DOI:
10.1039/D1AY01151K
(Paper)
Anal. Methods, 2021,
13, 4220-4227
An optimised small-scale sample preparation workflow for historical dye analysis using UHPLC-PDA applied to Scottish and English Renaissance embroidery†
Received
6th July 2021
, Accepted 12th August 2021
First published on 7th September 2021
Abstract
A sample preparation workflow for historical dye analysis requiring less sample has been developed. Samples as small as 0.01 ± 0.005 mg have been successfully analysed and high percentage recoveries (>85%), more automation and shorter preparation time have been achieved using filtration by centrifugation and only one manual transfer. The optimised workflow based on 96 well plates together with the shorter UHPLC method developed makes dye analysis data collection faster from unprocessed sample to result, facilitating the creation of larger datasets and application of chemometric approaches. The method was evaluated on 85 samples from 12 dye sources (RSD < 5.1%, n = 5) as well as 22 samples from a 17th century embroidered stomacher from the National Museums Scotland (NMS) collection.
Introduction
Historical dye analysis is an important field within heritage science as it not only informs curators and conservators how to best care for and display the textiles in museum collections but can also give a greater understanding of the materials and manufacturing techniques available in the past.1–3 Dye analysis is challenging due to the small sample sizes, low concentrations of dyestuffs, degradation, and complex dye mixtures often present in historical samples, which thus require sensitive analytical techniques.4–6 Micro-destructive techniques such as ultra-high performance liquid chromatography (UHPLC) and mass spectrometry (MS) are the two main analytical methods used due to their sensitivity, but their major disadvantage is that the sample is consumed during analysis.7,8 Since the objects analysed are often of great cultural significance, it is vital that any new sample preparation methods minimise the amount needed for meaningful results.
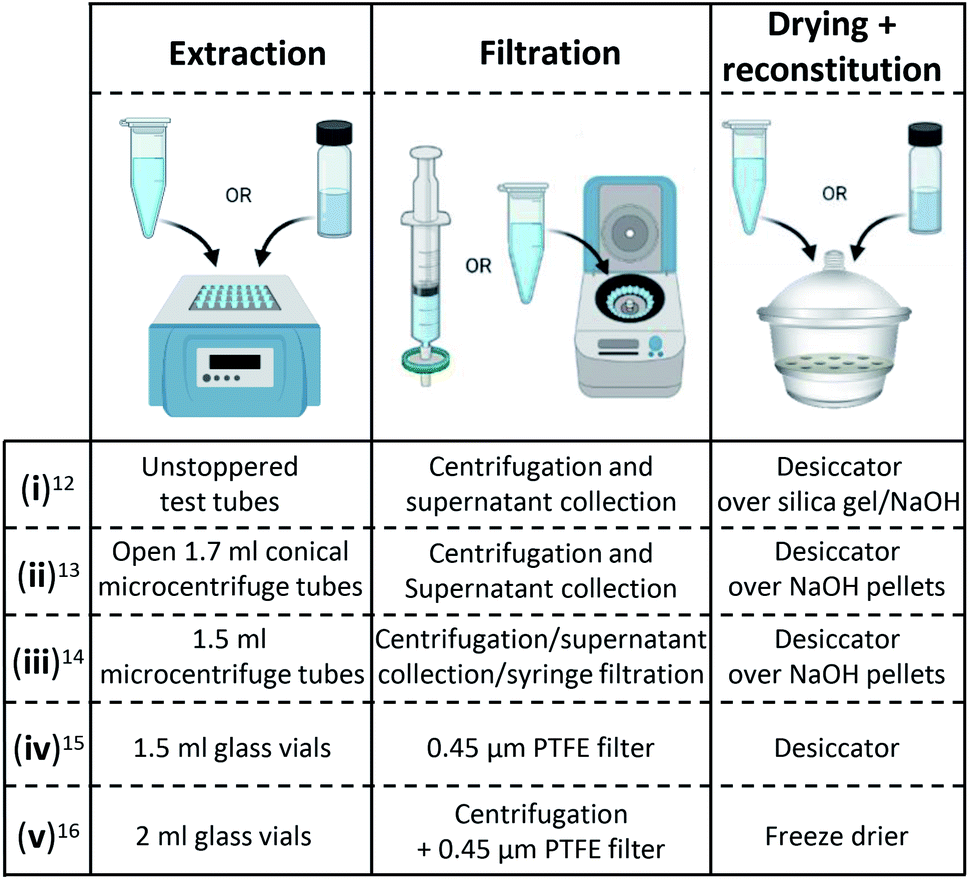
The focus of many dye analysis method papers in recent years has been on the extraction solvent and its impact on various dyestuffs and their markers;9–11 very few studies have focussed on the sample preparation workflow itself. Current sample preparation methods for LC and MS dye analysis12–16 (Fig. 1) follow a set of fundamental steps:6 extraction of the dye molecule from the fibre or mordant; filtration; drying; and reconstitution. These workflows are both labour- and resource-intensive, relying heavily on the preparation and analysis of individual samples.11,13,14 As a result, fewer studies in the field are underpinned by replicate analysis as compared to similar studies in biology and forensic science that face the same challenges. Filtration by centrifugation is routinely used in these fields; in proteomic investigations it is utilised in filter-aided sample preparation (FASP) protocols,17–19 and in forensics, it has been in use for DNA samples since the mid-90s.20,21 A workflow based on 96 well plates and filtration by centrifugation was developed to integrate these more automated bulk approaches into the dye analysis field (Fig. 2). The developed approach requires less sample, and the introduced error is reduced; making it possible to collect larger data sets and employ more in-depth statistical analysis than is currently available for natural dye data. Some studies using a chemometric approach, such as applying multivariate data analysis and principal component analysis, have been published within historical dye analysis in recent years,22–24 but these are still fairly uncommon. The transfer of similar workflows to those used in biology and forensic science into the dye analysis field would thus not only give rise to increased time efficiency and data replication but also the possibility of utilising modern data mining techniques.
 |
| | Fig. 1 Selection of sample preparation workflows for LC/LC-MS used in the dye analysis field. Images created with https://www.BioRender.com. | |
 |
| | Fig. 2 Developed sample preparation workflow for LC/LC-MS. Images created with https://www.BioRender.com. | |
Experimental methods
Instrumentation
Extraction was carried out using a Stuart Block heater (SBH2000D) with a 96 well plate aluminium insert. Filtration was carried out using a Thermo Scientific Megafuge 8 centrifuge and M10 Microplate swinging bucket rotor. All analyses were performed on a Waters Acquity UPLC™ system comprising the Waters Binary Gradient Manager with Waters Sample Manager incorporating a Waters Column Heater with sample detection by a Waters PDA detector (210–800 nm). Data were collected by Waters Empower 3 software and processed with Origin 9.5 (OriginLab, Northampton, MA, USA).
Materials
The standards fisetin (1), sulfuretin (2), luteolin (3), genistein (4), apigenin (5), chrysoeriol (6), diosmetin (7), alizarin (8) and prunetin (9) were obtained from Extrasynthese. Dyed reference samples were used to evaluate the method. These included silk and wool samples dyed with weld, dyer's greenweed (DGW), madder and brazilwood as well as wool samples dyed with oak galls and woad on weld, together with silk samples dyed with young fustic and safflower prepared during the COST G8 project “non-destructive testing and analysis of museum objects” (FP5)25 and the Monitoring of Damage to Historic Tapestries project (MODHT) (FP5, EC contract number EVK4-CT-2001-00048).26,27 Additional wool samples of young fustic, turmeric, kermes and logwood from the NMS scientific lab were also used. Oxalic acid (OA), dimethyl sulfoxide (DMSO), methanol and water (HPLC grade) were purchased from Honeywell and formic acid (FA) (98–100%) was obtained from Merck Millipore. Additionally, well plates (AB-2800, Thermo Scientific), filter plates (350 μL, 0.2 μm WWPTFE, Acroprep Adv, Pall Corporation), 96 well plate silicone sealing mats (Starlab) and Zone-free™ Sealing Films for 96 well plates were used. The developed method was applied to a range of reference materials dyed with natural dyestuffs at the National Museums Scotland (NMS) science laboratory as well as an embroidered Renaissance stomacher (Acc. No. A.1962.1067) from the NMS collection.
Extraction
Weighed samples (typically 0.01–0.2 mg ± 0.005 mg) were placed on a 96 well plate. 50 μL MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3COCH3:H2O:oxalic acid (OA) (1 M aq) (30:30:40:1 v/v/v/v)14,28 was added to each sample and the well plate covered with a silicone cover. The well plate was heated on a heat block at 60 °C for 30 min and left to cool to room temperature. The samples were then pipetted into a filtration plate. For samples suspected to contain indigoid dyestuffs, a second extraction was performed; 25 μL DMSO was added to each well and the well plate covered with a silicone cover. The well plate was heated on a heat block at 80 °C for 15 min10 and left to cool to room temperature before the DMSO solvent was transferred to a second filtration plate.
:CH3COCH3:H2O:oxalic acid (OA) (1 M aq) (30:30:40:1 v/v/v/v)14,28 was added to each sample and the well plate covered with a silicone cover. The well plate was heated on a heat block at 60 °C for 30 min and left to cool to room temperature. The samples were then pipetted into a filtration plate. For samples suspected to contain indigoid dyestuffs, a second extraction was performed; 25 μL DMSO was added to each well and the well plate covered with a silicone cover. The well plate was heated on a heat block at 80 °C for 15 min10 and left to cool to room temperature before the DMSO solvent was transferred to a second filtration plate.
Filtration
Following oxalic acid extraction14,28.
The filtration plate was placed over a receiving 96 well plate and the set-up centrifuged at 1500 rpm (300 × g) for 5 min. The samples were washed with 25 μL MeOH:H2O 1:1 v/v twice and centrifuged at 1500 rpm (300 × g) for 5 min after each wash.
Following DMSO extraction10.
The filtration plate was placed over a receiving 96 well plate and the set-up centrifuged at 1500 rpm (300 × g) for 7.5 min. The samples were then covered with sealing film and either analysed immediately by UHPLC or stored in the fridge until analysis.
Drying and reconstitution
The receiving plate with the filtered OA extracts was placed in a desiccator over P2O5 and dried completely (ca. 2–3 h). The extracts were then reconstituted using 25 μL 0.1% formic acid (FA) in MeOH:H2O 1:1 v/v or MeOH:H2O 1:3 v/v before UHPLC analysis.
UHPLC method
The UHPLC method developed used a Cortex BEH C18 1.6 μm column, 90 Å, 75 × 2.1 mm (length × i.d.) with an in-line filter. The total run time was 13.92 min at a flow rate of 400 μL min−1 and the column was maintained at 45 ± 1 °C. A binary solvent system was used; A = H2O + 0.05% FA and B = MeOH + 0.05% FA. The elution program was isocratic for 1.17 min (77A:23B) then a linear gradient from 1.17 min to 10.92 min (10A:90B) before recovery of the initial conditions over 0.5 min and equilibration over 2.5 min. The method was evaluated using a solution of standards 1–9 at seven concentrations ranging between 0.1 μg mL−1 and 20 μg mL−1 to evaluate the repeatability, resolution, linearity, limit of detection and limit of quantification. The repeatability was found to be <0.013 min (n = 6) for all chromophores. All compounds used in the solution were completely separated, except for 6 and 7 which have a Rs value of 1.17 (Fig. 3). However, these regioisomeric methylated compounds are difficult to fully separate even on longer chromatographic methods,29 so the level of separation obtained was considered acceptable. The limit of detection (LoD) and limit of quantification (LoQ) was calculated by the signal-to-noise-method, where LoD was defined as needing to be 3.3 larger than the noise and LoQ as 10 larger than the noise. The noise was defined as the average signal obtained in blank samples (n = 3) around the retention time (±0.25 min) of each analytical peak.30,31 The LoD for compounds 1–9 were determined to be in the range of 0.11 ± 0.03 (4) to 0.44 ± 0.07 μg mL−1 (5) and LoQ in the range of 0.32 ± 0.03 (4) to 1.33 ± 0.07 μg mL−1 (5).
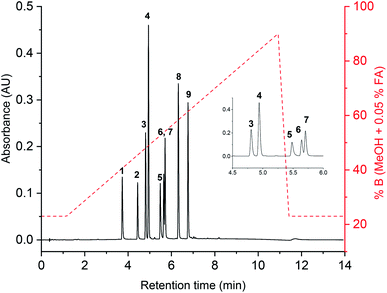 |
| | Fig. 3 Chromatogram at 254 nm of compound 1–9 (20 μg mL−1) with gradient overlayed in red. Zoomed in inset in top right corner shows extent of co-elution of 6 and 7. | |
Results and discussion
Extraction
Several extraction protocols using various mild extraction solvents have been published in recent years.9,32–34 In this study, the milder OA extraction method using MeOH:CH3COCH3:H2O:OA (1 M aq) (30:30:40:1 v/v/v/v) solution was chosen due to its high yield relative to other mild extraction solvents,14,35 its preservation of the glycosides of both weld and madder species11,14 and its low toxicity. The OA extraction was followed by a DMSO extraction for any dyestuff that cannot be extracted with an aqueous solution, e.g. vat dyes such as indigo. The effect of the order of extractions was investigated using triplicates of green MODHT references dyed with woad on weld as well as MODHT references dyed with weld on woad. No differences in peak area μg−1 were found. The order of OA then DMSO extraction was thus chosen so a direct comparison of the OA result between samples not requiring DMSO extraction (e.g. yellow) and samples needing DMSO extraction (e.g. green) could be made. The extracts were not combined in the drying step to reduce the number of transfers and make it possible to use the same method preparation for LC analyses as well as more DMSO-sensitive techniques such as MS.
Filtration
Filtration linearity.
The other factor tested for the filtration step was the linearity of the volume loss across the concentrations. Many dye analysis studies use relative peak areas, and it has recently been suggested that the ratio of aglycone:glycoside for individual dye compounds is important for species identification.11,16,22,39 It is therefore important that the sample preparation method does not affect the relative ratio of the chromophores in the sample at any concentration at the chosen wavelength of the analysis. To test the linearity of the volume loss, the peak area of the unfiltered vs. the peak area of the filtered was plotted and the intercept fixed at 0 (Fig. 4, middle). All chromophores show linearity of volume loss (R2 = 0.9932–0.9985), meaning that the volume loss is consistent across all concentrations. This level of linearity demonstrates that the sample preparation method is unlikely to impact the relative aglycone:glycoside ratio of the tested dyestuffs at any concentration, meaning that this ratio can be used in further research. Overall, the percentage recoveries of compounds 1–9 at 20 and 0.5 μg mL−1 show an average of 93% (range 87–100%) with an average relative standard deviation of 7.7% (range 3.8–13%) and all compounds show a linear volume loss (>0.9932). This means that the filtration step can be used in dye analysis studies with confidence.
Solubility
The last factor that will influence the efficiency of the sample preparation method is the reconstitution solvent. High solubility of all dyestuffs in the reconstitution solvent is necessary to increase the concentration of the analyte and thus minimise the sample size required. The reconstitution solvent must thus be chosen with care and ideally be MS compatible and show low toxicity. The last point is the reason DMSO but not DMF was tested. The solubility power of four different solvents (DMSO, 10% DMSO in MeOH:H2O 1:1 v/v, MeOH:H2O 1:1 v/v and 0.1% FA in MeOH:H2O 1:1 v/v) were tested by the drying of 50 μL unfiltered solution of compounds 1–9 (20 μg mL−1) in a desiccator and reconstitution at two different volumes (25 μL, 50 μL) for all four solvents. The experiment was performed in triplicate and the peak areas compared to each other and a control of 50 μL of the initial, unfiltered solution of compounds 1–9 (Fig. 4, right). Attempts to decrease the reconstitution volume further to 10 μL were found to lack reproducibility. The increase in concentration seen for all solvents in the 25 μL run (ca. ×2) compared to the control sample highlights the impact of the reconstitution step and 25 μL was thus chosen as the reconstitution volume. The error of all solvents for all standards were <5%, so 0.1% FA in MeOH:H2O 1:1 v/v was chosen as the reconstitution solvent based solely on its better recovery. In some instances, particularly with the flavonoid glycosides, the peaks obtained were not symmetrical with any of the solvents tested and instead MeOH:H2O 1:3 v/v was found to be the reconstitution solvent that gave the most symmetrical peaks.
Bulk experiment
To test the method's efficiency on a variety of natural dyestuffs as well as the bulk capacity of the method, a total number of 85 samples dyed with 12 dyestuffs were simultaneously prepared and analysed following the procedure in the experimental. To see if the fibre had any impact on the analysis, 5 of the dyestuffs were investigated on both wool and silk. The relative percentage peak area of the key component of each of the dyestuffs is plotted and tabulated in Fig. 5C to show the repeatability of the method. Madder (Rubia tinctorum L.) on silk is used as an example (Fig. 5A and B) to show how the relative percentage peak areas were calculated. The peaks in the chromatograms were integrated using ApexTrack in Empower3™. For the detection: the liftoff and threshold percentages were both set at 0.5% and for the integration: the minimum area and height of the smallest chromophore of interest of each dyestuff was set as the integration limit (in the case of madder this was lucidin primeveroside (Fig. 5A and B)). The chromatograms were monitored at the various wavelengths that are commonly used for dyestuff identification (table, Fig. 5C). The OA extracts for all samples were used except for the investigation of indigotin in woad on weld, which used the DMSO extract. Woad on weld was used instead of woad alone so the chromophores present in weld could be seen for comparison. For these samples, the peak areas of the yellow compounds were extracted at 350 nm and the peak area of indigotin was extracted at 630 nm and these were used to calculate the relative percentage and variability of indigotin in the samples. For all samples, the peak areas of the chromophores were normalised to 100% and the average relative percentage peak area of the key chromophore (n = 5) was used in Fig. 5C. The characterising component was chosen based on the literature and is not always the most abundant, explaining the variety in percentage areas found for each dyestuff. The repeatability of the method was shown by the low standard deviation seen across all reference samples, with the highest standard deviation being 4.2%. One exception, turmeric (5.1%) can be explained by the partial co-elution of curcumin with bisdemethoxycurcumin and demethoxycurcumin, which are all abundant in turmeric extracts.40 No major difference in repeatability between the wool (Fig. 5C(a–j)) and silk (Fig. 5C(k–q)) can be seen, meaning that the method is efficient on both fibre types. The low standard deviations also suggest that the method can be used on all dye classes: vat dyes (woad on weld), direct dyes (safflower and turmeric) and mordant dyes (all others). The complete analysis of the 85 samples took <24 h including all the UHPLC analyses which were run overnight. This highlights the advantage of the method when a large number of samples is available. The low standard deviations also show the benefit of a more automated approach, meaning that less error is introduced overall.
 |
| | Fig. 5 (A) Chromatogram of madder on silk (Rubia tinctorum L.) monitored at 430 nm. (B) Average relative% peak areas of chromophores (n = 5) present in the chromatograms of madder on silk with the integration minimum area and height set on the lucidin primeveroside in each individual chromatogram. (C) The average relative% peak area of the key chromophore of each reference dyestuff used. The dyestuff, fibre, key chromophore, relative% peak area, standard deviation and wavelength used can be seen in the table. | |
Case study
The method has been applied to historical samples taken from a collection of Scottish and English Renaissance embroideries housed at the NMS. Here, 22 samples from a stomacher with embroidery made in Scotland or England around 1600 (Acc. No. A.1962.1067, Fig. 6) will be used as a case study. The embroidery encapsulates the skill and taste on the British Isle during the 17th century, which can be seen as one of the greatest periods for embroidery in Scotland and England.41,42 The embroidery is made using yellow, green, brown, blue and pink silk threads in a repetitive, swirling design of flowers and pea pods. The flora is surrounded by a backdrop of sequins and gilded metal thread. The pea pods and some petals are expertly embroidered to give a 3-dimensional effect. Thus, the stomacher is an excellent example of the creativity and skill present in Renaissance Scotland and England which was needed to produce embroidered items of this quality.43,44 Visual inspection also shows that the embroidery was not originally made as a stomacher, but rather up-cycled to its current form later in the 17th century. Potentially, it was an embroidered jacket re-used when fashion changed. The object itself thus gives an idea of how people of the past interacted with textiles, indicating a similar mindset as the growing focus on up-cycling of clothes and more sustainable fashion in modern times. The analysed samples (0.01–0.07 ± 0.005 mg) included 2 yellow samples, 11 green samples, 1 brown sample, 2 blue samples and 6 pink samples. All 13 yellow and green samples show the presence of the flavones luteolin (3), apigenin (5), chrysoeriol (6) and related glycosidic compounds (Fig. 6A), which characterise weld as the dyestuff used for all yellow and green samples (+indigotin for the green). The pre-dominant use of weld is not unexpected for this time period in Scotland or England as recent studies on early English tapestries and Turkeywork table carpets ca. 1570–1620 similarly show extensive use of weld.16,45,46 The brown sample was identified as young fustic with traces of madder by the presence of the chromophores fisetin (1), sulfuretin (2), and residual amounts of alizarin (8) and purpurin (Fig. 6B). Young fustic was a lower quality yellow since it has poorer lightfastness than weld and was thus a cheaper alternative. There is analytical evidence that it was in use for the core fibre of metal threads during the Renaissance in European and English tapestries and it has also been found mixed with weld or DGW in small English tapestries to achieve orange-yellow hues.1,29,47 The dyestuff composition of the sample analysed in this study, however, show the use of young fustic alone rather than mixed with weld. Most of the pink silk samples were found to be weighted with ellagic acid48 and dyed with Mexican cochineal (>95% carminic acid and presence of dcII compound49) and trace amounts of madder identified by the presence of alizarin (8) and purpurin (Fig. 6C). The use of silk thread dyed with cochineal would have been an expensive material,50 which strengthens the importance of the object and a taste for specific shades of pink in Scotland and England at the time, as it also was observed in high quality early English tapestries from the Burrell Collection in Glasgow, UK.29,51 Finally, one pink silk yarn was found to contain safflower (Fig. 6D), identified by the presence of carthamin and the additional colourless markers Ct1–Ct4, whose structures still remain to be fully characterised.52 The use of safflower in such an early Scottish or English piece is quite remarkable as it has only been hitherto reported in a few items, including small English tapestries woven in England (1579–1625).29,51 Thus its use, in close combination with cochineal, in contemporary embroideries might correspond to a Scottish or English Renaissance workshop practice.29 The variety of dyestuffs found in the embroidery is thus consistent with the range of materials identified in early English tapestries and suggests access to similar resources. The identification of expensive dyes in the object also raises the question whether it was professionally or domestically produced as there were many highly skilled noble women working domestically and producing high quality items.41,53 Currently, professional and domestic work cannot be confidently distinguished but the identification of the dyestuffs present could help with the assignment and give an idea of what supplies were available to the professional as well as the domestic embroiderer. To answer those questions, larger historical datasets are needed. Overall, the variety of dyestuffs identified in this historical object, including direct dyes such as safflower and VAT dyes such as indigo, showcases the applicability of this new method to investigate historical textiles.
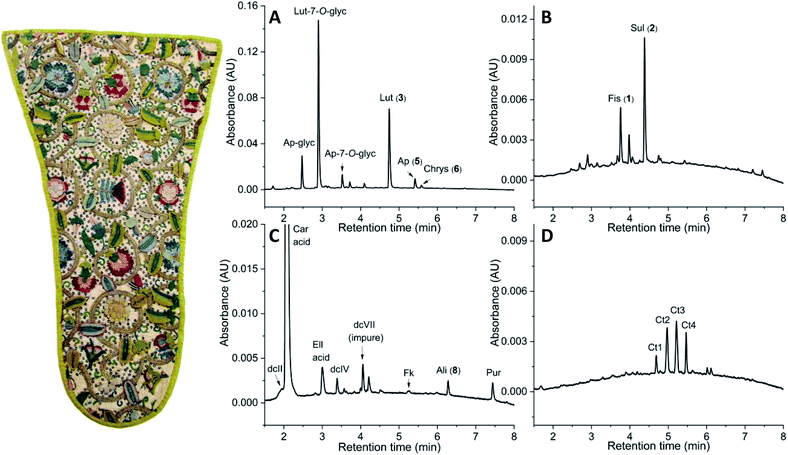 |
| | Fig. 6 Left: Embroidery designed with a range of coloured silk and metal threads, c. 1600 (Acc. No. A.1962.1067, ©National Museums Scotland). Right: (A) Chromatogram at 350 nm of sample containing weld characterised by markers: Ap-glyc = apigenin related glycoside, Lut-7-O-glyc = luteolin-7-O-glycoside, Ap-7-O-glyc = apigenin-7-O-glycoside, Lut (3) = luteolin, Ap (5) = apigenin, Chrys (6) = chrysoeriol. (B) Chromatogram extracted at 350 nm of sample containing young fustic characterised by markers: Fis (1) = fisetin, Sul (2) = sulfuretin. (C) Chromatogram extracted at 475 nm of sample containing Mexican cochineal, tannins and madder characterised by markers, dcII, Car acid = carminic acid, Ell acid = ellagic acid, dcIV, dcVII, Fk = flavokermesic acid, Ali (8) = alizarin, Pur = purpurin. (D) Chromatogram extracted at 300 nm of sample containing safflower characterised by markers: Ct1, Ct2, Ct3, Ct4. | |
Conclusions
An optimised sample preparation workflow and a UHPLC-PDA method for natural dye analysis have been developed. The method was shown to have a good percentage recovery and allows rapid acquisition of high-quality results from a smaller mass. Requiring only one transfer and with the filtration based on centrifugation, a smaller extraction volume (50 μL) is used and less error is introduced. The method was evaluated on 85 samples of 12 different dye sources simultaneously with the largest RSD being 5.1%. It was then successfully applied to 22 samples from a 17th century embroidered stomacher, in which weld, young fustic, cochineal, tannins, madder, safflower and indigo could be identified with confidence. The range of dyes found are consistent with the dye sources previously characterised in contemporary English tapestries, which suggests the availability of similar materials to both the embroiderer and tapestry maker in Renaissance Scotland and England. Regardless if the case study object was professionally or domestically produced, the expensive dyes used and the visual alteration of the embroidery into a stomacher at a later date, highlights the appreciation of the artistry, time and skill it took to produce and show how it has been a treasured possession throughout its history. More detailed conclusions are reliant on larger historical datasets, which this study has demonstrated can be obtained confidently with the developed sample preparation workflow combined with the shorter UHPLC method. The hope is therefore that the approach presented will be beneficial to the dye analysis field in the quest to address questions beyond merely dyestuff identification.
Author contributions
LT, ANH, CLM, ES designed the experiments. ES carried out the experiments. ES, HW sampled the historical objects. LT, ANH, ES undertook the data treatment. ES wrote the manuscript. LT, ANH, CLM, HW read and edited the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
ES, LT thank David Peggie (National Gallery London, UK) and Ester Ferreira (CICS TH Cologne, Germany) for the use of their dyed references from the NMS Scientific Lab, as well as Marei Hacke (Swedish National Heritage Board, Sweden) for the dyed references from the MODHT project and Irina Petroviciu (National Museum of Romanian History, Romania) for providing the dyed references from the COST G8 project. We also thank Witold Nowik (LRMH laboratory, France) for useful discussions on micro extraction. Financial assistance was provided through the Scottish Cultural Heritage Consortium AHRC CDP (studentship to ES, Grant ref. AH/S00176X/1).
References
-
D. Cardon, Natural Dyes: Sources, Traditions, Technology & Science, Archetype Books, London, 2007 Search PubMed.
-
J. H. Hofenk de Graaff, The Colourful Past, Archetype Books, London, 2007 Search PubMed.
- E. S. B. Ferreira, A. N. Hulme, H. McNab and A. Quye, Chem. Soc. Rev., 2004, 33, 329–336 RSC.
- M. Shahid, J. Wertz, I. Degano, M. Aceto, M. I. Khan and A. Quye, Anal. Chim. Acta, 2019, 1083, 58–87 CrossRef CAS PubMed.
- I. Degano, E. Ribechini, F. Modugno and M. P. Colombini, Appl. Spectrosc. Rev., 2009, 44, 363–410 CrossRef CAS.
- V. Pauk, P. Barták and K. Lemr, J. Sep. Sci., 2014, 37, 3393–3410 CrossRef CAS PubMed.
-
W. Nowik, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2013, DOI:10.1016/B978-0-12-409547-2.04724-7.
- I. Degano and J. La Nasa, Top. Curr. Chem., 2016, 374, 263–290 CrossRef PubMed.
- L. Ford, R. L. Henderson, C. M. Rayner and R. S. Blackburn, J. Chromatogr. A, 2017, 1487, 36–46 CrossRef CAS PubMed.
- D. Mantzouris, I. Karapanagiotis and C. Panayiotou, Microchem. J., 2014, 115, 78–86 CrossRef CAS.
- C. Mouri and R. Laursen, J. Chromatogr. A, 2011, 1218, 7325–7330 CrossRef CAS PubMed.
- J. Wouters, Stud. Conserv., 1985, 30, 119–128 CAS.
- X. Zhang and R. A. Laursen, Anal. Chem., 2005, 77, 2022–2025 CrossRef CAS PubMed.
- J. Wouters, C. M. Grzywacz and A. Claro, Stud. Conserv., 2011, 56, 231–249 CrossRef CAS.
- A. Manhita, T. Ferreira, A. Candeias and C. B. Dias, Anal. Bioanal. Chem., 2011, 400, 1501 CrossRef CAS PubMed.
- L. G. Troalen, A. S. Phillips, D. A. Peggie, P. E. Barran and A. N. Hulme, Anal. Methods, 2014, 6, 8915–8923 RSC.
- J. R. Wiśniewski, A. Zougman, N. Nagaraj and M. Mann, Nat. Methods, 2009, 6, 359–362 CrossRef PubMed.
- L. Switzar, J. van Angeren, M. Pinkse, J. Kool and W. M. A. Niessen, Proteomics, 2013, 13, 2980–2983 CrossRef CAS PubMed.
- J. Potriquet, M. Laohaviroj, J. M. Bethony and J. Mulvenna, PLoS One, 2017, 12, e0175967 CrossRef PubMed.
- M. R. Wilson, J. A. DiZinno, D. Polanskey, J. Replogle and B. Budowle, Int. J. Leg. Med., 1995, 108, 68–74 CrossRef CAS PubMed.
- L. Norén, R. Hedell, R. Ansell and J. Hedman, Invest. Genet., 2013, 4, 8 CrossRef PubMed.
- A. Serrano, M. M. Sousa, J. Hallett, J. A. Lopes and M. C. Oliveira, Anal. Bioanal. Chem., 2011, 401, 735–743 CrossRef CAS PubMed.
- C. Miguel, W. Jacobs, C. B. Dias, A. Manhita, T. Ferreira, A. F. Conde and A. Candeias, Eur. J. Sci. Theol., 2020, 16, 151–164 Search PubMed.
- P. Nabais, M. J. Mela, J. A. Lopes, M. Vieira, R. Castro and A. Romani, Heritage Sci., 2021, 9, 32 CrossRef CAS.
-
I. Petroviciu, Report for COST G8, ARTECH (FP6-506171), National Research Laboratory for Conservation and
Restoration, Bucuresti, 2005 Search PubMed.
-
M. Hacke, PhD thesis, University of Manchester, 2006.
-
A. Quye, K. Hallett and C. Herrero Carretero, Wrought in Gold and Silk: Preserving the Art of Historic Tapestries, NMS Enterprises Limited, Edinburgh, 2009 Search PubMed.
-
P. Guinot, PhD thesis, Universite de Lille 2, 2006.
-
L. G. Troalen, PhD thesis, University of Edinburgh, 2013.
-
Note for Guidance on Validation of Analytical Procedures: Text and Methodology, EMEA, London, 1995 Search PubMed.
- A. Villela, E. J. C. Van der Klift, E. S. G. M. Mattheussens, G. C. H. Derksen, H. Zuilhof and T. A. Van Beek, J. Chromatogr. A, 2011, 1218, 8544–8550 CrossRef CAS PubMed.
- K. Lech and M. Jarosz, Anal. Bioanal. Chem., 2011, 399, 3241–3251 CrossRef CAS PubMed.
- K. Lech and M. Jarosz, Anal. Bioanal. Chem., 2016, 408, 3349–3358 CrossRef CAS PubMed.
- I. Serafini, L. Lombardi, G. Vannutelli, C. Montesano, F. Sciubba, M. Guiso, R. Curini and A. Bianco, Microchem. J., 2017, 134, 237–245 CrossRef CAS.
- L. Vilanou, I. Karapanagiotis and Y. Chryssoulakis, Anal. Bioanal. Chem., 2009, 395, 2175–2189 CrossRef PubMed.
-
Bioanalytical Method Validation Guidance for Industry, FDA, Silver Spring, MD, 2018 Search PubMed.
- M. B.-C. de Bellaistre, W. Nowik, A. Tchapla and S. Heron, J. Chromatogr. A, 2011, 1218, 778–786 CrossRef PubMed.
- S. T. Talcott, J. E. Peele and C. H. Brenes, Food Res. Int., 2005, 38, 1205–1212 CrossRef CAS.
- C. Mouri and R. Laursen, Microchim. Acta, 2012, 179, 105–113 CrossRef CAS.
- J. Cheng, K. Weijun, L. Yun, W. Jiabo, W. Haitao, L. Qingmiao and X. Xiaohe, J. Pharm. Biomed. Anal., 2010, 53, 43–49 CrossRef PubMed.
-
G. Wingfield Digby, Elizabethan Embroidery, Faber and Faber, London, 1963 Search PubMed.
-
English Embroidery from the Metropolitan Museum of Art, 1580-1700, ed. A. Morrall and M. Watt, Yale University Press, New York, NY, 2008 Search PubMed.
-
L. Arthur, Embroidery 1600-1700 at the Burrell Collection, John Murray in association with Glasgow Museums, London, 1995 Search PubMed.
-
M. Brooks, English Embroideries of the Sixteenth and Seventeenth Centuries in the Collection of the Ashmolean Museum, Ashmolean Museum, Oxford, 2006 Search PubMed.
-
L. G. Troalen, A. N. Hulme, E. Cleland and L. Karafel, in Tapestries from the Burrell Collection, ed. V. Hamilton, K. Sleight, S. Pacitti and R. Quinton, Philip Wilson Publishers, London, 2017, Dye Sources used in English Workshops: An Analysis of Early Tapestries from the Burrell Collection, pp. 46–51 Search PubMed.
-
L. G. Troalen, R. Upton, J. Mulherron and A. N. Hulme, in Dyes in History and Archaeology 33/34, ed. J. Kirby, Archetype Publications, London, 2021, The Investigation of Dye Sources in English Turkeywork Carpets by UPLC-PDA Analysis, pp. 118–127 Search PubMed.
-
D. A. Peggie, PhD thesis, University of Edinburgh, 2006.
- M. Hacke, Stud. Conserv., 2008, 53, 3–15 CrossRef.
- D. A. Peggie, A. N. Hulme, H. McNab and A. Quye, Microchim. Acta, 2008, 162, 371–380 CrossRef CAS.
- J. Kirby, M. Spring and C. Higgitt, Natl. Gallery Tech. Bull., 2007, 28, 69–95 Search PubMed.
-
E. Cleland and L. Karafel, in Tapestries from the Burrell Collection, ed. V. Hamilton, K. Sleight, S. Pacitti and R. Quinton, Philip Wilson Publishers, London, 2017, English Tapestries, pp. 627–662 Search PubMed.
- J. Wouters, C. M. Grzywacz and A. Claro, Stud. Conserv., 2010, 55, 186–203 CrossRef CAS.
-
M. Swain, Historical Needlework: A study of Influences in Scotland and Northern England, Barrie & Jenkins Ltd, London, 1970 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ay01151k |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Lore G.
Troalen
a,
Lore G.
Troalen