
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA microfluidic system for monitoring glucagon secretion from human pancreatic islets of Langerhans†
Wesley J.
Eaton
 and
Michael G.
Roper
*
and
Michael G.
Roper
*
Department of Chemistry and Biochemistry, Florida State University, 95 Chieftain Way, Tallahassee, FL 32306, USA. E-mail: mroper@fsu.edu; Tel: +1-850-644-1846
First published on 26th July 2021
Abstract
Glucagon is a 29-amino acid peptide released from α-cells within pancreatic islets of Langerhans to help raise blood glucose levels. While a plethora of methodologies have been developed for quantitative measurement of insulin released from islets, such methods are not well developed for glucagon despite its importance in blood sugar regulation. In this work, a simple yet robust microfluidic device was developed for holding human pancreatic islets and perfuse them with glucose. The perfusate was collected into 2 min fractions and glucagon quantified using a homogeneous time-resolved Förster resonance energy transfer (TR-FRET) sandwich immunoassay. Simulation of fluid flow within the microfluidic device indicated the device produced low amounts of shear stress on islets, and characterization of the flow with standard glucagon solutions revealed response times within 2 fractions (<4 min). Results with human islets from multiple donors demonstrated either a “burst” of glucagon or a “sustained” glucagon release across the entire period of stimulation. The simplicity, yet robustness, of the device and method is expected to appeal to a number of researchers examining pancreatic islet physiology.
Introduction
Glucagon is a 29-amino acid hormone peptide derived from proglucagon that acts to increase blood glucose levels by initiating glycogenolysis and gluconeogenesis in the liver.1,2 As such, it acts as a counter regulatory hormone to insulin, and is essential for maintaining euglycemia during times of fasting. Glucagon is released from α-cells located in pancreatic islets of Langerhans. In human islets, α-cells make up ∼5–10% of the islet, with the insulin-secreting β-cells making up ∼70–80%.3 In type 2 diabetes, hyperglucagonemia is often observed,1,2 compounding the difficulties of glucose regulation. Despite the importance of glucagon, numerous questions exist into the mechanisms that govern its release.1,2,4Difficulties associated with measuring glucagon from islets in a time-resolved fashion hamper this understanding. A common way to measure glucagon secretion includes perfusion of a batch of islets followed by fraction collection of the perfusate and subsequent hormone quantitation performed with an enzyme-linked immunosorbent assay (ELISA) or radioimmunoassay.5,6 To increase automation and use fewer islets so that the dynamics of secretion can be observed, microfluidic devices have been widely implemented for in vitro cellular studies.7,8 These systems have been used in a number of studies with islets of Langerhans for improved control of dynamic conditions compared with traditional methods.9–17 Microfluidic devices provide a controlled cellular environment, easy fluid manipulation, automation, and low dilution connections; combined, these factors can lead to high temporal resolution measurements. For example, a microfluidic system was used to perfuse islets and “pseudoislets” to examine differences in secretion profiles of insulin and glucagon using an offline radioimmunoassay.16 In another example, an electrophoretic immunoassay was used to measure glucagon secretion online from 10 murine islets in response to a change in glucose from 16 to 1 mM.17 Although electrophoretic immunoassays have been widely used to measure insulin release from islets within microfluidic systems,10,12,18–21 they have not been reported as often with glucagon. We have developed a noncompetitive assay for glucagon using an aptamer,22 but a complicated fabrication procedure of the necessary microfluidic system limits the widespread applicability of the system.23
The previously-described methods employ heterogeneous assays that require separation of the bound and free components; conversely, homogeneous assays do not require a separation step and have been developed for insulin24,25 and other peptides24 released from islets; one example is a time-resolved Förster resonance energy transfer (TR-FRET) based assay for glucagon.26 The TR-FRET assay has a 2 pg mL−1 reported limit of detection, which would enable glucagon release from only a few islets, possibly allowing release dynamics to be observed.
In this report, we describe initial steps towards the implementation of this TR-FRET assay into a microfluidic based system. A relatively simple microfluidic system is used to house 10–25 human islets and glucose is perfused at varying levels to stimulate glucagon release. Fractions from the perfusate are collected every 2 min and glucagon release is quantified using the TR-FRET assay. We anticipate that the simplicity of the setup combined with the high sensitivity of the assay will enable multiple researchers to use this platform for gaining insight into glucagon secretion dynamics, which could in turn lead to better understanding of type II diabetes and further the development of improved therapeutics.
Materials and methods
Chemicals and reagents
Polydimethylsiloxane (PDMS) prepolymer (Sylgard 184) was obtained from Dow Corning (Midland, MI). Dextrose was obtained from Fisher Scientific (Pittsburgh, PA). The TR-FRET glucagon assay was obtained from Cisbio (Walthan, MA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless noted otherwise. All solutions were made with ultrapure DI water (NANOpure Diamond System, Barnstead International, Dubuque, IA).A balanced salt solution (BSS) was used for islet experiments which contained 125 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 2.4 mM CaCl2, 25 mM tricine, and brought to pH 7.4 before addition of 0.1% BSA. Different glucose concentrations were added as described in the text, and the BSS was filtered with a 0.2 μm nylon syringe filter (Pall Corporation, Port Washington, NY) prior to delivery to the microfluidic system.
Islets of Langerhans
Human islets were purchased from Prodo Laboratories Inc. (Aliso Viejo, CA) from donors who had not been diagnosed with diabetes. Human islet samples (85–95% pure) were incubated for a minimum of 1 day in complete Prodo Islet Media Standard PIM(S) at 37 °C and 5% CO2 upon delivery.18 Human islet samples were obtained from deidentified cadaveric organ donors and, therefore, were exempt from Institutional Review Board approval. Donor characteristics are provided in Table S1.†Microfluidic device
The microfluidic device was fabricated using methods described previously.27 Briefly, conventional soft lithography was used to make a 400 × 200 μm (width × depth) channel in PDMS. The access holes at the ends of the channel were made using a 0.508 mm diameter titanium nitride hole punch (SYNEO, Angleton, TX). A hole for loading islets was made in the middle of the channel length using a 400 μm diameter punch (Welltech, Taichung, Taiwan). The PDMS was irreversibly bonded to a 25 × 75 × 1 mm (width × length × thickness) glass coverslip (VWR, Randor PA) after plasma oxidation of both pieces. The glass coverslip formed the bottom of the channels.To maintain the temperature of the islet chamber at 37 °C, a thermofoil heater (Omega Engineering, Inc., Stamford, CT) was placed underneath the microfluidic device and a thermocouple sensor was applied adjacent to the islet chamber on top. A controller (Omega Engineering) was used to maintain the temperature at 36.5 ± 0.5 °C. To perform perfusion, the outlet from two syringe pumps were connected via a junction to the inlet access hole of the device. The total flow rate into the microfluidic device was maintained at 5 μL min−1. Perfusate was collected via a 10 cm length of 0.508 mm i.d. Tygon tubing connected to the outlet. Fractions were collected from this tube every 2 min into a new 200 μL Eppendorf tube.
To load islets, the device was filled with BSS containing 20 mM glucose and a number of islets were introduced into the islet loading port with the exact number given in the text and figure captions. The islet loading port was then covered with PCR tape and the input and output tubing were removed. The device was placed in a 37 °C, 5% CO2 incubator for 10 min to allow islets to settle to the bottom and attach to the glass surface. At the end of the 10 min, the device was removed from the incubator, the tubing reattached, and flow initiated with BSS containing 20 mM glucose. The islets were allowed to equilibrate to the flow for 15 min prior to fractions being collected. The glucose level delivered to the islets was varied by adjusting the syringe pump flow rates as described in the text. At the end of the experiment, all fractions were centrifuged to remove air bubbles, pipetted into a 96-well plate, and assayed as described below.
Islets were removed from the device and lysed to measure the total glucagon content as described.28 Briefly, all islets were removed from the device by aspiration and placed into a 200 μL tube. The tube was centrifuged and a 10 μL pipette was used to transfer all islets to a new tube. A 90 μL volume of acid–ethanol mixture (750 mL of 95% ethanol; 15 mL of 5 M HCl; 235 mL of H2O) was added and the tube sonicated for 5 min.29 The lysate was kept at −20 °C prior to analysis if not read immediately. For analysis, 10 μL of the lysate solution was diluted to 200 μL with BSS. To ensure the concentration of the lysate was within the calibration curve, three dilutions were then performed: 10 μL of the 200 μL lysate solution was diluted to 100 μL with BSS; 10 μL of this newly made solution was diluted to 100 μL using BSS; 10 μL of this solution was then removed and diluted to 100 μL with BSS. 10 μL of each of these three solutions was added to the 96-well plate, along with the perfusion fractions, and assayed using TR-FRET.
TR-FRET glucagon assay
In addition to the fractions collected from the perfusion and the lysate, each 96-well plate (HTRF 96-well low volume white plate) had three replicates of 2000, 1000, 500, 250, 125, 62.5, 31.25, 15.6 and 0 pg mL−1 standard glucagon solutions made in BSS. Reagents from the TR-FRET glucagon assay kit (10 μL) were pipetted into each well using a multi-channel pipette. The well plate was covered with film and incubated 12–14 h at room temperature in the dark. A plate reader (SpectraMax iD5, Madison, WI) was used to measure the TR-FRET according to the manufacturer's protocol. Briefly, excitation light (360 nm) was on for 50 μs, followed by a 100 μs delay, and a 600 μs recording time of the two FRET channels (620 and 665 nm). This timing protocol was repeated every 2 ms for 100 cycles.The average FRET signal from the 0 pg mL−1 standard solution was subtracted from all FRET measurements. FRET ratios are presented as the ratio of the emission at 665 nm to that at 620 nm multiplied by 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Calibration curves were generated by averaging the FRET ratio from the three replicates of each standard glucagon solution and plotting these values vs. the concentration of glucagon.
000. Calibration curves were generated by averaging the FRET ratio from the three replicates of each standard glucagon solution and plotting these values vs. the concentration of glucagon.
Finite element analysis
The proposed microfluidic system featuring islets was developed in a finite element simulation software (COMSOL Multiphysics v4.3, COMSOL Inc., Stockholm, Sweden). Flow rates were solved using the incompressible Navier–Stokes equation with a no slip boundary condition on solid surfaces; islets were modeled as porous spheres.30 All simulation parameters are given in Table S2.†Data analysis
To quantify glucagon, the FRET ratio from each fraction was converted to glucagon concentration (pg mL−1) using the calibration from the same plate with the amount of glucagon (pg) then determined. The lysate dilution which fell within the calibration curve was used to determine the amount of glucagon (pg) in the total lysate by accounting for the various dilutions. Secretion traces are presented as percent glucagon released, which was determined by calculating the percent of glucagon in each fraction with respect to the total glucagon (the sum of the lysate and all fractions).28 The number of islets used in each experiment are provided in the text or figure caption, but the islet equivalents were not calculated.Results
The measurement of glucagon secreted from pancreatic islets of Langerhans is typically carried out with macro-perfusion systems and offline ELISAs. Development of microfluidic systems to hold islets and deliver stimulants would provide an additional level of control and automation. Homogeneous assays for measurement of glucagon would decrease analysis times by reducing the number of steps for detection. Here, we have coupled a straightforward PDMS microfluidic device for islet perfusion with fraction collection and subsequent glucagon quantification with a TR-FRET homogeneous assay. We anticipate the simplicity of the system will enable other researchers to use similar methods to perform more complex experiments.Glucagon assay
A commercially available TR-FRET assay was used for quantitation of glucagon. In this system, a “sandwich assay” is used with different fluorophores attached to antibodies recognizing different epitopes. One of the antibodies was labeled with a lumi4-terbium cryptate donor (λex = 340 nm, λem = 620 nm), and the other was labeled with a d2 red acceptor (λex = 620 nm, λem = 665 nm). FRET between the two fluorophores confirmed the presence of the hormone and the ratio of intensities from 665 and 620 nm allowed for accurate quantitation. An advantage of this assay is the long-lived fluorescence lifetime of the donor (∼1–2 ms), which allowed for the two emission channels to be measured 100 μs after the excitation light was turned off, enabling low background measurements.Standard glucagon solutions were subjected to measurement with this system and resultant calibration curves of FRET ratio versus glucagon concentration showed a reproducible linear response (Fig. 1). The relative standard deviations of the points ranged from 1 to 12%, and the calculated limit of detection (LOD) was 15 pg mL−1. These results demonstrated good reproducibility and a sensitivity compatible with concentrations of glucagon typically released from islets.16,17 These values are similar to those reported for commercial ELISA systems, but without the necessity for wash steps, although an overnight (12–14 h) incubation period was required for producing high quality calibration curves in our hands. Shorter incubation times were attempted but resulted in higher RSD and/or lower linearity. Nevertheless, the LOD was sufficient to proceed with the development of the microfluidic system.
 | ||
| Fig. 1 Calibration of TR-FRET assay. A representative calibration of the TR-FRET assay is shown. The points are the average of 3 replicates and the error bars are ±1 standard deviation. The inset shows the lower region of the curve. The plot was fit with a linear regression line y = 2.6659x (r2 = 0.999). | ||
Microfluidic system
Once the glucagon assay was confirmed to produce LOD sufficient for islet measurements, a microfluidic device was designed to hold islets, perfuse glucose, and allow for fraction collection. The goals of the device were to have simple construction and operation, yet still provide a rapid response from islets housed in the device. Additionally, the tissue needed to be easy to load and recover from the device. These goals were achieved using a PDMS/glass microfluidic device that contained a single flow channel with a central loading port to add and remove islets (Fig. 2A). After adding islets, the loading port was sealed with PCR tape to keep solution within the device. | ||
| Fig. 2 Microfluidic system. (A) A 3D drawing of the PDMS/glass microfluidic device used in this report. The outputs of two syringe pumps were coupled at a tee (not shown) and input into the inlet port of the device. Islets were loaded through the central loading port and covered with a piece of PCR film. Tubing at the outlet (not shown) was used to collect fractions of the perfusate every 2 min. (B) An image of the microfluidic device loaded with human islets was used to mimic the placement and size distribution in a finite element model. (C) The picture in (B) was used to build a finite element model of fluid flow in the device. The image is from the simulation and shows a composite view from a slice of the fluid velocity taken in the middle of the channel and a surface plot of the shear stress on the islets. The scale bar at the bottom of the image is for both the fluid velocity and shear stress. The range for shear stress is 0.0–75 mPa, and the range for velocity is 0.0–1.25 mm s−1. | ||
To perform perfusion, the output from two syringes driven by syringe pumps were connected with a T-junction and then to the inlet port on the microfluidic device. The tubing was inserted into the hole punched in the PDMS, creating an interface that did not require sealant or glue. The ratio of the flow rates from the two syringe pumps was then varied to adjust the concentration of stimulant into the device while the total flow rate was maintained at a constant 5 μL min−1.
To ensure that the experimental conditions would not be detrimental to the islets, a 3-dimensional finite element simulation was used to model the fluid flow. To mimic experimental conditions as closely as possible, 15 islets were placed in a device and a photograph was obtained (Fig. 2B) to guide the sizes and locations of islets in the simulation. The islets ranged from ∼100–250 μm in diameter and they were modeled as porous spheres30 with a porosity of 0.1 and permeability of 10−15 m2. The flow rate into the device was 5 μL min−1 and other model parameters are provided in Table S2.† The linear fluid velocity (mm s−1) through the device is shown in Fig. 2C and indicated that the islets influenced the flow of the liquid in the channel. To determine how the flow conditions may affect the islets, the shear stress on the islet surfaces were calculated. In Fig. 2C, the islet surfaces have the shear stress overlaid and indicated a maximum of ∼71 mPa on the largest islet in the simulation, slightly below the physiological shear stress range.31 These results indicated that the design would not produce damagingly high levels of shear to the islets.
Upon finalization of the simulation, the dynamic response of the microfluidic system was determined by perfusing step changes of glucagon though the device. To perform the perfusion, syringe 1 was filled with 3 mL of BSS and syringe 2 was filled with 3 mL BSS buffer containing 1200 pg mL−1 glucagon. The device was flushed with syringe 1 for 15 min prior to the start of the experiments to condition the device. For the next 40 min, flow from syringe 1 and syringe 2 was alternated for 10 min each. Fig. 3 shows these results with the glucagon delivery profile shown as the red line, and the glucagon measured as the black points. Based on the volumes of the inlet and outlet tubing and the channel, the time required for glucagon to travel from the T-junction into the fraction collection tubes was 5.25 min. This time was subtracted from the glucagon delivery profile (red line in Fig. 3) and demonstrates that the glucagon measured in the fractions mirrored the delivery profile upon this correction. The average response time, defined as the time required to increase or decrease the signal from 10% to 90% of the final signal was 2 fractions or 4 min. Additionally, we did not observe a significant difference in the TR-FRET signal during subsequent perfusions of either glucagon (10–20 min vs. 30–50 min) or BSS (from 0–10 min vs. 20–30 min), which indicates that any non-specific adsorption in the device was undetectable.
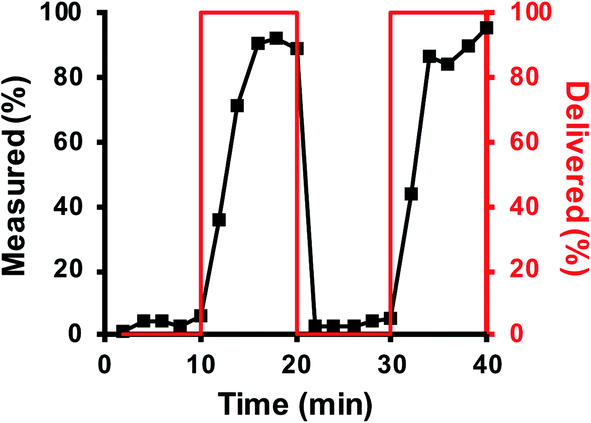 | ||
| Fig. 3 Flow dynamics. Low (0 pg mL−1) and high (1200 pg mL−1) glucagon concentrations were delivered to the device through the syringe pumps in a pattern shown by the red line (corresponding to the right y-axis). The flow through the device was fractionated every 2 min and measured using the TR-FRET assay. The amount of glucagon in each fraction is shown by the data points and corresponds to the left y-axis. | ||
Glucagon measurements
Following method development of the system with standard glucagon solutions, a batch of human islets were loaded into the device. Initial experiments were performed to ensure islets would not respond to variations from changing the syringe pump flow rates. To perform these control experiments, both perfusion syringes were filled with BSS containing 20 mM glucose. The first syringe pump delivered solution to the device for 10 min, after which the flow was switched to the second syringe pump for the next 20 min. After this time, the flow was switched back to the first syringe for the last 20 min. As shown in Fig. S1,† the islets did not respond to these changes in the solutions with a significant change in the glucagon secreted. This protocol was repeated a total of 3 times, each replicate with different islets.Upon confirming that the switch in perfusion flows did not induce changes in glucagon release, we then tested the ability to measure glucagon secretion in response to glucose changes. To perform these experiments, islets were placed in the microfluidic device, allowed to adhere for 10 min, and perfused with 20 mM glucose in BSS. After this period of basal release (usually 10–15 min), the glucose level was decreased to 1 mM. For some experiments, the glucose concentration was held at 1 mM for the remainder of the experiment (n = 3); in other experiments, the glucose concentration was increased back to 20 mM (n = 4). Due to the high variability in islet composition, secretion amounts were normalized to the total glucagon content in the islet population as described in the Materials and methods. The glucose time curve was also adjusted by 5.25 min to account for the volume of the device and tubing.
Qualitatively, most experiments exhibited one of two patterns of release, either a burst of glucagon secretion followed by a decline in the rate of release, or they showed a sustained level of release during the entire stimulation. Fig. 4 shows a representative trace from each of these groups. Fig. 4A illustrates a “burst” pattern of glucagon released (black points) in response to different glucose concentrations (red). These results were from a batch of 30 islets from donor 4. This “burst” pattern was observed for five experiments: one experiment from islets from donor 3, three experiments with islets from donor 4, and one experiment from donor 5. Fig. 4B shows a “sustained” pattern of glucagon release from a batch of 45 islets from donor 4. This pattern was observed for two of the experiments, with one each from donor 3 and 4. In one experiment (with a batch of islets from donor 3), a change in glucagon release was not observed. Fig. S2† shows all perfusion responses from each set of islets tested. At this time, it is unknown why some experiments showed one secretion profile versus another, although a previous report shows similar “burst” profiles with 10 islets.17
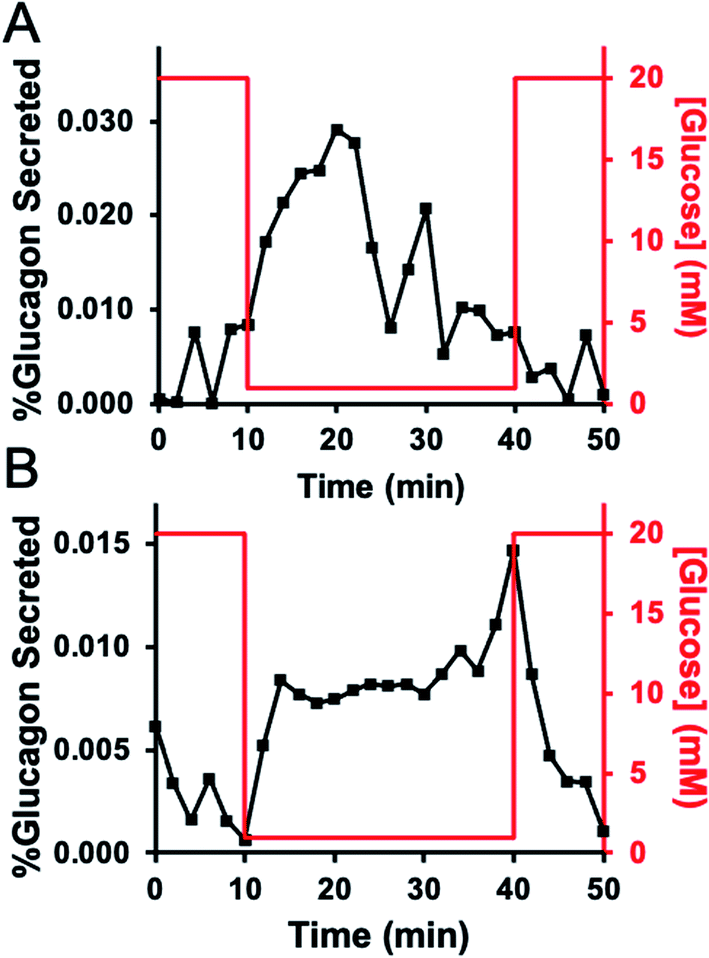 | ||
| Fig. 4 Representative glucagon secretion profiles. In both traces, the measured glucagon levels are shown by the black points and correspond to the left y-axis, while the glucose profile delivered to the device is shown as a red line and corresponds to the right y-axis. (A) A representative trace from a group of 30 islets from donor 4 that released in a “burst” pattern. (B) A representative group of 45 islets from donor 4 that released glucagon in a “sustained” pattern. | ||
The percent glucagon release during stimulation with high and low glucose was quantified for all experiments (n = 8) and normalized to the stimulation period. The results are shown in the scatter plot in Fig. 5 with the average shown as the horizontal bar and the error bars corresponding to ±1 SD. The average (±SD) percent glucagon release during stimulation with 1 mM glucose was ∼7-fold higher at 0.007 ± 0.003% min−1 compared to that measured at 20 mM glucose, 0.001 ± 0.001% min−1. A 1-tailed Student's t-test indicated that these results were significantly different (p = 0.002) for the two glucose conditions tested.
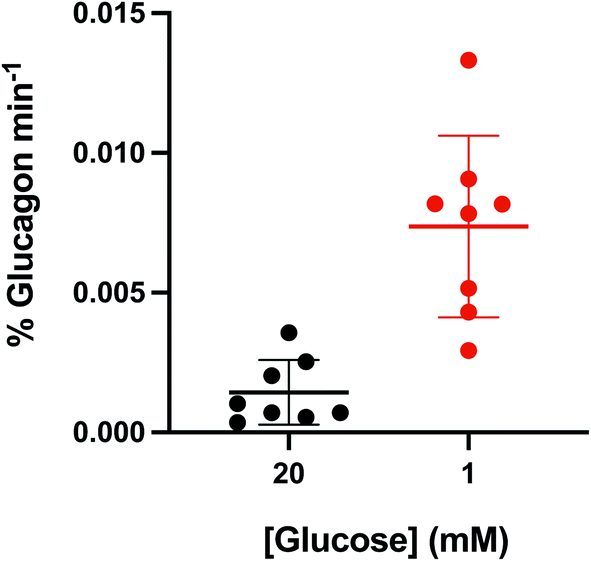 | ||
| Fig. 5 Average glucagon levels. The total glucagon secretion levels normalized over the collection time (total glucagon min−1) for all traces shown in Fig. S2† were averaged and shown as a function of glucose level. The difference in the amount of glucagon released during 1 mM glucose was significantly higher than that at 20 mM glucose (p = 0.002, 1-tailed Student's t-test). Average is shown as the thick horizontal line with error bars equal to ±1 standard deviation. | ||
Finally, in some initial experiments, the islet glucagon content after the experiment was not measured because the method was being developed. In these experiments, calculation of the percent glucagon released was not performed, but the traces are shown in Fig. S3.† Because the total glucagon content was not measured, these traces were omitted from statistical analysis and are shown here to demonstrate that other experiments from different donors also showed the secretion profiles mentioned above. Three experiments exhibited a “burst” profile, four showed the “sustained” release, and two did not show much release upon stimulation with low glucose.
Conclusions
Information regarding glucagon release from human islets of Langerhans is much less comprehensive relative to that for insulin secretion. The combination of a simple and robust microfluidic system with a TR-FRET assay described here is expected to provide a relatively simple glucagon measurement method while still providing valuable information regarding the release of this peptide hormone. For example, it could be used to further examine “burst” versus “sustained” secretion profiles. With the LOD of this assay, we approximate that release from about 5 islets could be measured, which may allow the dynamics of release to be observed more clearly. Finally, the dimensions of the microfluidic device are amenable to fabrication by other means, including milling or hot embossing in other materials, which could enable simple and rapid high throughput production while a mechanical system could be used to automate the fraction collection process. Because the TR-FRET assay can be performed without washing steps, the sensitivity and simplicity of the system are ideal for those investigating the release of glucagon.Author contributions
Wesley J. Eaton: investigation, validation, writing – original draft and editing. Michael G. Roper: investigation, resources, funding acquisition, and writing – review and editing.Conflicts of interest
The authors declare no conflicting interest.Acknowledgements
This work was supported in part by grants from the National Institutes of Health, R01 DK080714, and using resources and/or funding provided by the NIDDK-supported Human Islet Research Network (HIRN, RRID:SCR_014393; https://hirnetwork.org; UC4 DK116283 to MGR).References
- A. Lund, J. I. Bagger, M. Christensen, F. K. Knop and T. Vilsbøll, Curr. Diabetes Rep., 2014, 14, 555 CrossRef PubMed.
- H. Y. Gaisano, P. E. MacDonald and M. Vranic, Front. Physiol., 2012, 3, 349 CrossRef PubMed.
- A. Pisania, G. C. Weir, J. J. O’Neil, A. Omer, V. Tchipashvili, J. Lei, C. K. Colton and S. Bonner-Weir, Lab. Invest., 2010, 90, 1661–1675 CrossRef PubMed.
- P. Gilon, J. Mol. Biol., 2020, 432, 1367–1394 CrossRef CAS PubMed.
- C. Li, C. Liu, I. Nissim, J. Chen, P. Chen, N. Doliba, T. Zhang, I. Nissim, Y. Daikhin, D. Stokes, M. Yudkoff, M. J. Bennett, C. A. Stanley, F. M. Matschinsky and A. Naji, J. Biol. Chem., 2013, 288, 3938–3951 CrossRef CAS PubMed.
- R. J. Bevacqua, X. Dai, J. Y. Lam, X. Gu, M. S. H. Friedlander, K. Tellez, I. Miguel-Escalada, S. Bonàs-Guarch, G. Atla, W. Zhao, S. H. Kim, A. A. Dominguez, L. S. Qi, J. Ferrer, P. E. MacDonald and S. K. Kim, Nat. Commun., 2021, 12, 2397 CrossRef CAS PubMed.
- A. M. Schrell, N. Mukhitov, L. Yi, X. Wang and M. G. Roper, Annu. Rev. Anal. Chem., 2016, 9, 249–269 CrossRef PubMed.
- M. G. Roper, Anal. Chem., 2016, 88, 381–394 CrossRef CAS PubMed.
- F. R. Castiello, K. Heileman and M. Tabrizian, Lab Chip, 2016, 16, 409–431 RSC.
- M. G. Roper, J. G. Shackman, G. M. Dahlgren and R. T. Kennedy, Anal. Chem., 2003, 75, 4711–4717 CrossRef CAS PubMed.
- J. S. Mohammed, Y. Wang, T. A. Harvat, J. Oberholzer and D. T. Eddington, Lab Chip, 2009, 9, 97–106 RSC.
- J. F. Dishinger and R. T. Kennedy, Anal. Chem., 2007, 79, 947–954 CrossRef CAS PubMed.
- C. J. Easley, J. V. Rocheleau, W. S. Head and D. W. Piston, Anal. Chem., 2009, 81, 9086–9095 CrossRef CAS PubMed.
- L. A. Godwin, M. E. Pilkerton, K. S. Deal, D. Wanders, R. L. Judd and C. J. Easley, Anal. Chem., 2011, 83, 7166–7172 CrossRef CAS PubMed.
- G. Lenguito, D. Chaimov, J. R. Weitz, R. Rodriguez-Diaz, S. A. K. Rawal, A. Tamayo-Garcia, A. Caicedo, C. L. Stabler, P. Buchwald and A. Agarwal, Lab Chip, 2017, 17, 772–781 RSC.
- J. T. Walker, R. Haliyur, H. A. Nelson, M. Ishahak, G. Poffenberger, R. Aramandla, C. Reihsmann, J. R. Luchsinger, D. C. Saunders, P. Wang, A. Garcia-Ocaña, R. Bottino, A. Agarwal, A. C. Powers and M. Brissova, JCI Insight, 2020, 5, e137017 CrossRef PubMed.
- J. G. Shackman, K. R. Reid, C. E. Dugan and R. T. Kennedy, Anal. Bioanal. Chem., 2012, 402, 2797–2803 CrossRef CAS PubMed.
- B. Bandak, L. Yi and M. G. Roper, Lab Chip, 2018, 18, 2873–2882 RSC.
- A. R. Lomasney, L. Yi and M. G. Roper, Anal. Chem., 2013, 85, 7919–7925 CrossRef CAS PubMed.
- L. Yi, X. Wang, R. Dhumpa, A. M. Schrell, N. Mukhitov and M. G. Roper, Lab Chip, 2015, 15, 823–832 RSC.
- L. Yi, B. Bandak, X. Wang, R. Bertram and M. G. Roper, Anal. Chem., 2016, 88, 10368–10373 CrossRef CAS PubMed.
- L. Yi, X. Wang, L. Bethge, S. Klussmann and M. G. Roper, Analyst, 2016, 141, 1939–1946 RSC.
- W. Leng, K. Evans and M. G. Roper, Anal. Methods, 2019, 11, 5768–5775 RSC.
- E. Heyduk, M. M. Moxley, A. Salvatori, J. A. Corbett and T. Heyduk, Diabetes, 2010, 59, 2360–2365 CrossRef CAS PubMed.
- X. Li, J. Hu and C. J. Easley, Lab Chip, 2018, 18, 2926–2935 RSC.
- T. Fougeray, A. Polizzi, M. Régnier, A. Fougerat, S. Ellero-Simatos, Y. Lippi, S. Smati, F. Lasserre, B. Tramunt, M. Huillet, L. Dopavogui, L. Smith, C. Naylies, C. Sommer, A. Benani, J. T. Haas, W. Wahli, H. Duez, P. Gourdy, L. Gamet-Payrastre, A.-F. Burnol, N. Loiseau, C. Postic, A. Montagner and H. Guillou, bioRxiv, 2021 DOI:10.1101/2021.02.05.430014.
- A. G. Adams, R. K. M. Bulusu, N. Mukhitov, J. L. Mendoza-Cortes and M. G. Roper, Anal. Chem., 2019, 91, 5184–5190 CrossRef CAS PubMed.
- J.-C. Henquin, Mol. Metab., 2019, 30, 230–239 CrossRef CAS PubMed.
- M. J. Donohue, R. T. Filla, D. J. Steyer, W. J. Eaton and M. G. Roper, J. Chromatogr. A, 2021, 1637, 461805 CrossRef CAS PubMed.
- A. L. Glieberman, B. D. Pope, J. F. Zimmerman, Q. Liu, J. P. Ferrier, Jr, J. H. R. Kenty, A. M. Schrell, N. Mukhitov, K. L. Shores, A. B. Tepole, D. A. Melton, M. G. Roper and K. K. Parker, Lab Chip, 2019, 19, 2993–3010 RSC.
- K. S. Sankar, B. J. Green, A. R. Crocker, J. E. Verity, S. M. Altamentova and J. V. Rocheleau, PLoS One, 2011, 6, e24904 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ay00703c |
| This journal is © The Royal Society of Chemistry 2021 |