformamide (DMF), dimethyl sulfoxide (DMSO), tetrahydrofuran, diethyl ether, hexane, and acetone were of analytical grade, whereas phosphate buffered saline (PBS) was of ultrapure grade. Escherichia coli (ATCC 25922), Pseudomonas aeruginosa (ATCC 15692), Staphylococcus aureus (ATCC 25923), Staphylococcus epidermidis (ATCC 12228), methicillin-resistant S. aureus (MRSA, ATCC BAA-44), and standard 3T3 mouse fibroblast cells were purchased from American Type Culture Collection (ATCC, Manassas, VA). Fresh whole blood of Wistar Hannover rats was purchased from InVivos Pte. Ltd (Singapore).2.2. Synthesis of azido-functionalized chitosan
Chitosan was dialyzed against deionized water (molecular weight cutoff, MWCO 3.5 kDa) for 3 days and lyophilized to obtain chitosan with MW > 3.5 kDa. After presorting, chitosan (5 g, 31 mmol glucosamine units) was dissolved in 200 mL of MSA. 6-Bromohexanoyl chloride (800 mg, 3.75 mmol) was dissolved in 25 mL of MSA and was added dropwise to the CS solution under vigorous stirring. The reaction was allowed to proceed overnight at room temperature. The solution was precipitated and washed in excess acetone, dialyzed against deionized water for 3 days (MWCO 1 kDa), and lyophilized to obtain alkyl bromide-functionalized chitosan (CS-Br, brownish fluffy solid, yield 74%).CS-Br (3.9 g, ∼24 mmol glucosamine units, ∼2 mmol alkyl bromide groups) was dissolved in 100 mL of DMSO, followed by the addition of sodium azide (390 mg, 6 mmol). The suspension was stirred vigorously and heated to 100 °C under a reflux condenser overnight. After being cooled to room temperature, 50 mL of deionized water was added. The solution was dialyzed against deionized water for 3 days (MWCO 1 kDa) and lyophilized to obtain azido-functionalized chitosan (CS-N3, brownish fluffy solid, yield 79%).
2.3. Synthesis of cyclooctyne-terminated Nε-carbobenzoxy-L-lysine polymer
N ε-Carbobenzoxy-L-lysine N-carboxyanhydride (CbzLys-NCA, 306.32 g mol−1) monomer was prepared according to the previous method.28 Subsequently, ring-opening polymerization of the CbzLys-NCA was carried out (Fig. S1, ESI†). DBCO-NH2 (55.3 mg, 0.2 mmol) was dissolved in 5 mL of DMF and degassed with argon for 15 min. CbzLys-NCA (2.5 g, 8 mmol) was dissolved in 20 mL of DMF, degassed with argon for 15 min, and transferred via syringe to the DBCO-NH2 solution under stirring. The mixture was further degassed with argon for 30 min and sealed tightly. The reaction was allowed to proceed at room temperature for 24 h. The mixture was reprecipitated twice in 200 mL of diethyl ether and dried under reduced pressure to obtain Nε-carbobenzoxy-L-lysine polymer (CbzPLL), a pale yellowish solid, yield 91%.CbzPLL (2 g, ∼7.7 mmol of lysine repeat unit) was dissolved in 30 mL of trifluoroacetic acid. Hydrobromic acid (4-fold mol of lysine repeat unit) was added to the solution. The deprotection was allowed to proceed under stirring at room temperature for 3 h. The solution was precipitated in diethyl ether, dialyzed against deionized water for 3 days (MWCO 1 kDa, and lyophilized to obtain cyclooctyne-terminated α-poly(L)lysine (PLL, white cotton-like solid, yield 89%).
2.4. Synthesis of citraconyl-capped chitosan-polylysine copolymer and electrostatic nanomicelles
Click conjugation of CS-N3 and PLL was carried out via strain-promoted azide–alkyne cycloaddition (Fig. S2†). CS-N3 (1.05 g, ∼0.3 mmol) and PLL (1.6 g, ∼0.3 mmol) were dissolved separately in 15 mL of deionized water. The two solutions were quickly mixed and the click reaction was allowed to proceed under vigorous stirring at room temperature for 24 h. The mixture was dialyzed against deionized water for 3 days and lyophilized to obtain chitosan-polylysine copolymer (CS-PLL, pale brownish solid, yield 57%).CS-PLL (1.05 g, ∼0.12 mmol, ∼4.8 mmol of primary amine) was dissolved in 20 mL of deionized water and adjusted to pH 8.5 using 3 M NaOH solution. CA (1.3 mL, 14.4 mmol) was gradually added dropwise to the CS-PLL solution under vigorous stirring. During the addition, the reaction mixture was maintained at pH 8.5 using 3 M NaOH solution. The reaction was allowed to proceed overnight at room temperature. The mixture was then dialyzed against deionized water at pH 8.5 for 3 days to obtain an aqueous solution of citraconyl-capped chitosan-polylysine copolymer (CS-PLL-CA), which was stored at 4 °C for further use.
The CS-PLL/CA nanomicelles (NMs) were formed by electrostatic self-assembly. CS-PLL (532 mg, 0.06 mmol) was dissolved in deionized water and adjusted to pH 8.5 using 3 M NaOH. This CS-PLL solution and the CS-PLL-CA solution (containing 806 mg, 0.06 mmol of CS-PLL-CA) was gradually mixed dropwise in a flask under stirring. The mixture was maintained at pH 8.5 during self-assembly. The mixture was then dialyzed against deionized water at pH 8.5 for 3 days to obtain an aqueous solution of CS-PLL/CA NMs, which was stored at 4 °C for further use.
2.5. Characterization
Chemical structures of the compounds in deuterated solvents were characterized by 1H NMR spectroscopy on a Bruker ARX 500 MHz spectrometer. Dynamic light scattering and zeta potential of the compounds in aqueous solution at pH 8.5 and 22 °C was measured on a Malvern ZEN3600 zetasizer. Fluorescence analyses were performed on a Shimadzu RF-6000 spectrofluorophotometer. Transmission electron microscopy (TEM) images were obtained on a JEOL JEM-2100F field emission TEM.2.6. Hydrolysis study of citraconic amide of the CS-PLL-CA copolymer
The kinetics of citraconic amide hydrolysis was monitored using the fluorescamine protocol. An aqueous solution of CS-PLL-CA copolymer (5 mg mL−1, 50 μL) was added into 450 μL of citric acid-sodium phosphate buffer at pH 7.4, 6.0, or 5.0. The solutions were agitated in an orbital shaker at 37 °C for 24 h. At each predetermined time interval, 10 μL aliquots were taken and diluted into 1 mL of borate buffer (0.1 M, pH 9.3) to quench the hydrolysis. This mixture was incubated with 10 μL of florescamine solution in N,N-dimethylformamide (2 mg mL−1) at room temperature for 10 min. Its fluorescence intensity at 470 nm was measured under an excitation wavelength of 375 nm. The positive control (100% of exposed amine) was determined by applying this fluorescamine method on the solution after incubation of the CS-PLL-CA copolymer in 0.01 M HCl overnight. The negative control (0% of exposed amine) was determined from the fluorescamine in blank buffer solution.2.7. Antimicrobial assays of the CS-PLL/CA NMs
Bacteria were cultured to a mid-log phase in the respective growth media at 37 °C according to ATCC protocols, and diluted to 2 × 105 colony forming units (CFU) mL−1 in Mueller-Hinton broth (MHB). Stock solutions of sample compounds were incubated in PBS (pH 7.4 and 5.0) at a concentration of 512 μg mL−1 for 24 h. They were serially diluted 2-fold in PBS, and 100 μL of each dilution was placed in a 96-well plate (Greiner Bio-one, Germany). Then, 100 μL of bacterial suspension was added to each compound solution. The plate was incubated at 37 °C overnight and observed by the naked eye. Bacterial growth made the suspension appear cloudy, while the suspension with no bacterial growth remained clear. The lowest concentration of the compound that inhibited the growth of bacteria was recorded, the minimum inhibitory concentration (MIC). The absorbance at 600 nm wavelength was measured as optical density on a Multiskan GO 1510 UV microplate spectrophotometer (Thermo Scientific, Waltham MA). Subsequently, 100 μL of the suspension in the wells with no visible growth was spread on a Mueller-Hinton agar and incubated at 37 °C overnight to observe the viability of the bacteria. The lowest concentration of the compound that killed 99.9% of the initially inoculated bacteria was recorded, the minimum bactericidal concentration (MBC).For the live–dead fluorescence and time-kill bactericidal assays, 100 μL of compound solutions at MBC were placed in a 96-well plate. Then, 100 μL of bacterial suspension (2 × 105 cfu mL−1) in MHB was added to each compound solution, and the plate was incubated. At each predetermined time, 100 μL of the suspension in each well was spread on a Mueller-Hinton agar and incubated at 37 °C overnight to observe the viability of the bacteria. In addition, after the plate was incubated for 4 h, 20 μL of the suspension in each well was spread on a glass slide and stained with LIVE/DEAD BacLight solution for 15 min. The live (green) and dead (red) bacterial cells on the glass slide were observed on a Nikon ECLIPSE Ti-U fluorescence microscope (Japan) under a green filter (excitation/emission of 470 nm/525 nm) and a red filter (excitation/emission of 535 nm/645 nm).
2.8. Bacterial membrane perturbation and liposome leakage assays
The DiSC3(5) released assay and NPN uptake assay were performed as described previously with some modifications. Bacteria of the mid-log growth-phase were resuspended at 2 × 106 CFU mL−1 in PBS containing 0.4 μM of DiSC3(5) or 10 μM of NPN. Then, 100 μL of bacterial suspension was added to each well of a black 96-well plate (Greiner Bio-One, Germany) and incubated at 37 °C for 1 h until the fluorescence level became stable. Subsequently, 100 μL of sample solution in PBS at various concentrations was added to each bacterial suspension and the plate was incubated at 37 °C for 1 h. The fluorescence intensities of DiSC3(5) (λex/λem at 610/710 nm) and NPN (λex/λem at 300/410 nm) in each well were measured on a Tecan Infinite M200 fluorescence microplate reader (Männedorf, Switzerland).A liposome leakage assay was performed to study the membrane destabilization. Briefly, 10 mg of DSPC was dissolved in 0.5 mL of chloroform, slowly evaporated in a rotary evaporator, and dried under reduced pressure to form a thin lipid layer. The lipid layer was hydrated with 2 mL of doubly-distilled water containing 40 mM of calcein, dispersed in an ultrasonic bath for 1 h, and dialyzed against doubly-distilled water (MWCO 1 kDa) for 3 days. The lipid concentration was diluted to 5 μM with PBS, and 100 μL of this liposome suspension was placed in each well of a black 96-well plate. Then, 100 μL of the serially-diluted sample solutions in PBS, was added to each liposome suspension. Blank PBS and 0.1%-v of Triton X-100 in PBS were added to the liposome suspension as negative and positive controls, respectively. The plate was incubated at 25 °C for 1 h and the fluorescence intensity was measured at λex/λem of 485/515 nm. The percentage of liposome damage was calculated as [(FS − FN)/(FP − FN)] × 100%, where FS, FN, and FP are the fluorescence intensity of the calcein leakage by the incubated sample, negative control, and positive control, respectively.
2.9. Determination of hemolytic activity
Sample compounds were serially diluted in 100 μL of PBS and placed in a 96-well plate. Then, 100 mL of fresh whole rat blood dispersion in PBS (8%-v) was added to each compound solution. The blood suspension was added to blank PBS as the negative control, and to PBS containing 0.1%-v of Triton X-100 as the positive control. The plate was incubated at 37 °C for 1 h. Each mixture was centrifuged at 1500 rpm for 10 min, and the optical absorbance of the supernatant was measured at 560 nm wavelength on a UV microplate spectrophotometer. The percentage of hemoglobin release was calculated as [(AS − AN)/(AP − AN)] × 100%, where AS, AN, and AP are the optical absorbance of the supernatant from the incubated sample, negative control, and positive control, respectively.2.10. Cytotoxicity assay
A metabolic MTT assay was carried out to determine the cytotoxicity of the sample compounds. 3T3 fibroblast cells were cultured and resuspended in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum, 1 mM L-glutamine, and 100 IU mL−1 penicillin. Then, 100 μL of the suspension containing 5000 cells was placed in each well of a 96-well plate. The plate was incubated in a humidified atmosphere of 95% air and 5% CO2 at 37 °C for 24 h. Sample compounds were incubated in PBS (pH 7.4 and 5.0) at a concentration of 1024 μg mL−1 for 24 h and serially diluted 2-fold in DMEM. The medium in each well containing the cells was replaced with 100 μL of the sample solutions, and the plate was incubated at 37 °C for 24 h. A nontoxic control experiment was carried out using the supplemented DMEM without any compound. The medium in each well was then replaced with 100 μL of MTT solution (0.5 mg mL−1 in supplemented DMEM). After additional incubation at 37 °C for 4 h, the supernatant was aspirated and 100 μL of dimethyl sulfoxide was added to dissolve the internalized formazan crystal. After 15 min, the optical absorbance at 600 nm wavelength was measured using a UV microplate spectrophotometer, and expressed as a percentage relative to the absorbance of the nontoxic control.2.11. Lactate dehydrogenase (LDH) release study
The CyQUANT LDH cytotoxicity assay kit contains lysis buffer, reaction mixture, and stop solution. Briefly, 100 μL of the 3T3 fibroblast suspension in DMEM containing 5000 cells was placed in each well of a 96-well plate and incubated in a humidified atmosphere of 95% air and 5% CO2 at 37 °C for 24 h. In the maximum LDH activity wells, the medium was replaced with 100 μL of fresh DMEM. In the spontaneous LDH activity wells, the medium was replaced with a solution of 90 μL of fresh DMEM and 10 μL of sterile ultrapure water. In the compound-treated LDH activity wells, the medium was replaced with 100 μL of serially-diluted sample solutions in DMEM. The plate was incubated in a humidified atmosphere of 95% air and 5% CO2 at 37 °C for 4 h. In the maximum LDH activity wells, 10 μL of the medium was aspirated out and 10 μL of the lysis buffer was added. The plate was further incubated in a humidified atmosphere of 95% air and 5% CO2 at 37 °C for 1 h. Then, 50 μL of each medium was transferred to a new 96-well plate, and 50 μL of the reaction mixture was added to each well. The plate was incubated in the dark at 25 °C for 30 min, and 50 μL of the stop solution was added to each well. The absorbance was measured on a UV microplate spectrophotometer at 490 and 680 nm wavelength. The LDH activity was determined by subtracting the 680 nm absorbance (background) from the 490 nm absorbance. The percentage of LDH leakage was calculated as [(LDHC − LDHS)/(LDHM − LDHS)] × 100%, where LDHC, LDHS, and LDHM are the compound-treated, spontaneous, and maximum LDH activity, respectively.3. Results and discussion
3.1. Synthesis and characterization of the CS-PLL/CA nanomicelles
Chitosan with molecular weight (Mw) of ∼3572 g mol−1 (∼22 saccharide units, Table S1†) was used as the scaffold due to its good water solubility and compatibility for biomedical therapy. At this molecular size, chitosan has reasonable endurance in acidic environments, and a suitable chain length for conjugation with the polypeptide, two important criteria in the subsequent steps to prepare nanomicelles (NMs). The O-acylation of chitosan with 6-bromohexanoyl chloride to introduce alkyl bromide functions was carried out by maintaining the salt formation of the primary amino groups with a strong protonating solvent (methanesulfonic acid, MSA). In this amine-protected environment, the substitution site with acyl group occurred mainly at the C-6 position and less at the C-3 position (Fig. 1, step (i)).29,30 Presumably in the MSA environment, the amine group of chitosan was strongly protonated into ammonium ion (–NH3+), and the substitution of this proton shield by the acyl group was thermodynamically unfavourable. To further ensure major substitution at the C-6 position, the acyl chloride was set as the limiting reactant in the molar-feed composition. The 1H NMR spectrum of the alkyl bromide-functionalized chitosan (CS-Br) was compared with the unmodified CS (Fig. 2a). The chemical shifts at 3.1, 3.5–4.1, and 4.8 ppm are due to glucosamine unit protons (Fig. 2a).31 New chemical shifts for CS-Br at 1.5, 1.7, 1.9, 2.5, and 3.4 ppm arise from the alkane protons of the hexanoyl linker, indicating successful acetylation of chitosan.32 The alkyl bromide of CS-Br was then converted to the azide group by nucleophilic substitution to obtain ‘clickable’ azide-functionalized chitosan (CS-N3) (Fig. 1, step (ii)). As compared to the FTIR spectrum of CS-Br (Fig. 2b), the CS-N3 spectrum shows an emergence of the azide stretching band at 2108 cm−1, which confirms that the bromide groups have been substituted by clickable azide groups.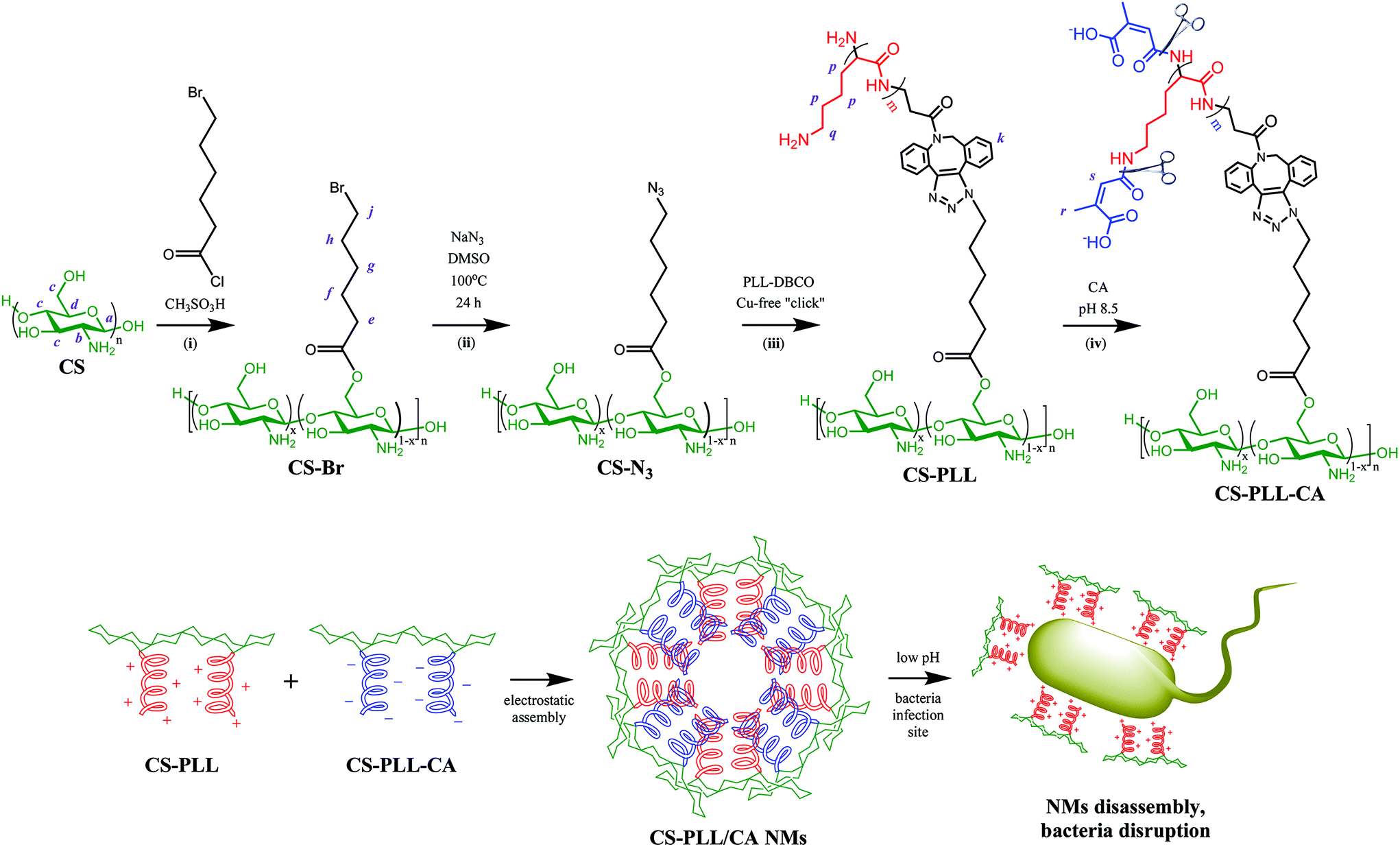 | ||
| Fig. 1 Synthesis of citraconyl-protected polypeptidosaccharides, self-assembly of nanomicelles (NMs), and pH-responsive disassembly of the NMs. | ||
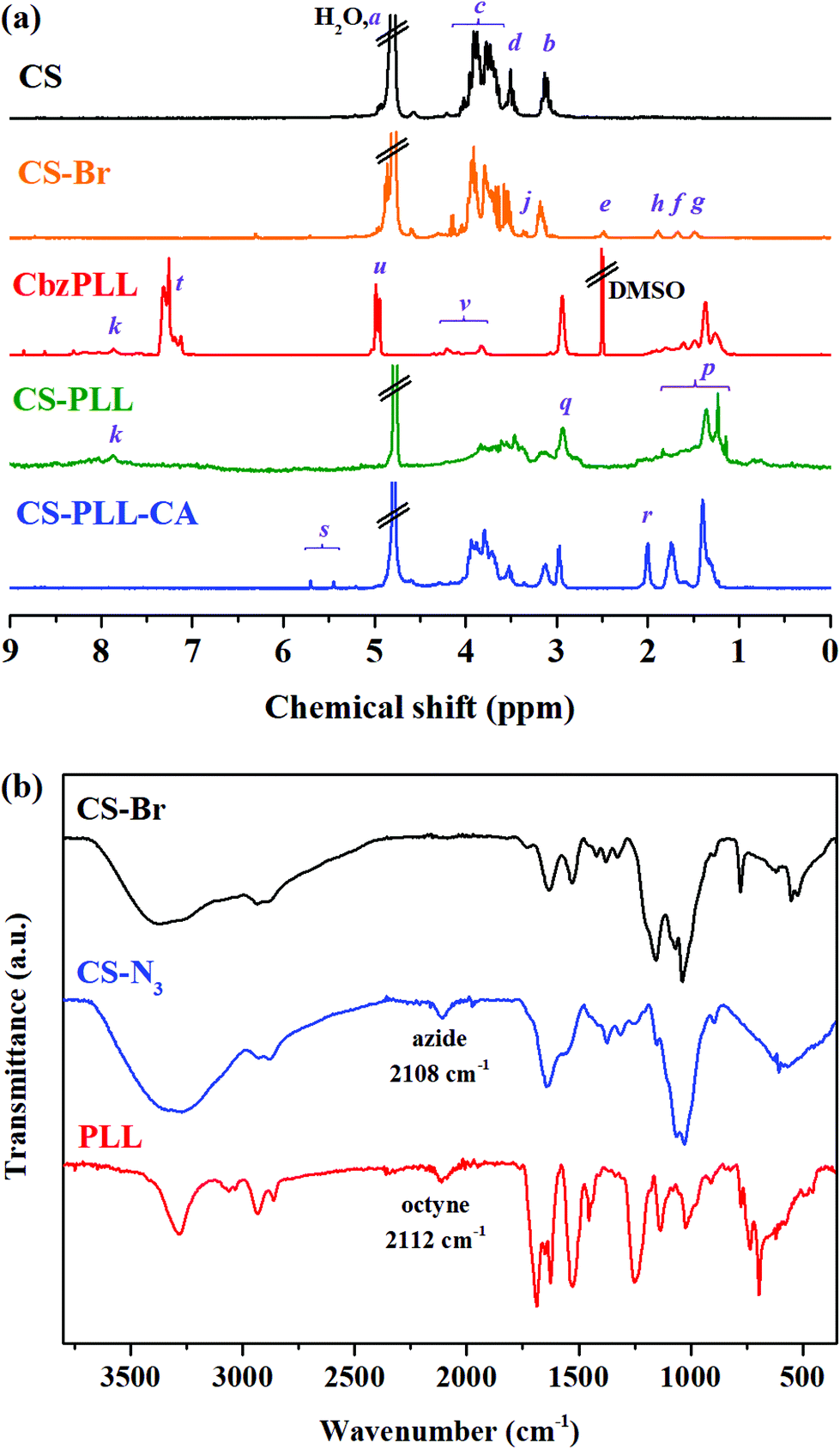 | ||
| Fig. 2 (a) 1H NMR spectra of the CS (black), CS-Br (orange), CbzPLL (red), CS-PLL (green), and CS-PLL-CA (blue) in the respective deuterated solvents, and (b) FTIR spectra of the CS-Br (black), CS-N3 (blue), and PLL (red). | ||
The cyclooctyne-terminated α-poly(L)lysine (PLL) was separately synthesized via ring-opening polymerization of Nε-carbobenzyloxy-L-lysine N-carboxyanhydride on a dibenzocyclooctyne-amine (DBCO-NH2) initiator, followed by acidic deprotection (Fig. S1†). Based on the peak integration ratio between the DBCO-NH2 initiator and the lysine unit in the 1H NMR spectrum (Fig. 2a), the CbzPLL polymer was estimated to have 40 repeat units. In the FTIR spectrum of PLL, the presence of a clickable octyne terminal was confirmed with the cyclooctyne stretch band at 2112 cm−1 (Fig. 2b). The PLL was then conjugated to the CS-N3via strain-promoted azide–alkyne ‘click’ cycloaddition to obtain chitosan-polylysine copolymer (CS-PLL) (Fig. S2†). The 1H NMR spectrum of CS-PLL contains the signature chemical shifts of glucosamine (3.3–4.1 ppm) and lysine (1.1–3.0 ppm) protons (Fig. 2a), indicating successful click conjugation of the CS-N3 and PLL. The PLL and CS-PLL polymers exhibited almost similar zeta potential values of 30.8 ± 3.2 and 30.3 ± 4.1 mV, respectively (Table S2†). Subsequent conversion of the cationic into anionic moieties was carried out by amidizing the primary amines on the lysine units of the CS-PLL with citraconic anhydride to obtain the citraconyl-capped chitosan-polylysine copolymer (CS-PLL-CA) (Fig. 1, step (iv)). As compared to the 1H NMR spectrum of CS-PPL, new chemical shifts characteristic of citraconyl amide at 2.0 and 5.5–5.7 ppm appear in the CS-PLL-CA spectrum (Fig. 2a), confirming the successful amidation of the primary amines.27 The citraconyl amide protection has made the CS-PLL-CA anionic with a zeta potential value of −23.2 ± 4.9 mV (Table S2†).
In the presence of a β-carboxylic acid terminal, amide bonds are acid labile and degradable at low pH.25,33 As such, the CS-PLL-CA copolymer exhibited a charge-reversal conversion from anionic carboxylic acid to cationic primary amine at low pH. The extent of degradation of citraconic amides was studied by incubating the CS-PLL-CA in buffers at different pH. The percentage of primary amine exposed as a result of the acid degradation was monitored using the amine-reactive fluorescamine dye. Fluorescamine is a spiro compound that is not fluorescent itself, but reacts with primary amines to form highly fluorescent pyrrolinone. In a buffer solution at pH 5.0, up to 99% of the citraconic amides were hydrolysed to form primary amines within 24 h (Fig. 3a). On the contrary, only 4% of the amides were hydrolysed after 24 h of incubation in the pH 7.4 buffer, demonstrating the pH-dependent charge-reversal conversion of the CS-PLL-CA copolymer. These observations are in agreement with the previous study on the degradation of β-carboxylic amides of the poly(N′-citraconyl-2-(3-aminopropyl-N,N-dimethylammonium)ethyl methacrylate) zwitterionic polymer.26
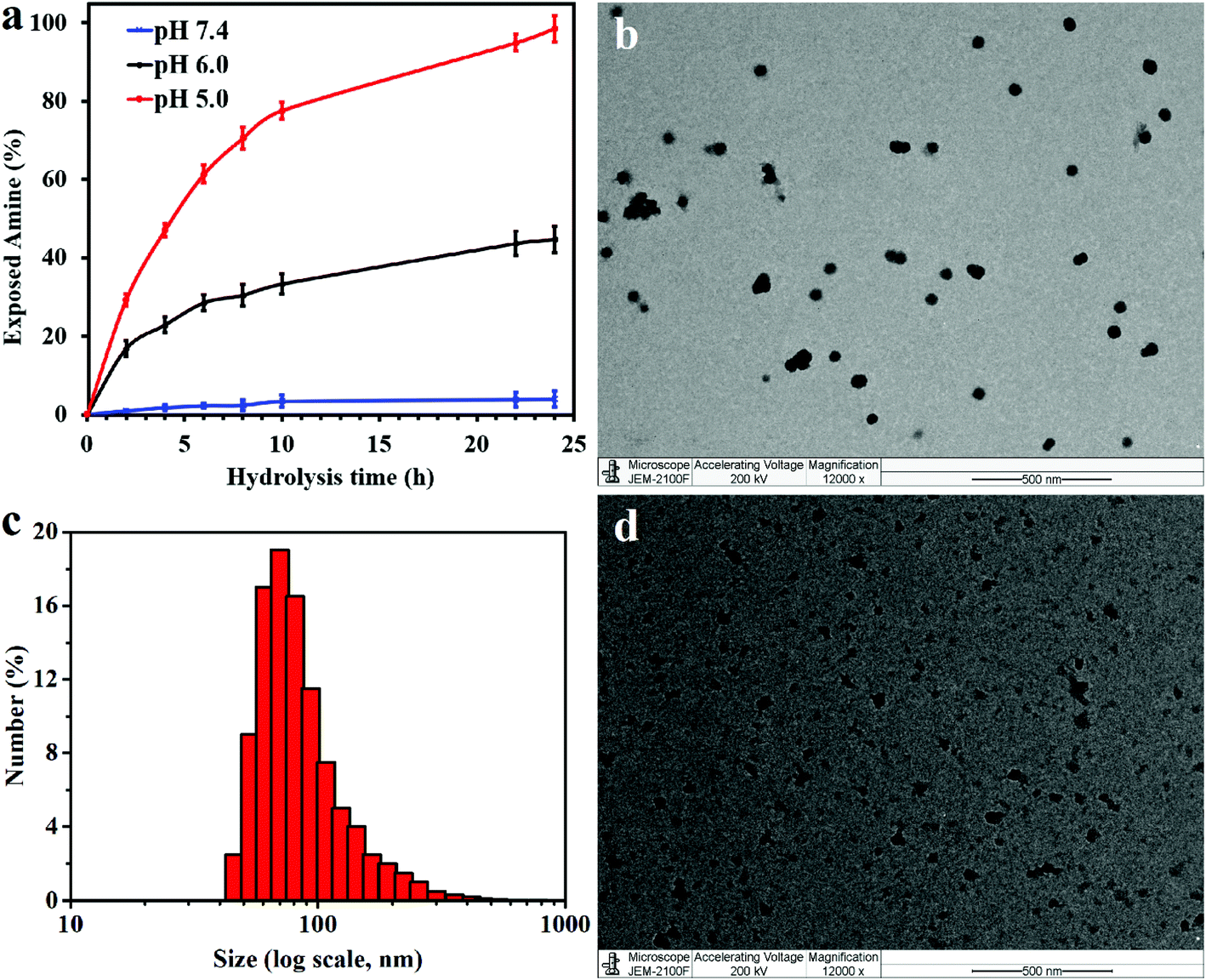 | ||
| Fig. 3 (a) Time-dependent degradation of citraconic amide of the CS-PLL-CA copolymer at different pH. Error bars indicate standard deviation obtained from three replicates. (b) TEM image and (c) hydrodynamic diameter (Dh) of the CS-PLL/CA NMs obtained from DLS, after incubation at pH 7.4 for 1 h. (d) TEM image of the CS-PLL/CA NMs after incubation at pH 5 for 1 h. Scale bars are 500 nm. | ||
The cationic CS-PLL and the anionic CS-PLL-CA copolymers were mixed at an equimolar ratio by dropwise addition under stirring to form the CS-PLL/CA nanomicelles (NMs) via electrostatic self-assembly. From the transmission electron microscopy (TEM) image, after incubation at pH 7.4 overnight, the CS-PLL/CA NMs exhibit well-defined size distributions with average diameters of about 55.2 ± 3.7 nm (Fig. 3b). From the dynamic light scattering (DLS) measurement, the hydrodynamic diameter of the CS-PLL/CA NMs was slightly bigger at about 76.4 ± 7.1 nm (Fig. 3c), due to the hydration effect of the particles. After incubation at pH 5.0 for 1 h, the CS-PLL/CA NMs underwent disassembly as shown in the TEM image (Fig. 3d) and the average particle size became 35.1 ± 25.3 nm. The DLS measurements show that the average particle size was 22.9 ± 9.5 nm after acidic incubation (Table S2†). Zeta potential value of the CS-PLL/CA NMs at pH 7.4 was 21.4 ± 5.2 mV, but after incubation at pH 5.0 the zeta potential increased to 29.7 ± 6.2 mV. The decreased particle size and the increased zeta potential results show that the citraconyl protection was hydrolyzed and the anionic CS-PLL-CA gradually became cationic CS-PLL, hence the NMs could not retain electrostatic interaction in the core and NMs disassembly occurred. Therefore, it is expected that the CS-PLL/CA NMs will remain stable and biocompatible by retaining their electrostatic interaction under physiological conditions (pH 7.4). However, they will expose the primary amines, which are readily protonated into cationic ammoniums, in the low pH environment of bacterial infection sites. Upon storage at 4 °C, and maintaining the pH at 8.5, the aqueous suspension of CS-PLL/CA NMs remained stable for 1 week.
3.2. Antimicrobial activity of the CS-PLL/CA NMs
The standard microplate dilution method was used to study the antimicrobial activity of the sample compounds towards various Gram-negative (E. coli and P. aeruginosa) and Gram-positive (S. aureus, S. epidermidis, and MRSA) bacteria.34 The PLL peptide shows minimum inhibitory concentration (MIC) values of 8–32 μg mL−1 towards the bacteria challenged (Table 1), which is consistent with the reported results.28 In comparison to the free peptide, the CS-PLL copolymer exhibited a lower antimicrobial activity towards the bacteria challenged, with MIC values about twice that of the free peptide (16–64 μg mL−1). The decrease in antimicrobial efficacy could probably be attributed to the incorporation of non-bactericidal chitosan scaffold. Upon protection with the citraconyl moieties, the CS-PLL-CA copolymer did not show any apparent antimicrobial activity, as no MIC value was observed up to the highest concentration tested (1024 μg mL−1). The result demonstrates thorough protection of the cationic amino moieties in the lysine units by the anionic citraconyl groups.| Sample | MIC, μg mL−1 | HC50, μg mL−1 | IC50, μg mL−1 | ||||
|---|---|---|---|---|---|---|---|
| E. coli | P. aeruginosa | S. aureus | S. epidermidis | MRSA | |||
| a The values were not observed up to the highest concentrations of compound tested (1024 μg mL−1). | |||||||
| PLL | 16 | 32 | 16 | 8 | 32 | >1024a | >18 |
| CS-PLL | 32 | 64 | 32 | 16 | 32 | >1024a | >28 |
| CS-PLL-CA | >1024a | >1024a | >1024a | >1024a | >1024a | >1024a | >1024a |
| CS-PLL/CA NMs, pH 7.4 | >1024a | >1024a | >1024a | >1024a | >1024a | >1024a | >1024a |
| CS-PLL/CA NMs, pH 5.0 | 32 | 64 | 32 | 16 | 32 | — | — |
The charge-reversal feature of the CS-PLL/CA NMs was investigated by pretreatment in PBS at different pH prior to the antibacterial assay. After pretreatment at pH 7.4 for 24 h, the CS-PLL/CA NMs did not exhibit apparent antimicrobial activity up to the highest concentration tested at 1024 μg mL−1. However, after pretreatment at pH 5.0 for 24 h, the CS-PLL/CA NMs displayed MIC values comparable to those of the CS-PLL copolymer, probably as a result of hydrolysis of the citraconic amides to regenerate the native primary amines. This result suggests that the CS-PLL/CA NMs can undergo the charge reversal and burst out their cationic core without losing their antimicrobial potency. The antimicrobial effect of CS-PLL/CA NMs after hydrolysis treatment was similar to that of the CS-PLL copolymer, implying that nearly all fractions of cytraconyl moieties were fully cleaved after the treatment. According to the fluorescamine assay, treatment of CS-PLL-CA copolymer in citrate buffer at pH 5.0 for 24 h could liberate 99% of primary amines as compared to the control treatment (Fig. 3a). The minimum bactericidal concentration (MBC, Table S3†) values of the free peptide, chitosan–peptide conjugate and NMs were consistent with their respective MIC values, which further confirm their efficacy in eradicating the pathogens.
The two-colour live/dead fluorescence kit containing SYTO9 and propidium iodide (PI) was used to assess the antibacterial effect of the compounds.35 SYTO 9 is a membrane-permeable green-fluorescent dye which stains both viable and nonviable bacteria. PI is a red-fluorescent dye that is rejected from viable bacteria by a membrane pump. Due to its higher affinity for nucleic acids, PI can outcompete SYTO9 when both dyes coexist. Gram-negative E. coli were incubated with each sample compound at the respective MBC value for 4 h. The control bacteria suspension shows that nearly the whole population was viable (appearing green, Fig. 4a). After incubation with the PLL and CS-PLL polymers, a large amount of dead bacteria as seen by the red fluorescence can be observed in the total population (Fig. 4b and c). After incubation with the CS-PLL-CA polymer, the amount of live bacteria exceeded that of dead bacteria due to the non-bactericidal effect of the negatively charged polymer (Fig. 4d). Similarly, a large amount of live bacteria can be observed after incubation with CS-PLL/CA NMs at pH 7.4 (Fig. 4e), because the cationic bactericidal segments are located inside the NM cores and are neutralized with the counter polyanionic segments. After incubation with CS-PLL/CA NMs at pH 5.0, a large amount of dead (red) bacteria appeared (Fig. 4f), because the NMs decomposed upon citraconyl-amide cleavage and spilled out the cationic bactericidal core which killed the bacteria.
 | ||
| Fig. 4 Fluorescence micrographs of live (green) and dead (red) E. coli in (a) blank control, and after incubation with the (b) PLL, (c) CS-PLL, (d) CS-PLL-CA, (e) CS-PLL/CA NMs at pH 7.4, and (f) CS-PLL/CA NMs at pH 5.0, with incubation period for 4 h at 37 °C. Scale bar is 50 μm. | ||
We studied the inner membrane depolarization using 3,3′-dipropylthiadicarbocyanine iodide (DiSC3(5)) dye. Accumulation of DiSC3(5) on cells with hyperpolarized cytoplasmic membranes of intact bacteria can self-quench its fluorescence. If the membrane potential or integrity is broken, the dye gets discharged and restores fluorescence.36 We also studied the outer membrane permeabilization using N-phenyl-1-naphthylamine (NPN), a nonpolar hydrophobic molecule that accumulates on the compromised membrane and exhibits strong fluorescence in the hydrophobic environment of the lipid membrane.37 An intact bacterial outer membrane can exclude NPN by a pump mechanism, where the dye will only show weak fluorescence in aqueous medium.38 Upon bacterial incubation with the PLL peptide, the fluorescence intensities of DiSC3(5) and NPN showed proportional dependency on the peptide concentration, demonstrating its potency to compromise bacterial membrane (Fig. 5, red line). In comparison to the PLL peptide, the CS-PLL copolymer inflicted lower cytoplasmic membrane depolarization and outer membrane permeabilization towards bacteria, because of the inclusion of the non-bactericidal chitosan scaffold (Fig. 5, black line). With pretreatment at pH 7.4, the CS-PLL/CA NMs showed only minor damage to the bacterial membrane up to the highest concentration tested (1024 μg mL−1) (Fig. 5, blue line). With pretreatment at pH 5.0, the CS-PLL/CA NMs exhibited cytoplasmic membrane depolarization and outer membrane permeabilization comparable to the CS-PLL copolymer (Fig. 5, green line). These results confirm that the CS-PLL/CA NMs could regenerate their antimicrobial efficacy at low pH, such as in the bacterial infection sites via membrane disruption.
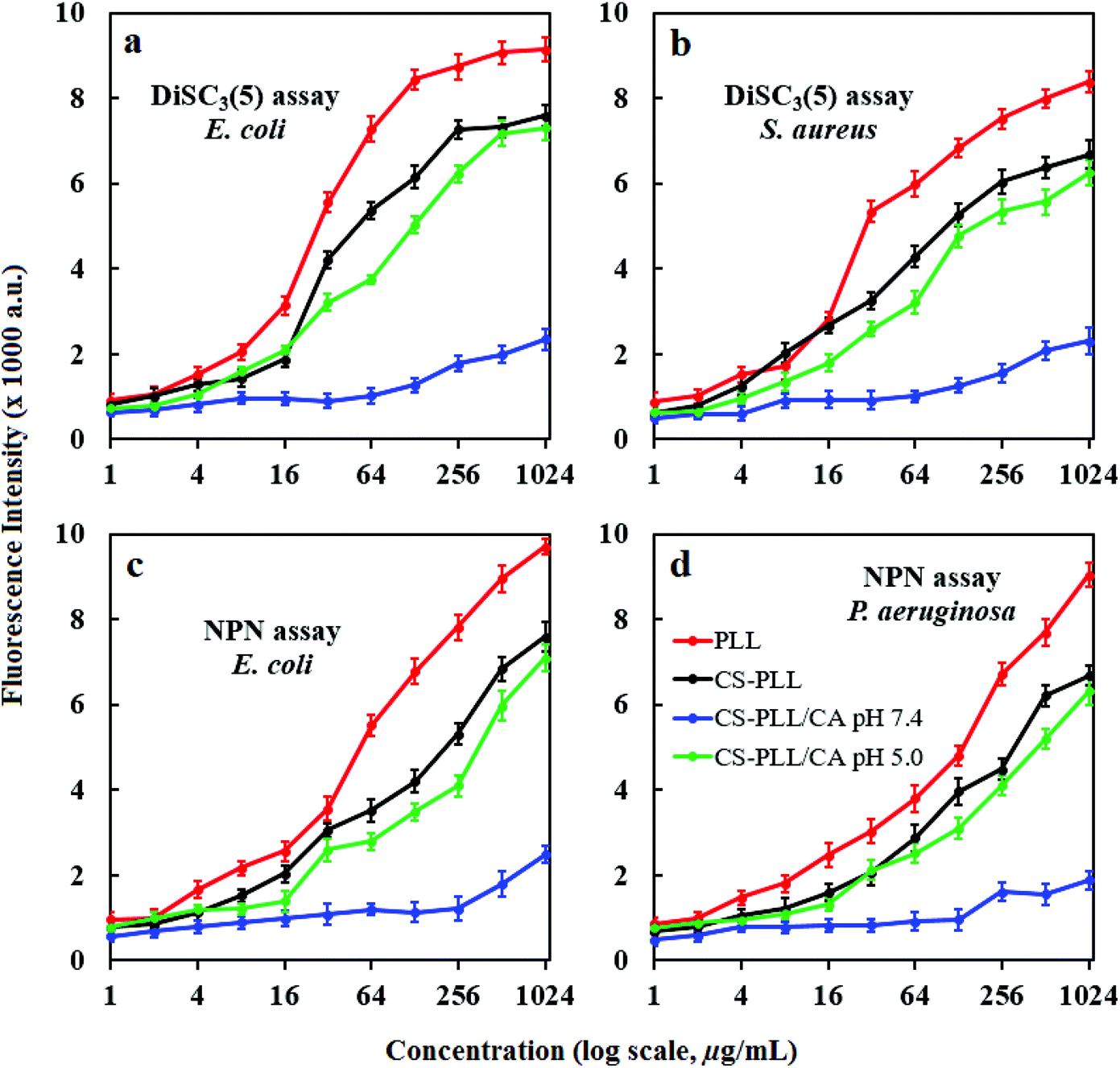 | ||
| Fig. 5 (a and b) DiSC3(5) release and (c and d) NPN uptake on (a, c and d) Gram-negative and (b) Gram-positive bacteria after incubation with the sample compounds in PBS for 1 h. Error bars indicate standard deviations obtained from three replicates. | ||
3.3. Hemotoxicity and cytotoxicity of the CS-PLL/CA NMs
The hemolytic activity of the compounds toward rat whole blood was determined as it is a toxicity parameter of antimicrobial agents, so the concentration of a compound which induces lysis of 50% of the red blood cells (HC50 value) was measured. The linear PLL did not show apparent hemolytic activity, and no HC50 value was recorded up to the highest concentration tested at 1024 μg mL−1 (Fig. 6a, red line). This result is in agreement with the previous report that linear polylysine up to a concentration of 10 mg mL−1 was non-hemolytic toward human erythrocytes.39 The excellent hemocompatibility of PLL was presumably attributed to the lack of a hydrophobic domain, as hydrophobic groups are known to be responsible for causing a hemolytic effect. Likewise, the CS-PLL and CS-PLL-CA polymers, as well as CS-PLL/CA NMs also showed remarkable hemocompatibility up to 1024 μg mL−1. Furthermore, the CS-PLL showed less hemolytic activity than the free PLL due to the incorporation of the biocompatible chitosan segment (Fig. 6a, black line). Among all the tested compounds, the CS-PLL-CA showed the lowest hemolytic activity, indicating that the citraconyl-amide protection was effective to shield the cationic amine moieties.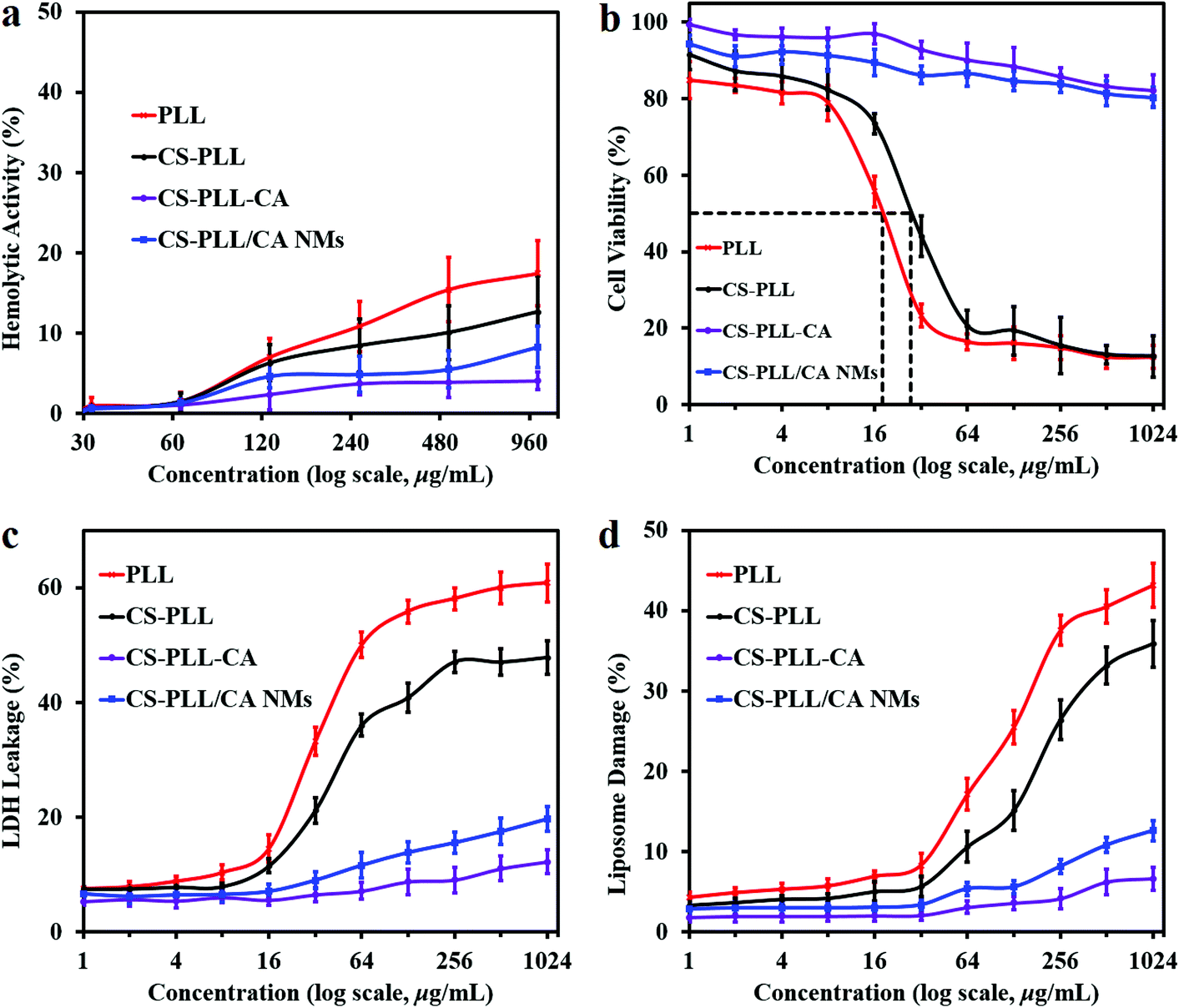 | ||
| Fig. 6 (a) Hemolytic activity of the samples after incubation with rat whole blood for 1 h. (b) Relative cell viability of the 3T3 fibroblasts after incubation with the samples for 24 h. (c) LDH leakage from 3T3 fibroblasts after incubation with the samples in DMEM for 5 h. (d) Calcein leakage from DSPC liposomes after incubation with the samples in PBS for 1 h. Error bars indicate standard deviation obtained from three replicates. | ||
Cytotoxicity of the sample compounds towards mouse 3T3 fibroblast cells as the standard mammalian model was determined by measuring the half maximal inhibitory concentration (IC50) using the metabolic methylthiazolyldiphenyl-tetrazolium bromide (MTT) protocol. The PLL peptide recorded an IC50 value at 18 μg mL−1 (Table 1), consistent with the previous report.28 The CS-PLL copolymer showed lower cytotoxicity than the free peptide, presumably due to the inclusion of biocompatible chitosan (Fig. 6b, Table 1). The CS-PLL copolymer showed a higher IC50 value at 28 μg mL−1 as compared to that of the PLL peptide, indicating a lower fraction of cationic lysine residues. The CS-PLL-CA copolymer did not show any indication of cytotoxicity up to the highest concentration tested at 1024 μg mL−1, indicating that the cytraconyl protection was effective to shield the amine moieties. Similar to the CS-PLL-CA copolymer, the CS-PLL/CA NMs also did not show apparent cytotoxicity up to the concentration of 1024 μg mL−1. Albeit in the presence of the CS-PLL copolymer in the NMs, the cationic lysine residues were located in the core and protected by the chitosan scaffold on the shell. These results indicate that the CS-PLL/CA NMs remain stable and retain their anionic protection characteristics under physiological conditions.
The toxicity of the uncomplexed cationic polymers were corroborated by the lactate dehydrogenase (LDH) assay.40 Viable cells produced only negligible LDH leakage at an amount similar to that of the spontaneous LDH activity. In general, the CS-PLL copolymer induced lower LDH leakage from 3T3 fibroblasts as compared to the free PLL peptide (Fig. 6c), presumably due to the mass incorporation of the cytocompatible chitosan which reduced the fraction of cationic lysine residues. On the other hand, the CS-PLL-CA copolymer did not show significant LDH leakage up to the highest concentration tested at 1024 μg mL−1. This result demonstrated that the cytraconyl protecting group was effective in reducing cell membrane perturbation by the amino moieties of the peptide. In comparison to the PLL peptide and CS-PLL copolymer, the CS-PLL/CA NMs only induced negligible LDH leakage up to the concentration of 1024 μg mL−1, demonstrating that the NMs remain stable and retain their anionic protection characteristics under physiological conditions. Therefore, the electrostatic formation of NMs and charge-reversal protection are a promising strategy to improve the therapeutic selectivity of antimicrobial peptides. The true potential of CS-PLL/CA NMs lies on the on-demand charge conversion, whereby they remain cytocompatible under physiological pH and turn bactericidal in a low pH environment, such as at the bacterial infection sites.
The membrane disruption ability of the polymers was also measured with calcein leakage from 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) liposomes. Lipids are important constituents of eukaryotic cell membranes. Interaction of the sample compounds with the cell membrane was investigated using calcein leakage from liposome vesicles as a model of the phospholipid bilayer. An increase in concentration of the antimicrobial compounds resulted in higher damage to liposome vesicles (Fig. 6d), due to the presence of a larger amount of cationic charge to destabilize the liposome membranes. The PLL peptide showed dose-dependent liposomal leakage due to the pore-forming ability of the cationic amino moieties toward cell membranes. At the same concentration, the CS-PLL copolymer induced lower liposomal damage than the PLL peptide, because the incorporation of chitosan scaffold had reduced the fraction of cationic lysine residues, resulting in lower destabilization of phospholipid membrane. The CS-PLL-CA copolymer only induced negligible damage towards liposome vesicles at neutral pH, indicating thorough protection of the amino moieties by the citraconyl groups. The CS-PLL/CA NMs also inflicted low liposome leakage up to the highest concentration tested at 1024 μg mL−1, demonstrating that the NMs remain stable and retain their anionic protection characteristics under physiological conditions.
4. Conclusion
We have shown that the cationic conjugate of α-poly(L)lysine side-grafted onto a chitosan backbone (CS-PLL) can be altered into nanomicelles (NMs) to achieve ultralow toxicity and ‘on-demand’ bactericidal activity. The cationic segments of CS-PLL were fixed on the NMs core by electrostatic interaction with the anionic segments of citraconyl amide-protected CS-PLL (CS-PLL-CA), while the chitosan segments formed the NMs corona. The NMs are stable in the physiological pH and exhibit high hemo- and cytocompatibility, because the cationic segments are neutralized by counterions and hidden in the NMs core by the biocompatible chitosan corona. But at pH 6 and below, the amide-protection of CS-PLL-CA can undergo self-hydrolysis and revert to the CS-PLL form, causing the NMs to lose their electrostatic stability and subsequently ‘decompose’ to spill out their cationic segments core. As a result, the liberated cationic CS-PLL can interact and kill a broad spectrum of Gram-positive and Gram-negative bacteria via membrane disintegration. This pH-triggered system is favourable because bacterial infection sites in the body usually show localized acidity as a result of the combined actions of bacterial metabolism and host immune response, and the pH can decrease to as low as 5.5. Thus, the electrostatic NMs represent a ‘smart’ delivery system that can retain the cationic polypeptide in a nontoxic mode at physiological pH, and auto-switch to bactericidal mode at the infection sites. This system opens a pathway to explore various combinations of cationic antimicrobial agents and anionic polymers in future designs to achieve optimal switchable balance between nontoxicity and bactericidal effects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded and supported by the Singapore Ministry of Education Tier 3 Grants (MOE2013-T3-1-002 and MOE2018-T3-1-003), ASTAR RIE2020 Advanced Manufacturing and Engineering (AME) IAP-PP Specialty Chemicals Programme Grant (No. A1786a0032), and NTU NAFTEC Funding.References
- G. A. Roth, D. Abate, K. H. Abate, S. M. Abay, C. Abbafati and C. J. L. Murray, Lancet, 2018, 392, 1736–1788 CrossRef.
- Centers for Disease Control and Prevention (CDC), Antibiotic resistance threats in the United States, 2019, DOI:10.15620/cdc:82532.
- K. Smerkova, K. Dolezelikova, L. Bozdechova, Z. Heger, L. Zurek and V. Adam, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1636 CAS.
- M. Duan, J. Gu, X. Wang, Y. Li, R. Zhang, T. Hu and B. Zhou, Ecotoxicol. Environ. Saf., 2019, 180, 114–122 CrossRef CAS.
- C. Tedijanto, S. W. Olesen, Y. H. Grad and M. Lipsitch, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E11988–E11995 CrossRef CAS.
- P. A. Smith, M. F. T. Koehler, H. S. Girgis, D. Yan, Y. Chen, Y. Chen, J. J. Crawford, M. R. Durk, R. I. Higuchi, J. Kang, J. Murray, P. Paraselli, S. Park, W. Phung, J. G. Quinn, T. C. Roberts, L. Rougé, J. B. Schwarz, E. Skippington, J. Wai, M. Xu, Z. Yu, H. Zhang, M.-W. Tan and C. E. Heise, Nature, 2018, 561, 189–194 CrossRef CAS.
- J. L. Fox, Nat. Biotechnol., 2013, 31, 379–382 CrossRef CAS.
- J. Wang, C. Lu, Y. Shi, X. Feng, B. Wu, G. Zhou, G. Quan, X. Pan, J. Cai and C. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 18363–18374 CrossRef CAS.
- M. Zhou, Y. Qian, J. Xie, W. Zhang, W. Jiang, X. Xiao, S. Chen, C. Dai, Z. Cong, Z. Ji, N. Shao, L. Liu, Y. Wu and R. Liu, Angew. Chem., 2020, 59, 6412–6419 CrossRef CAS.
- E. F. Palermo, K. Lienkamp, E. R. Gillies and P. J. Ragogna, Angew. Chem., Int. Ed., 2019, 58, 3690–3693 CrossRef CAS.
- P. Yang, P. Pageni, M. A. Rahman, M. Bam, T. Zhu, Y. P. Chen, M. Nagarkatti, A. W. Decho and C. Tang, Adv. Healthcare Mater., 2019, 8, 1800854 CrossRef.
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2015, 44, D1087–D1093 CrossRef.
- R. E. W. Hancock and H.-G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS.
- I. M. Chiu, B. A. Heesters, N. Ghasemlou, C. A. Von Hehn, F. Zhao, J. Tran, B. Wainger, A. Strominger, S. Muralidharan, A. R. Horswill, J. B. Wardenburg, S. W. Hwang, M. C. Carroll and C. J. Woolf, Nature, 2013, 501, 52–57 CrossRef CAS.
- M. I. Halpin-Dohnalek and E. H. Marth, J. Food Prot., 1989, 52, 267–282 CrossRef CAS.
- C. Korupalli, C.-C. Huang, W.-C. Lin, W.-Y. Pan, P.-Y. Lin, W.-L. Wan, M.-J. Li, Y. Chang and H.-W. Sung, Biomaterials, 2017, 116, 1–9 CrossRef CAS.
- L. Pichavant, C. Bourget, M.-C. Durrieu and V. Héroguez, Macromolecules, 2011, 44, 7879–7887 CrossRef CAS.
- I.-B. Kim, A. Dunkhorst, J. Gilbert and U. H. F. Bunz, Macromolecules, 2005, 38, 4560–4562 CrossRef CAS.
- X. Ding, A. Wang, W. Tong and F.-J. Xu, Small, 2019, 15, 1900999 CrossRef.
- H. Deng, X. Zhao, J. Liu, L. Deng, J. Zhang, J. Liu and A. Dong, J. Mater. Chem. B, 2015, 3, 9397–9408 RSC.
- Y. Wang, S. Lv, M. Deng, Z. Tang and X. Chen, Polym. Chem., 2016, 7, 2253–2263 RSC.
- H. Yi, P. Liu, N. Sheng, P. Gong, Y. Ma and L. Cai, Nanoscale, 2016, 8, 5985–5995 RSC.
- J.-Z. Du, X.-J. Du, C.-Q. Mao and J. Wang, J. Am. Chem. Soc., 2011, 133, 17560–17563 CrossRef CAS.
- Y.-Y. Yuan, C.-Q. Mao, X.-J. Du, J.-Z. Du, F. Wang and J. Wang, Adv. Mater., 2012, 24, 5476–5480 CrossRef CAS.
- Z. Zhou, Y. Shen, J. Tang, M. Fan, E. A. Van Kirk, W. J. Murdoch and M. Radosz, Adv. Funct. Mater., 2009, 19, 3580–3589 CrossRef CAS.
- P. Liu, G. Xu, D. Pranantyo, L. Q. Xu, K.-G. Neoh and E.-T. Kang, ACS Biomater. Sci. Eng., 2018, 4, 40–46 CrossRef CAS.
- D. Pranantyo, P. Liu, W. Zhong, E.-T. Kang and M. B. Chan-Park, Biomacromolecules, 2019, 20, 2922–2933 CrossRef CAS.
- D. Pranantyo, L. Q. Xu, Z. Hou, E.-T. Kang and M. B. Chan-Park, Polym. Chem., 2017, 8, 3364–3373 RSC.
- D. Pranantyo, L. Q. Xu, E.-T. Kang and M. B. Chan-Park, Biomacromolecules, 2018, 19, 2156–2165 CrossRef CAS.
- H. Sashiwa, N. Kawasaki, A. Nakayama, E. Muraki, N. Yamamoto, H. Zhu, H. Nagano, Y. Omura, H. Saimoto, Y. Shigemasa and S.-i. Aiba, Biomacromolecules, 2002, 3, 1120–1125 CrossRef CAS.
- Y. Belabassi, J. Moreau, V. Gheran, C. Henoumont, A. Robert, M. Callewaert, G. Rigaux, C. Cadiou, L. Vander Elst, S. Laurent, R. N. Muller, A. Dinischiotu, S. N. Voicu and F. Chuburu, Biomacromolecules, 2017, 18, 2756–2766 CrossRef CAS.
- D. Zhao, H. Zhang, W. Tao, W. Wei, J. Sun and Z. He, Biomater. Sci., 2017, 5, 502–510 RSC.
- T. Zhou, X. Zhou and D. Xing, Biomaterials, 2014, 35, 4185–4194 CrossRef CAS.
- CLSI, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, Clinical and Laboratory Standards Institute, Wayne, PA, 11th edn, 2018 Search PubMed.
- D. Pranantyo, L. Q. Xu, K.-G. Neoh, E.-T. Kang, Y. X. Ng and S. L.-M. Teo, Biomacromolecules, 2015, 16, 723–732 CrossRef CAS.
- R. C. Anderson, R. E. W. Hancock and P.-L. Yu, Antimicrob. Agents Chemother., 2004, 48, 673–676 CrossRef CAS.
- J. L. Grace, A. G. Elliott, J. X. Huang, E. K. Schneider, N. P. Truong, M. A. Cooper, J. Li, T. P. Davis, J. F. Quinn, T. Velkov and M. R. Whittaker, J. Mater. Chem. B, 2017, 5, 531–536 RSC.
- L. Johnson, H. Mulcahy, U. Kanevets, Y. Shi and S. Lewenza, J. Bacteriol., 2012, 194, 813–826 CrossRef CAS.
- E. H. H. Wong, M. M. Khin, V. Ravikumar, Z. Si, S. A. Rice and M. B. Chan-Park, Biomacromolecules, 2016, 17, 1170–1178 CrossRef CAS.
- D. Fischer, Y. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121–1131 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Polymerization and ‘click’ reaction schemes, 1H NMR spectra, modification degree of the chitosan derivatives, molecular weight, polydispersity index, zeta potential, particle diameter, and bactericidal activity of the compounds. See DOI: 10.1039/d0bm01382j |
| This journal is © The Royal Society of Chemistry 2021 |