DOI:
10.1039/D0BM00971G
(Paper)
Biomater. Sci., 2021,
9, 116-123
Nuclear delivery of dual anti-cancer drugs by molecular self-assembly†
Received
13th June 2020
, Accepted 3rd August 2020
First published on 7th September 2020
Abstract
Nanomedicines generally suffer from poor accumulation in tumor cells, low anti-tumor efficacy, and drug resistance. In order to address these problems, we introduced a novel nanomedicine based on dual anti-cancer drugs, which showed good cell nuclear accumulation properties. The novel nanomedicine consisted of three components: (1) dual anti-cancer drugs, 10-hydroxycamptothecin (HCPT) and chlorambucil (CRB), whose targets are located in the cell nucleus, (2) a nuclear localizing dodecapeptide, PMI peptide (TSFAEYWNLLSP), which could activate p53 by binding with MDM2 and MDMX located in the cell nucleus, and (3) an efficient self-assembling tripeptide FFY. Our nanomedicine exhibited enhanced cellular uptake and nuclear accumulation properties, thus achieving an excellent anti-cancer capacity both in vitro and in vivo. Our study will provide an inspiration for the development of novel multifunctional nanomaterials for cancer diagnosis and therapy.
Introduction
Nanomaterials of self-assembling peptides have been used in diverse fields, including anti-cancer drug delivery,1–3 immune modulation,4–6 cell culture7–9 and tissue engineering.10–12 However, their application in anti-cancer drug delivery has attracted increasing research attention because cancer is at present one of the most threatening diseases in the world.13–25 Nanomedicines have shown great potential in the delivery of anti-cancer drugs because of their enhanced cellular uptake capacity, possibly due to the enhanced permeation and retention (EPR) effect.26–32 However, it has been reported that less than 1% of the total dose of nanomedicines on average can accumulate in tumor cells.33 Thus, new strategies to enhance the therapeutic efficacy and tumor/cancer cell accumulation of nanomedicines must be developed. A drug amphiphile containing two different drugs has been applied for synergistic anti-cancer therapy.34,35 In addition, strategies capable of increasing nuclear accumulation of anti-cancer drugs should also be investigated because the targets of many anti-cancer drugs are located within the nucleus.36–40
p53, a widely studied tumor suppressor gene, can prevent the transformation of normal cells into malignant cells. MDM2 and MDMX are two crucial regulators of the transcriptional activity of the p53 gene and the stability of the p53 protein, respectively. Inhibition of MDM2 and MDMX can effectively activate p53, thus inhibiting the growth of tumor cells.41–44 Recent studies have reported that a dodecapeptide ligand (TSFAEYWNLLSP, PMI) exhibited a high affinity for MDM2 and a relatively weaker affinity for MDMX, and it possessed the ability to enter the cell nucleus. Based on this information, we opted to construct nanomedicines consisting of a combination of dual anti-cancer drugs, a self-assembling peptide, and a PMI peptide, and studied their nuclear accumulation and anti-cancer properties.45
Materials and methods
Materials
The HepG2 cell line was maintained in our laboratory. The HepG2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. FBS, DMEM, and penicillin/streptomycin were purchased from Gibco Corporation. The Fmoc-amino acids were purchased from J&K Chemicals or GL Biochem (Shanghai). N,N-Diisopropylethylamine (DIEA) was obtained from Energy Chemical (Shanghai). CCK-8 was purchased from Solarbio. All chemical reagents and solvents were obtained from commercial sources and used without further purification. Six-week-old mice were used for the experiments and all ethical rules of the Animal Association were strictly followed.
Synthesis of peptides
The peptide derivatives CRB-K (HCPT) FFYG-PMI (Comp. 1), CRB-K (Nap) FFYG-PMI (Comp. 2), HCPT-K (Nap) FFYG-PMI (Comp. 3), CRB-K (HCPT) FFYG-scrambled PMI (Comp. 4), and CRB-K (HCPT) GGGG-PMI (Comp. 5) were prepared by standard Fmoc solid-phase peptide synthesis (SPPS) using 2-chlorotrityl chloride resin and the corresponding amino acids with side chains protected by N-Fmoc. O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) was used as a coupling reagent. After CRB or HCPT–glutaric acid coupling to the last amino acid, excess reagents were removed by washing with N,N′-dimethylformamide (DMF) four times followed by washing with dichloromethane (DCM) four times. Finally, to cleave the peptide from the resin, 95% trifluoroacetic acid containing 2.5% trimethylsilane and 2.5% H2O was used. The mixture was then evaporated using a rotary evaporator and anhydrous diethyl ether was used to sediment the peptide. In order to couple Nap or HCPT with the –NH2 groups of lysine, Nap or HCPT–GA and the purified peptide were mixed in a ratio of 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and stirred in DMF overnight. HBTU was used as a coupling reagent, and the pH was adjusted within the range of 8.5–9.0.
:1 and stirred in DMF overnight. HBTU was used as a coupling reagent, and the pH was adjusted within the range of 8.5–9.0.
Transmission electron microscopy
The microstructures in solution or hydrogels were observed using a transmission electron microscope by the negative staining technique. First, 10–15 μL of the solution was loaded onto 200 mesh copper grids coated with a continuous thick carbon film; uranyl acetate was used as a negative stain, and deionized water was used to rinse the grid. Finally, the grids were allowed to dry in a desiccator overnight.
Critical aggregation concentration (CAC)
The CAC values of different peptide derivatives were determined by dynamic light scattering (DLS). Solutions containing different concentrations of compounds were tested, and the light scattering intensity was recorded for analysis. Lower CAC values represent a better assembly capacity.
Laser scanning confocal microscopy
The HepG2 cells were incubated in a glass bottom cell culture dish at a density of 3 × 105 cells per dish. After 24 h of incubation, DMEM containing 200 μM of the compound was added to the cell culture dish and incubated. After 4 h, the medium was removed and the cells were washed with 1× phosphate buffered saline (PBS) thrice. The cells were then stained with 1.0 μg mL−1 of Red Dot for 10 min at 37 °C in the dark. After that, the cells were rinsed thrice with PBS and imaged using a laser scanning confocal microscope (Leica TSC SP5) at the same voltage (λexc = 405 nm for HCPT, and λexc = 633 nm for Red Dot).
Cell viability analysis and determination of IC50
To test tumor cell inhibition in vitro, HepG2 cells were seeded in 96-well plates at a density of 6 × 103 cells per well and incubated with DMEM at 37 °C for 24 h. After that, the culture medium was removed and fresh culture medium containing different peptide derivatives, CRB or HCPT, was added. For HCPT Comp. 1, and Comp. 3–5, the initial concentration was 50 μM and the final concentration was 0.195 μM. For CRB and HPCT Comp. 2, the initial concentration was 800 μM and the final concentration was 3.125 μM. After 48 h of incubation, 10 μL CCK-8 solution (5 mg mL−1) was added to each well and incubated at 37 °C for another 4 h. Then, the CCK-8 solution was removed and 100 μL of DMSO was added to stop the reaction and dissolve the purple formazan; the optical density of the solution was measured at 450 nm using a microplate reader (Bio-Rad iMarkTM, USA). The cytotoxicity assay was performed thrice. To ensure reproducibility in determining the IC50 of different drugs, we analyzed their cytotoxicity with 9 concentration gradients for three separate experiments and averaged all the results.
In vivo evaluation of antitumor activity
All animal experimental protocols were performed in accordance with the ethical guidelines approved by the Animal Care and Research Committee of Nanjing Medical University. All animals were kept in the Animal Core Facility of Nanjing Medical University (ACFNMU, Nanjing, Jiangsu, China). The animal experiments in this study were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University with an affidavit of Approval of Animal Ethical and Welfare number of IACUC 1905032. We inoculated female Balb/c nude mice with 5 × 106 HepG2 cells in the mammary fat pad. When the tumor size reached ∼75 mm3, the mice were randomly divided into eight treatment groups with five mice in each group and injected through the caudal vein with the drug dose shown in Table S1.† The drugs were injected at an interval of 3 days, and the i.v. injection was stopped after 6 days. Tumor growth was recorded with a caliper every two days over a period of 21 days. The tumor volume was calculated using the formula: length × width2/2. The weight of the mice was also monitored during the treatment.
H&E staining
H&E staining was performed to observe the degree of apoptosis. All visible tumors were isolated from HepG2 tumor-bearing Balb/c nude mice at day 21. The histology of tumors was visualized using hematoxylin and eosin staining. The hematoxylin solution is alkaline and it stains the chromatin and ribosomes in the cytoplasm violet blue. Eosin is an acidic solution that stains the cytoplasm and extracellular matrix red. The degree of tissue apoptosis can be roughly observed by the atrophy of the nucleus.
TUNEL staining
To evaluate the apoptosis of the tumor tissue, TUNEL staining was performed using an in situ cell death detection kit according to the manufacturer's instruction (Roche Diagnostics GmbH, Penzberg, Germany). Blue fluorescence from DAPI represents the nucleus and green fluorescence from FITC represents apoptotic cells. Bright green fluorescence indicates more apoptosis while weaker green fluorescence indicates less apoptosis.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5. All data are represented as mean ± standard error of mean (SEM), and differences among the groups were determined using Student's t-test or one-way analysis of variance (ANOVA). *p < 0.05, **p < 0.01 and ***p < 0.001 were used to show statistical significance.
Results and discussion
Molecular design and synthesis
For the development of the nanomedicine, we chose two anti-cancer drugs, 10-hydroxycamptothecin (HCPT) and chlorambucil (CRB), because the targets of both are located in the nucleus. In order to enhance the peptide assembly capacity, we introduced the tripeptide FFY to the PMI peptide because the dipeptide FF and the tripeptide FFY have been widely used to generate supramolecular nanomaterials. Therefore, we designed and synthesized CRB-K (HCPT) FFYG-PMI (Comp. 1), CRB-K (Nap) FFYG-PMI (Comp. 2, without HCPT), HCPT-K (Nap) FFYG-PMI (Comp. 3, without CRB), CRB-K (HCPT) FFYG-scrambled PMI (Comp. 4, with the scrambled PMI peptide), and CRB-K (HCPT) GGGG-PMI (Comp. 5, without the FFY peptide) (Fig. 1). Comp. 1 was a dual drug-based nanomedicine containing both PMI and FFY peptides; Comp. 2 and Comp. 3 were single drug-based nanomedicines; Comp. 4 contained the disrupted PMI peptide; and Comp. 5 possessed a less efficient self-assembling capacity due to the absence of the FFY peptide.
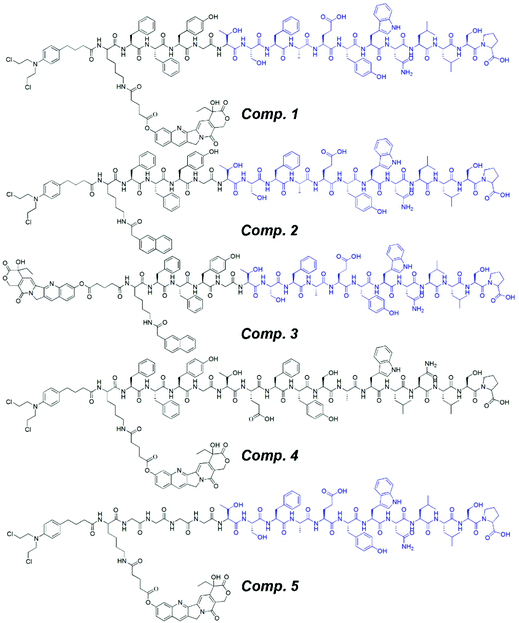 |
| | Fig. 1 Chemical structures of CRB-K (HCPT) FFYG-PMI (Comp. 1), CRB-K (Nap) FFYG-PMI (Comp. 2), HCPT-K (Nap) FFYG-PMI (Comp. 3), CRB-K (HCPT) FFYG-scrambled PMI (Comp. 4), and CRB-K (HCPT) GGGG-PMI (Comp. 5). | |
These compounds were synthesized using a combination of solid-phase peptide synthesis and solution-phase organic synthesis (Fig. 2); the synthesis process was easy and straightforward. For Comp. 1, we first obtained the peptide derivative CRB-KFFYG-PMI by solid-phase peptide synthesis using 2-chloride resin. We then prepared HCPT–glutaric acid N-hydroxyl succinimide (NHS)-activated ester, which was coupled to the free amine on the side chain of K in CRB-KFFYG-PMI to afford the title compound. The pure compound was obtained by high-performance liquid chromatography (HPLC). Other compounds were synthesized and purified using similar procedures.
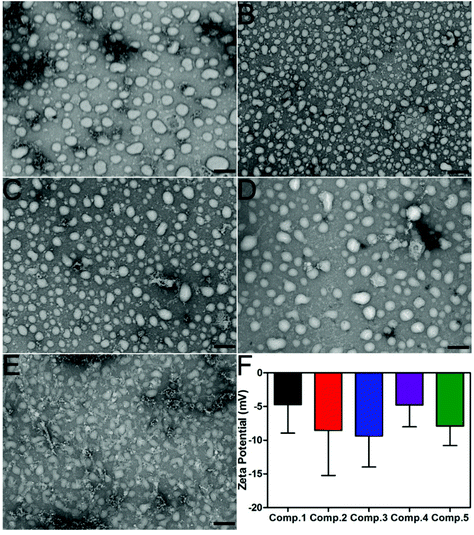 |
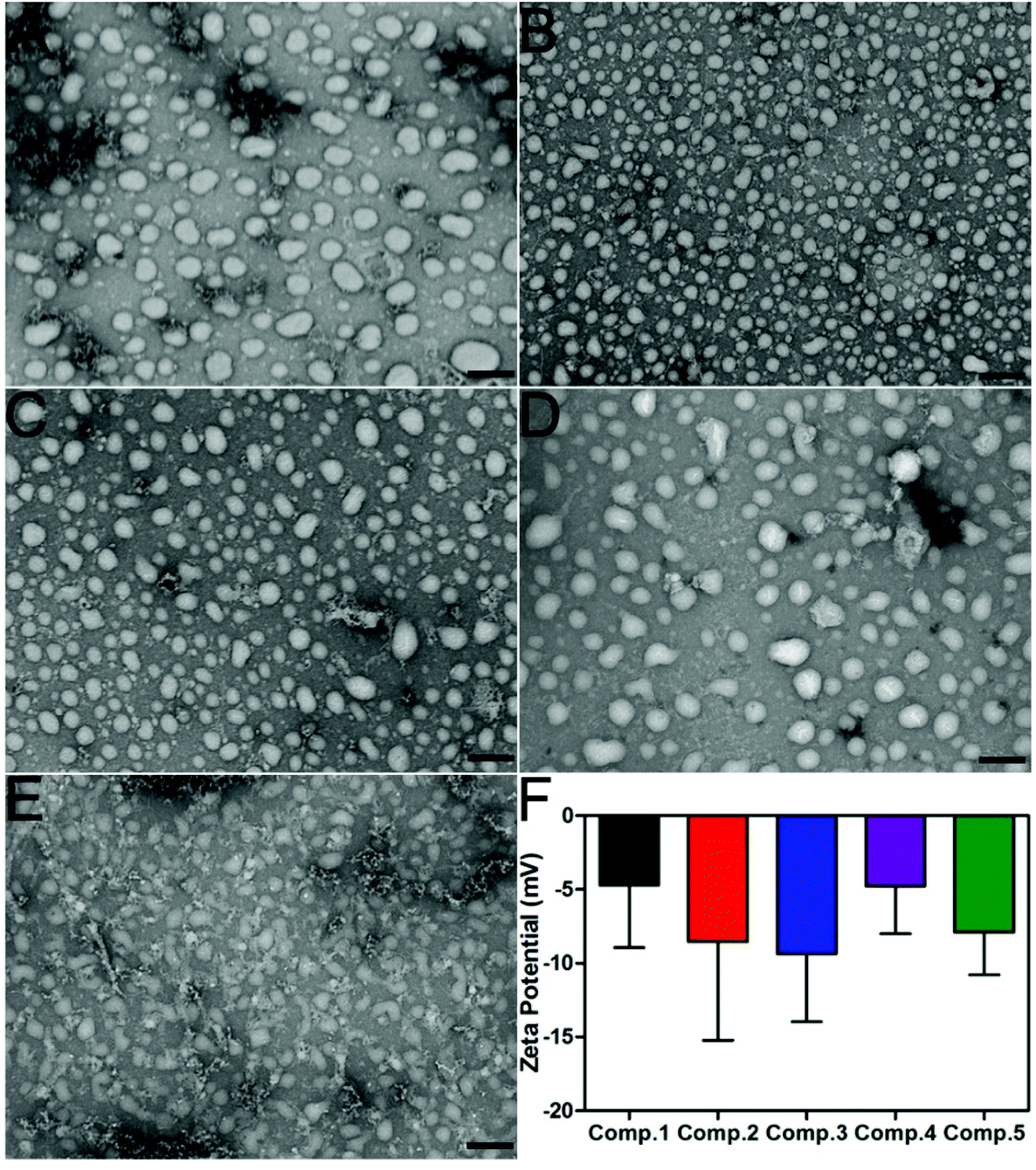
| | Fig. 2 (A–E) TEM images of nanoparticles formed by Comp. 1, Comp. 2, Comp. 3, Comp. 4, and Comp. 5 in PBS supplemented with 1% DMSO. (F) The Z-potential of Comp. 1–5 in PBS. Scale bar = 200 nm. | |
Self-assembly behavior
Comp. 1–5 could form transparent and clear solutions in PBS supplemented with 1% DMSO via the heating–cooling process, the pH of which was adjusted to 7.4–7.5 using concentrated Na2CO3 solution (Fig. S7†). Comp. 1–5 would form precipitates after overnight incubation while DMSO could increase their solubility and self-assembly. The critical aggregation concentration (CAC) values of Comp. 1–5 were approximately 75.90, 60.97, 50.04, 82.23, and 106.29 μM, respectively, suggesting that Comp. 1–4 showed similar assembly performances, while Comp. 5 without FFY showed a weaker self-assembly property. We then used transmission electron microscopy (TEM) to characterize the morphology of nanomaterials in the solutions (0.2 wt%). Fig. 2A–D indicate that Comp. 1–4 assembled into nanoparticles with different sizes, approximately 90–170, 50–90, 50–110, 50–160, and 60–90 nm in diameter for Comp 1, 2, 3, and 4, respectively. For Comp. 5, we observed nanospheres with a 60–80 nm diameter as well as amorphous structures in the sample, which indicated the relatively weaker self-assembly property of Comp. 5. Zeta potentials of the nanomaterials in the solutions of Comp. 1–5 were −4.715, −8.54, −9.36, −4.77, and −7.91 mV, respectively (Fig. 2F).
Enhanced cellular uptake and nuclear accumulation
To test whether the nanomaterials could assist the entry of anti-cancer drugs into the nucleus, we investigated the subcellular localization of HCPT in HepG2 cells using confocal laser scanning microscopy (CLSM). The cell nucleus was stained red by Red Dot, and HCPT was represented by green fluorescence. As shown in Fig. 3C–E, we observed weak green fluorescence in the cytosome and rarely in the nucleus of the HepG2 cells 4 h after treatment with 200 μM of HCPT alone. For Comp. 1 and Comp. 3 treated cells, we observed bright green fluorescence in the cytosome, suggesting enhanced cellular uptake of the compounds. We also observed moderate green fluorescence in the nucleus, indicating significant nuclear accumulation of the compounds. Comp. 4- and Comp. 5-treated cells showed moderate green fluorescence in the cytosome but weak fluorescence in the nucleus. These observations clearly suggest the importance of the self-assembling peptide FFY and the nuclear-targeting PMI peptide in the cellular uptake and nuclear accumulation properties of the nanomaterials. The enhanced cellular uptake and nuclear accumulation properties might play a vital role in better inhibition of cancer cells and anti-tumor behavior of the nanomedicines as compared to free anti-cancer drugs.
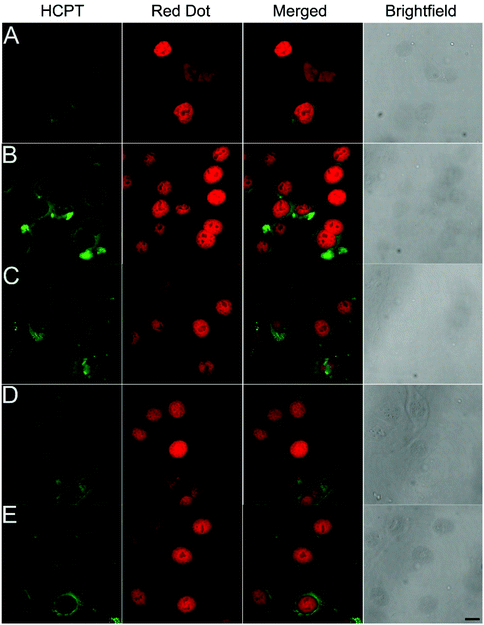 |
| | Fig. 3 Confocal laser scanning microscopy images of HepG2 cells after 4 h of treatment with 200 μM of HCPT (A), Comp. 1 (B), Comp. 3 (C), Comp. 4 (D), and Comp. 5 (E) (scale bar represents 25 μm). | |
Enhanced cancer cell inhibition capacity of nanomaterials
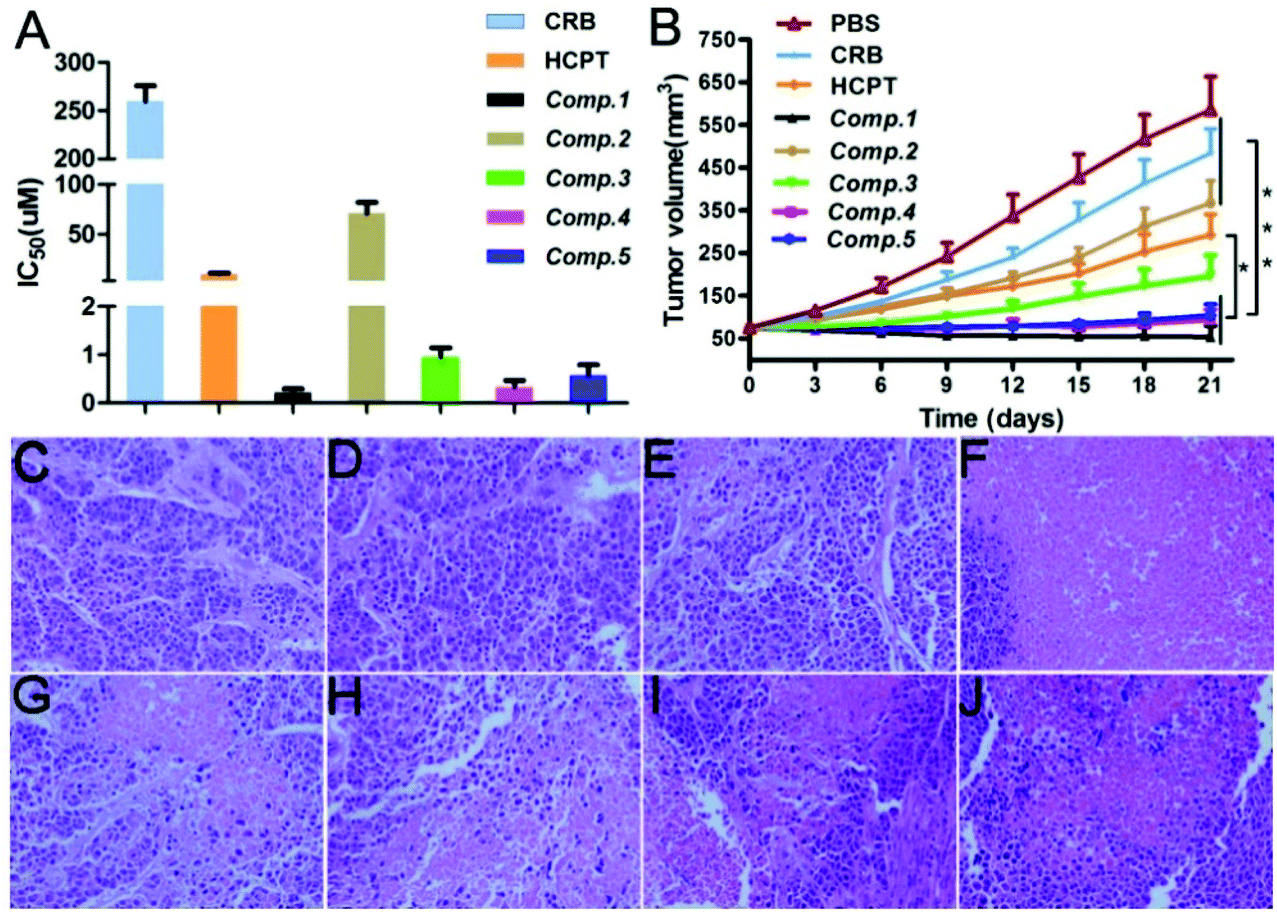
We assessed the HepG2 cell inhibition capacity of the different nanomaterials. As shown in Fig. 4A, the IC50 value of CRB was above 250 μM, and that of HCPT was about 9.88 μM. The incorporation of the self-assembling tripeptide FFY and the PMI peptide led to an enhancement of the HepG2 cell inhibition capacity of the free drug, as evidenced by the IC50 value of Comp. 2 and Comp. 3, which was 70.79 and 0.94 μM, respectively. Comp. 1 possessed the best HepG2 cell inhibition capacity, with an IC50 value of 0.21 μM. For Comp. 4 with the scrambled PMI peptide and Comp. 5 without the self-assembling tripeptide FFY, the IC50 value was about 0.47 and 0.57 μM, respectively. These observations further demonstrate the importance of molecular self-assembly, the nuclear-targeting PMI peptide, and dual-drug systems in enhancing the cancer cell inhibition capacities of nanomaterials.
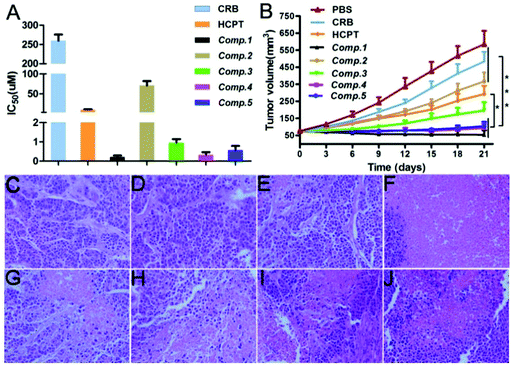 |
| | Fig. 4 (A) Cell viability of HepG2 cells treated with CRB, HCPT, and Comp. 1–5 (n = 3), and (B) tumor inhibition effects of CRB, HCPT, and Comp. 1–5 for HepG2 tumor (n = 5). (C–J) H&E staining of the cells treated with PBS, CRB, HCPT, and Comp. 1–5. Scale bar = 100 μm. The data were analyzed by one-way ANOVA. | |
Enhanced anti-tumor effects of nanomaterials in vivo
We evaluated the tumor inhibition capacity of different nanomaterials in vivo using a HepG2 tumor inhibition assay. When the volume of the tumor reached approximately 75 mm3, we injected different nanomaterials through the tail vein at the safety dosage shown in Table S1† every three days (total administration time = 3). We observed that the tumors in the Comp. 1 group grew more slowly as compared to that in other groups. The final tumor volume was about 34.1 mm3 and decreased to 6.1% compared to the untreated group (Fig. 4B). The final tumor volume of mice receiving CRB, HCPT, Comp. 2, Comp. 3, Comp. 4, and Comp. 5 was about 403.8, 233, 303.1, 144.3, 67.8, and 84.65 mm3, respectively. These results together with the in vitro cancer cell inhibition assay results clearly suggest that nanomedicines of Comp. 1 have the best anti-cancer property.
H&E staining
We used hematoxylin and eosin (H&E) staining to evaluate the degree of cancer cell apoptosis in tumors after treatment with different compounds. The tumors were isolated from HepG2 tumor-bearing Balb/c nude mice at day 21, and then the assay was performed. Hematoxylin stained the nucleus purple and eosin stained the cytoplasm pink. For the samples, a pink area without a purple nuclear structure indicated the apoptosis of tumor cells after treatment. As shown in Fig. 4C–J, we observed dense nuclei and uniform cytoplasm for the PBS group tumor tissues, suggesting weak apoptosis of cancer cells (Fig. 4C). For the samples in the CRB and HCPT groups (Fig. 4D and E), we found similar observations, suggesting that administration of a single anti-cancer drug does not lead to significant apoptosis of cancer cells. The Comp. 2 and Comp. 3 group tissues (Fig. 4G and H) showed much smaller amounts of purple nuclear structures and more pink areas as compared to the PBS group tissues. These observations indicated that the incorporation of the self-assembling tripeptide FFY and the PMI peptide to the free drugs would result in an enhanced tumor inhibition capacity. The combination of dual anti-cancer drugs led to further enhanced cancer cell apoptosis, as indicated by the results in Fig. 4H and I. For tumor tissues in the Comp. 1 group, we observed nearly all pink areas and hardly purple nuclear structures, suggesting significant cancer cell apoptosis in the sample. These observations clearly demonstrate that the combination of the self-assembling tripeptide FFY, the PMI peptide, and dual anti-cancer drugs would lead to the development of nanomedicines with significantly enhanced anti-cancer capacity.
TUNEL assay
We performed the TUNEL assay to monitor the degree of cancer cell apoptosis in tumors. DAPI was used to stain the nucleus blue, while the green fluorescence from FITC represented apoptotic cells. As shown in Fig. 5A–H, the PBS group tissues showed no green fluorescence, indicating that no apoptosis occurred. The CRB and HCPT group tissues (Fig. 5B and C) showed weak green fluorescence, suggesting that the administration of a single anti-cancer drug slightly induced cancer cell apoptosis. In contrast, the Comp. 2 and Comp. 3 group tissues showed bright green fluorescence, suggesting that the nanomedicines consisting of the self-assembling tripeptide FFY and the PMI peptide significantly induced cancer cell apoptosis. For the Comp. 4 and Comp. 5 group tissues, we observed much brighter green fluorescence, suggesting that the dual anti-cancer nanomedicines led to enhanced cancer cell apoptosis. However, as compared to the Comp. 4 and Comp. 5 groups, the intensity of green fluorescence in the Comp. 1 group was weaker due to extensive cell necrosis in addition to cell apoptosis, which led to weaker cell staining (Fig. 5D). These results suggest that the combination of dual anti-cancer drugs, the self-assembling FFY tripeptide, and the nuclear localizing PMI peptide will lead to the development of nanomedicines with enhanced anti-cancer capacity.
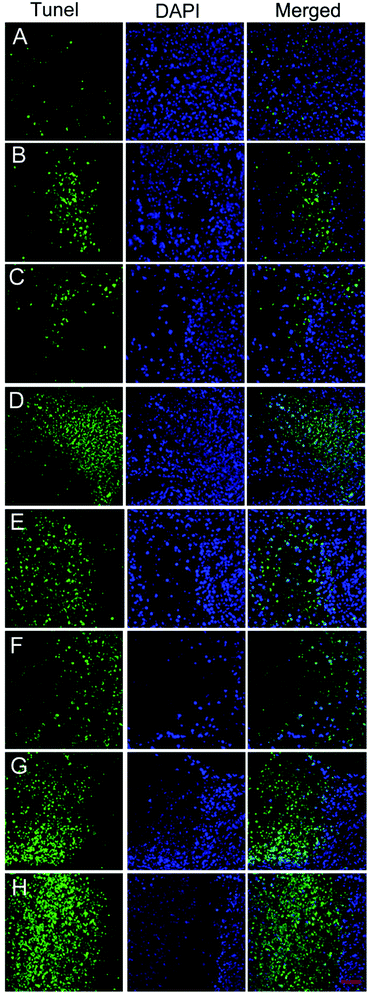 |
| | Fig. 5 (A–H) TUNEL staining of tumor tissues treated with PBS, CRB, HCPT, and Comp. 1–5, respectively. Scale bar = 50 μm. | |
Conclusions
Our study, in combination with other reports on peptide self-assembly, demonstrates that a self-assembling peptide is a universal platform for constructing multifunctional nanomedicines. In this study, we constructed a nanomedicine by integrating an efficient self-assembling tripeptide FFY, a nuclear localizing PMI peptide, and dual anti-cancer drugs HCPT and CRB. The nanomedicine could efficiently deliver dual anti-cancer drugs into the cell nucleus, and significantly inhibit cancer cells both in vitro and in vivo. We believe that our study will stimulate the development of novel nanomedicines for counteracting issues such as low anti-tumor efficacy and drug resistance of general methodologies during chemotherapy.
Conflicts of interest
The authors have declared that no competing interests exist.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81972768, 81270553, 81300363, 81521004 and 81572262), Young and middle-aged academic leaders of Jiangsu University Blue Project 2019, Support Plan 2019 for excellent young and middle-aged teachers of Nanjing Medical University, Fund of State Key Laboratory of Reproductive Medicine, Nanjing Medical University (SKLRM-K201706), Jiangsu Youth Medical Talents (QNRC2016580), Jiangsu Province's Key Provincial Talents Program (ZDRCA2016028), and 333 high class “Talented Man Project” (BRA2016516).
References
- J. Chen, J. Ding, Y. Wang, J. Cheng, S. Ji, X. Zhuang and X. Chen, Adv. Mater., 2017, 29, 1701170 CrossRef.
- G. Liang, Z. Yang, R. Zhang, L. Li, Y. Fan, Y. Kuang, Y. Gao, T. Wang, W. W. Lu and B. Xu, Langmuir, 2009, 25, 8419–8422 CrossRef CAS.
- F. Zhao, M. L. Ma and B. Xu, Chem. Soc. Rev., 2009, 38, 883–891 RSC.
- F. Azmi, A. A. H. Ahmad Fuaad, M. Skwarczynski and I. Toth, Hum. Vaccines Immunother., 2014, 10, 778–796 CrossRef CAS.
- Z. Luo, Q. Wu, C. Yang, H. Wang, T. He, Y. Wang, Z. Wang, H. Chen, X. Li and C. Gong, Adv. Mater., 2017, 29, 1601776 CrossRef.
- Y. Wen and J. H. Collier, Curr. Opin. Immunol., 2015, 35, 73–79 CrossRef CAS.
- H. Liu, Y. Hu, H. Wang, J. Wang, D. Kong, L. Wang, L. Chen and Z. Yang, Soft Matter, 2011, 7, 5430–5436 RSC.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS.
- C. Yang, D. Li, Z. Liu, G. Hong, J. Zhang, D. Kong and Z. Yang, J. Phys. Chem. B, 2012, 116, 633–638 CrossRef CAS.
- W.-J. Chung, J.-W. Oh, K. Kwak, B. Y. Lee, J. Meyer, E. Wang, A. Hexemer and S.-W. Lee, Nature, 2011, 478, 364–368 CrossRef CAS.
- Y. Hu, H. Wang, J. Wang, S. Wang, W. Liao, Y. Yang, Y. Zhang, D. Kong and Z. Yang, Org. Biomol. Chem., 2010, 8, 3267–3271 RSC.
- S. I. Stupp, Nano Lett., 2010, 10, 4783–4786 CrossRef CAS.
- P. Chen, Y. Ma, Z. Zheng, C. Wu, Y. Wang and G. Liang, Nat. Commun., 2019, 10, 1–10 CrossRef.
- Z. Feng, X. Han, H. Wang, T. Tang and B. Xu, Chem, 2019, 5, 2442–2449 CAS.
- Z. Feng, T. Zhang, H. Wang and B. Xu, Chem. Soc. Rev., 2017, 46, 6470–6479 RSC.
- H. Guo, F. Li, W. Xu, J. Chen, Y. Hou, C. Wang, J. Ding and X. Chen, Adv. Sci., 2018, 5, 1800004 CrossRef.
- P. Huang, Y. Gao, J. Lin, H. Hu, H.-S. Liao, X. Yan, Y. Tang, A. Jin, J. Song and G. Niu, ACS Nano, 2015, 9, 9517–9527 CrossRef CAS.
- C. Liang, L. Zhang, W. Zhao, L. Xu, Y. Chen, J. Long, F. Wang, L. Wang and Z. Yang, Adv. Healthcare Mater., 2018, 7, 1800899 CrossRef.
- W. Ma, A. G. Cheetham and H. Cui, Nano Today, 2016, 11, 13–30 CrossRef CAS.
- C. G. Pappas, R. Shafi, I. R. Sasselli, H. Siccardi, T. Wang, V. Narang, R. Abzalimov, N. Wijerathne and R. V. Ulijn, Nat. Nanotechnol., 2016, 11, 960 CrossRef CAS.
- H. Su, W. Zhang, H. Wang, F. Wang and H. Cui, J. Am. Chem. Soc., 2019, 141, 11997–12004 CrossRef CAS.
- H. Wang, Z. Feng and B. Xu, Chem. Soc. Rev., 2017, 46, 2421–2436 RSC.
- Q. Yao, F. Lin, X. Fan, Y. Wang, Y. Liu, Z. Liu, X. Jiang, P. R. Chen and Y. Gao, Nat. Commun., 2018, 9, 1–9 CrossRef.
- Y. Yuan, L. Wang, W. Du, Z. Ding, J. Zhang, T. Han, L. An, H. Zhang and G. Liang, Angew. Chem., Int. Ed., 2015, 54, 9700–9704 CrossRef CAS.
- J. Zhou, X. Du and B. Xu, Angew. Chem., Int. Ed., 2016, 55, 5770–5775 CrossRef CAS.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- U. Prabhakar, H. Maeda, R. K. Jain, E. M. Sevick-Muraca, W. Zamboni, O. C. Farokhzad, S. T. Barry, A. Gabizon, P. Grodzinski and D. C. Blakey, Cancer Res., 2013, 73, 2412–2417 CrossRef CAS.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS.
- L. Zhang, C. Jiang, F. Zeng, H. Zhou, D. Li, X. He, S. Shen, X. Yang and J. Wang, Biomater. Sci., 2020, 8, 2255–2263 RSC.
- D. Yu, N. Zhang, S. Liu, W. Hu, J. J. Nie, K. Zhang, B. Yu, Z. G. Wang and F. J. Xu, ACS Mater. Lett., 2020, 2, 550–556 CrossRef CAS.
- S. Ma, W. Song, Y. Xu, X. Si, S. Lv, Y. Zhang, Z. Tang and X. Chen, Nano Lett., 2020, 20, 2514–2521 CrossRef CAS.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. Chan, Nat. Rev. Mater., 2016, 1, 1–12 Search PubMed.
- A. G. Cheetham, P. Zhang, Y. A. Lin, R. Lin and H. Cui, J. Mater. Chem. B, 2014, 2, 7316–7326 RSC.
- W. Ma, H. Su, A. G. Cheetham, W. Zhang, Y. Wang, Q. Kan and H. Cui, J. Controlled Release, 2017, 263, 102–111 CrossRef CAS.
- Y. Cai, H. Shen, J. Zhan, M. Lin, L. Dai, C. Ren, Y. Shi, J. Liu, J. Gao and Z. Yang, J. Am. Chem. Soc., 2017, 139, 2876–2879 CrossRef CAS.
- W. H. Chen, G. F. Luo and X. Z. Zhang, Adv. Mater., 2019, 31, 1802725 CrossRef.
- L. Pan, Q. He, J. Liu, Y. Chen, M. Ma, L. Zhang and J. Shi, J. Am. Chem. Soc., 2012, 134, 5722–5725 CrossRef CAS.
- E. C. Sutton, C. E. McDevitt, J. Y. Prochnau, M. V. Yglesias, A. M. Mroz, M. C. Yang, R. M. Cunningham, C. H. Hendon and V. J. DeRose, J. Am. Chem. Soc., 2019, 141, 18411–18415 CrossRef CAS.
- R. Tang, M. Wang, M. Ray, Y. Jiang, Z. Jiang, Q. Xu and V. M. Rotello, J. Am. Chem. Soc., 2017, 139, 8547–8551 CrossRef CAS.
- B. Hu, D. M. Gilkes and J. Chen, Cancer Res., 2007, 67, 8810–8817 CrossRef CAS.
- M. Liu, C. Li, M. Pazgier, C. Li, Y. Mao, Y. Lv, B. Gu, G. Wei, W. Yuan and C. Zhan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14321–14326 CrossRef CAS.
- M. Liu, M. Pazgier, C. Li, W. Yuan, C. Li and W. Lu, Angew. Chem., Int. Ed., 2010, 49, 3649–3652 CrossRef CAS.
- M. Pazgier, M. Liu, G. Zou, W. Yuan, C. Li, C. Li, J. Li, J. Monbo, D. Zella and S. G. Tarasov, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4665–4670 CrossRef CAS.
- C. Liang, X. Yan, R. Zhang, T. Xu, D. Zheng, Z. Tan, Y. Chen, Z. Gao, L. Wang and X. Li, J. Controlled Release, 2020, 317, 109–117 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) is available. See DOI: 10.1039/d0bm00971g |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Wenzhou
Ding‡
ab,
Guoyong
Han‡
ab,
Wei
You
ab,
Wen
Gao
a,
Hongbing
Shen
b,
Jinhai
Tang
a,
Qiyun
Tang
*a and
Xuehao
Wang
*ab
*ab,
Wenzhou
Ding‡
ab,
Guoyong
Han‡
ab,
Wei
You
ab,
Wen
Gao
a,
Hongbing
Shen
b,
Jinhai
Tang
a,
Qiyun
Tang
*a and
Xuehao
Wang
*ab