
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A relative quantitation method for measuring DNA methylation and hydroxymethylation using guanine as an internal standard
Krisztina
Németh
ab,
Katalin
Mészáros
c,
Borbála
Szabó
d,
Henriett
Butz
cd,
Tamás
Arányi
ef and
Pál T.
Szabó
 *b
*b
aInstitute of Chemistry, Eötvös Loránd University, Pázmány Péter stny. 1/A, H-1117 Budapest, Hungary
bMS Metabolomics Research Group, Centre for Structural Study, Research Centre for Natural Sciences, Eötvös Loránd Research Network, Magyar tudósok krt. 2, H-1117 Budapest, Hungary. E-mail: szabo.pal@ttk.hu
cHereditary Tumours Research Group, Eötvös Loránd Research Network, Semmelweis University, Szentkirályi u. 46, H-1088 Budapest, Hungary
dDepartment of Laboratory Medicine, Semmelweis University, Bókay János u. 53-54, H-1089 Budapest, Hungary
eInstitute of Enzymology, Research Centre for Natural Sciences, Eötvös Loránd Research Network, Magyar tudósok krt. 2, H-1117 Budapest, Hungary
fDepartment of Molecular Biology, Semmelweis University, Tűzoltó u. 37-47, H-1094 Budapest, Hungary
First published on 16th September 2021
Abstract
Global DNA methylation and hydroxymethylation play an important role in gene expression. They can be connected with several diseases. The modification status could be a biomarker to determine the status of disease. A fast, easy and accurate liquid chromatography – tandem mass spectrometry method has been developed for the precise quantitation of 5-methylcytosine and 5-hydroxymethylcytosine. Formic acid was used for the hydrolysis of the DNA strand resulting in nucleobases. These polar hydrolysis products were separated on a normal phase column using reversed phase eluents in inverse gradient mode. Multiple reaction monitoring was applied to achieve high selectivity and sensitivity for the quantitation. A new relative quantitation model was developed by using guanine, as an internal standard, present in samples. The new method was successfully validated with excellent accuracy and precision values in the range of 0.005–0.5% for 5hmC and 1–15% for 5mC. The main advantages of this quantitation method are that, due to relative quantitation, calibration curves can be used without reacquiring the calibration points and no additional isotope labeled internal standards are required. The method was tested to identify the concentrations of 5mC and 5hmC in various sample types. The lowest level of DNA sample required in the case of 0.005% 5hmC is 0.5 μg.
1. Introduction
DNA methylation is one of the most important epigenetic modifications and occurs mainly in CpG dinucleotides.1 This modification takes place at the 5-position of cytosine, forming 5-methylcytosine (5mC).2 It plays an important role in many biological processes, such as transcription, genome stability and embryogenesis.3 Aberrant methylation can alter genomic imprinting,4 transcription5 and chromosomal stability.4 It has been associated with many types of cancers and also non-neoplastic diseases.1 Enzymatic oxidation of 5-methylcytosine results in 5-hydroxymethylcytosine (5hmC).6 This modification is very important in the DNA demethylation process7 and it is critical in embryonic development, cellular differentiation,8 in the regulation of gene expression and transcriptional activity as well.2 An abnormal level of 5hmC can lead to many disorders, such as cardiovascular diseases, neurological modification,9,10 or cancer.11 As indicators of the degree of methylation and hydroxymethylation, 5mC and 5hmC could be biomarkers of diseases such as cancers.Different analytical methods are used for analyzing the methylation levels of DNA. These methods start with the hydrolysis of DNA bonds. The hydrolysis could be achieved enzymatically or with a strong acid.12 The enzymatic method results in nucleotides or nucleosides, while acidic hydrolysis results in free bases. It is important to note that the samples need to be RNA free because the hydrolysis products of DNA and RNA are mainly identical.13
Among the techniques used for studying DNA methylation, bisulfite sequencing is used as the “gold standard”, but it has limitations; for example, it is time-consuming, needs a large amount of DNA, and is not really feasible for the quantitation of 5hmC.14 There are a wide variety of analytical techniques for the analysis of the hydrolysis products of DNA. Nucleosides can be analyzed by chemiluminescence, capillary electrophoresis, thin layer chromatography,15 and gas chromatography.16 An alternative approach is high performance liquid chromatography,13,17 which can be used for measuring the amount of nucleosides and free bases as well. Chromatographic separation is very difficult because of the high polarity of these compounds. Mainly reversed phase (RP) chromatography13,17–22 and, in some cases, hydrophilic interaction liquid chromatography (HILIC)17,23 techniques were established. Appropriate separation is challenging. The HILIC method needs a longer equilibration time, while the molecules have little retention on reversed phase columns and can elute with the void volume. Ion-pairing modifiers used in mobile phases for better separation can help in retaining these polar components on the RP column. These modifiers are, however, not compatible with commonly used mass spectrometric detectors.
To quantitate 5mC and 5hmC, mainly absolute calibration is used with isotope-labeled cytosine 13C15N2.13,23–25 This calibration model requires the measurement of the calibration sample points every time resulting in more sample preparation and a longer measurement time. Another popular approach is to determine the ratio of the molar quantities of the compound to the sum of cytosine-based molecules;21,26,27 however, only a few articles have reported the use of deoxyguanosine (dG) for quantitation.20,22
Our work on the identification of DNA methylation started several years ago. We reported a study where we identified the concentration of 5mC using an Rx-SIL column.28 The sensitivity of the mass spectrometer was not enough to use that method for the analysis of 5hmC. Later we reported a study using a more sensitive mass spectrometer capable of measuring 5hmC as well.29 That method used cytosine as an internal standard making data processing more complex. Our goal in this study was to finalize our findings on the identification of the concentrations of 5mC and 5hmC and arrive at an easy, cheap, fast and reliable LC-MS/MS method. The optimization of the sample preparation (hydrolysis, evaporation, and reconstitution), chromatographic separation and mass spectrometric detection is shown. A new calibration model using guanine from DNA samples as an internal standard is also presented. The final method has been validated and used for the identification of the methylation levels in biological samples.
2. Experimental
2.1. Materials and reagents
5-Methyl-dCTP (2′-deoxy-5-methylcytidine 5′-triphosphate, 5mdC) and 5-hydroxymethyl-dCTP (2′-deoxy-5-hydroxymethylcytidine 5′-triphosphate, 5hmdC) were purchased from Zymo Research (Irvine, CA, USA) and dNTP (deoxynucleotide triphosphates) mix was purchased from Promega Corporation (Madison, WI, USA). Acetonitrile (ACN, MeCN, gradient grade) and formic acid (FA, >99%) were obtained from VWR International (Fontenay-sous-Bois, France). Water was produced by a Milli-Q Purification System from Millipore (Burlington, MA, USA).2.2. Instrumentation and conditions
A QTRAP 6500 triple quadrupole – linear ion trap mass spectrometer equipped with a Turbo V Source (Sciex, MA, USA) and an Agilent 1100 Series HPLC (Agilent, CA, USA) was used for LC-MS/MS analysis. Data acquisition was performed using Analyst software 1.6.3, and the data were analyzed using MultiQuant software 2.1 (Sciex, MA, USA). Chromatographic separation was carried out on an Agilent Zorbax Rx-SIL column (250 × 4.6 mm, 5 μm, Kromat Ltd, Hungary). Water containing formic acid at 0.1 v/v% (eluent A) and acetonitrile containing formic acid at 0.1 v/v% (eluent B) were used for separation. The flow rate was 1 ml min−1 and 10 μl of the samples were injected. The column temperature was ambient, and samples were kept at 10 °C in an autosampler during the acquisition. The following inverse gradient program was used, where the water is the stronger eluent: A/B 15/85(6)-3-90/10(2)-0.5-15/85(6.5). Electrospray ionization was performed in positive mode. Optimized source conditions in mass spectrometric measurements were as follows: spray voltage 4000 V, evaporation temperature 550 °C, curtain, evaporation, and drying gases 40, 45, and 50 au (arbitrary units), respectively. The MS/MS was operated under multiple reaction monitoring (MRM) mode with nitrogen as collision gas the value of which was set at “high” instrument unit. The MRM transitions and analyzer parameters can be found in Table 1.| Analyte | MRM transitions (Da) | Dwell time (ms) | Declustering potential (DP, volts) | Entrance potential (EP, volts) | Collision energy (CE, volts) | Collision cell exit potential (CXP, volts) |
|---|---|---|---|---|---|---|
| G | 152/110 | 25 | 100 | 10 | 40 | 15 |
| 152/135 | 25 | 50 | 10 | 15 | 12 | |
| 5mC | 126/83 | 25 | 60 | 9 | 17 | 15 |
| 126/108 | 25 | 60 | 9 | 25 | 12 | |
| 5hmC | 142/81 | 80 | 50 | 9 | 30 | 10 |
| 142/124 | 35 | 80 | 5 | 20 | 14 |
2.3. Samples and DNA isolation
Four different types of cells were used as biological samples. They were purchased from American Type Culture Collection (ATCC). We used two different immortalized cell lines (Homo sapiens and Rattus norvegicus). The human cell lines were HL60 (CCL-240) and K562 (CCL-243); the rat cell lines were RC-4 B/C (CRL-1903) and GH3 (CCL-82.1).All types of cells were cultured following the ATCC recommendations. The decitabine (5-aza-2′-deoxycytidine) agent was used as a DNA methyltransferase (DNMT) inhibitor on rat and human cells. The rat and human cell preparations were carried out following the DNeasy® Blood & Tissue Handbook (Qiagene, Germany) and by the Puregene (158745; Qiagen Germany) protocol, respectively.
2.4. Preparation of stock solutions and calibration standards
The expected 5mC and 5hmC levels were 1–15% and 0.005–0.5% of the amount of C in the samples, respectively.The dNTP mix contained 10 mM concentrations of dATP, dCTP, dGTP, and dTTP, as well. It was diluted with water to get a stock solution at a concentration of 125 μM.
The concentration of the 5mdC standard was 10 mM. It was diluted with water to a concentration of 100 μM. This 5mdC stock solution (100 μM) was further diluted with water to reach the working solutions of 5, 15, 25, 35, 50, 60, and 75 μM. Calibration solutions were prepared by mixing 80 μl dNTP stock solution and 20 μl of each 5mdC working solution. 10 μl from each 5mdC calibration solution was taken into a 2 ml crimped glass vial for hydrolysis to get the calibration points. The concentrations of the calibration points of 5mC are 1, 3, 5, 7, 10, 12 and 15 μM, containing 100 μM dNTP (including dGTP). This resulted in final 5mC/G concentrations of 1, 3, 5, 7, 10, 12 and 15%, respectively.
100 mM 5hmdC standard was diluted into two stock solutions with concentrations of 100 nM and 10 μM, respectively. Working solutions of 25 and 50 nM were prepared from the 100 nM stock solution, while working solutions of 100 nM, 250 nM, 1 μM and 2.5 μM were prepared from the 10 μM stock solution. 20 μl of each 5hmdC working solution was mixed with 80 μl dNTP stock solution resulting in calibration solutions. 10 μl of each 5hmdC calibration solution was crimped in a 2 ml glass vial for hydrolysis. The final concentrations of 5hmC are 5, 10, 20, 50, 200 and 500 nM, containing 100 μM dNTP (including dGTP) in each. This results in 0.005, 0.01, 0.02, 0.05, 0.2 and 0.5% 5hmC/G.
2.5. Sample preparation
Control and treated cell lines were used for test measurements.A cell solution containing 1 μg DNA or 10 μl of calibration solutions was hydrolyzed by adding 50 μl formic acid (>99%) to them in a 2 ml crimped vial. The samples were kept at 140 °C for 90 min. After they cooled to room temperature, the samples were evaporated under nitrogen. The residue was reconstituted in 70 μl acetonitrile–water (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v) containing 0.1 v/v% formic acid.
:50 v/v) containing 0.1 v/v% formic acid.
2.6. Optimization of the parameters of the sample preparation
Different parameters were investigated in order to achieve the best yield of hydrolysis and reach the best LLOQ (lower limit of quantitation) values. Three different volumes of formic acid (50, 100, and 200 μl) at different hydrolysis temperatures (70, 100, 120, 140, and 200 °C) and hydrolysis times (45, 90, 135, and 180 min) were applied. The samples were evaporated when left in open air, and by using nitrogen or compressed air. Four different reconstituting eluents (acetonitrile, water, acetonitrile:water 1:1 containing 0.1 v/v% formic acid, and acetonitrile:water 9:1 containing 0.1 v/v% formic acid) were tested with three different volumes (70, 100, and 2 × 50 μl).
2.7. Method validation
The method was validated based upon linearity, the lower limit of detection (LLOD), the lower limit of quantitation (LLOQ), accuracy and precision, matrix effects and stability. Linearity was measured using a 7-point and 6-point calibration curve for 5mC and 5hmC, respectively. LLOD and LLOQ values were required to be calculated only in the case of 5hmC with the signal-to-noise ratio greater than 3 and 10, respectively. The concentration of 5mC in the biological DNA samples is much higher than the LLOQ of the method, so the LLOD and LLOQ identification was irrelevant. Accuracy and precision were assessed by the analysis of calibration standards within the same day and on 5 different validation days, with 2 parallel sample preparations and 2 different injection volumes. The matrix effect was studied by comparing the shape of the signal obtained by continuously infused 5mC and 5hmC solutions using a T-splitter when acetonitrile, water and a pooled sample were injected. The pooled sample is a mix of several prepared and measured biological (GH3 and Rc-4 B/C cells) samples, without specified concentrations. It was used for system suitability tests and for stability measurements. Post-preparation stability was evaluated by comparing the peak area ratios with pooled samples under different storage conditions: short term stability at 10 °C for 3 h, and long term stability at freeze – thaw cycles 3 times. Further stability measurements at different temperatures were not necessary to be performed. The percentage of methylation and hydroxymethylation was determined by calculating the area ratio of 5mC or 5hmC to G ([5mC]/[G] and [5hmC]/[G]).3. Results and discussion
3.1. MS optimization
Several criteria should be fulfilled to have a sensitive method. The most important of these is the sensitivity of the mass spectrometer. When our DNA methylation study started, a medium category mass spectrometer was only available in the laboratory. Our efforts were relegated to the quantitation of 5mC because the instrument was unable to reach the required sensitivity level for 5hmC. There are, however, other parameters that can be optimized resulting in even better LOD and LOQ values. MS optimization gives the best ion formation in the ion source and in the collision cell and the best ion transmission for ions to reach the detector. In this study, we are focusing on G, 5mC and 5hmC of which 5hmC is the most critical due to its extremely low concentration in DNA samples.An automatic MRM optimization protocol was performed on each target molecule by using direct infusion of stock solutions into the mass spectrometer. The two most intense fragment ions were selected for the quantitative and qualitative determination for each component. The source parameters such as the spray voltage, source gas values and the temperature were set to get the highest signal for the 5hmC molecule. The dwell times were adjusted to get a final cycle time suitable for the chromatography and to get the highest sensitivity for 5hmC. For this reason, the standard 25 ms dwell time was increased to 80 and 35 ms in the case of quantifier and qualifier MRM transitions of 5hmC, respectively. The final cycle time was 245 ms including the pause times.
The differences in the concentrations of 5hmC and G might be 4 orders of magnitude. The mass spectrometer should have a dynamic range of at least this. Under our initial conditions, guanine gave a quite high signal. The intensity of G depends on the amount of the initial DNA. The increase of the signal of G can cause detector saturation changing the ion ratios and making our method useless. To eliminate this effect, the MRM parameters of guanine were changed and moved from the ideal values resulting in a lower signal but still linear with the change of concentration.
3.2. HPLC optimization
The separation of the extremely polar hydrolysis products of DNA degradation was a substantial challenge. Different chromatographic conditions including stationary and mobile phases were tested to find the optimal system for quantitation. The HILIC technique is known to be dedicated for the separation of polar compounds, where a normal phase column is used for the separation with a polar mobile phase, which is usually modified with buffers. Therefore, different HILIC columns (Waters Halo Penta-HILIC, Phenomenex Luna HILIC) and eluents (methanol, ammonium-formate) have been tested, but none of them were robust and accurate enough. Then we tried a normal phase Rx-SIL HPLC column from Agilent. The Rx-SIL column is a totally porous silica column capable of separating basic, neutral and acidic samples. Normal and reversed phase eluents can also be used as mobile phases. It is important to mention that ammonium ions as a commonly used eluent modifier should be avoided in the case of the Rx-SIL column. Ammonium ions can modify the surface of the stationary phase resulting in asymmetric broad analyte peaks. In our experiments, water and acetonitrile, both containing 0.1 v/v% formic acid, were used. These are commonly used eluents in reversed phase chromatography where the acetonitrile is the stronger eluent. In the classical gradient mode, the acetonitrile concentration increases during the run. In our method, the same eluents were used but the aqueous one is the stronger eluent, whose concentration increased during the run. The method using classical reversed phase eluents in gradient mode and having an aqueous eluent as the stronger eluent is called the inverse gradient method. Although our method seems like a HILIC technique, the gradient program and the mobile phases used are not typical of classical HILIC methods.We tried to do fine adjustments in the gradient profile to get better retention and separation of the nucleobases of interest. A long isocratic part was set as the initial condition with 15% eluent A. This part is followed by a linear gradient increasing the aqueous eluent to 90%. This is the optimal region of separation of the compounds. Due to the gradient delay of the system, 5mC and 5hmC elute at the end of this linear gradient part. The retention times of 5mC and 5hmC are close to each other; however, no suppression or any effects could be observed during modeling experiments, which could be modifying the peak intensities measured separately. The washing and equilibration parts of the method were adjusted to get the system back to the initial conditions. The overall run time of the method was 18 min. To obtain better separation we tried to increase the column temperature and used gradient elution with different slopes, but these changes did not result in better selectivity. The sensitivity of a method, in general, depends on the injected volume. The higher the volume injected, the higher the sensitivity. Different injection volumes (5, 10 and 35 μl) were tested. The increase in the injected volume is good for the detection of 5mC and 5hmC but might have an opposite effect for the component with a high concentration. Peak broadening and distortion were observed in the case of 35μl injection. We decided to have smaller injection volumes (5 and 10 μl) for standards and biological samples as well.
The measured extracted ion chromatograms of the nucleobases with the final settings can be seen in Fig. 1. Intensity values are in counts per second (cps).
 | ||
| Fig. 1 Extracted ion chromatograms of (a) G, (b) 5mC and (c) 5hmC. The retention times are 7.4; 10.01; 10.07 min, respectively. Sharp and symmetric peaks are obtained especially in the case of 5mC and 5hmC. | ||
3.3. Optimization of sample preparation
A sensitive and reliable method requires the optimization of not only the MS and chromatographic conditions but also the sample preparation. At the earlier stage of our methylation study, we tried both enzymatic and acidic hydrolysis; but later on, we decided to use the acidic one. The acidic hydrolysis was faster, easier and more reliable. Several optimization steps have been performed to get the best hydrolysis conditions. We tested the volume of the formic acid used, the hydrolysis time and temperature, the evaporation conditions and finally the reconstituting conditions (the volume and solvent composition). We found that a higher temperature helped in the hydrolysis, but increasing the temperature can cause the cleavage of the nucleobases. The efficiency of the hydrolysis was tested by mixing nucleosides and free bases (C + G, dC + G, C + dG, and dC + dG) and the samples were hydrolyzed at different temperatures for different times. The hydrolysis products were then compared to see the efficiency of the hydrolysis (the nucleoside peaks should be eliminated) and the stability of free nucleobases. No further degradation was observed in the case of free nucleobases and complete hydrolysis was found in the case of nucleosides.When an extremely high temperature is applied for the hydrolysis, the liquid samples evaporate forming a large volume of vapour increasing the pressure in the reservoir. This elevated pressure can cause an explosion and the acid vapor can burn the skin. Safety is the most important in scientific work so an absolutely safe reactor should be found. Several vials were tested, and the 2 ml crimped vial was found to be the safest for our experiments. It has a volume high enough to keep vapors inside without an explosion and the septum can safely release the overpressure without explosion of the glass vial itself in case of emergency. The evaporation conditions have been tested by applying different gases and gas flow. The nitrogen flow was found to be the best for the evaporation of the liquid because due to the flow the evaporation time is short (half an hour) and no oxidative side reaction could be found in the case of 5mC. 5mC can be oxidized forming 5hmC when air is applied. Leaving in open air resulted in a longer evaporation time and a higher level of 5hmC compared to compressed air evaporation.
The final step of the sample preparation is the reconstitution of the evaporated residues. Different solvent compositions and final volumes were tested and acetonitrile:water 1:1 containing 0.1 v/v% formic acid was found to be the best reconstituting eluent in a volume of 70 μl. The optimized sample preparation parameters are summarized in Section 2.4.
3.4. Quantitation model
The classical method of calibration is to compare the intensity of a given compound in the unknown sample with that of the known calibration standard solution. This method gives absolute concentration values. An internal standard is usually used to make the method more precise and robust by compensating for the intensity changes caused by different effects such as ion suppression or a contaminated ion source. When an internal standard is used the intensity ratios of the sample and internal standard are plotted as a function of the concentration ratios of the sample and the internal standard. While in the study of DNA methylation, the level of 5mC or 5hmC is reported as a percentage of total C, this model is said to be relative quantitation. At the earlier stage of our methylation study, we used C as an internal standard and the exact concentrations of 5mC and 5hmC could be calculated in a complicated manner taking into account that the total C is the sum of the 5mC, 5hmC and the residue of C in the sample. This is because a small part of the total C from the sample is present as 5mC and 5hmC during DNA modifications.In this work, we used G, as an internal standard. It is known that the molar ratio of G and C in DNA samples is equal so using guanine as an internal standard, the results can be obtained from the calibration curve without any calculation or conversion. The other advantage of this method is that while guanine is always present in DNA samples no additional internal standard is required. Based on intensity ratios, it was found that the system is independent of the absolute concentration and the area ratios are not influenced by the dilution or the injection volume. This means that it is not necessary to know the exact amount of the initial DNA in the sample, and the calibration curve measured once can be used without re-acquiring the calibration points day by day.
Although free nucleobases are available, for a better similarity of the experimental conditions to the biological samples, nucleosides were used for the calibration. The same hydrolysis protocol was performed on the standard samples as on DNA samples. The relative calibration method can be applied only in the case where the change of intensities is proportional to the changes of concentration and this relationship is linear. Mass spectrometers are known to be sensitive detectors where the signal is linear with the concentration, especially at a low concentration level. However, intensity changes can occur due to the dimer formation or detector saturation in a high concentration range. While the guanine concentration is quite high in the DNA samples, we had to check the linearity of the MS in the region of interest. Triplicate measurements have been performed on a dilution series by measuring the area of the guanine peak as a function of the concentration and were found to be linear in the range of 4 orders of magnitude. The logarithmic result of these experiments is shown in Fig. 2.
 | ||
| Fig. 2 Logarithmic plot of the peak area vs. concentration of the guanine standard (released by acid hydrolysis from a pure solution of dGTP). The intensities vary linearly with the concentration in the entire range. The method can only be applied in the range where the concentration–area plot is linear. | ||
Similar linear results were obtained in the case of 5mC and 5hmC. The next important thing is to check the effect of dilution on the area ratios. One calibration point (10% 5mC/G) was selected and a series of dilutions was applied by a factor of 2 and the samples gave the same intensity ratio values. This experiment proved that the area ratios are not influenced by the dilution. The effect of the injected volume on the intensity ratios was studied by injecting different volumes (5 and 10 μl) of samples. No significant difference was observed indicating that the identification of area ratios is independent of the actual injected volumes.
3.5. Method validation
 | ||
| Fig. 3 Calibration curves of 5mC and 5hmC. A zoomed region in the case of 5hmC/G shows the lower concentration points. | ||
Linear regression analysis revealed adequate accuracy and precision (relative standard deviation, RSD) of the calculated concentration for both analytes at each concentration level (Table 2). The weighting was 1/concentration for 5hmC and no weighting was used for 5mC. LLOD and LLOQ values were only necessary to calculate for 5hmC because concentrations of the other two analytes (5mC, and G) were much above the reachable LLOD and LLOQ. The detection and quantitation limits for 5hmC were found to be 1.43 fmol and 7.14 fmol on the column, respectively. They were calculated from the concentration data of the calibration points, meeting the LLOD and LLOQ criteria (2.7.), respectively.
| Analyte | Statistical variable | Theoretical concentration (n = 9) | ||||||
|---|---|---|---|---|---|---|---|---|
| 5mC/G% concentration | ||||||||
| 1 | 3 | 5 | 7 | 10 | 12 | 15 | ||
| 5mC | Mean | 1.0 | 3.0 | 5.1 | 7.1 | 9.9 | 12.0 | 15.1 |
| Accuracy (%) | 99.1 | 100.2 | 101.3 | 101.0 | 99.0 | 99.6 | 100.6 | |
| RSD (%) | 3.0 | 1.7 | 1.4 | 1.4 | 2.3 | 1.8 | 1.5 | |
| Analyte | Statistical variable | Theoretical concentration (n = 9) | |||||
|---|---|---|---|---|---|---|---|
| 5hmC/G% concentration | |||||||
| 0.005 | 0.01 | 0.02 | 0.05 | 0.2 | 0.5 | ||
| 5hmC | Mean | 0.005 | 0.010 | 0.020 | 0.050 | 0.199 | 0.501 |
| Accuracy (%) | 98.5 | 100.7 | 100.5 | 99.5 | 99.5 | 100.2 | |
| RSD (%) | 2.0 | 2.0 | 4.5 | 1.8 | 3.5 | 2.6 | |
 | ||
| Fig. 4 The signal changes of 5mC after chromatographic injection of (a) ACN, (b) H2O and the (c) biological sample. No significant matrix effect can be observed in the expected retention time region. | ||
| Analyte | Statistical variable | Intra-day (n = 9) | Inter-day (n = 9) |
|---|---|---|---|
| 5mC | Mean (5mC/G%) | 0.652 | 0.66 |
| Accuracy (%) | 99.0 | 99.6 | |
| RSD (%) | 0.5 | 3.0 | |
| 5hmC | Mean (5hmC/G%) | 0.0051 | 0.0049 |
| Accuracy (%) | 101.1 | 98.6 | |
| RSD (%) | 1.0 | 4.0 |
3.6. Biological sample measurements
The newly developed and validated method was tested on different types of biological samples. The aim of this test was to compare the results obtained with the classical method using an isotope labeled internal standard and with the new calibration model. The 5mC and 5hmC values were measured in various cell lines before and after DNMT inhibitor treatment. The same cell lines and sample preparation protocol were used as applied previously.29,30 Rat cells and the human cells were prepared freshly and kept at −80 °C after treatment and before measurements. Cells were treated with DMSO (referred to as control) and decitabine, a known inhibitor of C-5 DNA methyltransferases. The possible RNA contamination of DNA was measured using a spectrophotometer. There were three different biological replicates with three different technical replicates; all of them were isolated from different culture plates. Each sample contains 1 μg of DNA and the prepared samples were measured three times. The mean and standard deviation (SD) values of the results are presented on the graphs.The summary of the biological sample measurements is shown in Fig. 5. All cell data (5mC and 5hmC) were normalized to DMSO. The same tendency was obtained for all cell lines as found in our earlier studies, with demethylation resulting in lower 5mC levels. In the case of human cell lines, the changes of 5mC and 5hmC levels were the same as described previously.29 In rat cell lines, the 5hmC level decreased in Rc-4 B/C cells and increased in GH3 cells, as found earlier.30 In rat samples, we could reproduce the previous results when the classical calibration method was applied using an isotope labeled standard.
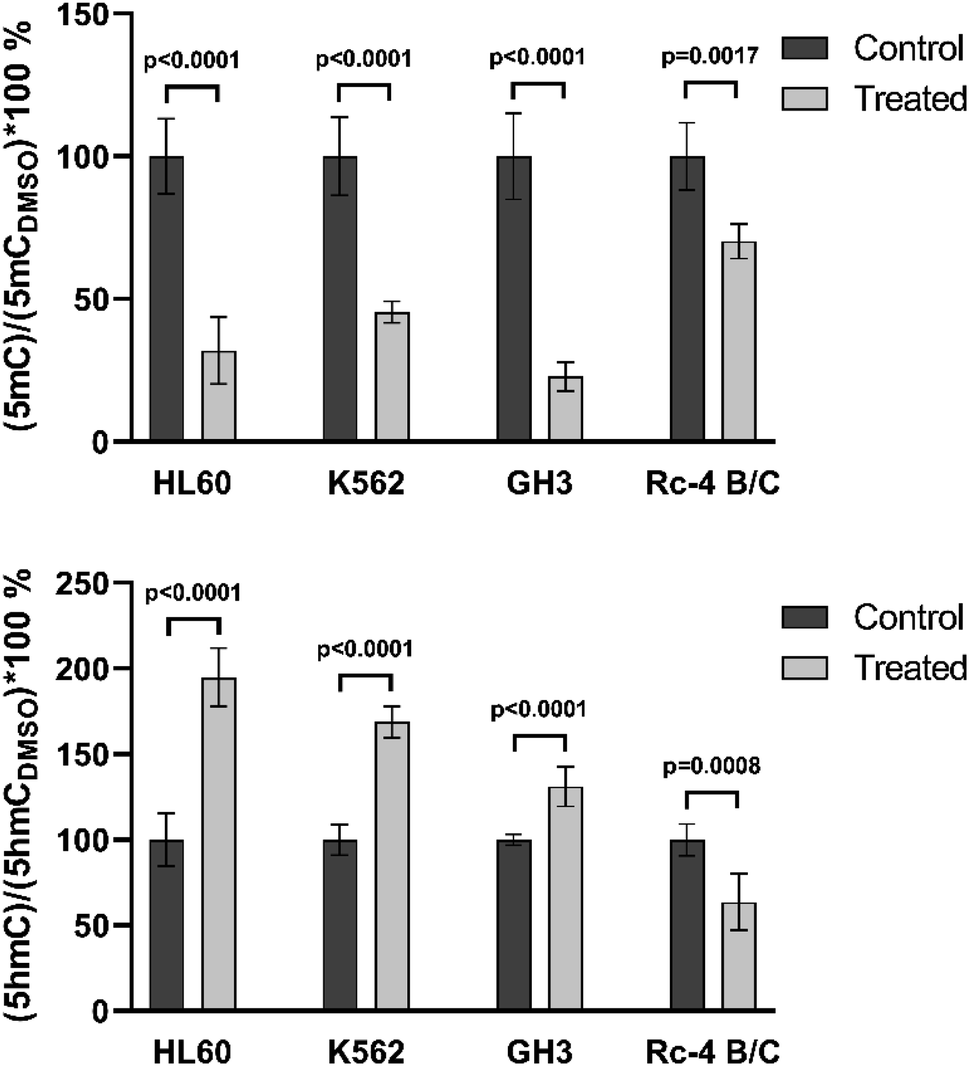 | ||
| Fig. 5 Application of the method on biological samples. It can be seen that the control and treated samples could easily be differentiated for 5mC and 5hmC. | ||
The analytical precision of the method was tested by measuring the control samples of two cell lines (HL60 and K562). There were four biological samples and each sample was measured on 3 different days. It can be seen that the analytical precision of the measurements is much better than the biological accuracy. For this reason, it is recommended to use at least 3 parallel biological samples for accurate 5mC and 5hmC identification. The results of the precision measurements are summarized in Fig. 6.
 | ||
| Fig. 6 Analytical precision study. The results of the parallel measurements of controls (HL60 and K562 cell lines). | ||
4. Conclusions
A sensitive and simple method was developed and validated for the DNA methylation study. Relative quantitation was performed using guanine, as an internal standard, which is in situ present in each sample. The advantages of this new method are it is not necessary to know the exact initial DNA concentration; no expensive isotope-labeled internal standards are required because guanine is always in the samples; the dilution of the sample does not influence the final result. The main advantage is, however, that the same calibration can be used on different days for a long time; no day-by-day calibration is required. The method is suitable for identifying the concentrations of 5mC and 5hmC in a range of 1–15% and 0.005–0.5%, respectively. The required minimum DNA sample depends on the 5hmC content. The LOD of 5hmC was found to be 1.43 fmol on a column. The method was tested many times on biological samples and was able to differentiate between the control and treated samples. The developed method is robust and accurate and can be used in high-throughput analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Higher Education Institutional Excellence Programme of the Ministry of Human Capacities in Hungary, within the framework of the Molecular Biology thematic program of Semmelweis University to Attila Patócs. This work was supported by a NKFIH K_132695 grant awarded to Tamás Arányi and Pál T. Szabó, and NKFI FK 135065 to Henriett Butz. Henriett Butz's work is supported by Bolyai Research Fellowship of Hungarian Academy of Sciences. The authors acknowledge the financial support and a critical review of the manuscript by Attila Patócs. The authors thank Lilla Krokker for helping in repeated sample preparation.References
- H. Shi, M. X. Wang and C. W. Caldwell, Expert Rev. Mol. Diagn., 2007, 7, 519–531 CrossRef CAS PubMed.
- H. Wu and Y. Zhang, Genes Dev., 2011, 25, 2436–2452 CrossRef CAS PubMed.
- D. Schübeler, Nature, 2015, 517, 321–326 CrossRef PubMed.
- T. Jursch, C. Miskey, Z. Izsvák and Z. Ivics, Mobile DNA, 2013, 4, 1–11 CrossRef PubMed.
- S. B. Baylin and J. E. Ohm, Nat. Rev. Cancer, 2006, 6, 107–116 CrossRef CAS PubMed.
- D. R. Grayson and A. Guidotti, Neuropsychopharmacology, 2013, 38, 138–166 CrossRef CAS PubMed.
- R. M. Kohli and Y. Zhang, Nature, 2013, 502, 472–479 CrossRef CAS PubMed.
- R. Straussman, D. Nejman, D. Roberts, I. Steinfeld, B. Blum, N. Benvenisty, I. Simon, Z. Yakhini and H. Cedar, Nat. Struct. Mol. Biol., 2009, 16, 564–571 CrossRef CAS PubMed.
- A. Shukla, M. Sehgal and T. R. Singh, Gene, 2015, 564, 109–118 CrossRef CAS PubMed.
- T. Jiang, L. Zong, L. Zhou, Y. Hou, L. Zhang, X. Zheng, H. Han, S. Li, W. Zhang, J. Zhang, C. Deng, Y. Jia and C. Zhao, Psychiatry Res., 2017, 257, 497–500 CrossRef CAS PubMed.
- J. Jeschke, E. Collignon and F. Fuks, Curr. Opin. Genet. Dev., 2016, 36, 16–26 CrossRef CAS PubMed.
- M. S. Rocha, R. Castro, I. Rivera, R. M. Kok, Y. M. Smulders, C. Jakobs, I. T. De Almeida and H. J. Blom, Clin. Chem. Lab. Med., 2010, 48, 1793–1798 CAS.
- R. M. Kok, D. E. C. Smith, R. Barto, A. M. W. Spijkerman, T. Teerlink, H. J. Gellekink, C. Jakobs and Y. M. Smulders, Clin. Chem. Lab. Med., 2007, 45, 903–911 CAS.
- S. G. Jin, S. Kadam and G. P. Pfeifer, Nucleic Acids Res., 2010, 38, 1–7 CrossRef PubMed.
- Z. Taleat, K. Mathwig, E. J. R. Sudhölter and L. Rassaei, TrAC, Trends Anal. Chem., 2015, 66, 80–89 CrossRef CAS.
- Y. Tang, X. D. Gao, Y. Wang, B. F. Yuan and Y. Q. Feng, Anal. Chem., 2012, 84, 7249–7255 CrossRef CAS PubMed.
- L. Rastegar, H. Mighani and A. Ghassempour, Anal. Biochem., 2018, 557, 123–130 CrossRef CAS PubMed.
- X. Wang, Y. Suo, R. Yin, H. Shen and H. Wang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 1647–1652 CrossRef CAS PubMed.
- P. Si-Yuan, Z. Jie, T. Mei-Ping, W. Zhan-Lin and S. He-Qing, Chin. J. Anal. Chem., 2012, 40, 1201–1206 Search PubMed.
- X. Li and A. A. Franke, Anal. Chim. Acta, 2011, 703, 58–63 CrossRef CAS PubMed.
- T. Le, K. P. Kim, G. Fan and K. F. Faull, Anal. Biochem., 2011, 412, 203–209 CrossRef CAS PubMed.
- S. E. Dwi Putra, C. Neuber, C. Reichetzeder, B. Hocher and B. Kleuser, Cell. Physiol. Biochem., 2014, 33, 945–952 CrossRef PubMed.
- X. Ye, L. Zhang, B. Chen, J. Li, Q. Yang, Q. Huang, J. Zhang, Y. Gao, Z. Li and C. Cai, Talanta, 2017, 169, 136–140 CrossRef CAS PubMed.
- L. Zhang, L. Zhang, K. Zhou, X. Ye, J. Zhang, A. Xie, L. Chen, J. X. Kang and C. Cai, J. Biomol. Screening, 2012, 17, 877–884 CrossRef PubMed.
- J. J. Zhang, L. Zhang, K. Zhou, X. Ye, C. Liu, L. Zhang, J. Kang and C. Cai, Anal. Biochem., 2011, 413, 164–170 CrossRef CAS PubMed.
- L. Rastegar, H. Mighani and A. Ghassempour, Anal. Biochem., 2018, 557, 123–130 CrossRef CAS PubMed.
- Z. Liu, J. Wu, Z. Xie, S. Liu, P. Fan-Havard, T. H. M. Huang, C. Plass, G. Marcucci and K. K. Chan, Anal. Biochem., 2009, 391, 106–113 CrossRef CAS PubMed.
- Y. Yamagata, P. Szabó, D. Szüts, C. Bacquet, T. Arányi and A. Páldi, Epigenetics, 2012, 7, 141–145 CrossRef CAS PubMed.
- B. Vető, P. Szabó, C. Bacquet, A. Apró, E. Hathy, J. Kiss, J. M. Réthelyi, F. Szeri, D. Szüts and T. Arányi, FEBS Open Bio, 2018, 1–9 Search PubMed.
- B. Szabó, K. Németh, K. Mészáros, N. Szücs, S. Czirják, L. Reiniger, H. Rajnai, I. Krencz, K. Karászi, L. Krokker, A. Patócs and H. Butz, J. Clin. Endocrinol. Metab., 2020, 105, 2015–2026 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |