
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn-column internal standardisation as an alternative calibration strategy for speciation analysis: feasibility demonstration through analysis of inorganic As in rice†
Panayot
Petrov
 *,
Simon
Cowen
and
Heidi
Goenaga-Infante
*,
Simon
Cowen
and
Heidi
Goenaga-Infante
LGC Ltd, National Measurement Laboratory, Queens Road, Teddington, TW11 0LY, UK. E-mail: Panayot.Petrov@lgcgroup.com
First published on 7th July 2021
Abstract
Species-specific isotope dilution mass spectrometry (SS-IDMS) has been the calibration method of choice for high accuracy speciation analysis because it can correct for detector sensitivity drifts, matrix effects, and analyte loss during sample preparation and analysis. However, in many cases SS-IDMS calibration is either not applicable (e.g. for monoisotopic elements) or not feasible (e.g. limited by the cost and availability of like-for-like isotopically enriched species). The work presented here demonstrates the potential of a novel on-column species-specific internal calibration approach, which is based on the chromatographic injection of the same species of the analyte as the internal standard (IS), after the sample injection. It can compensate for on-column analyte losses and signal drift and can be applied with any detector capable of recording time-resolved data, provided that enough species resolution can be achieved. The feasibility of this novel calibration strategy for accurate quantitative elemental speciation in complex matrices is demonstrated here through the analysis of inorganic arsenic in rice. An expanded uncertainty (k = 2) of <10% was obtained for a mass fraction range of 60 to 300 μg kg−1 inorganic-As (i-As) in dry rice products. The method is currently used for the certification of i-As in baby food matrices to support Commission Regulation (EU) 2015/1006 in regard to the maximum levels of i-As in foodstuffs.
Introduction
Obtaining high accuracy speciation results from complex samples in the absence of relevant quality control and reference materials represents a remaining challenge.1–4 The method of standard addition (MSA) is a calibration approach that can correct for matrix effects, known to cause bias in the speciation results. However, when a significant drift is superimposed, the results obtained by the typical MSA calibration are substantially biased. As a result, a number of correction approaches for matrix effects and sensitivity drift have been tested over the years but with limited practical applicability.5 When an inductively coupled plasma mass spectrometer (ICP-MS) is used as a detector, internal standardisation (IS) is the preferred calibration approach for routine analysis6 and isotope dilution (ID) mass spectrometry for high accuracy analytical determinations. However, it is well known that not only the element of choice but also the particular choice of species is crucial to obtain accurate results with IS calibration.4 The same requirement applies to the isotopically enriched standards used with IDMS calibration. Unfortunately, in some cases the species specific IDMS (SS-IDMS) approach is not applicable5 due to (a) the monoisotopic nature of some elements (e.g. with arsenic speciation, excluding radioactive isotopes), (b) the unavailability of isotopically enriched species (more analytes than available isotopes) or (c) similar natural isotopic abundances for specific elements (e.g. 51% 79Br and 49% 81Br). For the latter the approach is still applicable but a large amount of the enriched standard would be required to perform accurate quantification, strongly impacting the price of analysis. An additional economic consideration is the price of the synthesis of a compound enriched in one specific isotope. This is often a practical factor favouring the selection of a lower order calibration approach, such as internal standardisation with other species. However, the issue with IS calibration is that other species, even from the same element, do not necessarily have the same chemical, electrochemical, or physicochemical behaviour in general, to secure efficient correction for matrix and drift effects in all cases.4 In many applications the choice of internal standard has been empirical and has consisted of fortification of the samples and standards with a number of frequently used, or even analyst favourite, internal standards while seeking an agreement in the final mass fractions of the analytes. Clearly, such approach does not exclude the possibility of the results in agreement being equally wrong. In contrast to total elemental determinations, for speciation analysis a smaller number of species are usually applied as internal standards. Fundamentally, there is no limitation for the number of elements that can be used as internal standards with multi-element detectors like ICP-MSs or ICP-OESs, provided that (1) the IS does not react with the sample components or the mobile phase and (2) the detector is fast and sensitive enough to record the transient signal with sufficient frequency and stability.The two approaches used for internal standardisation in speciation analysis are continuous IS and discrete IS.7 A schematic diagram summarising the type of IS by point of introduction is presented in the ESI† (Diagram 1). The former (continuous IS) relies on constantly supplying a suitable and fixed amount and flow of element(s), which is merged with the analyte flow before reaching the detector. The disadvantage of this approach is the same as discussed above for total element determination, i.e. the difference in the behaviour of elements/species used as IS and the species of interest.8–10 The second approach (discrete IS) has several modifications, depending on how the IS has been introduced (e.g. within each sample as a surrogate standard or post-column). Details on the different types of internal standardisation approach are readily available in the literature.11–14 In summary, the post-column set-up includes a switching valve, positioned on the sample flow line, after the separation module (e.g. chromatographic column). An injection loop is filled with IS, which is then injected into the effluent. The two main types of discrete post column internal standardisation then are (1) IS injection immediately after the sample injection within a couple of seconds delay, needed for baseline estimation,7 and (2) IS injection after the elution of the strongest retained species. In both cases, the advantage of this approach compared to continuous IS is that the same element and even the same species can used as IS. It is possible to have more than one species as an internal standard by using an array of switching valves or even performing IS with MSA.15 For example, one of the IS species could be injected at the beginning of the separation and one at the end, although IS injections can be done so to elute at other retention times where no analytes are present. However, many researchers prefer to use only one species with ICP-MS detection, assuming equal species behaviour irrespective of their different volatility and therefore transport efficiency to the plasma. With more and more reports on species dependent ICP-MS sensitivity being published,16 the preferred option has shifted towards the use of an “average” internal standard consisting of species with sensitivity in-between the sensitivities of the other species of interest and if feasible, with similar chemical behaviour. Alternatively, the most abundant species or the species of particular scientific interest can be used as IS to minimise the bias from the inadequate IS correction. By employing these strategies, alone or in combination with MSA calibration, many of the known sources of bias can be efficiently mitigated. These include moderately sized low frequency noise (e.g. small sensitivity drift) and some matrix effects.4,9,17 Yet, all of the above being post-column corrections, they do not correct for any adverse effects accompanying the separation process. When samples with complex matrices are analysed, some of the target species can be either significantly more strongly retained on the separation column or eluted earlier compared to the respective standards. Additionally, peak splits have been reported. These have led to large ambiguity in species identification. Moreover, quality controls based on mass balance calculations will be blind to such adverse effects. As a consequence, the species-specific on-column losses are not compensated for by the common IS approaches and may negatively bias the results for the species of interest.16
In order to minimise the impact of the discussed potential sources of bias on the accuracy and precision of the speciation data, we propose a novel on-column species-specific internal standardisation approach as an alternative to SS-IDMS, the former being particularly attractive for mono-isotopic elemental species. With this approach, the drift effect is monitored and corrected for on a sample-to-sample basis. Both the analyte and internal standard are the same species, thus minimising the issues related with different species behaviours. The feasibility of this novel calibration strategy for accurate quantitative elemental speciation in complex matrices is investigated here through the analysis of i-As in rice samples in support of EU Commission Regulation 2015/1006.18 The method accuracy was assessed through analysis of certified reference materials and by recovery experiments on spiked samples. A full uncertainty budget indicating the main contributing factors has been estimated and reported. Moreover, the capability of the method to quantify more than one species under non-isocratic conditions was investigated through simultaneous determination of i-As and Methyl Arsonate (MA) in a standard mixture submitted to the same preparation procedure as the rice sample.
Experimental
Authors' note: The instruments' makes and models presented in this work are for example purposes only. Similar performance can possibly be achieved with instrumentation produced by other manufacturers.General set-up for on-column IS
In this work the potential of a novel on-column calibration approach is demonstrated through the analysis of i-As in rice extracts by employing anion-exchange liquid chromatography for the species separation and ICP-MS for their quantification. The instrumental setup schematics are presented in Fig. 1. | ||
| Fig. 1 Instrumental setup for on-column IS calibration. (A) The sample is injected into the column while the loop is filled with IS. After a user defined delay (time buffer) the valve is switched to position (B), injecting the IS into the column. | ||
With this setup, the analyst can set the injection time and therefore precisely position the IS peak within the chromatogram for each particular speciation analysis. Since the valve is connected before the separation column, the operator should ensure that its operational pressure exceeds the separation column backpressure. This is a clear difference with the post-column IS approach, for which low-pressure valves can be used. Such a high pressure valve can be integrated inside the liquid chromatography (HPLC) compartment or be positioned just before the analytical column. Finally, the mass fraction of the analyte in the sample is calculated based on eqn (1) below:
 | (1) |
Eqn (1) is used for the calculation of the analyte mass fraction via on-column internal standardisation with a linear calibration curve, where CS is the mass fraction of the analyte, a and b are the slope and intercept of the built linear calibration curve and AS and AIS are the peak areas of the analyte and internal standard, respectively. The calibration curve is built once, in the beginning of the analysis, to calculate parameters a and b using linear regression (Y vs. X). For this purpose, the ratios of the peak areas of calibration standards to the injected (see Fig. 1) internal standards peak areas (ratio plotted on axis Y) for each calibration standard level are plotted against the respective calibration standard concentrations (plotted on axis X). By using MS Excel© array function Linest for Y vs. X, a and b are calculated. More details are presented in the Results and discussion section.
Detector parameters and settings
For the determination of i-As in rice, a tandem ICP-MS (Agilent 8800 ICP-MS/MS, Agilent Technologies, Tokyo, Japan), operated by the Agilent MassHunter software package (Version C.01.05, 2019), was used for the data collection and manual peak integration. The ICP-MS parameters are summarized in Table 1.| RF power | 1600 W |
| Carrier Ar flow rate | 850 mL min−1 |
| Makeup/dilution gas | 280 mL min−1 |
| Sample/skimmer cones | Ni/Ni |
| Spray chamber temp. | 2 °C |
| Reaction gas (flow) | O2 (30% setting; ∼3 mL min−1) |
| Data acquisition mode | TRA |
| Points per spectral peak | 1 |
| Acquisition mode | Tandem MS |
| Monitored mass-shift | 75As–75As16O (m/z 75–>91) |
| Integration time | 0.3 to 1.0 s (HPLC method dependent) |
The acquired data (as peak areas) were processed using laboratory developed MS Excel™ spreadsheets. Two different types of nebuliser have been used depending on the required flow; Micromist (Crawford Scientific, Strathaven, Scotland) performed better with lower signal RSDs for flows below 900 μL min−1 and Conikal (Crawford Scientific) was used for flows of 1 mL min−1 and above.
HPLC parameters and settings
An Agilent 1260 Infinity Bio-inert HPLC system (Agilent Technologies, Cheadle, UK), consisting of a HiPs autosampler (G5667A) and bio-inert pump (G5611A), was used for the determination of i-As in rice extracts. The sample was injected through the autosampler 6-port valve and directed to a strong anion exchange column (AEC) where i-As species separation was completed. The chromatographic conditions and the separation method are shown in Table 2. The listed alternative parameters provide six fold quicker separation at the expense of slightly impaired arsenic species resolution, particularly for the As(III)/DMA pair. However, when As(III) is converted to As(V), as is done in this work, these were the preferable separation conditions (please refer to the Results and discussion section, below).| AEC column | PRP-X100, 250 mm × 4.1 mm id × 10 μm (PRP-X100, 50 mm × 4.1 mm id × 5 μm) |
| Separation mode | Isocratic |
| Eluent | 37 mM (55 mM) NH4HCO3 in 1% v/v CH3OH, pH(9) |
| Eluent flow | 0.8 mL min−1 (1 mL min−1) |
| Column temperature | 22 ± 2 °C |
| Autosampler temperature | 22 ± 2 °C |
| Sample throughput rate | 1.5 (7.5) per hour |
A bio-inert valve (600 bar, 6-ports, two positions) was installed on automatic valve driver 1290 Infinity model G1170A (both obtained from Agilent Technologies, Cheadle, UK) and used for the on-column IS injection. The valve was equipped with sampling loops made of PEEK tubing with nominal volumes between 20 and 1000 μL. The internal standard (stored in a 250 mL PTFE container, see Fig. 1) was delivered through a secondary medium pressure pump (model P 4.1S, Knauer, Northampton, UK) with a nominal flow rate of 20 μL min−1. The IS container lid was pierced at two positions to (1) allow fitting of the 1/16′′ IS pump inlet tubing, along with (2) another small (1/16′′) hole for pressure equilibration. The plumbing schematics are presented in Fig. 1.
Reagents and sample preparation
Unless otherwise stated all reagents are of analytical grade quality or better, and were prepared using 18.2 MΩ cm UHP water (Elga, Veolia, High Wycombe, UK).Concentrated nitric acid UpA grade (mass fraction w(HNO3) ≥ 63%, density of ρ(HNO3) = 1.4 g mL−1) and hydrogen peroxide UpA grade (w(H2O2) ≥ 30%) were both purchased from Romil (Cambridge, UK). A mixture of 2% v/v HNO3 and 1% v/v H2O2 in water was then used for the extraction of i-As from 0.5 g rice sample (90 °C for 4 hours). The extracts were filtered through 0.22 μm syringe filters (33 mm, PES, Starlab Ltd, Milton Keynes, UK) and neutralised with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v diluted ammonia (32%, HiPerSolv, VWR, Leighton Buzzard, UK) to a pH of 9.0 ± 0.5. Ammonium bicarbonate (Fluka, Sigma-Aldrich, Gillingham, UK) was dissolved in water (concentrations detailed in Table 2) and the pH was adjusted to 9.0 ± 0.2 with 1:1 (v/v) ammonia. The pH was measured using a FisherBrand Hydrus 300 pH meter (Fisher Scientific, Leicestershire, UK). Methanol (Romil low metal, Spa, Romil, Cambridge, UK) was finally added to the solution to a final concentration of 1% (v/v). The solution was used both as a mobile phase and diluent, and for internal standard preparation for the on-column IS calibration. The total dilution factor for both the samples and the matrix CRMs (see below) was 40 (1 + 39 w/w ±2%). Sodium Arsenate CRM 7912-a (NMIJ, Tsukuba, Japan) and sodium arsenate dibasic heptahydrate (99.995% trace metal basis, Sigma-Aldrich, Dorset, UK) were used for the preparation of the calibration standard and IS, respectively. Certified for i-As reference materials of white rice (ERM BC211, 124 ± 11 μg kg−1, k = 2) and brown rice (NMIJ 7532-a, 298 ± 8 μg kg−1, k = 2) were both purchased from LGC Ltd (LGC, Teddington, UK) and used as quality control samples for the demonstration of the method trueness. Baby rice food products (ground rice >99%) were purchased from local grocery stores in the UK. The moisture content of these samples was determined by drying a 0.5 g subsample in an oven at 103 ± 2 °C until constant weight, following the procedure described in the ERM BC211 certificate.
:1 v/v diluted ammonia (32%, HiPerSolv, VWR, Leighton Buzzard, UK) to a pH of 9.0 ± 0.5. Ammonium bicarbonate (Fluka, Sigma-Aldrich, Gillingham, UK) was dissolved in water (concentrations detailed in Table 2) and the pH was adjusted to 9.0 ± 0.2 with 1:1 (v/v) ammonia. The pH was measured using a FisherBrand Hydrus 300 pH meter (Fisher Scientific, Leicestershire, UK). Methanol (Romil low metal, Spa, Romil, Cambridge, UK) was finally added to the solution to a final concentration of 1% (v/v). The solution was used both as a mobile phase and diluent, and for internal standard preparation for the on-column IS calibration. The total dilution factor for both the samples and the matrix CRMs (see below) was 40 (1 + 39 w/w ±2%). Sodium Arsenate CRM 7912-a (NMIJ, Tsukuba, Japan) and sodium arsenate dibasic heptahydrate (99.995% trace metal basis, Sigma-Aldrich, Dorset, UK) were used for the preparation of the calibration standard and IS, respectively. Certified for i-As reference materials of white rice (ERM BC211, 124 ± 11 μg kg−1, k = 2) and brown rice (NMIJ 7532-a, 298 ± 8 μg kg−1, k = 2) were both purchased from LGC Ltd (LGC, Teddington, UK) and used as quality control samples for the demonstration of the method trueness. Baby rice food products (ground rice >99%) were purchased from local grocery stores in the UK. The moisture content of these samples was determined by drying a 0.5 g subsample in an oven at 103 ± 2 °C until constant weight, following the procedure described in the ERM BC211 certificate.
Results and discussion
Evaluation of method performance with As standard solutions
The on-column IS calibration follows the same calculation procedure for species quantification as the other, typical, internal standardisations (see eqn (1)). The ratios of the signals of the calibration standards to internal standards, both as peak areas, are used to build a linear calibration, and those of the samples to internal standards, to calculate the analyte mass fraction. In the example below, the actual chromatographic analyte is the As(V) species. Since the extraction of the arsenic species from rice samples is performed in the presence of 1% (v/v) H2O2 at elevated temperature (see the Reagents and sample preparation section), the As(III) species oxidise completely to the measured As(V), and this peak effectively represents the total i-As in the sample. Therefore, for these analyses the calibration is based on arsenate standards only. Fig. 2 shows overlaid chromatograms of a series of As(V) calibration standards and a blank, obtained with the on-column injection setup and ICP-MS/MS detector. In this case (for demonstration purposes only) the IS concentration was chosen to match the highest calibration standard concentration. | ||
| Fig. 2 Overlaid chromatograms of a series of As(V) calibration standards measured with on-column IS. | ||
The ratio between the As(V)-analyte peak area (obtained from the calibration standard) and As(V) peak area (obtained from the internal standard; As(V)-IS) is then plotted against the calibration standard concentration and a typical calibration curve is built. The analyte concentrations are calculated from the obtained slope and intercept. Using this calibration strategy, the calibrations were linear in the range 25 ng kg−1 to 25 μg kg−1, at the instrument, with the squared Pearson coefficient (r2) exceeding 0.9998. If raw peak areas were used instead of on-column IS, the measurement accuracy would be significantly impaired due to the presence of drift. As an example, in one particular case when the sensitivity drift reached approximately 15% over the course of analysis, the linearity of the external calibration was significantly impaired and this led to lower r2 (0.998) and further biased results up to 15%.
Fig. 3 presents the signal variation with time from 50 injection pairs of 20 μL 2.5 μg kg−1 As(V) standard and 2.5 μg kg−1 As(V) internal standard represented with blue and red markers, respectively. The ratio between the analyte and IS (green triangles) was plotted against time and no statistically significant time dependence was found, although as evident from the figure the signal intensity drifted by more than 15% from its initial value, similarly to the discussed example above. The analyte to IS peak area ratio distribution had no significant deviation from the normal Gaussian profile. The slope of this ratio with time was in the order of 10−4 per injection when a linear fitting was used, which demonstrates excellent drift correction and no linear time trends. The RSD ratio was below 1% in this case. Discussion on the significance of this value can be found in the uncertainty estimation section below.
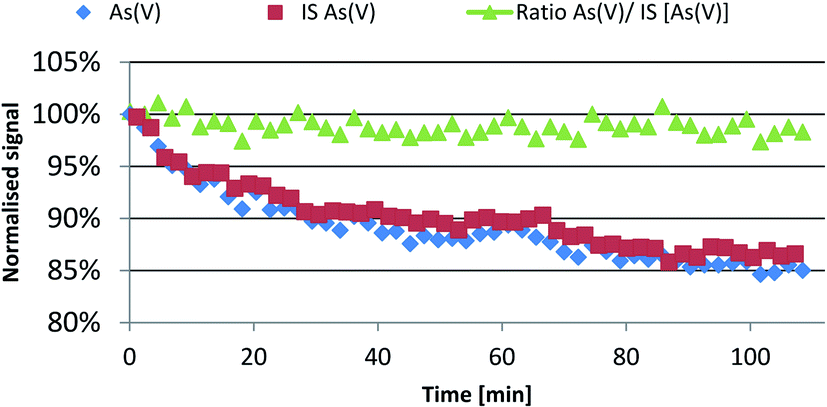 | ||
| Fig. 3 Efficient correction of significant sensitivity drift by the on-column internal standardisation approach. | ||
Similarly to SS-IDMS calibration, the value of the analyte-to-internal standard ratio can affect the accuracy of the quantitative speciation data. In this work we have investigated this effect within the range from 1:4 to 4:1. The relative standard recoveries of reagent blanks spiked with As(V) standards were between 99.5% and 100.9% (ESI Fig. 1†). The latter results confirm that an exact analyte-to-IS signal matching is not essential to achieve calibration uncertainties in this range (1%), which also agrees with the findings by Yuzuru and Matsuda14 who obtained similar uncertainty using conventional IS calibration. The described order of injections of sample and IS in this work can be swapped and the IS could be injected before the sample. This would allow for mixing of the IS with the non-retained components of the sample matrix providing similar (or potentially even better) correction of the matrix effects. Additionally, this approach would be beneficial if there is a co-eluting peak at the analyte's tail but not a fronting one (resolution R > 2 requirement, see section Feasibility of application to more than one As species, below) expanding the applicability of the approach to more complex samples. Unfortunately, most of the commercial LC control software do not provide an easy option for valve control prior to the sample injection (see Fig. 1) and therefore this approach has not been further pursued.
Measurement uncertainty estimation
| u2(V) = u2(T) − u2(S) − u2(I) | (2) |
Eqn (2) is used for the calculation of the sample and internal standard injection volume uncertainties, where u(S) and u(I) are the standard uncertainties of the ICP-MS signal and integration, respectively, and u(T) is the combined, total uncertainty. Since the u(Dr) component is not accounted for in the equation above, it should be noted that it is still present in the calculated u(V). As for the relative u(S) and u(I), values of 0.45% and 0.09% have been obtained. These uncertainty sources are uncorrelated; therefore, covariance terms do not need to be included. Based on this, u(V) was calculated to be 0.625% and 0.627% for the sample and for the internal standard injections, respectively. These values are sufficiently low and the u(Dr) contribution can therefore be ignored.
| i-As fraction [μg kg−1] | Sample/IS area ratios | Standard recoveries | Relative calibration uncertainty |
|---|---|---|---|
| 0.1 | 0.0612 | 101.2% | 22.2% |
| 0.5 | 0.2886 | 99.9% | 4.8% |
| 0.7 | 0.4300 | 99.2% | 3.2% |
| 1.0 | 0.5814 | 100.0% | 2.4% |
| 5.0 | 2.9213 | 100.3% | 0.5% |
| 7.7 | 4.4796 | 99.9% | 0.3% |
| 10.1 | 5.8993 | 100.0% | 0.3% |
For verification purposes, 150 injections (within 3 batches with 50 injections per batch) of the 2.5 μg kg−1 As(V) standard were performed and the signal ratio for the IS injections was calculated. The relative standard deviation of the ratios (and therefore of the final concentrations) varied between 0.5% and 1.5% per batch, which is in agreement with the expected relative uncertainty between 2.4% and 0.5% for 2.5 μg kg−1 As (Table 3, above). As expected, getting closer to the 0.025 μg kg−1 limit of detection results in increased measurement uncertainty. Although the calibration approach proposed here provides accurate results where other calibration strategies might fail, it is worth noting that on-column or any other type of IS calibration is not always a necessity for performing accurate measurements. The calibration uncertainty with IS would always be higher, or at least equal to the linear regression one, when there are no drift and matrix effects to compensate for. With IS calibration two uncertainties from peak area integrations are combined for each measurement vs. only one if simple external calibration is used. With the method described in this work this effect was below 1% increase in the calibration uncertainty in case of on-column IS calibration vs. typical external calibration. However, if there is a drift present the contribution of the uncertainty of the IS peak area estimation becomes negligible compared to the improved accuracy (0.5% compared to up to 15% bias, as observed with our experiments).
| Material code | A | B | C | D | E |
|---|---|---|---|---|---|
| Natural i-As content [μg kg−1] | 61 | 116 | 97 | 111 | 269 |
| i-As spike recovery [%] | 104.6–105.4 | 99.9–101.4 | 100.2–101.8 | 103–105 | 97.8–98.7 |
A typical chromatogram of a baby food rice extract is presented in Fig. 4. To confirm the quantitative conversion of As(III) to As(V), and therefore representativeness of the As(V) to the i-As in the sample, within each batch of rice samples a random sample was spiked with As(III) before extraction. In all cases the species was quantitatively recovered as As(V), confirming the complete species oxidation within the matrix. Additionally, two matrix reference materials (white rice ERM-BC211 and brown rice NMIJ 7532a, certified for i-As) were extracted and analysed along with the rice samples. The relative i-As recoveries from both CRMs were quantitative (between 97% and 107%) and within the certified measurement uncertainties, demonstrating the accuracy of both the extraction procedure and the newly proposed calibration approach (t-test used in both cases). The relative expanded measurement uncertainty for the whole method (k = 2) was about 10% or lower in the range 60 to 300 μg kg−1 i-As in dry rice products. This includes the contribution of the materials' homogeneity and within-run and between-run uncertainty components. The on-column calibration expanded relative uncertainty was between 2% and 3% (k = 2) with ≤1.5% sample/IS peak area ratio relative uncertainty (k = 2, see the section on calibration performance above).
 | ||
| Fig. 4 Example chromatogram from the analysis of i-As in baby rice food by on-column IS calibration. | ||
 | ||
| Fig. 5 Chromatogram for simultaneous determination of MA and As(V) in a standard mixture by on-column IS utilising isocratic elution. The dashed lines show the analyte/IS pair peaks for the quantified species. | ||
The applicability of the method to gradient elution chromatography is demonstrated in Fig. 6 where the same two species have been quantified with different amounts of carbon in the eluent (and therefore in ICP-MS plasma) at the time of their elution – 37 mM NH4HCO3 (eluent A) and 55 mM NH4HCO3 (eluent B), both pH adjusted to 9.0 (±0.2). The separation method involved 5 min isocratic 100% A, followed by a linear increase to 100% B in 20 min and 5 min column equilibration (100% A) before the next sample injection. The analytes [MA and As(V)] and the respective internal standards [MA IS and As(V) IS] were of the same concentration (2.5 μg kg−1 each) but in this case, as expected the species signals showed different sensitivities due to the different carbon loads in the plasma. The MA peak has smaller peak area than the MA IS peak and the As(V) peak has smaller peak area than that of As(V) IS, in contrast to observations under isocratic conditions (see Fig. 2) where sensitivities are equal. However, the analyte/IS ratio remained constant with time, e.g. throughout the batch, and the instrument drift was efficiently compensated for. Standards recoveries close to 100% (±2%) were obtained.
 | ||
| Fig. 6 Chromatogram for simultaneous determination of MA and As(V) in a standard mixture by on-column IS utilising gradient elution. The dashed lines show analyte/IS pair peaks for the quantified species. | ||
An additional “stress-test” was performed under the same conditions but introducing 5% MeOH in water in between samples to induce stronger drift. Although under such conditions the plasma became unstable and the peak areas increased for both species (MA and As(V)), their ratio to the respective IS species remained constant (Fig. 7a and b in the ESI†).
Conclusions
A novel calibration strategy for high accuracy speciation analysis based on on-column internal standard injection is described for the first time. As the IS species are the same as the target analytes the method offers an attractive alternative to IDMS calibration to compensate for large signal drifts, matrix effects and potential on-column losses with unique prospects for determination of mono-isotopic elemental species. Another advantage of the proposed calibration approach is that it can be employed with any detector that can measure time-resolved signals. Moreover, multiple elements and species can be simultaneously quantified with this approach provided that a good chromatographic selectivity is achieved. However, the success of on-column IS is hampered when the resolution between the analyte and the following and preceding peaks is insufficient (R < 2) as in such cases the chromatographic separation of the IS is also compromised. The developed calibration strategy has been demonstrated to be suitable for high accuracy speciation analysis of inorganic As in a rice based matrix with relatively expanded measurement uncertainty for the whole method (k = 2) of 10% or lower. Respectively, the relative uncertainty from the on-column calibration was between 2% and 3% (k = 2) in the range 60 to 300 μg kg−1 i-As in dry rice products. The proposed method is currently applied to the certification of reference materials in support of current EU legislation for the maximum amount of inorganic arsenic in rice and rice products.Author contributions
Funding acquisition: Heidi Goenaga-Infante, Panayot Petrov. Project administration: Heidi Goenaga-Infante, Panayot Petrov. Resources: Heidi Goenaga-Infante, Panayot Petrov. Supervision: Heidi Goenaga-Infante, Panayot Petrov. Writing – original draft: Panayot Petrov, Simon Cowen, Heidi Goenaga-Infante. Writing – review & editing: Panayot Petrov, Heidi Goenaga-Infante. Software: Simon Cowen. Conceptualization: Panayot Petrov. Data curation: Panayot Petrov, Simon Cowen. Methodology: Panayot Petrov. Formal analysis: Simon Cowen, Panayot Petrov, Heidi Goenaga-Infante. Investigation: Panayot Petrov. Validation: Panayot Petrov, Simon Cowen. Visualization: Panayot Petrov.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work described in this paper was performed at the UK National Measurement Laboratory at LGC Ltd and was funded by the UK government Department for Business, Energy & Industrial Strategy (BEIS).References
- J. P. Vicente, J. E. Romero and S. C. Broch, Validation of Analytical Methods Based on Chromatographic Techniques: An Overview, in Analytical Separation Science, Wiley-VCH Verlag GmbH & Co. KGaA, 1st edn, 2015, vol. 14, pp. 1–52 Search PubMed.
- B. Magnusson and U. Örnemark, EURACHEM Guide: The Fitness for Purpose of Analytical Methods: A Laboratory Guide to Method Validation and Related Topics, issued in, 1998 Search PubMed.
- S. L. Ellison and M. Thompson, Analyst, 2008, 133, 992–997 RSC.
- J. Kang, L. A. Hick and W. E. Price, Rapid Commun. Mass Spectrom., 2007, 21, 4065–4072 CrossRef CAS PubMed.
- A. K. Hewavitharana, N. S. A. Kassim and P. N. Shaw, “Standard addition with internal standardisation as an alternative to using stable isotope labelled internal standards to correct for matrix effects—Comparison and validation using liquid chromatography- tandem mass spectrometric assay of vitamin D”, J. Chromatogr. A, 2018, 1553, 101–107 CrossRef CAS PubMed.
- E. Stokvis, H. Rosing and J. H. Beijnen, Rapid Commun. Mass Spectrom., 2005, 19, 401–407 CrossRef CAS PubMed.
- K. M. Kubachka, N. V. Shockey, T. A. Hanley, S. D. Conklin and D. T. Heitkemper, Arsenic Speciation in Rice and Rice Products Using High Performance Liquid chromatography Inductively Coupled Plasma-Mass Spectrometric Determination, FDA: Version Draft 1.1, November 2012 Search PubMed.
- H. Stahnke, T. Reemtsma and L. Alder, Anal. Chem., 2009, 81, 2185–2192 CrossRef CAS PubMed.
- H. W. Liao, G. Y. Chen, M. S. Wu, W. C. Liao, I. L. Tsai and C. H. Kuo, J. Chromatogr. A, 2015, 1375, 62–68 CrossRef CAS PubMed.
- J. Rossmann, L. D. Renner, R. Oertel and A. E. Armouche, J. Chromatogr. A, 2018, 1535, 80–87 CrossRef CAS PubMed.
- P. Haefelfinger, J. Chromatogr. A, 1981, 218, 73–81 CrossRef CAS.
- P. Araujo, F. Couillard, E. Leirnes, K. Ask, A. Bøkevoll and L. Frøyland, J. Chromatogr. A, 2006, 1121, 99–105 CrossRef CAS PubMed.
- K. D. Altria and H. Fabre, Chromatographia, 1995, 40, 313–320 CrossRef CAS.
- Y. Hayashit and R. Matsuda, Anal. Sci., 1995, 11, 389–400 CrossRef.
- J. P. Jesus, C. A. Suárez, J. R. Ferreira and M. F. Giné, Talanta, 2011, 85, 1364–1368 CrossRef CAS PubMed.
- J. Feldmann, A. Raab and E. M. Krupp, Anal. Bioanal. Chem., 2018, 410, 661–667 CrossRef CAS PubMed.
- P. Kościelniak, M. Wieczorek, J. Kozak and M. Herman, Anal. Lett., 2011, 44, 411–430 CrossRef.
- Commission Regulation (EU), 2015/1006, Official Journal of the European Union, 2015, vol. 161, pp. 14–16 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ay00699a |
| This journal is © The Royal Society of Chemistry 2021 |