
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanomedicines and microneedles: a guide to their analysis and application
Rachel E.
Sully
 *abc,
Colin J.
Moore
d,
Hemda
Garelick
b,
Eriketi
Loizidou
b,
Adrian G.
Podoleanu
c and
Vladimir
Gubala
a
*abc,
Colin J.
Moore
d,
Hemda
Garelick
b,
Eriketi
Loizidou
b,
Adrian G.
Podoleanu
c and
Vladimir
Gubala
a
aMedway School of Pharmacy, Universities of Greenwich and Kent, Anson Building, Central Avenue, Chatham, ME4 4TB, UK. E-mail: res91@kent.ac.uk
bDepartment of Natural Sciences, Middlesex University, The Burroughs, Hendon, London NW4 4BT, UK
cSchool of Physical Sciences, University of Kent, Ingram Building, Canterbury, Kent CT2 7NH, UK
dDrug Discovery and Development, Istituto Italiano di Tecnologia, Via Morego 30, 16136 Genova, Italy
First published on 27th July 2021
Abstract
The fast-advancing progress in the research of nanomedicine and microneedle applications in the past two decades has suggested that the combination of the two concepts could help to overcome some of the challenges we are facing in healthcare. They include poor patient compliance with medication and the lack of appropriate administration forms that enable the optimal dose to reach the target site. Nanoparticles as drug vesicles can protect their cargo and deliver it to the target site, while evading the body's defence mechanisms. Unfortunately, despite intense research on nanomedicine in the past 20 years, we still haven't answered some crucial questions, e.g. about their colloidal stability in solution and their optimal formulation, which makes the translation of this exciting technology from the lab bench to a viable product difficult. Dissolvable microneedles could be an effective way to maintain and stabilise nano-sized formulations, whilst enhancing the ability of nanoparticles to penetrate the stratum corneum barrier. Both concepts have been individually investigated fairly well and many analytical techniques for tracking the fate of nanomaterials with their precious cargo, both in vitro and in vivo, have been established. Yet, to the best of our knowledge, a comprehensive overview of the analytical tools encompassing the concepts of microneedles and nanoparticles with specific and successful examples is missing. In this review, we have attempted to briefly analyse the challenges associated with nanomedicine itself, but crucially we provide an easy-to-navigate scheme of methods, suitable for characterisation and imaging the physico-chemical properties of the material matrix.
Introduction
Invisible to the naked eye, nano-sized drug delivery systems (DDSs) showed promise in revolutionizing the treatments of many conditions and diseases that were considered effectively untreatable. They showed great potential in improving the efficacy of existing drugs and conventional therapy. New generations of nanomedicines such a dendrimers, liposomes, synthetic and bio-inspired nanoparticles (NPs), nanoclusters, nanorods and nanoplates1–6 have been introduced as drug carriers, due to their small size and high volume-to-surface ratios, aiming to evade the body's natural defence mechanisms. However, the reality is that the translation of promising NPs from the benchtop to the marketplace is extremely challenging and has led to a drive to better understand bio–nano interactions and to develop strategic frameworks that can efficiently test potential toxicological effects induced by NP exposure.One area that has been highly studied has been the interaction of NPs with complex biological fluids (e.g. blood and serum) and the formation of biomolecular coatings on particle surfaces that can govern the NP's biological fate. This surface fouling is attributed to the adsorption of biomolecules (mainly protein) around NP surfaces and is referred to as the ‘protein corona’. The corona is composed of two layers: ‘hard’ and ‘soft’.7 A hard corona is a near-monolayer of tightly bound biomolecules to the NP surface (though not irreversibly bound). The ‘soft’ corona surrounds this hard corona layer and is composed of rapidly exchanging and more loosely associated biomolecules. The hard corona on the NP surface is believed to be the layer that interacts with cells8,9 and, consequently, has begun to be regarded as defining the ‘bio–nano interface’ that can result in unpredicted biological interactions10,11 Considering the continuously growing popularity of NPs in bioapplications12–14 like nanomedicine, there has been increasing importance placed on assessing NP interactions with complex biological fluids. For example, Tenzer et al.15 showed that a corona can form in 30 seconds on silica NPs. This poses a challenge to nanomedical applications as NP surfaces are often engineered to recognise specific receptors on cell surfaces to impart particle targeting. For example, it has been shown that corona formation on biomolecule-functionalised NPs can lead to a loss of targeting capabilities16,17 or can even contribute to particle biodistribution.18 However, as the understanding of corona formation increases, there have been reports on the manipulation of the hard corona composition to modulate biomolecular19 and cellular interactions.20
With the wealth of research on the corona over the past decade it is evident that a variety of factors play a role in its formation (temperature of incubation, time of incubation, and the total mass of biomolecules in the exposure media) and also the analytical methods used for its characterization.21 A 2019 article by Lynch and co-workers highlighted such characterisation challenges and suggested the best practices for experimental design for studying corona–nanomaterial interactions and the subsequent reporting of results.22 Several regulatory bodies have also highlighted the relevance of the corona, including the US Food and Safety Authority23 and the European Food and Safety Authority,24 and have reported the importance of evaluating the toxicological impact of nanomaterials due to the acquisition of new, corona-derived biological identities.25 Nonetheless, regulatory gaps and standardised approaches to the characterisation of the biomolecular corona remain in nanomedicine despite the acceptance of its importance.
The emergence of the importance of the biomolecular corona is indicative of the fact that the use of nanomaterials in biological applications continues to present new challenges and considerations for nanomedicine. The standardisation and identification of knowledge gaps that could accelerate the translation of nanomedicines into useable therapies is therefore required. However, a lack of robust datasets and regulatory experience with innovative nanomaterials means that knowledge gaps to achieve this goal exist.
Halamoda-Kenzaoui et al.26 attempted to tackle this problem by conducting a gap analysis based on information released by regulatory scientists and made four observations where standardisation is required in nanomedicine:
(1) Methods for assessing.
• drug loading/release from nanomaterials.
• protein corona formation.
• interactions of nanomaterials with the immune system.
• nanomaterial accumulation in biological tissues.
(2) Guidance for assessing the comparability of methods used to evaluate nanomedically relevant endpoints.
(3) Reference materials relevant to nanomedicine.
(4) Identification and subsequent recognition of standards developed in other sectors.
It is also important to consider safe-by-design (SbD) as a useful tool for assessing the potential hazards associated with a given nanomedicine. The purpose of SbD is to assess the safety of a nano-enabled product as early as possible in the innovation process, the aim of which is to reduce side effects on human health and environmental impacts through alterations in nanomaterial designs. This encourages safety throughout the lifecycle of a nanomaterial for a given intended use. SbD therefore differs from conventional risk assessment processes that typically only consider safety once a product is fully developed.27
‘Grouping’ is one approach to streamline the identification of potential hazardous side effects associated with newly developed nanomaterials, i.e. utilising similar physicochemical characteristics and associated hazard endpoints to establish potential risk to health (or the environment). For example, a structure–toxicity paradigm has identified long, biopersistent, fibrous nanomaterials as hazardous upon pulmonary exposure.28 It can therefore be hypothesised that newly developed nanomaterials with a ‘high aspect ratio’ (i.e. physicochemical characteristic: shape) may be ‘grouped’ as potentially toxic/carcinogenic in pulmonary exposure scenarios. Such hypotheses can then be tested using structured approaches based on standardised physicochemical and toxicological methods.29,30
These approaches can be useful to establish the potential hazards of nanomedicines early in the innovation process. However, general SbD strategies are typically stage-gate based and, according to Schmutz et al.,31 are not reflective of the ‘drug discovery and development’ field of nanomedicine and through the GoBioNanoMat consortium, SbD methodical approaches were adapted to nanomedicine applications (Fig. 1). Unlike general SbD approaches, the nanomedicine SbD approach uses iterations (Fig. 1, red arrows), whereby knowledge can be built to better understand the physicochemical properties and biological impacts of developed nanomedicines. The nanomedicine SbD approach also incorporates key manufacturing considerations like batch-to-batch uniformity testing, stability monitoring and adherence regulatory requirements. However, the nanomedicine approach only sets out guidelines to develop safe nanomedicines and does not present guidance for evaluating the efficacy of the therapies. Nonetheless, such nanomedicine-specific developments are what is required to ensure that the undoubted potential of nanomaterials in medicine can be realised and pushes the field in an important direction by establishing framework stakeholders (innovators, manufacturers, and regulators) required to successfully translate nanomedicines into useable, cost effective therapies. These considerations should be to the forefront of any microneedle-based nanomedical application.
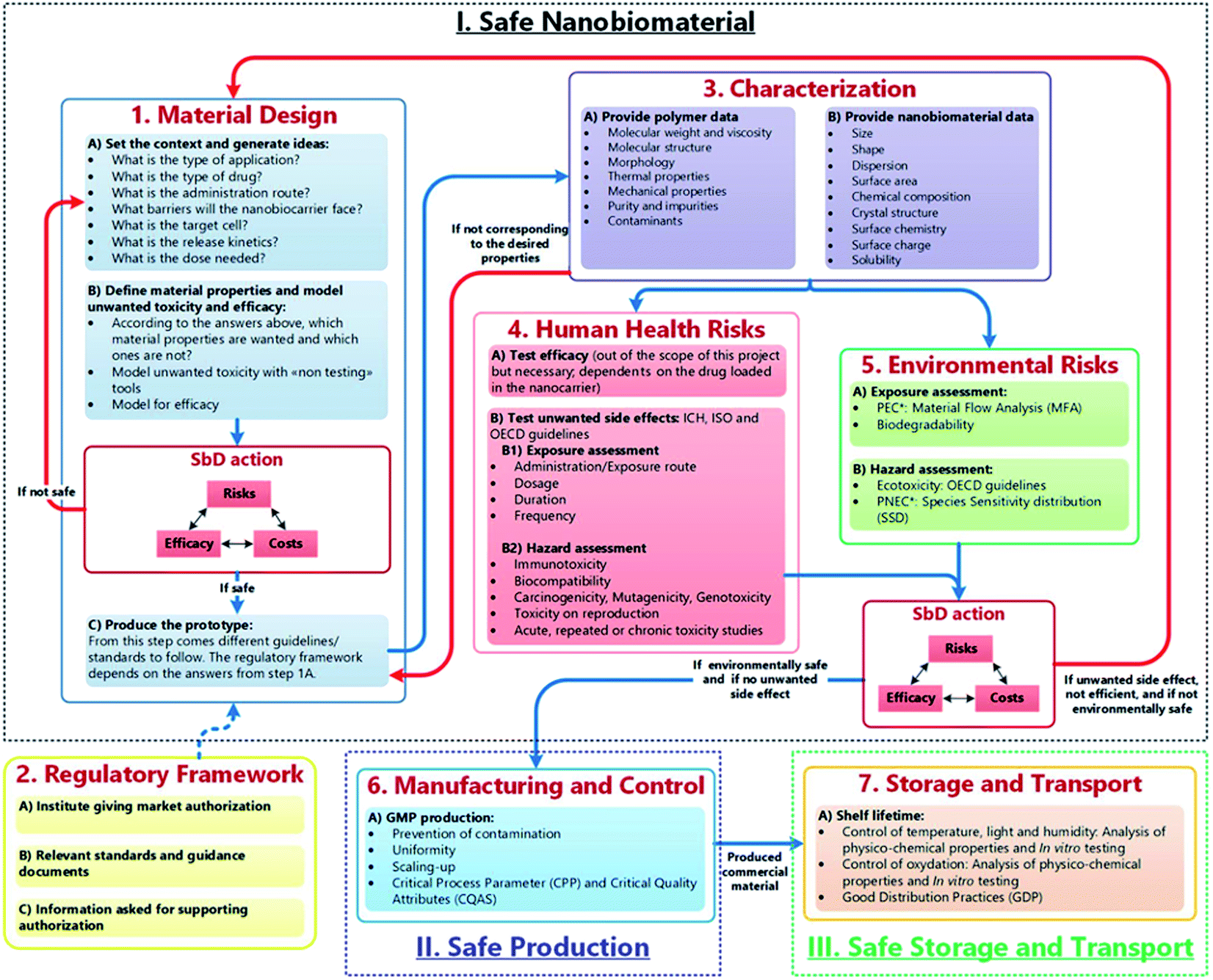 | ||
| Fig. 1 The safe-by-design approach for the development of nanomaterials for medical applications. Blue arrows refer to the flow of bio–nanomaterials from design to storage to transport. Red arrows allow for feedback/redesign whenever the bio–nanomaterial is unsafe or inefficient.28 (M. Schmutz, O. Borges, S. Jesus, G. Borchard, G. Perale, M. Zinn, Ä. A. J. A. M. Sips, L. G. Soeteman-Hernandez, P. Wick and C. Som, Front. Bioeng. Biotechnol., 2020, 8, 1–7. Copyright (2003) National Academy of Sciences, U. S. A.) | ||
The advantages of using engineered nanomaterials for medicinal purposes have been discussed extensively in many comprehensive reviews.32–40 However, despite the massive interest in nanomedicine, effectively all the developed concepts or commercially available products have been formulated as solutions and suspensions, aimed to be injected into the systemic circulation. Unfortunately, that brings about one major challenge that has not been fully addressed yet – nanomaterials have a tendency to aggregate/agglomerate in solution.41 Aggregation is observed when dispersed molecules collide to form aggregates,42 which are ‘an assembly of particles exhibiting an identifiable collective behaviour’,43 and the rate depends on the frequency of collisions and the probability of cohesion.44,45 Although agglomeration is similar in that it is defined as a process in which dispersed particles assemble46 by growing together or partial fusion,47 it refers to stronger bonds forming between the agglomerates.43 The aggregation/agglomeration phenomena, illustrated in Fig. 2,48 pose significant challenges for the translation of nanomedicine DDSs from the laboratory bench to a clinical product. Pristine NPs, such as ZnO and TiO2, have been topically applied on the skin for more than a decade, mostly for cosmetic purposes. However, despite their inherently small size, NPs cannot freely penetrate through the skin's outermost layer, the stratum corneum, due to the size-exclusion dependency of the skin.49 Fortunately, this is where the two separate concepts, microneedles (MNs) and nanomaterials, can be exploited together in order to convert the promise presented more than 20 years ago into a reality.
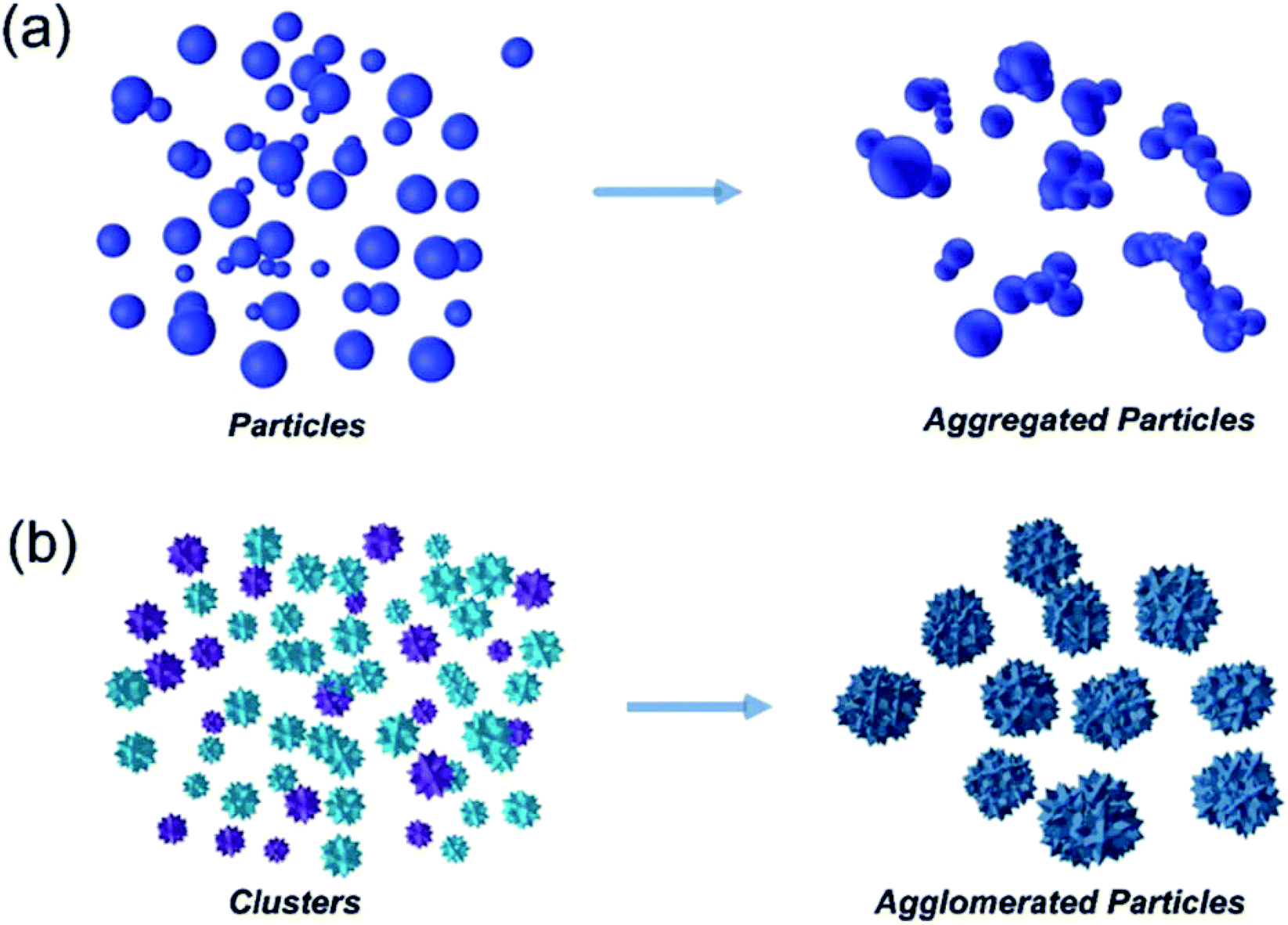 | ||
| Fig. 2 Diagram showing the mechanism difference between aggregation (a) and agglomeration (b).48 | ||
One significant milestone that offers a possibly elegant solution to the MN–NP delivery hypothesis came in 2003, when the Prausnitz research group50 were among the first to fabricate dissolvable microneedles (DMNs). Their pioneering work has prompted scientists to ask many relevant research questions, one of which is particularly interesting: could the formulation of nanoparticles inside dissolvable microneedles finally solve the aggregation/agglomeration problems? Many interesting and comprehensive reviews giving praise51–55 but also critique56,57 to the plethora of functional NP designs have been presented. More recently, it has been established that NPs can be stabilised and released from hydrogel-like materials, thus effectively preventing aggregation.58,59
In the first part of this review, we will specifically focus on DMNs, fabricated from biodegradable matrices, which provides a suitable environment for NPs, facilitating the delivery of NPs through the skin into the body. Importantly though, in the second part of the review we will provide a comprehensive overview of the analytical tools associated with MNs and NPs, with specific and successful examples. To the best of our knowledge, as no one has done before, we have attempted to provide an easy-to-navigate scheme of methods, capable of analysing the size-dependent biological function of NPs and their drug-targeting abilities and toxicological response. Importantly, this review will provide a clear strategy of all the analytical steps scientists need to consider in order to fabricate new drug delivery systems that might shape the future of home and community-based healthcare.
Microneedles
The skin covers approximately 16% of the body and comprises several layers of epithelial, mesenchymal, glandular and neurovascular components, approximately 2 mm thick.60 Skin has many functions but the most prominent is the protective function from environmental stressors such as infectious organisms, pollution, radiation from the sun, etc. People have been applying a wide variety of substances on their skin for thousands of years, mostly for therapeutic and cosmetic purposes. Since the official approval of the first transdermal system for systemic drug delivery in 1979 for the treatment of motion sickness, several generations of transdermal drug delivery products have been explored.61 The advances in microfabrication in the mid-1990's enabled large scale research exploration of a new technology for transdermal drug delivery – microneedles.62 Researchers use the terminology microneedles and microneedle arrays to refer to arrays of individual microneedles and often refer to a single microneedle as a needle. For this review, when the term microneedles (MNs) is used, this refers to an array of individual microneedles, whereas microneedle (MN) refers to an individual needle. When the abbreviation DMNs is used, this refers specifically to dissolvable microneedle arrays.A MN has been defined as ‘a canula which is either solid or hollow with an approximate length ranging between 50 and 900 μm and an external diameter of 300 μm’.63 However, this is not entirely accurate as some MNs are over 900 μm in length, i.e. for cosmetological applications, and can have varying base diameters. For example, Dermarollers® are very common, clinically used MNs for cosmetic procedures and can range in needle length from 0.5–3 mm.64–68 Depending on the length of the MN, penetration into the dermis can be achieved (Fig. 3b), by-passing the stratum corneum barrier of the skin, e.g. during collagen induction procedures where the needle length is 1.5 mm.69 The stratum corneum prevents certain sized materials from passing through, i.e. those over 2 nm in size or 500 daltons in molecular weight.70 Therefore, the delivery of pharmaceuticals, whether in vesicles or not, is challenging without assistance. The use of microneedles can overcome this size-dependent barrier.49 For this reason, combining microneedle and nanoparticle technologies is an attractive idea to improve drug delivery and the administration of therapies. MNs were actually patented for the first time in 1976,71,72 where the structure of an array was claimed to enable controlled drug release from a reservoir through the stratum corneum, but it took another 25 years for them to attract significant interest. Many variations of this transdermal delivery concept have since appeared. As illustrated in Fig. 4b, the interest in this new drug delivery technology is still exponentially growing – more than 1100 papers have been published on the topic of microneedles just in the past 10 years. Fabricated as low-cost patches, they are simple for patients to apply73 and have been trialled to deliver a wide range of substances, such as low molecular weight drugs, protein drugs, cosmetics, biotherapeutics, and biomacromolecules, targeting vaccine delivery to antigen-presenting cells in the skin.39,40,71,74–85
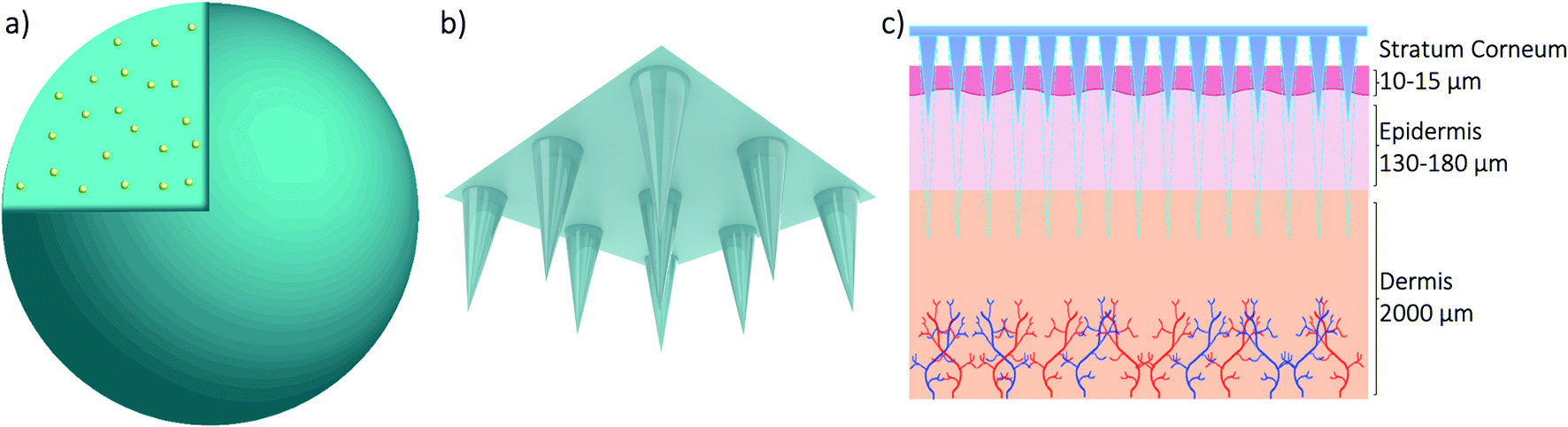 | ||
| Fig. 3 3D models of a nanoparticle with drug cargo (a) and a microneedle array (b) and a diagram showing the application of the microneedle array through skin (c). | ||
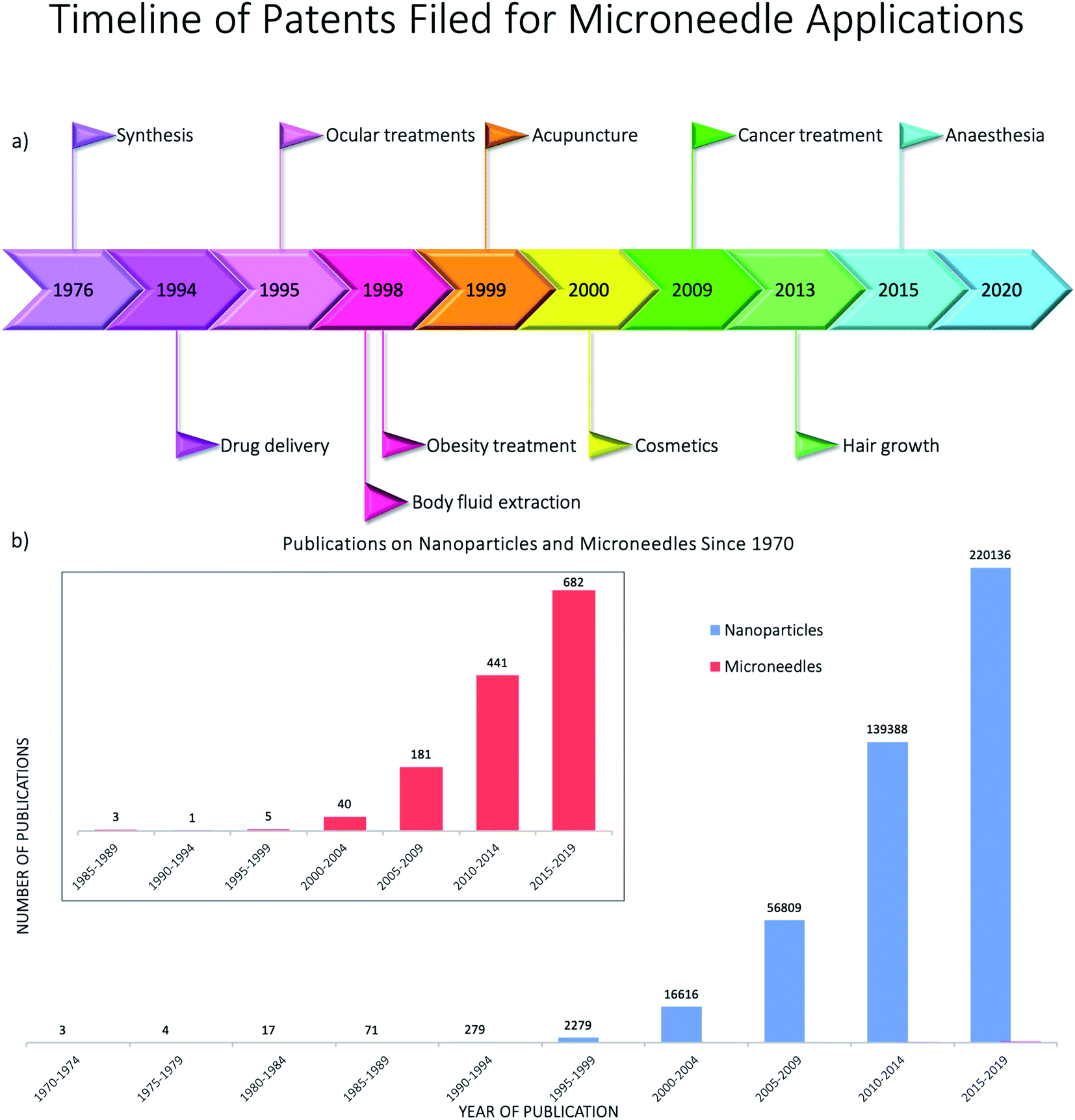 | ||
| Fig. 4 (a) Timeline showing the years in which the first patents were filed for the main microneedle applications using Espacenet5 – the patents were found using the advanced search of the title and abstract with the keyword microneedle and then refining the dates to five year periods from 1970–2020; (b) publications of papers on nanoparticles and microneedles since 1970 from Scopus – the keywords “nanoparticles” and “microneedles” were searched separately and the following document types were excluded: for microneedles – conference review; note; letter; short survey; undefined; for nanoparticles – note; short survey; letter; conference review; retracted; erratum; abstract report; undefined – before averaging the number of publications for five year periods. | ||
The potential of DMNs to be used as transdermal DDSs was first demonstrated when Park et al.50 synthesised the first biodegradable MNs. They fabricated DMNs from polyglycolide (PGA), polylactide (PLA) and poly lactide-co-glycolide (PLGA) and successfully encapsulated two model drugs into them – calcein as a model for low molecular weight drugs and bovine serum albumin (BSA) as a model for high molecular weight drugs. They demonstrated the ability of DMNs to insert into skin and release their cargo.
Early research into DMNs was mainly about perfecting their shape, as the geometry and size of DMNs, as well as the matrix materials used, play a vital role in how well they penetrate the skin. In terms of geometry, spherical needle tips have been shown to have a higher fraction force than square needle tips, suggesting that they can withhold more stress.86 Loizidou et al.87 (Fig. 5)87,88 studied hexagonal, square and triangular DMNs and predicted by simulations that the hexagonal DMNs would withstand higher stress until buckling occurs. It was suggested that the geometry and aspect ratio affect the mechanical properties of DMNs and the penetration depth. Although these studies can help provide an insight into what shaped DMNs would be best to use, this can also be affected by other geometrical factors, such as the needle length, centre-to-centre spacing and base diameters. The best shape can also be affected by the material matrix used. A detailed analysis of the MN shape is presented by Nava-Arzaluz et al.89 The analysis of DMNs is carried out using a variety of techniques, ranging from simple imaging techniques, such as light microscopy, to more advanced techniques, such as optical coherence tomography (OCT). The aim of this review is to provide an outline of the data each technique can produce and how it is beneficial. The aim is to guide researchers in the decision of what techniques work best for their specific DMNs.
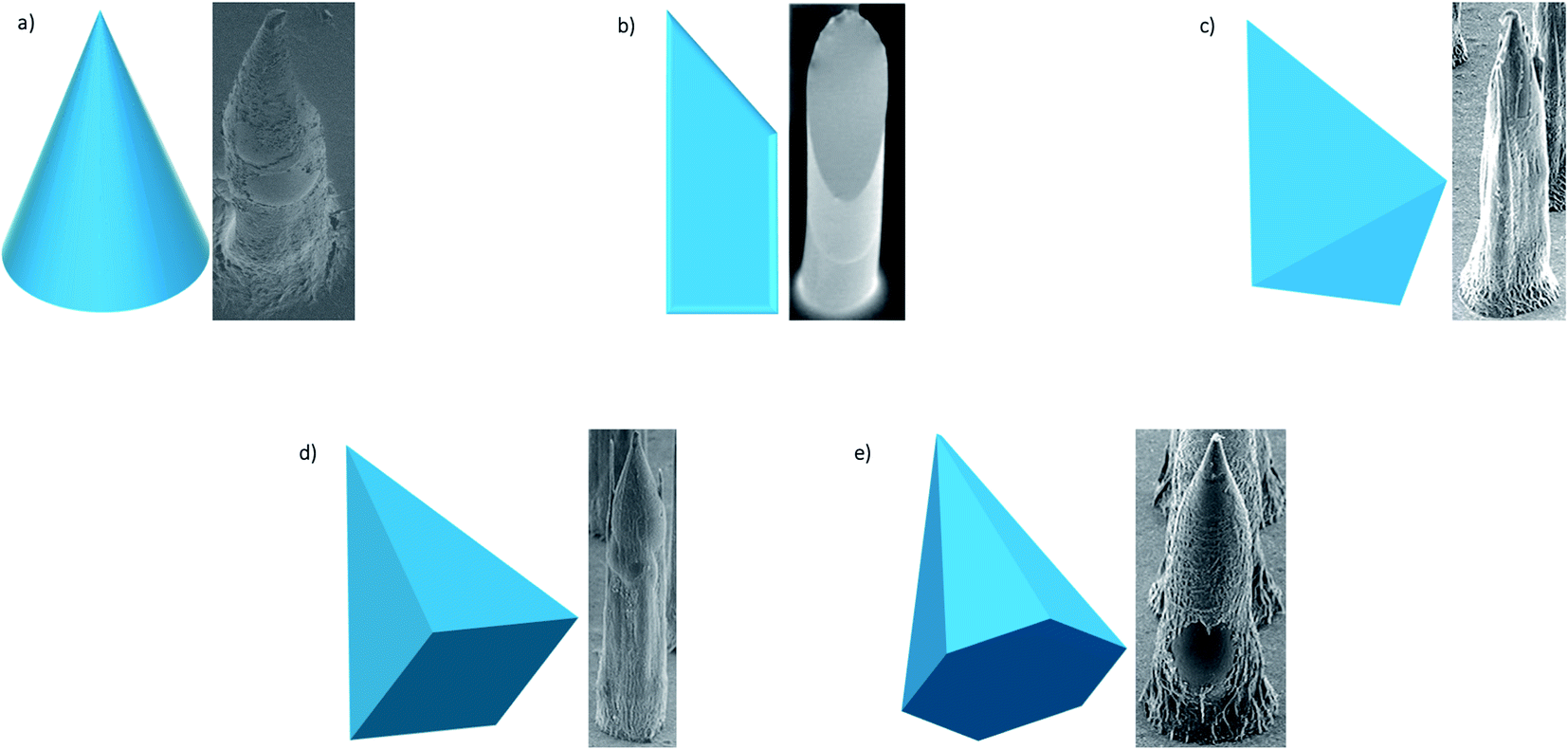 | ||
| Fig. 5 SEM images of different shapes of microneedles most frequently used – conical; beveled tip; triangular-based pyramid; square-based pyramid; hexagonal-based pyramid (adapted from Loizidou et al.87 and McAllister et al.88). | ||
One of the main potential and earliest applications for DMNs is a new way to administer vaccinations, such as influenza,90–95 measles,96 polio,97 hepatitis B (HBV)98 and tetanus toxoid99 vaccines. One of the most recent studies into using DMNs as a way to administer vaccinations is for COVID-19, which is an ongoing pandemic. Kim et al.100 have shown the potential use of a recombinant spike (S) protein as the vaccine. They have also demonstrated the benefits of DMN delivery over subcutaneous delivery. First, they developed three types of MERS-S1 vaccines from the native coronavirus structure, which all showed strong reactivity and antibody production. These vaccines were then formulated into DMN arrays of 10 × 10 obelisk-shaped needles of height 750 μm and base diameter 225 μm. The matrix used to fabricate these arrays was carboxymethyl cellulose (CMC) by a spin-drying technique, i.e. centrifugation. In vivo studies were carried out to compare the antibody response of the different types of vaccines and determine whether the delivery route effected the response. In terms of an immediate antibody response, for subcutaneous injection, only the vaccine with an adjuvant showed an antibody response, whereas for DMNs, an antibody response was seen regardless of whether an adjuvant was present or not, and it was a significantly higher response than that for subcutaneous injection. They observed that the subcutaneously injected vaccines did not appear to produce an antibody response until two weeks after a booster injection, and even when a response occurred, it was significantly lower than that seen for vaccines administered by DMNs. A long-term immune response was observed in 23 and 55 weeks for both subcutaneously injected and DMN injected vaccines. Subcutaneous injection showed the same or increased antibody levels as in week 6; however DMNs showed significantly higher levels, suggesting the potential for long-term immune responses by DMN vaccination. The sterilisation of the vaccine did not appear to affect it. Although it can be concluded that these vaccines, whether in combination with DMN delivery or adjuvant and subcutaneous delivery, have great potential as COVID vaccines, especially now that this particular strain has been sequenced. However, it is too early to predict whether humans will have the same responses.
Other potential applications of DMNs for medical purposes, include intraocular delivery,101 diagnostic tools,102 delivery of certain medications103,104 and cancer therapy.105 One of the most interesting studies was the development of an effervescent patch for long-term contraception using levonorgestrel (LNG). Recently, DMNs consisting of PLGA and diglyme/water (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) needles with LNG encapsulated attached to a backing plate of polyvinylpyrrolidone (PVP) were studied.106 When the patch is inserted into the skin, the interstitial fluid penetrates the patch and begins to dissolve the backing plate. Sodium bicarbonate and citric acid from within the backing plate react to give off effervescence, causing the connection between the backing plate and needles to become weakened and break. This all occurs in a span of 10 seconds. The backing plate can then be removed, leaving the needles to slowly dissolve over time giving a sustained release of LNG. The DMNs were 10 × 10 arrays of conical shaped needles with a height of 600 μm, base diameter of 150 μm and tip diameter of 10 μm. The LNG release profile was first studied in phosphate buffered saline (PBS) showing a sustained release of LNG for over one month. This was repeated in vivo using rats and displayed the same findings. The ability of these DMNs to insert and produce effervescence in humans was confirmed using DMNs without the LNG encapsulated and water-soluble needles in women of reproductive age. It was concluded that these DMNs provide a rapid, efficient way of administering contraception that releases the hormones and maintains the required plasma concentration for over one month.
:5) needles with LNG encapsulated attached to a backing plate of polyvinylpyrrolidone (PVP) were studied.106 When the patch is inserted into the skin, the interstitial fluid penetrates the patch and begins to dissolve the backing plate. Sodium bicarbonate and citric acid from within the backing plate react to give off effervescence, causing the connection between the backing plate and needles to become weakened and break. This all occurs in a span of 10 seconds. The backing plate can then be removed, leaving the needles to slowly dissolve over time giving a sustained release of LNG. The DMNs were 10 × 10 arrays of conical shaped needles with a height of 600 μm, base diameter of 150 μm and tip diameter of 10 μm. The LNG release profile was first studied in phosphate buffered saline (PBS) showing a sustained release of LNG for over one month. This was repeated in vivo using rats and displayed the same findings. The ability of these DMNs to insert and produce effervescence in humans was confirmed using DMNs without the LNG encapsulated and water-soluble needles in women of reproductive age. It was concluded that these DMNs provide a rapid, efficient way of administering contraception that releases the hormones and maintains the required plasma concentration for over one month.
MNs have been used to enhance the permeation of NP drug vesicles (Fig. 6) for over a decade now, as discussed in previous reviews,39,89 and these were first done using solid MNs followed by topical application, hollow MNs delivering NP solutions and coated MNs. Drug delivery using DMNs was widely studied by encapsulating the drug directly into the DMN matrix; however protecting these drug molecules in NPs before encapsulation into DMNs was not demonstrated until around the 2010s.
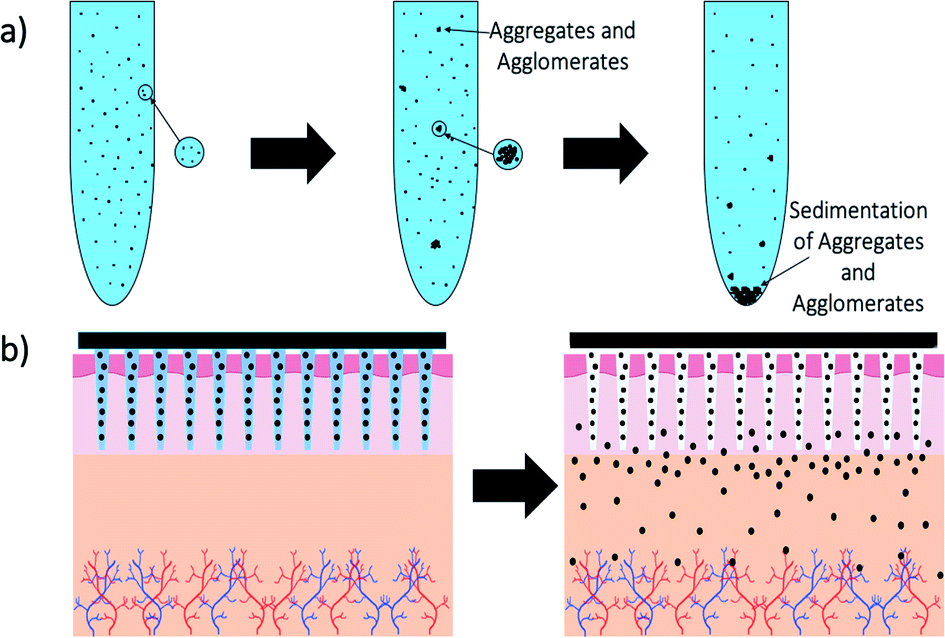 | ||
| Fig. 6 Schematic showing the process of microneedle application and dissolution of needles into skin – (a) schematic of how nanoparticles aggregate and agglomerate over time in solution with eventual sedimentation; (b) diagram of how dissolvable microneedles stabilise and then release nanoparticles from their matrix. | ||
Ovalbumin (OVA), as a model antigen, was encapsulated into PLGA-NPs and delivered using Gantrez DMNs to observe antitumour and antiviral immune responses.107 The DMNs consisted of 19 × 19 needles of 600 μm height. In vivo studies of mice skin using OCT showed successful insertion of DMNs without mechanical failure and rapid dissolving of DMNs to release their cargo. However, when trying to determine whether dendritic cells were capable of taking up PLGA-NPs, it was seen that the majority of the NPs released from the DMNs localised to the insertion site. As the OVA-NPs were smaller than 100 nm in size, they were significantly less likely to be spontaneously removed from the body. Not only did the encapsulation of OVA into NPs promote sustained delivery, it also increased their stability in DMNs compared to antigens. In vivo studies showed that one dose of OVA by DMNs produced tumour killing cells, suggesting that this delivery method may be effective for immunotherapy strategies, especially as it induced immunity against a viral challenge using a recombinant Sendai virus. Later studies by this group supported these findings and demonstrated that DMN vaccination can lead to memory responses.108
Wound healing and possible infection are the major concerns in the medical field. Xu et al.109 investigated the ability of DMNs to enhance the drug delivery of antimicrobials. Some antimicrobials (chloramphenicol (CAM) specifically) can cause anaemia as they heighten cytotoxicity. In the above study, CAM was therefore incorporated into gelatin NPs, to reduce this cytotoxic effect. The NPs were characterised by dynamic light scattering (DLS) and transmission electron microscopy (TEM), showing their size to be 197.6 nm and 131.6 nm respectively. DLS also showed a zeta potential of −12.6 mV, which is above the literature value of good stability at charges more negative than −30 mV, suggesting NP instability.41 The encapsulation and loading efficiencies of the NPs, as well as their release, were studied using high-performance liquid chromatography (HPLC). It was observed that CAM was released by gelatinase in a controlled manner. To test the cytotoxicity of the NPs, free CAM, plain NPs and CAM-NPs were incubated with fibroblast cells and CCK-8 assays were run. They showed reduced cytotoxicity of the CAM-NPs in comparison to free CAM. Once these NPs had been fully characterised, they were encapsulated into arrays made from PVP. The DMNs were imaged by scanning electron microscopy (SEM) and fluorescence microscopy, showing that the CAM-NPs were distributed in the tips. When the DMNs were inserted into the biofilms, their structure was disrupted allowing the dye (used as a model drug) to diffuse into the biofilm matrix. It was concluded that these DMN arrays containing CAM-NPs allowed for local and specific drug release within biofilms, suggesting the potential of DMNs for the treatment of bacterial biofilms in wounds.
MNs, although a relatively novel research concept, have gained great interest due to their potential for transdermal drug delivery, with over 54 clinical trials currently being evaluated.110 However, there are still challenges to overcome before their translation to clinical use. One of the main challenges is the dosage and activation of immune cells beneath the skin to benefit the diffusion of the cargo.110 If the required dose needs to be on a larger scale, then the size of the MNs will need to be adjusted. This may lead to continuous re-application of MNs to achieve the desired dosage. This can bring about its own challenges, in terms of affecting the characteristics (Table 1)111,112 of the MNs, as well as safety concerns.
| Characteristics | Applications | Challenges |
|---|---|---|
| • Fracture force – ability to resist insertion force | • Vaccination | • Dosage – repeatability |
| • Penetration depth – required depth of tissue for desired application | • Biopharmaceutical's delivery – proteins; nucleic acids; cells; tissues | • Size of the patch – higher the dose, larger the patch |
| • Dimensions – length 1–900 μm; base width 50–300 μm; tip diameter 1–50 μm | • Drug delivery – pharmaceuticals; insulin; birth control; cancer therapy | • Variability in skin – skin layer thickness; skin hydration; skin age |
• Margin of safety = 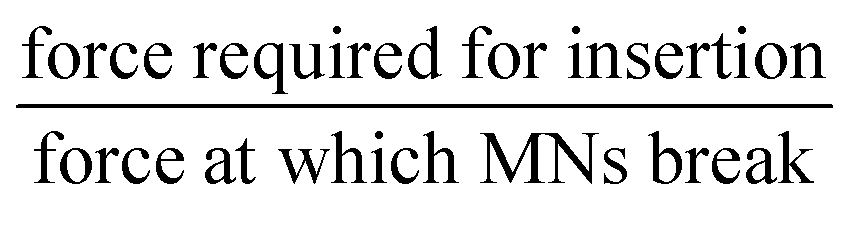 |
• Diagnostics – sensing; sample collection | • Manufacturability – how to fabricate on a large scale with pharmaceuticals encapsulated |
| • Effect of the length on pain – as the length increases, pain increases | • Cosmetics – wrinkle treatments; acupuncture; acne treatment; scar treatment | • Patient compliance – educating patients; monitoring of patients |
| • Transepidermal water loss – evaluation of skin before and after MN application | • Biocompatibility – most made from sugars but are these biocompatible on this larger scale; cytotoxicity studies | |
| • Biological safety testing – cytotoxicity studies |
Another potentially significant challenge of MNs is their manufacturability.110 Although MNs for cosmetic applications have already been successfully manufactured, e.g. Dermarollers®, manufacturing patches that contain therapeutics could be difficult due to the much more stringent regulatory and quality control requirements. For example, can the drug possibly cause structural and mechanical defects in the arrays? This also raises additional questions of safety of MNs – will there be long-term damage to skin from repetitive application? Is the distribution of cargo from MNs accurate?
The desire of microneedles is the ability to improve patient compliance and assist in at-home applications. However, there are several challenges to be addressed. The patient may need to be educated in how to correctly apply microneedles, to ensure that a clinically relevant dose has been delivered. If the patient needs to apply more than one patch, medical staff or even insurers may need to monitor this, by use of sensors within the patches, or by contacting the patient on a regular basis. Although the benefit of MNs for transdermal delivery of therapeutics is clear and understandable, potential can only be realised after very careful evaluation of each step as this exciting technology makes its way from laboratory benches to the clinic. For further discussion of the characteristics, applications and prospects of microneedles, the reader is directed to various reviews on these topics.110–114
As shown by the current research into DMNs, they have great potential as transdermal drug delivery systems for nanomedicines. However, for their use in the medical field, there needs to be robust evidence of how to carry out their analysis. Currently there are over 25 techniques that can be used for the analysis of DMNs alone or with nanomedicines encapsulated, varying from common techniques that can be performed in almost any laboratory, to those that are specialised and found at specific labs only. To the best of our knowledge, there is no literature reviews on the analysis of DMNs or NPs encapsulated into DMNs. Therefore, we have created a flowchart (Fig. 7) followed by a detailed description of each technique to allow the interested reader to determine what techniques are most suitable to analyse particular DMNs.
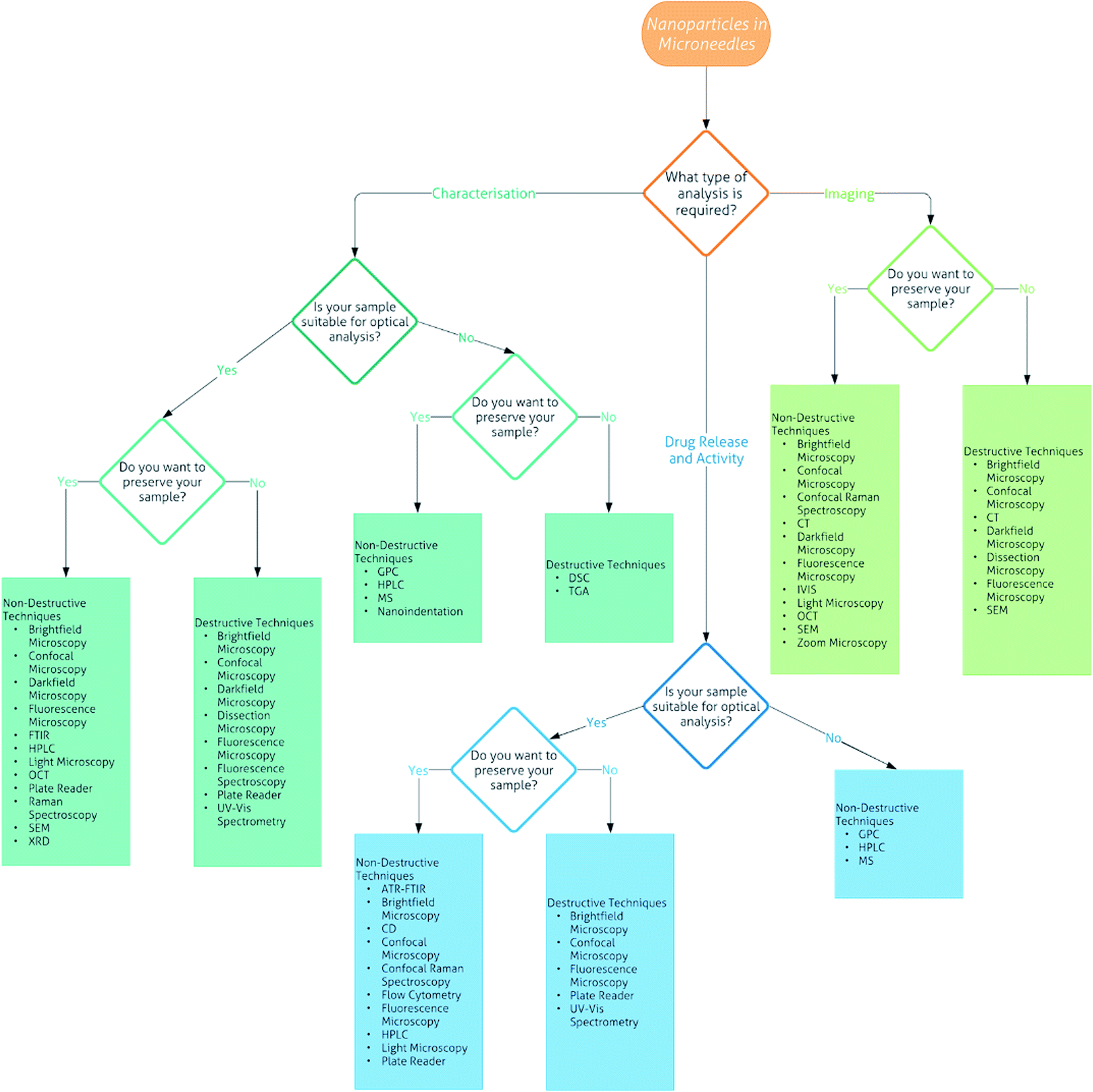 | ||
| Fig. 7 Flowchart of the analytical techniques that can be used for the characterisation, imaging and drug release and activity of nanoparticles in microneedles. | ||
Analytical techniques
Definitions
• Characterisation – this refers to the shape and size of the arrays as well as the penetration depth and fracture force of the needles.• Non-destructive – this refers to when a sample can be recovered without physical or chemical change after analysis.
• Destructive – this refers to when a sample cannot be recovered without physical or chemical change after being analysed, either due to the technique being destructive, or if doping or coating of the sample is required to be visible for particular techniques.
• Coating – this refers to when an array, particularly the needles, is coated with a chemical, such as gold, to be visible for particular techniques.
• Doping – this refers to chemically bonding a dye or other chemical to the nanoparticles or array matrix in order to be visible for particular techniques.
Characterisation
DMNs made from sugar glass can be characterised using DSC. Martin et al.119 created sugar glass films and analysed them by DSC, where the sugar glass films were heated between 20 and 220 °C, at increments of 10 °C min−1. The DSC spectrum showed peaks at 101 and 214 °C as well as an endothermic region between 130 and 150 °C for the trehalose anhydrous (TRA) film. The DSC spectrum of unprocessed TRA was compared to the TRA film spectrum and showed only a single peak at 200 °C when the trehalose crystal melted. The trehalose dihydrate (TRD) film also showed similar peaks to the TRA film, indicating that both TRD and TRA solutions form similar sugar glass. For the sucrose (SUC) film, a peak was observed at 191 °C when the SUC crystal melted. The combination of both TRA and TRD with SUC was then studied by DSC, both at 75:25% w/w% of TRA–SUC and TRD–SUC. Both showed similar spectra with a peak at 97 °C and a poorly defined endothermic region at 130–170 °C. This further supported the conclusion that TRA and TRD once dehydrated form sugar glass of a similar nature.
DSC can also be used to determine the glass transition temperature (Tg) of DMNs which can determine their stability and moisture content over time, for example by increasing the temperature to 200 °C in 10 °C min−1 increments and observing the Tg of fresh DMNs in comparison to that of stored DMNs.120 When disruptions start to occur in the heat flow versus temperature curve, then this represents the Tg. Kolluru et al.120 found that over storage time, the Tg of DMNs increased from 48 °C to 59 °C, suggesting that the residual moisture content decreases over time. The stability of the DMNs appeared to be better at lower temperatures of 5 °C and 25 °C in comparison to that at 40 °C, which may be due to this higher temperature being closer to the Tg.
Kolluru et al.120 used TGA to determine the residual moisture content of their DMNs fabricated from 35% w/v% fish gelatin and 10% w/v% D-sorbitol. These arrays consisted of 10 × 10 needles with a height of 650 μm. TGA was used to analyse DMNs and study the effect lyophilisation and drying temperature had on the residual moisture content of the arrays. They observed that the residual moisture content depended on the drying temperature and sometimes lyophilisation. After storage for one week at 40 °C an equilibrium state was seen. At 5 °C, the residual water content was highest in comparison to drying at 25 °C and 40 °C. However, lyophilisation reduced this moisture content for air drying at 5 °C but not for any other temperatures. It was concluded that as the storage temperature increased, the DMN moisture content decreased due to the matrix becoming more amorphous.
As Bhatnagar et al.105 demonstrated, fluorescence spectrometry can be used to determine concentrations, specifically of drug cargo (doxorubicin (DOX) and docetaxel (DTX)) in DMNs fabricated from PVP and polyvinyl acetate (PVA). A calibration curve was plotted for known concentrations of DOX and then drug released from dissolved DMNs was determined. The concentration of DOX loaded into the DMNs was measured using an excitation wavelength of 475 nm and emission wavelength of 595 nm and showed a drug loading mass of 621.8 μg in DOX-MNs and 533.5 μg in DOX–DTX-MNs. Fluorescence spectrometry was then used to study the DMN dissolution and produce release profiles. It was seen that DOX was completely released from DOX–DTX-MNs within five minutes after application and 15 minutes for DOX-MNs. Ex vivo skin permeation studies in Franz diffusion cells were carried out and at regular intervals, 300 μL aliquots were taken and the DOX and DTX concentrations were observed using fluorescence spectrometry and HPLC respectively. Fluorescence spectrometry showed that 67.8% of DOX permeated the skin within 48 hours after DOX-MN application, whereas a high release of 73.1% was observed for DOX–DTX-MNs. Tape stripping was used to measure the amount of drug within the stratum corneum and it was found that 3.6% was present for the DOX-MNs. To measure the amount of DOX within the epidermis, the skin sample was homogenised, and drug was extracted showing that 21.9% of DOX was present for DOX-MNs. Similar results were seen for the DOX–DTX-MNs. Both the release and skin permeation studies showed that DOX was released from the various DMNs faster than DTX, which was studied by HPLC and is described later.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N groups respectively. DOX has characteristic bands at 3222 cm−1 (N–H amine), 2884 cm−1 (C–H), 1725 cm−1 (CO) and 1277 cm−1 (C–O), whereas DTX has characteristic bands at 3420 cm−1 (N–H), 1685 cm−1 (ester group) and 700 cm−1 (N–H). Although the characteristic peaks can be seen for each drug alone in polymer solution or the DMNs, when combined, these characteristic peaks cannot be seen.
O and C–N groups respectively. DOX has characteristic bands at 3222 cm−1 (N–H amine), 2884 cm−1 (C–H), 1725 cm−1 (CO) and 1277 cm−1 (C–O), whereas DTX has characteristic bands at 3420 cm−1 (N–H), 1685 cm−1 (ester group) and 700 cm−1 (N–H). Although the characteristic peaks can be seen for each drug alone in polymer solution or the DMNs, when combined, these characteristic peaks cannot be seen.
DMNs can be characterised by XRD, as well as analysing how storage affects the polymer matrix. Kolluru et al.120 investigated the use of a DMN patch, consisting of 35% w/v% fish gelatin and 10% w/v% D-sorbitol, for the vaccination of polio. XRD was used to analyse the changes in the matrix structure of the DMNs. The measurements were made in a θ–2θ mode with a Cu Kα source at 45 kV and 40 mA. Chips of the DMN matrix showed a peak at 20°, which suggests that the matrix is amorphous due to the DMNs being mainly composed of gelatin from the polymer matrix and maltodextrin which the antigen was constituted into. Freshly made DMNs also showed the presence of a peak at around 42°, which suggests that a crystalline phase was also present due to D-Sorbitol. However, over storage time, this crystalline peak decreased in height suggesting that the DMNs became more amorphous over time as residual water evaporates further.
A similar system, a Cu Kα source at 45 kV and 40 mA, was used to study DOX and DTX loaded DMNs and analysis showed peaks present for the drugs and matrices used in fabrication in all samples.105 Another way to use XRD is to characterise NPs before encapsulating them into DMNs, like Yan et al.127
Imaging
CMC–TRD dissolvable MN arrays were studied as potential transdermal DDSs for human growth hormone (hGH).132 The arrays consisted of needles of height 600 μm, base diameter 300 μm and tip diameter 10 μm. These arrays were imaged using brightfield microscopy and showed a fully formed array, with sharp tipped needles. In vivo studies were carried out in hairless rats by five different administration methods – placebo of CMC only; hGH only; hGH solution mixed with CMC; hGH-MNs; hGH-MNs after 3 months of storage – and plasma hGH concentration was monitored. It was observed that the hGH concentration initially rapidly increased within the first hour and then sharply decreased over the next 6 hours. This trend was seen for all five methods; however the absolute concentrations and bioavailability differed. After euthanasia, brightfield microscopy was used to study the skin reactions of the mice. The skin was biopsied and histologically sectioned into 1 μm thick pieces. They were then stained with hematoxylin and eosin before viewing. The DMN arrays of CMC and CMC–TRD showed slight erythema of the skin, whereas subcutaneous injections showed no erythema. However, all skin reactions seen were not severe enough to cause safety or cosmetic concern.132 This study shows the different ways in which brightfield microscopy can be used to image arrays or the effects of arrays on tissue.
Earlier we briefly discussed how Bhatnagar et al.105 encapsulated anti-cancer drugs into DMNs. As DOX is fluorescent, it is visible under darkfield microscopy. The pyramidal shape of the needles can be seen distinctly, and the sharp needle tips are identifiable. The fluorescence appears uniformly spread through the needle tips and backing plate, suggesting the uniform distribution of DOX within the DMN matrix.
An example study that used stereomicroscopy to image both skin after DMN insertion, after histology to determine the penetration depth and after drug delivery, was carried out by Sullivan et al.135 They treated porcine skin with the fabricated PVP-MNs, in a 10 × 10 array, and then stained the skin post-insertion so the sites of insertion could be identified when the skin surface was imaged. The microscopy images showed that 100 needles entered the skin and when the array was imaged after, no needles were broken or deformed, suggesting that their PVP-MNs had enough mechanical strength to withstand insertion into skin. The PVP-MNs were loaded with β-galactosidase, which is enzymatically active and when stained with X-gal, gives a blue colour. Porcine skin was treated with PVP-MNs containing β-galactosidase and when histologically sectioned and imaged, a blue dye was seen, showing that not only do the PVP-MNs successfully deliver β-galactosidase, but they also do not alter the enzymatic activity of this drug in the process. Mazzarra et al.136 also visualised the application site of their DMNs using stereomicroscopy by loading their DMNs with a dye and allowing them to dissolve into the skin, releasing the dye. The samples were then histologically sectioned into 50 μm thick sections before being imaged.
Xu et al.109 used fluorescence microscopy when fabricating CAM-NPs encapsulated into PVP-MNs for wound healing. They imaged their 15 × 15 arrays to observe whether the needles were fully formed and how well distributed the NPs were within the DMN matrix. The images showed nice sharp needles of pyramidal shape with fluorescence present only in the needle tips. To determine whether their PVP-MNs containing CAM-NPs were capable of overcoming the barrier of biofilms on wounds, they used sulforhodamine B as a model drug within the DMNs and imaged the biofilms after the application of the array. The images showed the disruption of the biofilms after PVP-MN application and some dye had diffused into the biofilm matrix, suggesting that CAM-NPs would be successfully delivered to wounds through the biofilms if administered by DMNs. These images were compared to the images of topically applied sulforhodamine B solution, which showed no biofilm disruption, further supporting this conclusion.109
Wang et al.130 used fluorescence microscopy to observe the cellular expression of cells treated with up-conversion nanoparticles (UCNPs), with or without encapsulation into HA-MNs. The cells were stained with a nuclear stain before imaging to show successful delivery and gene expression.
Another way to use fluorescence microscopy is to measure the drug loading of DMNs, whether using a dye as a model drug139,140 or a fluorescent drug itself.102 It can also be used to observe the penetration of DMNs and drug release to the skin surface, as well as after histology to observe fluorescent drug molecules as deep as the dermis.102
There are various types of confocal microscopy, for example confocal laser scanning microscopy (CLSM). This type of confocal microscopy allows optical sectioning and depth selectivity of the sample. In CLSM, the light source is a laser which passes through an aperture and is focused by the objective lens before reaching the sample. Emitted photons are collected and the image is built up pixel-by-pixel.142 CLSM has been used widely for the analysis of DMNs including characterisation,105 dissolution studies99,101,140 and cell internalisation of drug molecules from DMNs.130
Another type is reflectance confocal microscopy (RCM) which uses a diode laser, giving off monochromatic and coherent light, which passes through a beam splitter and then an optical lens and onto the skin. The light penetrates the skin, illuminating an area of tissue, and any light reflected from this point passes back through the lens and is focused by a pinhole into the photodetector.143
The ability of strengthening CMC-MNs by incorporating LDH-NPs into the matrix was discussed briefly earlier.127 They used both types of confocal microscopy described above to observe the penetration and drug delivery of CMC-MNs. Fluorescein isothiocyanate (FITC)–dextran, which was used as a model drug, was mixed with a solution of CMC with LDH-NPs. RCM was used to image the porcine and human skin, which had been treated with DMNs, at an excitation wavelength of 785 nm. Uniform penetration of the stratum corneum was observed for insertion into porcine skin with LDH-NP loaded CMC-MNs, to a depth of 71 μm. However, insertion by CMC-MNs without LDH-NPs did not show uniform insertion, as only central needles penetrated the skin to a depth of 46 μm. This trend was also seen in human skin, showing a penetration depth of 64 μm and 39 μm for CMC-MNs with and without LDH-NPs respectively. These data suggest that LDH-NPs strengthen the CMC-MNs. The same samples were then imaged by CLSM to observe where the model drug had been released. For this the tissue was stained with a nuclei stain and then excited at both 405 nm and 488 nm to visualise the stain and FITC–dextran respectively. The payload was seen to have been delivered vertically and some had even diffused within the skin after only five minutes. RCM was also used to image the DMNs after certain time periods penetrating into skin. It was seen that almost all of the DMNs had dissolved after only one minute in the skin. These results show the potential of using various types of confocal microscopy for the imaging of not only the skin, but also the DMNs themselves.
UCNPs were fabricated into HA-MNs and their morphology was studied using SEM.130 The HA-MNs were studied both in the presence and absence of UCNPs, showing pyramidal 10 × 10 arrays in both cases. The height of the needles was found to be 1000 μm and the morphology did not change when UCNPs were encapsulated.
Other studies have used SEM for the characterisation of DMNs, mainly in terms of shape and morphology,109,127 and some require various ways to make their DMNs visible, i.e. sputter coating with gold.119
Although Abramson demonstrated the ability to use CT to image DMNs in situ, Loizidou et al.87 demonstrated imaging DMN arrays themselves, as well as in vitro on porcine skin. The CT scanner used had an adjustable voltage between 20 and 50 kV and a current up to 800 μA. They compared different geometry DMNs and their penetration depth by CT scan, observing that square and triangular DMNs had greater penetration than hexagonal DMNs. The process of CT scan imaging can be non-invasive when imaging tissue; however when imaging the DMN arrays themselves, they may need to be coated.
This technique was widely developed for ophthalmology and steadily penetrated other medical imaging fields as well as being extended to non-destructive testing. There are already several non-destructive testing reports of using OCT to image microneedles.
Thakur et al.101 employed a swept-source Fourier domain system at a wavelength of around 1305 nm, scan width of 2 mm, scan rate of 15 B-scans per second and resolutions of 7.5 μm laterally and 10 μm vertically to image fabricated PVP-MNs in skin. They used these images to determine the penetration depth of DMNs in ocular tissue. Lee et al.151 used a similar wavelength system, however at an A-scan rate of 100 kHz and power 30 mW, to visualise PVA–PVP-MNs that they had fabricated. The visualisation of Gantrez DMNs, with photosensitiser NPs encapsulated, in human volunteers was accomplished by Donnelly et al.139 and the penetration of DMNs was measured to be at a depth of 460 μm. However, no information on the system used was given in the paper.
Over the past three years, several research groups reported OCT systems applied to visualise the dissolution of DMNs and the release of cargo from them. Chen et al.152 fabricated gelatin/CMC patches containing R6G dye and imaged the dissolution of the dye. A year later a LUMI device that deposits DMNs into the small intestines to release insulin was fabricated.147 The DMNs were imaged in situ at a wavelength of 1.3 μm once deposited by the device and their dissolution was observed. Another study used OCT to compare hydrogel-forming MNs and DMNs in terms of the penetration depths and dissolution of OVA.153 The potential of using OCT to track contrast agents released from HA-MNs was demonstrated by Wang et al.130 These contrast agents were made from UCNPs with siRNA encapsulated.
Although OCT has been used to image DMNs themselves or in vitro in tissue samples, few have used OCT to image DMNs in situ. Quinn et al.154 used a handheld topical OCT system to image DMNs whilst inserted into human participant arms. This system consisted of a swept-source at 1305 nm which imaged to approximately 2 mm depth in skin with 7.5 μm lateral and 10 μm axial resolution. The 11 × 11 MN matrix was composed of Gantrez and polyethylene glycol (PEG), with conical needles of height 600 μm. This study154 evaluated the human variability in applying DMNs to skin, where the insertion depths for each age group were calculated and compared between self-application and researcher application. For 20–30 year olds, no significant difference in insertion depths was seen. However, in the over 65 s group, the insertion depth from researcher application was significantly lower than that of self-application, which may be due to researchers being wary of the age of the participants and therefore being softer on application. There were no significant differences seen in the pore widths between the self-administration by both age groups and of all ages, and all applications. This study is an example of how OCT can be used for characterisation101,130 in terms of penetration depth of DMNs.
The above findings demonstrate that OCT can be a useful tool for both the characterisation of DMNs and the real time monitoring of their application. However, for OCT to expand as an imaging technique within the field of microneedles, a uniform evaluation of the OCT parameters is essential for DMN characterisation and calibration of these parameters is required. For example, OCT parameters for ophthalmology were calibrated by the National Institute for Health Research in the UK and National Institutes of Health in the US. The parameter calibrations should include wavelength, power, and resolution, as well as how these parameters affect DMNs.
A system that combines bioluminescence and fluorescence imaging in 3D, providing tomographic images of internal structures, has recently been produced.160,161 Termed an In Vivo Imaging System (IVIS), this can also allow non-invasive imaging of living organisms in real time. This technique is a relatively new development and was used by McCaffrey et al.162 to image mice that had been administered DNA loaded NPs in PVP-MNs. These DMNs were applied to both ears of mice and imaged for bioluminescence 24 and 48 hours after administration. Unfortunately, the mice did not show any bioluminescence; however luciferase production was seen in the livers and kidneys at both time points. Another study observed the delivery of molecular beacons by UCNPs encapsulated into DMNs using the fluorescence imaging feature on the IVIS.130
A system similar to this has been used by Dadkhah et al.163 to image tissue. It combines OCT, photoacoustic (PA) microscopy, optical doppler tomography and confocal fluorescence microscopy to give functional, molecular and structural information.155 While an OCT signal is created by scattering (leading to morphology characterisation), the PA signal is created by absorption (leading to functionality characterisation). Advances in technology, using the same optical source for both OCT and PA, allows an easier integration of OCT with PA.164
Goswami et al.165 used an OCT/SLO system to image nanoparticles in mice retina. They used an SLO system with a wavelength of 488 nm as the light source and a wavelength of 685 nm to photoactivate the NPs. The OCT system used had a bandwidth of 132 nm, wavelength of 860 nm and power of 600 μW. This study shows the possibility of using multimodality systems to image NPs in vivo once they have been delivered by the DMNs.
Osseiran et al.166 used a system that combined corneometry, which is used to measure the hydration of superficial layers of the skin, and coherent Raman scattering (CRS) to study the structure and function of the stratum corneum of human skin. The CRS consisted of a dual-output laser source, with one output being tuneable from 680–1300 nm and the other being kept constant at 1040 nm. The combined system was used to observe how topically applied compounds act on skin and measure their uptake.
These studies demonstrate the potential of multimodality systems as imaging techniques, with the combinations often leading to quantitative analysis as well as qualitative analysis.
Drug release and activity
Choi et al.167 demonstrated the potential of using DMNs, fabricated from CMC–TRD for the needles and SUC–PVA for the backing plate, as DDSs for vaccination with HBV. They used ELISPOT analysis at 450 nm to observe T-cell responses, by administering HBV either by intramuscular (IM) injection or DMNs. A higher T-cell response was seen for DMN vaccination compared to IM vaccination immediately after administration and from five weeks post-booster vaccination. This demonstrates another way that plate readers can be used for studying drug activity.
Plate readers have mainly been used to analyse assays, such as the CCK-8 assay used to determine the cytotoxicity of materials to fibroblast cells. The cytotoxicity of CAM-NPs was observed at 450 nm and a viable cell number was determined.109 The ELISA assay is used to quantify peptides, proteins, antibodies and hormones. Edens et al.97 used the ELISA assay to detect the poliovirus antibodies at 450 nm.
They can also be used to determine drug concentration, for example the concentration of FITC–dextran from the release medium at an excitation of 493 nm and an emission of 520 nm.101 Plate readers can also be used during characterisation to determine the concentrations of drugs within DMNs.
Aditya et al.169 used UV-Vis spectroscopy to observe the dissolution of collagen–PVP-MNs doped with trypan blue, a model drug. The DMNs were dissolved in deionised (DI) water or 0.8% NaCl and UV-Vis was used to measure the dye release every 20 seconds. The DMNs readily dissolved, releasing trypan blue into the medium, in both DI and NaCl. However, it was found to be faster in DI water than in NaCl. They also observed a relationship between the height of the needle and the loading capacity where the longer the needle, the more dye it could encapsulate.
UV-Vis spectroscopy was also used to study the stability of β-galactosidase encapsulated into sugar glass at an absorbance of 405 nm. The study showed that although the enzyme lost activity over storage time, this loss was significantly reduced by storage in sugar glass.119
Kochhar et al.171 fabricated DMNs from a poly(ethylene glycol)diacrylate (PEGDA) matrix encapsulating lidocaine. In vitro drug release studies showed that the amounts of lidocaine released were lower than that encapsulated. Therefore, they used ATR-FTIR to observe whether there are any interactions between the PEGDA matrix and lidocaine.172 They fabricated PEGDA with lidocaine films and compared it to the PEGDA solution with and without lidocaine, and found that the N–H peak seen in pure lidocaine in pre-polymer solution shifted and broadened in the PEGDA film spectrum. They concluded from these results and comparison to Cui et al.'s173 results that the lidocaine is possibly hydrogen bonded with the PEGDA matrix, preventing drug release.172 The disappearance of the CC peaks in the polymerised PEGDA film, compared to liquid PEGDA, confirmed the cross-linking and polymerisation of PEGDA in the films.
Chen et al.116 used tandem MS, at a flow rate of 0.3 mL min−1, to analyse the plasma concentration of mice as part of their in vivo studies on whether GO-doping of DMNs is practical for transdermal drug delivery. Mice with melanoma tumours were administered an anti-melanoma drug HA15 by three different methods – GO–HA15-MNs; HA15-MNs; subcutaneous injection of HA15. They were also compared to two controls of saline DMNs and GO-MNs. Immediately after every treatment, all mice were irradiated with a near-IR laser for 10 minutes, no matter what group they were in. The saline MNs and GO-MNs showed no tumour growth inhibition. One and three hours after the administration, both GO–HA15-MN and HA15-MN mice showed significantly lower plasma HA15 concentrations than the mice that were subcutaneously injected. However, after 12 hours, this had drastically changed and the two drug DMN mice groups now had significantly higher plasma HA15 concentrations than the injection group. Although both drug DMN groups showed a plasma concentration profile of stable concentration followed by a slow decrease, the GO–HA15-MN mice plasma concentrations were higher than those of the HA15-MN mice at all time points.
LC/MS was used to quantify the concentration of dihydroergotamine mesylate (DHE) loaded into PVP-MNs. Tas et al.103 used LC/MS, at 25 °C and a flow rate of 0.5 mL min−1, to form a calibration curve of known DHE concentrations before running blood plasma from in vivo bioavailability studies to determine the amount of DHE in the mouse plasma. The mice were administered DHE by one of the three different methods – IV injection of DHE; subcutaneous injection of DHE and PVP-MNs loaded with DHE. The absolute and relative bioavailability of DHE administered via PVP-MNs was 72% and 97% respectively, which shows that DMNs have great potential to be used as transdermal DDSs instead of injections.
Ultraperformance MS was used by Li et al.106 to determine the concentration of LNG that is released from effervescent DMNs in vitro as an alternative form of contraception. The MS column was maintained at 50 °C and at a flow rate of 0.3 mL min−1. The DMNs were dissolved in PBS containing Tween 80 and varying concentrations of ethanol (0–25%) at 37 °C whilst being shaken. Aliquots of the release medium were collected at different time points and analysed by ultraperformance MS. There was no burst release observed, just a sustained release of 1.4% per day over at least 60 days. The presence of ethanol increased the burst release up to 25% on day 1 at a maximum ethanol concentration of 25%. No matter whether ethanol was present or not, or in what concentration, all studies showed that LNG was released over one month in all samples, which was the desired time frame.
Mazzara et al.136 loaded microparticles (MPs) containing OVA into DMNs and showed successful delivery of their cargo of OVA and other drugs to their targets through a sustained release. They used GPC to determine the mass of MPs that dissolved from the PVA–SUC-MNs by analysing the mass of MPs left in the patch after insertion into dorsal mouse skin in vivo for 20 minutes. The analysis showed that only 25% of the MPs in the array were delivered. GPC could also be used in a similar way for the characterisation of DMNs by quantifying the mass of drug NPs inside the whole DMN before use or for other drug release studies, such as Franz diffusion cell studies, by taking aliquots at regular intervals and measuring the mass of drug NPs present.
HPLC can be used in many ways to determine the concentration of a drug. Tas et al.103 used HPLC coupled with a fluorescence detector to determine DHE concentration, both during the characterisation of their PVP-MNs and drug release studies. The reverse phase column was kept at 35 °C and a flow rate of 1.2 mL min−1 was set. The fluorescence detector was set to an excitation wavelength of 280 nm and emission wavelength of 350 nm. For the characterisation, the PVP-MNs were dissolved in DI water at room temperature and analysed showing that 108% of the 50 μg DHE used in fabrication was present in the dissolution medium. They concluded that this showed that their fabrication process was successful for drug loading of DMNs. Their ex vivo-studies on porcine skin involved inserting the PVP-MNs for a period of time and at regular intervals, removing the DMNs and tape stripping the skin to remove residual DHE. The used PVP-MNs were dissolved in DI water and analysed by HPLC to determine the concentration of DHE left. The amount of drug delivered was found to increase over time whilst the PVP-MNs stayed in the skin, with 62% released after one minute and up to 79% after ten minutes. In vitro studies were carried out by dissolving the PVP-MNs loaded with DHE in DI water at 32 °C and removing aliquots at regular time intervals for analysis by HPLC. This study showed the desired rapid dissolution of DHE within two minutes of dissolution. HPLC was also used to assess DHE solubility with increasing PVP concentration, and a proportional relationship was observed.
Bhatnagar et al.105 showed the ability to use HPLC to determine the concentrations of drug cargo in DMNs fabricated from PVP and PVA. A calibration curve was plotted for known concentrations of DTX and then drug released from DMNs dissolved in PBS was determined. For these studies, the column was maintained at 25 °C with a flow rate of 1 mL min−1. The detector was set to an absorbance wavelength of 230 nm. The concentration of DTX loaded into the DMNs was 302.4 μg in DTX-MNs and 227.0 μg in DOX–DTX-MNs. The release profiles showed that only 90% of DTX was released within 15 minutes from both types of DMNs. Ex vivo skin permeation studies showed that only 33.6% and 26.7% of DTX from DTX-MNs and DOX–DTX-MNs respectively had permeated mouse skin within 48 hours. A lag in permeation was observed for DTX from DOX–DTX-MNs also.
As discussed earlier, Lee et al.131 studied the effect of the geometry and DMN matrix on mechanical strength, penetration and drug release. To measure protein stability, CD was carried out on untreated lysosomes, lysosomes from CMC-MNs one hour after fabrication and lysosomes from CMC-MNs two months after storage. There was no change of the lysosome secondary structure detected for all. However, although there was no enzyme activity loss after encapsulation into the CMC-MNs, there was a slight loss after two months of storage giving only 96% enzyme activity. As demonstrated, CD can be used to determine whether the activity of enzymes or proteins encapsulated into DMNs directly or into NPs before being encapsulated into DMNs is affected by the DMN matrices.
Zaric et al.107 used flow cytometry to asses surface marker expression triggered by OVA-NPs in poly(methyl vinyl ether) (PMVE)/maleic acid (MA)-MNs. First, they analysed dendritic cells that were treated with OVA-NPs and compared the results to those of untreated cells to demonstrate cellular uptake. Proliferation studies showed that the OVA-NPs were able to induce antigen-specific proliferation, which shows that the OVA-NPs are taken up and processed by dendritic cells, activating a T cell dose response. In vivo studies on mouse ears with plain PMVE/MA-MNs or PMVE/MA-MNs with plain NPs encapsulated were performed by flow cytometry and a localised inflammatory response was seen, which could be what initiates or enhances the immune response to vaccines delivered by DMNs. To determine whether the plain PLGA-NPs or tetramethylrhodamine (TRITC)-NPs were taken up by lymph nodes, the ear proximal draining lymph nodes were analysed 72 hours after DMN application. The data showed that TRITC-NPs were present in lymph nodes for dendritic cell-enriched suspensions; however for blood-derived dendritic cells, no uptake of TRITC-NPs was detected, suggesting that the drug cargo of DMNs stays localised to the vaccination site. When immunised with OVA-NPs in PMVE/MA-MNs, proliferation and an immune response were observed. These mice were then tumour challenged and flow cytometry analysis demonstrated an efficient antigen-specific response, as well as the presence of memory cells, suggesting that OVA vaccination through DMNs has potential. All flow cytometry was done using phycoerythrin staining of cells or tissue.107
Closing remarks
The interest in nanomaterials and their delivery into the human body in order to create a new generation of medicine is still unabated now, more than 30 years since the invention of scanning tunnelling microscopy (STM) and the subsequent development of atomic force microscopy (AFM). These two analytical tools caused the field of nanotechnology to expand and accelerate significantly, starting with the discovery of fullerenes and carbon nanotubes in the 1980s, leading to carbon dots and a myriad of nano-engineered objects in the early 2000s, many designed with the aim to specifically target cells or precise locations in the human body with minimum collateral exposure to other cells or tissues. Hundreds of thousands of articles were published, thousands of patents have been filed, and vast amounts of resources (financial and non-financial) were invested, yet the childish question ‘Are we there yet?’ still has one simple answer – ‘Not just yet’. The introduction of microneedles, which can even be 3D printed with very high resolution and accuracy, signals a new era for nanomedicine. Since nanotechnology progressed rapidly after the discovery of specific analytical tools (STM and AFM), in this review, we have provided a very comprehensive roadmap of analytical methods used to characterise the unique properties of nanomaterials and an exciting type of drug formulation – dissolvable microneedles. We believe that the specific analytical examples and the best laboratory practices mentioned in this review will serve as a useful guide and inspiration to many scientists working on next-generation drug delivery systems intended to be used in oncology; for the mass administration of vaccines; new therapeutic systems for inflammation; microbial infections; and to re-accelerate stem-cell research in tissue repair, just to name a few. Importantly though, this review might also offer useful information for researchers working in seemingly unrelated disciplines, producing nanopesticides and even reagents for biomedical diagnostics and testing.Conflicts of interest
There are no conflicts to declare.Abbreviations
| AFM | atomic force microscopy |
| ATR-FTIR | attenuated total reflectance Fourier transform infrared |
| BSA | bovine serum albumin |
| CAM | chloramphenicol |
| CD | circular dichroism |
| CLSM | confocal laser scanning microscopy |
| CMC | carboxymethyl cellulose |
| CRS | coherent Raman scattering |
| CT | computerised tomography |
| DDS | drug delivery system |
| DHE | dihydroergotamine mesylate |
| DI | deionised |
| DLS | dynamic light scattering |
| DMN | dissolvable microneedle array |
| DOX | doxorubicin |
| DSC | differential scanning calorimetry |
| DTX | docetaxel |
| FITC | fluorescein isothiocyanate |
| FTIR | Fourier transform infrared |
| GM-CSF | granulocyte-macrophage colony stimulating factor |
| GPC | gel permeation chromatography |
| GO | graphene oxide |
| HA | hyaluronic acid |
| HBV | hepatitis B vaccine |
| hGH | human growth hormone |
| HPLC | high performance liquid chromatography |
| IM | intramuscular |
| IR | infrared |
| IVIS | in vivo imaging system |
| LC/MS | liquid chromatography/mass spectrometry |
| LDH | layered double hydroxide |
| LNG | levonorgestrel |
| LUMI | luminal unfolding microneedle injector |
| MA | maleic acid |
| MN | individual microneedle |
| MNs | microneedle array |
| MP | microparticle |
| MS | mass spectrometry |
| m/z | mass-to-charge |
| NP | nanoparticle |
| OCT | optical coherence tomography |
| OVA | ovalbumin |
| PA | photoacoustic |
| PBS | phosphate buffered saline |
| PEG | polyethylene glycol |
| PEGDA | poly(ethylene glycol)diacrylate |
| PGA | polyglycolide |
| PLA | polylactide |
| PLGA | poly lactide-co-glycolide |
| PMVE | poly(methyl vinyl ether) |
| PVA | polyvinyl acetate |
| PVP | polyvinylpyrrolidone |
| RCM | reflectance confocal microscopy |
| S | spike protein |
| SbD | safe-by-design |
| SEM | scanning electron microscopy |
| SLO | scanning laser ophthalmoscopy |
| STM | scanning tunnelling microscopy |
| SUC | sucrose |
| TEM | transmission electron microscopy |
| T g | glass transition temperature |
| TGA | thermogravimetric analysis |
| TRA | trehalose anhydrous |
| TRD | trehalose dihydrate |
| TRITC | tetramethylrhodamine |
| UCNP | up-conversion nanoparticle |
| UV-Vis | ultraviolet-visible |
| XRD | X-ray diffraction |
Acknowledgements
We would like to acknowledge and thank Medway School of Pharmacy at the Universities of Kent and Greenwich for providing funding for this project and Middlesex University for the use of their facilities and materials.References
- S. Svenson, Curr. Opin. Solid State Mater. Sci., 2012, 16, 287–294 CrossRef CAS.
- S. Svenson, Eur. J. Pharm. Biopharm., 2009, 71, 445–462 CrossRef CAS PubMed.
- S. Svenson and A. S. Chauhan, Nanomedicine, 2008, 3, 679–702 CrossRef CAS PubMed.
- C. Sinico and A. M. Fadda, Expert Opin. Drug Delivery, 2009, 6, 813–825 CrossRef CAS PubMed.
- A.-M. Caminade and C.-O. Turrin, J. Mater. Chem. B, 2014, 2, 4055–4066 RSC.
- S. Liu, Z. Huang, F. Li, T. Yan, S. Fu, R. Tian, C. Hou, Q. Luo, J. Xu and J. Liu, Polym. Chem., 2019, 10, 3566–3570 RSC.
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- N. Li, S. Zeng, L. He and W. Zhong, Anal. Chem., 2010, 82, 7460–7466 CrossRef CAS PubMed.
- E. Mahon, A. Salvati, F. Baldelli Bombelli, I. Lynch and K. A. Dawson, J. Controlled Release, 2012, 161, 164–174 CrossRef CAS PubMed.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- L. Soddu, D. N. Trinh, E. Dunne, D. Kenny, G. Bernardini, I. Kokalari, A. Marucco, M. P. Monopoli and I. Fenoglio, Beilstein Arch., 2019, 0–33 Search PubMed.
- M. Ashrafizadeh, R. Mohammadinejad, S. K. Kailasa, Z. Ahmadi, E. G. Afshar and A. Pardakhty, Adv. Colloid Interface Sci., 2020, 278, 102123 CrossRef CAS PubMed.
- S. K. Kailasa, V. N. Mehta, J. R. Koduru, H. Basu, R. K. Singhal, Z. V. P. Murthy and T.-J. Park, Analyst, 2021, 146, 1489–1513 RSC.
- J. R. Koduru, S. K. Kailasa, J. R. Bhamore, K.-H. Kim, T. Dutta and K. Vellingiri, Adv. Colloid Interface Sci., 2018, 256, 326–339 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Åberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- P. M. Kelly, C. Åberg, E. Polo, A. O'Connell, J. Cookman, J. Fallon, Ž. Krpetić and K. A. Dawson, Nat. Nanotechnol., 2015, 10, 472–479 CrossRef CAS PubMed.
- S. Lara, F. Alnasser, E. Polo, D. Garry, M. C. Lo Giudice, D. R. Hristov, L. Rocks, A. Salvati, Y. Yan and K. A. Dawson, ACS Nano, 2017, 11, 1884–1893 CrossRef CAS PubMed.
- D. Maiolo, P. Bergese, E. Mahon, K. A. Dawson and M. P. Monopoli, Anal. Chem., 2014, 86, 12055–12063 CrossRef CAS PubMed.
- S. Wan, P. M. Kelly, E. Mahon, H. Stöckmann, P. M. Rudd, F. Caruso, K. A. Dawson, Y. Yan and M. P. Monopoli, ACS Nano, 2015, 9, 2157–2166 CrossRef CAS PubMed.
- C. Carrillo-Carrion, M. Carril and W. J. Parak, Curr. Opin. Biotechnol., 2017, 46, 106–113 CrossRef CAS PubMed.
- A. J. Chetwynd, K. E. Wheeler and I. Lynch, Nano Today, 2019, 28, 100758 CrossRef CAS PubMed.
- US FDA Center for Drug Evaluation and Research, Drug products, including biological products, that contain nanomaterials—guidance for industry, 2017 Search PubMed.
- A. Hardy, D. Benford, T. Halldorsson, M. J. Jeger, H. K. Knutsen, S. More, H. Naegeli, H. Noteborn, C. Ockleford, A. Ricci, G. Rychen, J. R. Schlatter, V. Silano, R. Solecki, D. Turck, M. Younes, Q. Chaudhry, F. Cubadda, D. Gott, A. Oomen, S. Weigel, M. Karamitrou, R. Schoonjans, A. Mortensen and EFSA Scientific Committee, EFSA J., 2018, 16(7), 5327 Search PubMed.
- SCENIHR, Opinion on the Guidance on the Determination of Potential Health Effects of Nanomaterials Used in Medical Devices, 2015 Search PubMed.
- B. Halamoda-Kenzaoui, U. Holzwarth, G. Roebben, A. Bogni and S. Bremer-Hoffmann, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 11, e1531 Search PubMed.
- C. Schwarz-Plaschg, A. Kallhoff and I. Eisenberger, Nanoethics, 2017, 11, 277–281 CrossRef.
- K. Donaldson, F. Murphy, A. Schinwald, R. Duffin and C. A. Poland, Nanomedicine, 2011, 6, 143–156 CrossRef CAS PubMed.
- V. Stone, S. Gottardo, E. A. J. Bleeker, H. Braakhuis, S. Dekkers, T. Fernandes, A. Haase, N. Hunt, D. Hristozov, P. Jantunen, N. Jeliazkova, H. Johnston, L. Lamon, F. Murphy, K. Rasmussen, H. Rauscher, A. S. Jiménez, C. Svendsen, D. Spurgeon, S. Vázquez-Campos, W. Wohlleben and A. G. Oomen, Nano Today, 2020, 35, 100941 CrossRef CAS.
- F. Murphy, S. Dekkers, H. Braakhuis, L. Ma-Hock, H. Johnston, G. Janer, L. di Cristo, S. Sabella, N. R. Jacobsen, A. G. Oomen, A. Haase, T. Fernandes and V. Stone, NanoImpact, 2021, 22, 100314 CrossRef.
- M. Schmutz, O. Borges, S. Jesus, G. Borchard, G. Perale, M. Zinn, Ä. A. J. A. M. Sips, L. G. Soeteman-Hernandez, P. Wick and C. Som, Front. Bioeng. Biotechnol., 2020, 8, 1–7 CrossRef PubMed.
- V. Gubala, L. J. Johnston, H. F. Krug, C. J. Moore, C. K. Ober, M. Schwenk and M. Vert, Pure Appl. Chem., 2018, 90, 1325–1356 CAS.
- H. A. E. Benson, J. E. Grice, Y. Mohammed, S. Namjoshi and M. S. Roberts, Curr. Drug Delivery, 2019, 16, 444–460 CrossRef CAS PubMed.
- K. Kuche, R. Maheshwari, V. Tambe, K.-K. Mak, H. Jogi, N. Raval, M. R. Pichika and R. Kumar Tekade, Nanoscale, 2018, 10, 8911–8937 RSC.
- Y. Chen, X. Feng and S. Meng, Expert Opin. Drug Delivery, 2019, 16, 847–867 CrossRef CAS PubMed.
- N. Dragicevic and H. Maibach, Adv. Drug Delivery Rev., 2018, 127, 58–84 CrossRef CAS PubMed.
- B. Iqbal, J. Ali and S. Baboota, Int. J. Dermatol., 2018, 57, 646–660 CrossRef PubMed.
- R. Jijie, A. Barras, R. Boukherroub and S. Szunerits, J. Mater. Chem. B, 2017, 5, 8653–8675 RSC.
- E. Larrañeta, M. T. C. McCrudden, A. J. Courtenay and R. F. Donnelly, Pharm. Res., 2016, 33, 1055–1073 CrossRef PubMed.
- M. Rabiei, S. Kashanian, S. S. Samavati, S. Jamasb and S. J. P. McInnes, J. Drug Targeting, 2020, 28, 356–367 CrossRef CAS PubMed.
- V. Gubala, L. J. Johnston, Z. Liu, H. Krug, C. J. Moore, C. K. Ober, M. Schwenk and M. Vert, Pure Appl. Chem., 2018, 90, 1283–1324 CAS.
- IUPAC, Aggregation (except in polymer science), in IUPAC Compendium of Chemical Terminology, IUPAC, Research Triagle Park, NC, DOI:10.1351/goldbook.AT07608, (accessed 5 May 2020).
- IUPAC, Aggregate in catalysis, in IUPAC Compendium of Chemical Terminology, IUPAC, Research Triagle Park, NC, DOI:10.1351/goldbook.A00184, (accessed 5 May 2020).
- IUPAC, Orthokinetic aggregation in colloids, in IUPAC Compendium of Chemical Terminology, IUPAC, Research Triagle Park, NC, DOI:10.1351/goldbook.O04336, (accessed 5 May 2020).
- IUPAC, Perikinetic aggregation in colloids, in IUPAC Compendium of Chemical Terminology, IUPAC, Research Triagle Park, NC, DOI:10.1351/goldbook.P04492, (accessed 5 May 2020).
- IUPAC, Agglomeration in polymer science, in IUPAC Compendium of Chemical Terminology, IUPAC, Research Triagle Park, NC, DOI:10.1351/goldbook.AT07607, (accessed 5 May 2020.
- G. Nichols, S. Byard, M. J. Bloxham, J. Botterill, N. J. Dawson, A. Dennis, V. Diart, N. C. North and J. D. Sherwood, J. Pharm. Sci., 2002, 91, 2103–2109 CrossRef CAS PubMed.
- Z. Wu, S. Yang and W. Wu, Nanoscale, 2016, 8, 1237–1259 RSC.
- M. Schneider, F. Stracke, S. Hansen and U. F. Schaefer, Dermatoendocrinol, 2009, 1, 197–206 CrossRef CAS PubMed.
- J.-H. Park, S. Davis, Y.-K. Yoon, M. R. Prausnitz and M. G. Allen, in The Sixteenth Annual International Conference on Micro Electro Mechanical Systems, 2003. MEMS-03 Kyoto. IEEE, IEEE, 2003, pp. 371–374 Search PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. D. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS PubMed.
- I. Slowing, J. Viveroescoto, C. Wu and V. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- B. E. Rabinow, Nat. Rev. Drug Discovery, 2004, 3, 785–796 CrossRef CAS PubMed.
- J. Kreuter, Int. J. Pharm., 2007, 331, 1–10 CrossRef CAS PubMed.
- H. F. Krug, Angew. Chem., Int. Ed., 2014, 53(46), 12304–12319 CAS.
- V. J. Venditto and F. C. Szoka, Adv. Drug Delivery Rev., 2013, 65, 80–88 CrossRef CAS PubMed.
- G. Giovannini, F. Kunc, C. C. Piras, O. Stranik, A. A. Edwards, A. J. Hall and V. Gubala, RSC Adv., 2017, 7, 19924–19933 RSC.
- G. Giovannini, P. Warncke, D. Fischer, O. Stranik, A. J. Hall and V. Gubala, Nanotoxicology, 2018, 12, 407–422 CrossRef CAS PubMed.
- J. D'Orazio, S. Jarrett, A. Amaro-Ortiz and T. Scott, Int. J. Mol. Sci., 2013, 14, 12222–12248 CrossRef PubMed.
- M. R. Prausnitz and R. Langer, Nat. Biotechnol., 2008, 26, 1261–1268 CrossRef CAS PubMed.
- Y.-C. Kim, J.-H. Park and M. R. Prausnitz, Adv. Drug Delivery Rev., 2012, 64, 1547–1568 CrossRef CAS PubMed.
- M. Ogundele and H. Okafor, J. Pharm. Res. Int., 2017, 18, 1–14 CrossRef.
- S. Bhatnagar, K. Dave and V. V. K. Venuganti, J. Controlled Release, 2017, 260, 164–182 CrossRef CAS PubMed.
- P. Serrano-Castañeda, J. J. Escobar-Chavez, I. M. Rodriguez-cruz, L. M. Melgoza and J. Martinez-Hernandez, J. Pharm. Pharm. Sci., 2018, 21, 73–93 Search PubMed.
- M. M. Badran, J. Kuntsche and A. Fahr, Eur. J. Pharm. Sci., 2009, 36, 511–523 CrossRef CAS PubMed.
- J.-H. Park, S.-O. Choi, S. Seo, Y. Bin Choy and M. R. Prausnitz, Eur. J. Pharm. Biopharm., 2010, 76, 282–289 CrossRef CAS PubMed.
- A. Kumar, Y. W. Naguib, Y. Shi and Z. Cui, Drug Delivery, 2014, 176, 1–7 CrossRef PubMed.
- J. A. Scott and A. K. Banga, Ther. Delivery, 2015, 6, 1089–1099 CrossRef CAS PubMed.
- J. D. Bos and M. M. H. M. Meinardi, Exp. Dermatol., 2000, 9, 165–169 CrossRef CAS PubMed.
- J. Yang, X. Liu, Y. Fu and Y. Song, Acta Pharm. Sin. B, 2019, 9, 469–483 CrossRef PubMed.
- M. S. Gerstel and V. A. Place, US Pat., US3964482A, 1971 Search PubMed.
- S. Kaushik, A. H. Hord, D. D. Denson, D. V. McAllister, S. Smitra, M. G. Allen and M. R. Prausnitz, Anesth. Analg., 2001, 92, 502–504 CrossRef CAS.
- R. Bajracharya, J. G. Song, S. Y. Back and H.-K. Han, Comput. Struct. Biotechnol. J., 2019, 17, 1290–1308 CrossRef CAS PubMed.
- M. L. Bookstaver, S. J. Tsai, J. S. Bromberg and C. M. Jewell, Trends Immunol., 2018, 39, 135–150 CrossRef CAS PubMed.
- Y. Hao, W. Li, X. Zhou, F. Yang and Z. Qian, J. Biomed. Nanotechnol., 2017, 13, 1581–1597 CrossRef CAS PubMed.
- L. F. Santos, I. J. Correia, A. S. Silva and J. F. Mano, Eur. J. Pharm. Sci., 2018, 118, 49–66 CrossRef CAS PubMed.
- J. H. Cary, B. S. Li and H. I. Maibach, Biomed. Microdevices, 2019, 21, 66 CrossRef PubMed.
- M. Kim, H. Yang, H. Kim, H. Jung and H. Jung, Int. J. Cosmet. Sci., 2014, 36, 207–212 CrossRef CAS PubMed.
- Y. Park, J. Park, G. S. Chu, K. S. Kim, J. H. Sung and B. Kim, Biotechnol. Bioprocess Eng., 2015, 20, 543–549 CrossRef CAS.
- Y. Park, K. S. Kim, M. Chung, J. H. Sung and B. Kim, J. Ind. Eng. Chem., 2016, 39, 121–126 CrossRef CAS.
- R. Soltani-Arabshahi, J. W. Wong, K. L. Duffy and D. L. Powell, JAMA Dermatol., 2014, 150, 68 CrossRef PubMed.
- W. Sun, M. Inayathullah, M. A. C. Manoukian, A. V. Malkovskiy, S. Manickam, M. P. Marinkovich, A. T. Lane, L. Tayebi, A. M. Seifalian and J. Rajadas, Ann. Biomed. Eng., 2015, 43, 2978–2990 CrossRef PubMed.
- B. M. Torrisi, V. Zarnitsyn, M. R. Prausnitz, A. Anstey, C. Gateley, J. C. Birchall and S. A. Coulman, J. Controlled Release, 2013, 165, 146–152 CrossRef CAS PubMed.
- X. Wu, H. Zhang, S. He, Q. Yu, Y. Lu, W. Wu, N. Ding, Q. Zhu, Z. Chen, Y. Ma and J. Qi, Int. J. Biol. Macromol., 2020, 150, 528–535 CrossRef CAS PubMed.
- P. Aggarwal and C. R. Johnston, Sens. Actuators, B, 2004, 102, 226–234 CrossRef CAS.
- E. Z. Loizidou, N. T. Inoue, J. Ashton-Barnett, D. A. Barrow and C. J. Allender, Eur. J. Pharm. Biopharm., 2016, 107, 1–6 CrossRef PubMed.
- D. V. McAllister, P. M. Wang, S. P. Davis, J.-H. Park, P. J. Canatella, M. G. Allen and M. R. Prausnitz, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13755–13760 CrossRef CAS PubMed.
- M. G. Nava-Arzaluz, I. Calderon-Lojero, D. Quintanar-Guerrero, R. Villalobos-Garcia and A. Ganem-Quintanar, Curr. Drug Delivery, 2012, 9, 57–73 CrossRef CAS PubMed.
- M. J. Mistilis, A. S. Bommarius and M. R. Prausnitz, J. Pharm. Sci., 2015, 104, 740–749 CrossRef CAS PubMed.
- L. Y. Chu, L. Ye, K. Dong, R. W. Compans, C. Yang and M. R. Prausnitz, Pharm. Res., 2016, 33, 868–878 CrossRef CAS PubMed.
- M. J. Mistilis, J. C. Joyce, E. S. Esser, I. Skountzou, R. W. Compans, A. S. Bommarius and M. R. Prausnitz, Drug Delivery Transl. Res., 2017, 7, 195–205 CrossRef CAS PubMed.
- E. Q. Littauer, L. K. Mills, N. Brock, E. S. Esser, A. Romanyuk, J. A. Pulit-Penaloza, E. V. Vassilieva, J. T. Beaver, O. Antao, F. Krammer, R. W. Compans, M. R. Prausnitz and I. Skountzou, J. Controlled Release, 2018, 276, 1–16 CrossRef CAS PubMed.
- S. P. Sullivan, D. G. Koutsonanos, M. del Pilar Martin, J. W. Lee, V. Zarnitsyn, S.-O. Choi, N. Murthy, R. W. Compans, I. Skountzou and M. R. Prausnitz, Nat. Med., 2010, 16, 915–920 CrossRef CAS PubMed.
- Y.-H. Chen, K.-Y. Lai, Y.-H. Chiu, Y.-W. Wu, A.-L. Shiau and M.-C. Chen, Acta Biomater., 2019, 97, 230–238 CrossRef CAS PubMed.
- C. Edens, M. L. Collins, J. L. Goodson, P. A. Rota and M. R. Prausnitz, Vaccine, 2015, 33, 4712–4718 CrossRef CAS PubMed.
- C. Edens, N. C. Dybdahl-Sissoko, W. C. Weldon, M. S. Oberste and M. R. Prausnitz, Vaccine, 2015, 33, 4683–4690 CrossRef CAS PubMed.
- M. B. Perez Cuevas, M. Kodani, Y. Choi, J. Joyce, S. M. O'Connor, S. Kamili and M. R. Prausnitz, Bioeng. Transl. Med., 2018, 3, 186–196 CrossRef CAS PubMed.
- S. P. Pattarabhiran, A. Saju, K. R. Sonawane, R. Manimaran, S. Bhatnagar, G. Roy, R. B. Kulkarni and V. V. K. Venuganti, AAPS PharmSciTech, 2019, 20, 257 CrossRef PubMed.
- E. Kim, G. Erdos, S. Huang, T. W. Kenniston, S. C. Balmert, C. D. Carey, V. S. Raj, M. W. Epperly, W. B. Klimstra, B. L. Haagmans, E. Korkmaz, L. D. Falo and A. Gambotto, EBioMedicine, 2020, 55, 102743 CrossRef PubMed.
- R. R. S. Thakur, I. A. Tekko, F. Al-Shammari, A. A. Ali, H. McCarthy and R. F. Donnelly, Drug Delivery Transl. Res., 2016, 6, 800–815 CrossRef CAS PubMed.
- D. Brambilla, S. T. Proulx, P. Marschalkova, M. Detmar and J.-C. Leroux, Small, 2016, 12, 1053–1061 CrossRef CAS PubMed.
- C. Tas, J. C. Joyce, H. X. Nguyen, P. Eangoor, J. S. Knaack, A. K. Banga and M. R. Prausnitz, J. Controlled Release, 2017, 268, 159–165 CrossRef CAS PubMed.
- Y. Li, F. Liu, C. Su, B. Yu, D. Liu, H.-J. Chen, D. Lin, C. Yang, L. Zhou, Q. Wu, W. Xia, X. Xie and J. Tao, ACS Appl. Mater. Interfaces, 2019, 11, 30575–30584 CrossRef CAS PubMed.
- S. Bhatnagar, N. G. Bankar, M. V. Kulkarni and V. V. K. Venuganti, Int. J. Pharm., 2019, 556, 263–275 CrossRef CAS PubMed.
- W. Li, J. Tang, R. N. Terry, S. Li, A. Brunie, R. L. Callahan, R. K. Noel, C. A. Rodríguez, S. P. Schwendeman and M. R. Prausnitz, Sci. Adv., 2019, 5, eaaw8145 CrossRef CAS PubMed.
- M. Zaric, O. Lyubomska, O. Touzelet, C. Poux, S. Al-Zahrani, F. Fay, L. Wallace, D. Terhorst, B. Malissen, S. Henri, U. F. Power, C. J. Scott, R. F. Donnelly and A. Kissenpfennig, ACS Nano, 2013, 7, 2042–2055 CrossRef CAS PubMed.
- M. Zaric, O. Lyubomska, C. Poux, M. L. Hanna, M. T. McCrudden, B. Malissen, R. J. Ingram, U. F. Power, C. J. Scott, R. F. Donnelly and A. Kissenpfennig, J. Invest. Dermatol., 2015, 135, 425–434 CrossRef CAS PubMed.
- J. Xu, R. Danehy, H. Cai, Z. Ao, M. Pu, A. Nusawardhana, D. Rowe-Magnus and F. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 14640–14646 CrossRef CAS PubMed.
- D. Li, D. Hu, H. Xu, H. K. Patra, X. Liu, Z. Zhou, J. Tang, N. Slater and Y. Shen, Biomaterials, 2021, 264, 120410 CrossRef CAS PubMed.
- P. Makvandi, M. Kirkby, A. R. J. Hutton, M. Shabani, C. K. Y. Yiu, Z. Baghbantaraghdari, R. Jamaledin, M. Carlotti, B. Mazzolai, V. Mattoli and R. F. Donnelly, Nano-Micro Lett., 2021, 13, 93 CrossRef CAS PubMed.
- S. Dugam, R. Tade, R. Dhole and S. Nangare, Future J. Pharm. Sci., 2021, 7, 19 CrossRef.
- M. Wang, L. Hu and C. Xu, Lab Chip, 2017, 17, 1373–1387 RSC.
- J. H. Jung and S. G. Jin, J. Pharm. Invest. DOI:10.1007/s40005-021-00512-4.
- Horiba, What is Raman Spectroscopy?, accessed 21 April 2020, https://www.horiba.com/en_en/raman-imaging-and-spectroscopy/.
- Y. Chen, Y. Yang, Y. Xian, P. Singh, J. Feng, S. Cui, A. Carrier, K. Oakes, T. Luan and X. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 352–360 CrossRef CAS PubMed.
- J. D. Menczel, L. Judovits, R. B. Prime, H. E. Bair, M. Reading and S. Swier, in Thermal Analysis of Polymers, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2008, pp. 7–239 Search PubMed.
- S. Mukherji and D. Mondal, in Medical Biosensors for Point of Care (POC) Applications, Elsevier, 2017, pp. 99–131 Search PubMed.
- C. J. Martin, C. J. Allender, K. R. Brain, A. Morrissey and J. C. Birchall, J. Controlled Release, 2012, 158, 93–101 CrossRef CAS PubMed.
- C. Kolluru, Y. Gomaa and M. R. Prausnitz, Drug Delivery Transl. Res., 2019, 9, 192–203 CrossRef CAS PubMed.
- A. W. Coats and J. P. Redfern, Analyst, 1963, 88, 906 RSC.
- P. Inc.
- K. R. Rajisha, B. Deepa, L. A. Pothan and S. Thomas, in Interface Engineering of Natural Fibre Composites for Maximum Performance, Elsevier, 2011, pp. 241–274 Search PubMed.
- ThermoFisher Scientific, FTIR Basics, accessed 21 April 2020, https://www.thermofisher.com/uk/en/home/industrial/spectroscopy-elemental-isotope-analysis/spectroscopy-elemental-isotope-analysis-learning-center/molecular-spectroscopy-information/ftir-information/ftir-basics.html.
- B. E. Warren, X-Ray Diffraction, Addison-Wesley Publishing Company Inc, Reading, 1st edn, 1990 Search PubMed.
- Montana State University Carlton College Science Education Resource Center, X-Ray Powder Diffraction (XRD), accessed 21 April 2020, https://serc.carleton.edu/research_education/geochemsheets/techniques/XRD.html.
- L. Yan, A. P. Raphael, X. Zhu, B. Wang, W. Chen, T. Tang, Y. Deng, H. J. Sant, G. Zhu, K. W. Choy, B. K. Gale, T. W. Prow and X. Chen, Adv. Healthcare Mater., 2014, 3, 555–564 CrossRef CAS PubMed.
- B. Bhushan and M. L. B. Palacio, in Encyclopedia of Nanotechnology, ed. B. Bhushan, Springer Netherlands, Dordrecht, 2012, pp. 1576–1596 Search PubMed.
- Microscope Master, Brightfield Microscopy, accessed 21 April 2020, https://www.microscopemaster.com/brightfield-microscopy.html.
- M. Wang, Y. Han, X. Yu, L. Liang, H. Chang, D. C. Yeo, C. Wiraja, M. L. Wee, L. Liu, X. Liu and C. Xu, Adv. Healthcare Mater., 2020, 9, 1900635 CrossRef CAS PubMed.
- J. W. Lee, J.-H. Park and M. R. Prausnitz, Biomaterials, 2008, 29, 2113–2124 CrossRef CAS PubMed.
- J. W. Lee, S.-O. Choi, E. I. Felner and M. R. Prausnitz, Small, 2011, 7, 531–539 CrossRef CAS PubMed.
- Nikon MicroscopyU, Darkfield Illumination, accessed 21 April 2020, https://www.microscopyu.com/techniques/stereomicroscopy/darkfield-illumination.
- Sciencing, Difference between Compound and Dissecting Microscopes, accessed 21 April 2020, https://sciencing.com/difference-between-compound-dissecting-microscopes-5576645.html.
- S. P. Sullivan, N. Murthy and M. R. Prausnitz, Adv. Mater., 2008, 20, 933–938 CrossRef CAS PubMed.
- J. M. Mazzara, L. J. Ochyl, J. K. Y. Hong, J. J. Moon, M. R. Prausnitz and S. P. Schwendeman, Bioeng. Transl. Med., 2019, 4, 116–128 CrossRef CAS PubMed.
- J. D. Kim, M. Kim, H. Yang, K. Lee and H. Jung, J. Controlled Release, 2013, 170, 430–436 CrossRef CAS PubMed.
- J. W. Lichtman and J.-A. Conchello, Nat. Methods, 2005, 2, 910–919 CrossRef CAS PubMed.
- R. F. Donnelly, D. I. J. Morrow, F. Fay, C. J. Scott, S. Abdelghany, R. R. T. Singh, M. J. Garland and A. David Woolfson, Photodiagn. Photodyn. Ther., 2010, 7, 222–231 CrossRef CAS PubMed.
- Y. Choi, S. G. Lee, J. H. Jeong, K. M. Lee, K. H. Jeong, H. Yang, M. Kim, H. Jung and S. Lee, Int. J. Nanomed., 2013, 9, 289 CrossRef PubMed.
- Olympus, Introduction to Confocal Microscopy, What is Confocal Laser Scanning Microscopy?, https://bitesizebio.com/19958/what-is-confocal-laser-scanning-microscopy/, (accessed 21 April 2020).
- Bio Bitesize, What is Confocal Laser Scanning Microscopy?, accessed 21 April 2020, https://bitesizebio.com/19958/what-is-confocal-laser-scanning-microscopy/.
- DermNet NZ, Reflectance Confocal Microscopy, accessed 21 April 2020, https://dermnetnz.org/topics/reflectance-confocal-microscopy/.
- Bruker, Confocal Raman Spectroscopy, accessed 21 April 2020, https://www.bruker.com/products/infrared-near-infrared-and-raman-spectroscopy/raman/confocal-raman.html.
- P. J. Caspers, G. W. Lucassen, H. A. Bruining and G. J. Puppels, J. Raman Spectrosc., 2000, 31, 813–818 CrossRef CAS.
- National Institute of Biomedical Imaging and Bioengineering, Computerised Tomography, accessed 21 April 2020, https://www.nibib.nih.gov/science-education/science-topics/computed-tomography-ct.
- A. Abramson, E. Caffarel-Salvador, V. Soares, D. Minahan, R. Y. Tian, X. Lu, D. Dellal, Y. Gao, S. Kim, J. Wainer, J. Collins, S. Tamang, A. Hayward, T. Yoshitake, H.-C. Lee, J. Fujimoto, J. Fels, M. R. Frederiksen, U. Rahbek, N. Roxhed, R. Langer and G. Traverso, Nat. Med., 2019, 25, 1512–1518 CrossRef CAS PubMed.
- A. G. Podoleanu, J. Microsc., 2012, 247, 209–219 CrossRef PubMed.
- J. G. Fujimoto, Nat. Biotechnol., 2003, 21, 1361–1367 CrossRef CAS PubMed.
- W. Drexler and J. G. Fujimoto, Optical Coherence Tomography: Technology and Applications, Springer, Berlin, Heidelberg, 2008 Search PubMed.
- I.-C. Lee, J.-S. He, M.-T. Tsai and K.-C. Lin, J. Mater. Chem. B, 2015, 3, 276–285 RSC.
- C.-H. Chen, V. Shyu and C.-T. Chen, Materials, 2018, 11, 1625 CrossRef PubMed.
- A. J. Courtenay, A. M. Rodgers, M. T. C. McCrudden, H. O. McCarthy and R. F. Donnelly, Mol. Pharm., 2019, 16, 118–127 CrossRef CAS PubMed.
- H. L. Quinn, C. M. Hughes and R. F. Donnelly, Drug Delivery Transl. Res., 2018, 8, 307–316 CrossRef CAS PubMed.
- A. Podoleanu, J. Izatt, B. Lumbroso, M. Pircher, R. Rosen and R. Weitz, Biomed. Opt. Express, 2019, 10, 2135 CrossRef PubMed.
- A. G. Podoleanu and D. A. Jackson, Electron. Lett., 1998, 34, 1088 CrossRef.
- A. G. Podoleanu, G. M. Dobre, R. Cernat, J. A. Rogers, J. Pedro, R. B. Rosen and P. Garcia, J. Biomed. Opt., 2007, 12, 014019 CrossRef PubMed.
- M. J. Marques, M. R. Hughes, K. Vyas, A. Thrapp, H. Zhang, A. Bradu, G. Gelikonov, P. Giataganas, C. J. Payne, G.-Z. Yang and A. Podoleanu, J. Biomed. Opt., 2019, 24, 1 Search PubMed.
- A. Bradu, L. Ma, J. W. Bloor and A. Podoleanu, J. Biophotonics, 2009, 2, 380–388 CrossRef PubMed.
- Newcast University, In Vivo Imaging System, accessed 21 April 2020, https://www.ncl.ac.uk/pivi/equipment/ivis/.
- Berthold, In Vivo Imaging Systems, accessed 21 April 2020, https://www.berthold.com/en/bioanalytic/products/invivo-imaging-systems/.
- J. McCaffrey, C. M. McCrudden, A. A. Ali, A. S. Massey, J. W. McBride, M. T. C. McCrudden, E. M. Vicente-Perez, J. A. Coulter, T. Robson, R. F. Donnelly and H. O. McCarthy, J. Controlled Release, 2016, 226, 238–247 CrossRef CAS PubMed.
- A. Dadkhah, J. Zhou, N. Yeasmin and S. Jiao, Biomed. Opt. Express, 2019, 10, 137 CrossRef CAS PubMed.
- M. Bondu, M. J. Marques, P. M. Moselund, G. Lall, A. Bradu and A. Podoleanu, Photoacoustics, 2018, 9, 21–30 CrossRef CAS PubMed.
- M. Goswami, X. Wang, P. Zhang, W. Xiao, S. J. Karlen, Y. Li, R. J. Zawadzki, M. E. Burns, K. S. Lam and E. N. Pugh, Biomed. Opt. Express, 2019, 10, 151 CrossRef CAS PubMed.
- S. Osseiran, J. Dela Cruz, S. Jeong, H. Wang, C. Fthenakis and C. L. Evans, Biomed. Opt. Express, 2018, 9, 6425 CrossRef CAS PubMed.
- Y. H. Choi, M. B. Perez-Cuevas, M. Kodani, X. Zhang, M. R. Prausnitz, S. Kamili and S. M. O'Connor, J. Infect. Dis., 2019, 220, 1926–1934 CrossRef CAS PubMed.
- J. Clark, UV-Visible Absorption Spectra, accessed 21 April 2020, https://www.chemguide.co.uk/analysis/uvvisible/theory.html.
- A. Aditya, B. Kim, R. D. Koyani, B. Oropeza, M. Furth, J. Kim and N. P. Kim, J. Drug Delivery Sci. Technol., 2019, 52, 618–623 CrossRef CAS.
- ThermoFisher Scientific, FTIR Sample Techniques: Attenuated Total Reflection (ATR), accessed 21 April 2020, https://www.thermofisher.com/uk/en/home/industrial/spectroscopy-elemental-isotope-analysis/spectroscopy-elemental-isotope-analysis-learning-center/molecular-spectroscopy-information/ftir-information/ftir-sample-handling-techniques/ftir-sample-handling-tec.
- J. S. Kochhar, W. J. Goh, S. Y. Chan and L. Kang, Drug Dev. Ind. Pharm., 2013, 39, 299–309 CrossRef CAS PubMed.
- J. S. Kochhar, J. J. Y. Tan, Y. C. Kwang and L. Kang, Microneedles for Transdermal Drug Delivery, Springer International Publishing, Cham, 2019 Search PubMed.
- Y. Cui and S. G. Frank, J. Pharm. Sci., 2006, 95, 701–713 CrossRef CAS PubMed.
- E. de Hoffmann, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2005 Search PubMed.
- Agilent Technologies Inc., Primer, 2015, pp. 1–32 Search PubMed.
- Y. Kazakevich, R. LoBrutto, Y. Liu, A. Vailaya, G. Chen, L.-K. Zhang, B. N. Pramanik, T. Patel, M. McBrien, D. B. Kassel, I. Kazakevich, R. Bakhtiar, T. K. Majumdar, F. L. S. Tse, R. Thompson, J. Etse, A. D. Jerkovich, R. V. Vivilecchia, R. M. Smith, Y.-H. Liu, M. V. S. Elipe, E. Kuesters, N. Grinberg, T. Burakowski and A. M. Stalcup, HPLC for Pharmaceutical Scientists, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2007 Search PubMed.
- A. Rodger and B. Norden, Circular Dichroism and Linear Dichroism, Oxford University Press, Oxford, 1997 Search PubMed.
- ThermoFisher Scientific, How A Flow Cytometer Works, accessed 21 April 2020, https://www.thermofisher.com/uk/en/home/life-science/cell-analysis/cell-analysis-learning-center/molecular-probes-school-of-fluorescence/flow-cytometry-basics/flow-cytometry-fundamentals/how-flow-cytometer-works.html.
- Abcam, Introduction to Flow Cytometry, accessed 21 April 2020, https://www.abcam.com/protocols/introduction-to-flow-cytometry.
| This journal is © The Royal Society of Chemistry 2021 |