
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe application of aptamer Apt-236 targeting PvpA protein in the detection of antibodies against Mycoplasma gallisepticum†
Ping
Fu‡
 a,
Feng
Wang‡
b,
Yunke
Zhang
b,
Xilan
Qiao
b,
Yuewei
Zhang
c,
Wenyan
Zhou
d,
Xinbo
Yan
b and
Wenxue
Wu
*b
a,
Feng
Wang‡
b,
Yunke
Zhang
b,
Xilan
Qiao
b,
Yuewei
Zhang
c,
Wenyan
Zhou
d,
Xinbo
Yan
b and
Wenxue
Wu
*b
aInstitute of Blood Transfusion, Chinese Academy of Medical Sciences, Chengdu 610052, China
bKey Laboratory of Animal Epidemiology and Zoonosis, College of Veterinary Medicine, China Agricultural University, Beijing 100083, China. E-mail: wuwenxue@cau.edu.cn
cBeijing QuantoBio Biotechnology Co. Ltd, Beijing 100176, China
dUniversity of Science and Technology Beijing, Beijing 100081, China
First published on 18th June 2021
Abstract
Mycoplasma gallisepticum (M. gallisepticum) is the primary agent of chronic respiratory disease causing important economic losses in the poultry industry. Compared to antibodies, aptamers used to diagnose M. gallisepticum have many advantages, such as being chemically, animal-free produced and easily modifiable without affecting their affinity. Herein, a single-stranded DNA (ssDNA) aptamer Apt-236 which can specifically bind to PvpA protein of M. gallisepticum with a Kd of 1.30 ± 0.18 nM was selected successfully. An indirect blocking ELAA (ib-ELAA) for M. gallisepticum antibodies detection was also developed using Apt-236, in which M. gallisepticum antibodies would block the binding-position of aptamers. Therefor positive sera would prevent color development whereas negative sera will allow a strong color reaction. The ib-ELAA was consistent with other three widely used assays in terms of the growth and decline of the antibody response to M. gallisepticum, and showed substantial agreement with the results obtained using a commercial ELISA kit in clinical chicken sera samples. Therefore, the ib-ELAA developed in this study was a new format for aptamer application and would be an alternative method for the surveillance of M. gallisepticum.
Introductions
Mycoplasma gallisepticum is the primary agent of chronic respiratory disease causing important economic losses in the poultry industry.1 Infection caused by M. gallisepticum has a wide variety of clinical manifestations, but the most important disease presentation is chronic respiratory disease in chickens and infectious sinusitis in turkeys, resulting in reduced meat and egg production.2M. gallisepticum infection occurs frequently in chickens of different ages, especially in the presence of co-infections, such as laryngotracheitis virus, influenza A virus, avian adenovirus and Escherichia coli. Co-infection will aggravate the development of the disease, causing serious damage.3–5Chronic respiratory disease in chickens caused by M. gallisepticum can be diagnosed based on clinical and pathological findings as well as with other tests/methods, including detection of specific antibodies [such as serum plate agglutination (SPA), hemagglutination inhibition (HI) and enzyme-linked immunosorbent assay (ELISA)],6,7 isolation of M. gallisepticum, or detection of specific DNA by the polymerase chain reaction (PCR).8,9 Although these methods have high sensitivity, some defects still exist. For example, the use of protein antibodies increases the cost of testing and requires strict reagent storage and reaction conditions.10 In addition, PCR needs complex nucleic acid extraction processes and special instruments.
Aptamers (also known as chemical antibodies) are single-stranded DNA or RNA that binds to a wide range of molecules with high specificity and affinity.11 Aptamers are isolated during an in vitro selection process called systematic evolution of ligands by exponential enrichment (SELEX),which was first reported in 1990.12 The application of aptamers in ELISA gives rise to an ELISA-derived assay called enzyme linked aptamer assay (ELAA).13 ELAA has been reported to be used in several different configurations, including direct, indirect, and sandwich assays for detection of the human vascular endothelial growth factor (VEGF), porcine reproductive and respiratory syndrome virus type II, ochratoxin A in wine, or dopamine.14,15 Compared to antibodies, aptamers are smaller in size, can be easily modified, are cheaper to produce, and can be generated against a wide array of target molecules.15
In this study, we developed an indirect blocking ELAA for M. gallisepticum antibody detection. Series of tests proved that this ib-ELAA had good sensitivity and specificity, consistent with other three widely used serological tests.
Materials and methods
Mycoplasma and virus strains
M. gallisepticum BJ44Tstrain (CVCC350, preserved in China Veterinary Culture Collection Center, Beijing, China) was grown in pleuropneumonia-like organism (PPLO) medium (BD Company, Franklin Lakes, NJ, USA) as described earlier.16Newcastle disease virus (NDV), influenza A H9N2, and infectious bronchitis virus (IBV) were cultured separately in chick embryo and harvested as chick embryo allantoic liquid.Sera samples
Twenty negative chicken sera samples were collected from SPF chicken. Twenty positive chicken sera samples were also collected from chicken naturally infected with M. gallisepticum, from which M. gallisepticum had been isolated in the nasal swabs using PPLO medium. Avian influenza subtype H5, and H7 positive control sera were purchased from the Harbin National Engineering Research Center of Veterinary Biologics (Harbin, China). IBV, Infectious bursal disease (IBDV), NDV, and Mycoplasma synoviae (M. synoviae) positive control sera were purchased from the China Institute of Veterinary Drug Control (Beijing, China). Various bacteria and virus species are listed in Table 1.| Strain | Source |
|---|---|
| a China Institute of Veterinary Drug Control, Beijing, China. b A gift from Professor Wang in China Agricultural University, China. c Field strain isolated from lungs of chicken having clinical symptoms of H9N2 in China. | |
| Staphylococcus aureus | CVCC1885a |
| Escherichia | CVCC3801a |
| Newcastle disease virus (NDV) | Field isolates (La sota)b |
| Influenza A H9N2 | A3/98c |
| Mycoplasma synoviae | CVCC385a, CVCC2960a |
| Infectious bronchitis virus (IBV) | Field isolatesb |
Three 4–6 weeks-old SPF chicken were immunized with 109 color change unit (CCU) per mL inactivated M. gallisepticum BJ44T strain (4 doses, at approximately 12 days intervals) supplemented with Freund's complete or incomplete adjuvant. Chicken sera samples were collected 11 times from the 6th day after the first immunization, and at approximately 7 day intervals.
Two hundred and thirty clinical chicken sera samples were randomly collected from chicken farms in China and stored at −20 °C.
Preparation of recombinant PvpA protein
In order to clone the encoding gene of PvpA protein of M. gallisepticum, a pair of primers was designed based on the gene sequence (Genbank DQ989519.2) as shown below.| MqPvpA-F: CGGGATCCATGGAGTTAAATAAATTAAAAAAACATAA |
| MqPvpA-R: CCGCTCGAGTTAATTTGGTCTTGGACCCATTG |
Amplified products were cloned into pEasy-T1 plasmid (Transgen, Beijing, China), followed by clone confirmation by sequencing and subcloning in pET-28a vector (Novagen, Darmstadt, Germany) to yield His-tag rMGA PvpA, and Transetta (DE3) Chemically Competent Cell (Transgen, Beijing, China) transformation and overexpression by induction with 1 mM IPTG at 37 °C for 6 h. Under native conditions, the recombinant protein was renatured and purified by nickel affinity chromatography (Qiagen, NY, USA). The purity and yield of the recombinant PvpA protein were evaluated by SDS-PAGE and the Bradford method according to two previously reported standard protocols in our laboratory.17,18
Oligonucleotide synthesis
The oligonucleotide library was synthesized as a single-stranded 80-mer with the following sequence: 5′-GCTGCAATACTCATGGACAG-(N)40-GTCTGGAGTACGACCCTGAA-3′, where the central N40 represents random oligonucleotides based on equal incorporation of A, G, C, and T in each position. The PCR primers were as follows:| Forward primer (FP17): 5′-GCTGCAATACTCATGGACAG-3′ |
| Reverse primer (BP17): 5-TTCAGGGTCGTACTCCAGAC-3′ |
The library and all primers were synthesized and HPLC-purified was provided by Sangon Inc (Shanghai, China).The ssDNA pools were denatured by heating at 95 °C for 10 min in 0.1 M phosphate buffer saline PBS (pH 7.4) and then immediately cooled on ice for 20 min.
Selection of DNA aptamer targeting PvpA protein
Purified PvpA protein was dissolved in 100 μl PBS, and coated on 96-well micro plates at 4 °C overnight. Then the plates were washed three times with PBS containing 0.05% Tween20 (PBST) and incubated with 200 μl 5% bovine serum albumin (BSA) in PBS for 2 h at 37 °C. They were washed three times again with PBST, and the initial ssDNA pools (12.7 nmol) were added to the wells coated by PvpA protein solution and incubated at 37 °C for 45 min. The unbound ssDNA was removed by washing three times with PBST. 100 μl elution buffer (20 mM Tris–HCl, 4 M guanidinium isothiocyanate, 1 mM dithiothreitol, pH 8.3) was added to each well and incubated for 10 min at 80 °C. After extraction with phenol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :chloroform:isoamyl alcohol at 25:24:1, the precipitate was dissolved in 60 μl ddH2O. PvpA-bound ssDNA was then amplified by the symmetry PCR method according to a previous report.17 In the second, fourth and fifth selection rounds, the anti-SELEX procedure was also included in the aptamer selection: the well was firstly coated with the anti-SELEX material, which was blocked with BSA the same as the wells coated with PvpA protein. The previous round ssDNA library was added into the anti-SELEX well and incubated for 30 min at 37 °C; the supernatant solution was collected and used for the selection of aptamers by which the ssDNA that could bind to the anti-SELEX material, BSA or the plate was screened out from the ssDNA library. The selection parameters for each round are shown in Table 2.
:chloroform:isoamyl alcohol at 25:24:1, the precipitate was dissolved in 60 μl ddH2O. PvpA-bound ssDNA was then amplified by the symmetry PCR method according to a previous report.17 In the second, fourth and fifth selection rounds, the anti-SELEX procedure was also included in the aptamer selection: the well was firstly coated with the anti-SELEX material, which was blocked with BSA the same as the wells coated with PvpA protein. The previous round ssDNA library was added into the anti-SELEX well and incubated for 30 min at 37 °C; the supernatant solution was collected and used for the selection of aptamers by which the ssDNA that could bind to the anti-SELEX material, BSA or the plate was screened out from the ssDNA library. The selection parameters for each round are shown in Table 2.
| SELEX round | PVPA protein (μg) | Incubating time (min) | Anti-SELEX |
|---|---|---|---|
| 1 | 100 | 45 min | — |
| 2 | 50 | 45 min | 200 μg cell lysates of Transetta (DE3), and 100 μl 1:2 diluted SPF serum |
| 3 | 25 | 30 min | 200 μg cell lysates of Mycoplasma synoviae |
| 4 | 10 | 30 min | 100 μl 1:5 diluted NDV chick embryo allantoic liquid (hemagglutination titer: 29) |
| 5 | 5 | 20 min | 50 μl 1:2.5 diluted H9N2 chick embryo allantoic liquid (hemagglutination titer: 29) and 50 μl 1:1.5 diluted IBV chick embryo allantoic liquid (hemagglutination titer: 29) |
Sequence analysis of the ssDNA pool
After five runs of selection described as above, the purified symmetric PCR products of the second and fifth run were high-throughput sequenced by Atomy Co., Ltd (China). Clustalx software was used to analyze the abundance and homology of the aptamer from two rounds.Determination of dissociation constants by i-ELAA
The affinities between aptamers and the PvpA protein were determined by i-ELAA with the following procedure: 96-well micro plates were coated with PvpA protein, and blocked with 5% BSA. 100 μl denatured bio-aptamers of different concentrations (100 nM, 50 nM, 20 nM, 10 nM, 5 nM, 2 nM, 1 nM, 0.5 nM, 0.25 nM, and 0 nM) were added into each well, and incubated for 45 min at 37 °C. Washing was performed three times with PBST, 100 μl 1:32000 HRP-SA was added and incubation was carried out at 37 °C for 45 min. Washing was performed three times with PBST, 100 μl substrate TMB (Sigma, U.S.A) was added and incubation was carried out for 10 min at 37 °C. Then 50 μl 2 M sulfuric acid was added to stop the reaction. The optical densities were read at 450 nm. A calibration curve was obtained using bio-aptamers with concentrations in the range of 0–100 nM. A saturation curve was obtained based on these data, and equation Y = BmaxX/(Kd + X) was used to calculate Kd according to GraphPad Prism 5.0. Y represented the mean value of OD450 nm, Bmax was the maximal value of OD450 nm, and X was the concentration of the bio-aptamer.
Establishing indirect blocking ELAA (ib-ELAA)
Micro plates (96 wells) were coated with 100 ng per well of PvpA protein in 0.1 M PBS at 4 °C overnight. The plates were washed with PBST three times and blocked with 200 μl 5% swine serum in PBST at 37 °C for 2 h. Next, 50 μl chicken serum and 50 μl 5% swine serum (50 μl PBS was added into two wells for use as the buffer control at the same time) were added into the plates; the plates were incubated at 37 °C for 45 min and washed with PBST three times. 100 μl 100 nM bio-Apt-236 were added into the plates and the plates were incubated at 37 °C for 45 min. Plates were then washed three times with PBST to remove the unbound material, HRP-SA (1:32000) was added, and incubation was carried out at 37 °C for 45 min. After four times of washing with PBST, 100 μl TMB (Sigma, Aldrich) was added for visualization. The reaction was stopped after 10 min at 37 °C by the addition of 50 μl per well 2 M sulfuric acid. The optical densities (OD) were read at 450 nm on a micro plate reader. The percent inhibition (PI) values were determined using the formula: PI (%) = (1 − OD450 nm of test serum/OD450 nm of aptamer control) × 100%. A total of 20 negative chicken sera and 20 positive chicken sera were used to determine the cut-off value between the positive and negative sera. The cut-off value was designed as the mean PI of negative sera +2 standard deviations (SD), which would ensure that 95% of the PI values for the negative sera fell within this range.
ib-ELAA for detecting M. gallisepticum antibodies in the sera collected from chicken immunized with M. gallisepticum and clinical farms
A total of 33 sera samples were collected from 3 chickens at 6, 16, 24, 31, 46, 61, 79, 102, 132, 164, and 192 days after immunization and detected by ib-ELAA, one commercial ELISA kit from IDEXX (Canada), homemade ELISA kit and hemagglutination inhibition assay (HI) established in our laboratory. The homemade ELISA kit was developed with PvpA as the coated antigen similar to the previous report,19 and the S/P: [OD450 nm (sample) − OD450 nm (negative control)]/[OD450 nm (positive control) − OD450 nm (negative control)] ≥ 0.67 was considered as positive. The HI assay were performed as described in Kleven's report,7 and the HI titer ≥4 was considered as positive. All animal research was approved by the Beijing Association for Science and Technology (approval ID SYXK (Beijing) 2007-0023) and complied with the guidelines of the Beijing Laboratory Animal Welfare and the Ethics of the Beijing Administration Committee of Laboratory Animals. All animal studies were also performed in accordance with the China Agricultural University Institutional Animal Care and Use Committee guidelines (ID: SKLAB-B-2010-003) and approved by the Animal Welfare Committee of China Agricultural University.A total of 230 chicken sera samples from a chicken farm in China were also detected by ib-ELAA, and the results were compared with those obtained using commercial i-ELISA kits from IDEXX (Canada).
Results and discussion
Expression of PvpA protein in E. coli
In the case of M. gallisepticum infections, PvpA, an integral membrane surface protein, is a potential diagnostic antigen of choice since this surface-exposed protein is accessible to the host immune response.1,20 The species-specific and immunogenic properties of the rPvpA336 protein as well as the potential use of the species-specific diagnostic prototype have clearly been demonstrated.1,21 Recently, enzymatic rapid immunofiltration assay prototype (ERIFA) and indirect ELISA based on recombinant PvpA have been developed for monitoring M. gallisepticum infections in the field and under limited laboratory conditions.1,19 To clone PvpA gene of M. gallisepticum strain BG44T, a pair of primers (MqPvpA-F and MqPvpA-R) was used. The amplified 1149 bp product was cloned, sequenced, and then cloned into plasmid PET28 (a+) and transferred into Transetta (DE3). Luckily, sequence analysis results showed that M. gallisepticum PvpA gene contained no UGA codons, which is treated as tryptophan according to the mycoplasma genetic code, but interpreted as stop codon in the E. coli expression system.17 As expected, approximately a 44 kDa protein band was analyzed by SDS-PAGE (Fig. 1). | ||
| Fig. 1 (a) PCR identification of PvpA gene: (M1) DM 2000 marker; (1) an amplified product of 1149 bp as expected was obtained. (b) SDS-PAGE analysis of PvpA protein in E. coli: (M2), prestained protein molecular weight marker (14–120 kDa); (3) total cell lysates of Transetta (DE3) with PET28 (a+)-PvpA gene plasmid; (4) an approximately 44 kDa purified PvpA protein band with a purity of 95%. | ||
In vitro selection of the ssDNA aptamer for the PvpA protein
In this study, microplates were also selected as the SELEX matrix as described by Rhodes22 with minor modifications.14 The initial library contained 80 nt ssDNA including 40-mer random nucleotide inserts, which meant the library was a pool of more than 1012 unique sequences theoretically and provided enough candidates for the selection of aptamers with higher affinity and specificity. The concentration of the PvpA protein, and the incubation time in the subsequent rounds of selection were decreased gradually to increase the probability of obtaining aptamers with higher affinity; meanwhile, different counter selection materials were also employed as anti-SELEX from the second round of selection to remove the nonspecific sequences to improve specificity (Table 2). As ssDNA generation is an important step in the aptamer selection procedure,23 two kinds of PCR were designed as described previously to generate enough ssDNA for the subsequent selection of aptamers.14After five rounds of selection, the second and fifth symmetry PCR products were high-throughput sequenced. In each round, more than 500000 sequences were obtained, and sorted by abundance. The top 400 sequences were found repeated many times, as listed in Table S1.† In addition, the enrichment of top 400 sequences was also analyzed by comparing the abundance between the second and fifth round (enrichment = abundance (%)fifth round/abundance (%)second round). With a larger enrichment value, the specific sequence was better enriched, which might be specific aptamer to PvpA with good affinity. The homology of the top 100 sequences with high abundance and sequences with a high enrichment value were analyzed using clustal X software and divided into seven families according to their homology (Fig. 2). In previous reports, to identify individual aptamers in SELEX, traditional single-clone Sanger sequencing of the enriched ssDNA pool is widely employed, by which up to 100 single aptamers were obtained.24,25 In this study, we adopted high-throughput sequencing technology reported previously11,25,26 to obtain 1137812 sequences in round two and 1638583 sequences in round five, avoiding the omission of the optimal aptamer.
 | ||
| Fig. 2 The homology analysis of 110 aptamer sequences. Family 1 contained 14 aptamers, family 2 contained 23 aptamers, family 3 contained 21 aptamers, family 4 contained 9 aptamers, family 5 contained 5 aptamers, family 6 contained 33 aptamers, family 7 contained 5 aptamers. | ||
Dissociation constants of the PvpA aptamers
According to results of the homology analysis, 20 aptamers from 7 families were randomly selected, and their dissociation constants (Kd) were analyzed using dose-dependent i-ELAA as reported previously by our group.14 Compared to other methods (e.g. fluorescence intensity, fluorescence anisotropy/polarization, UV-vis absorption, circular dichroism, surface plasmon resonance, isothermal titration calorimetry, and affinity capillary), i-ELAA was convenient, and specific, showed high-throughout and did not need expensive equipment.11,27 As listed in Table 3, all the 20 aptamers had nanomole per L level affinities, with Kd values ranging from 0.58 ± 0.10 nM to 4.4 ± 0.82 nM. As Apt-10 has the strongest binding affinity (Kd = 0.58 ± 0.10 nM) with PvpA, and Apt-236 has the highest enrichment value (=48.95, listed in Table S1†), both of them were good candidate aptamers for the following application studies. Two dimensional stem-loop structures of apt-10 and apt-236 were predicted using online software: http://mfold.rit.albany.edu/28 using 4 °C as a folding temperature, with 156 nM Na+ concentrations. As shown in Fig. 3, the two aptamers both had several loop or hairpin structures that represented clearly the accessible single-stranded regions, which were probably more energetically favorable for target binding versus an “induced fit” in double-stranded stem regions wherein hydrogen bonds between the nitrogen bases of the aptamers would be broken.29| Family | Aptamer | Random sequnces (5′–3′) | K d (nM) |
|---|---|---|---|
| 1 | Apt-2 | GGCGTAGCACTGTCCCATTCCTGACACTATCGCACAGTCG | 0.90 ± 0.15 |
| Apt-5 | GGCGTAGCACTGTCCCGTTCCTAACACTACCACACAGTCG | 1.14 ± 0.21 | |
| Apt-173 | GGCGTAGCACTGTCCCACTCCTGACACTATCGCACAGTCG | 0.92 ± 0.17 | |
| 2 | Apt-7 | GGAGCAAGGCGACCTGGCCCACAAGCAAACCCACTTCGCC | 1.55 ± 0.21 |
| Apt-21 | GGGACGGAACGGACCCTGCCAACACAAGCCGAATGTGCTC | 1.77 ± 0.39 | |
| Apt-57 | GCGAGGAACGAGCCTGGACACACATAATACGTCCGTTGGC | 0.84 ± 0.18 | |
| Apt-167 | GAGCAAGGCGACCTGGCCCACAAGCAAACCCACTTCGCCG | 0.82 ± 0.16 | |
| 3 | Apt-4 | GTGCGAAGCGCCTGGACTGCACATCCCATTCCTTCTACCG | 1.24 ± 0.29 |
| Apt-6 | GTGCGAAGCGCCTGGACTGTACATCCCACTCCCTCTACCG | 1.02 ± 0.19 | |
| Apt-236 | GTGCGAAGCGCCTGGACTGTACATCCTACTCCCTCTACCG | 1.30 ± 0.18 | |
| 4 | Apt-55 | CACGAAGGGCTGGCCAGGCTTCAGGGCTATACTCCAGACC | 1.07 ± 0.14 |
| 5 | Apt-10 | GGGAGGCGGGTGGGTCGTCGGGGTGCGTCGTTCTGTGTGG | 0.58 ± 0.10 |
| 6 | Apt-1 | GAGCAGCGCGACTGTCCGAGCCAATCCGCCATCTAGTGGT | 1.02 ± 0.14 |
| Apt-3 | GAGCAACGCGACTGTCTGAGCCAACCTGACATCTAGTGGT | 1.35 ± 0.24 | |
| Apt-8 | GAGCAACGCGACTGTCTGAACCAATCTGACATCAAGTGGT | 1.42 ± 0.36 | |
| Apt-9 | GAGCAACGCGACTGTCCGAGCCAATCCGCCATCTAGTGGT | 1.67 ± 0.46 | |
| Apt-53 | GAGCAACGCGACTGTCCGGACCAATCTGGCATCTGGTGGT | 1.24 ± 0.21 | |
| Apt-133 | GAGCAGTGCGACTGTCCGAGCCAATCCGCCATCTAGTGGT | 0.90 ± 0.09 | |
| Apt-153 | GAGCAACGCGACTGTCTGGACCAATCTGACATCTAGTGGT | 4.4 ± 0.82 | |
| 7 | Apt-54 | GACACGGTGCTGCAATACTCATGGACAGGAGCAACGCGAC | 2.31 ± 0.40 |
 | ||
| Fig. 3 Characteristics of two dominant candidate DNA aptamers. (a) Dissociation constant (Kd) determined by dose-dependent i-ELAA. Each data point represents the averaged results of three replicates; (b) secondary structures specific to the PvpA protein. | ||
Establishing indirect blocking ELAA (ib-ELAA)
Since Drolet reported the first enzyme-linked aptamer assay (ELAA) to detect the human vascular endothelial growth factor on microtiter plates in 1996,13 similar mixed assays have been set up on the surface of microbeads, in which aptamers were used in combination with antibodies or by replacing them completely.30–32 Different ELAA formats were exploited using either immobilized or labeled aptamers as capture or detecting reagents, such as direct ELAA, indirect ELAA, sandwich ELAA, competition ELAA, ect.32–34 In this study, we established an indirect blocking ELAA (ib-ELAA) for M. gallisepticum antibody detection using Apt-236, which led to a better result than Apt-10 (data not shown) in the pre-experiment. In the indirect blocking assay, immunoglobulins in positive sera blocked the antigenic epitopes binding to the PvpA aptamer, as depicted schematically in Scheme 1. Therefore, positive sera would prevent color development whereas negative sera will allow a strong color reaction. In 2014, an indirect competitive ELAA for M. bovis antibody detection was firstly reported by our group.14 It was reported that a significant fraction (1.5%) of the serum proteins is known to bind strongly to DNA.35 In the indirect competitive format, the interaction between the ssDNA aptamer and antigen (PvpA) might been inhabited by serum proteins. Thus, in this study, we modified the indirect competitive format into the indirect blocking format, in which the aptamer would not come into contact with serum protein directly. Serum was first incubated with PvpA, after washing; the serum components which did not combine with PvpA were removed, and then aptamers were added; aptamers would not come into contact with serum directly. It avoided the interference of aptamers by sera components, improving the specificity and repeatability of the assay significantly (a cutoff value of 45.4% PI was confirmed for the indirect competitive ELAA in the pre-experiment; five negative sera and other antisera of related pathogens were tested; the results are shown in the ESI Fig. S1†). | ||
| Scheme 1 Schematic mechanism of indirect blocking ELAA. | ||
After optimization, 20 SPF chicken sera were used to determine the cutoff value of percent inhibition (PI). These negative sera had a mean percent inhibition value of 4.2% with a standard deviation (SD) of 7.8%. As shown in Fig. 4a, all positive sera have PI values higher than 20%. The cutoff value of 20% PI could clearly distinguish between the negative and positive M. gallisepticum sera samples. Hence, the cutoff value of percent inhibition was set at 20% (mean + 2SD)36 to determine the status of sera samples in response to M. gallisepticum antibodies (Fig. 4a). The specificity of ib-ELAA for the detection of M. gallisepticum antibodies was also evaluated by cross-reactivity with antisera of other related pathogens. The percent inhibition values of these sera were calculated using the same method as described above. As shown in Fig. 4b, all the antisera of the other related pathogens showed PI values lower than 20%, which were considered as negative. This suggested that our ib-ELAA had no cross-activity with other antisera of related pathogens.
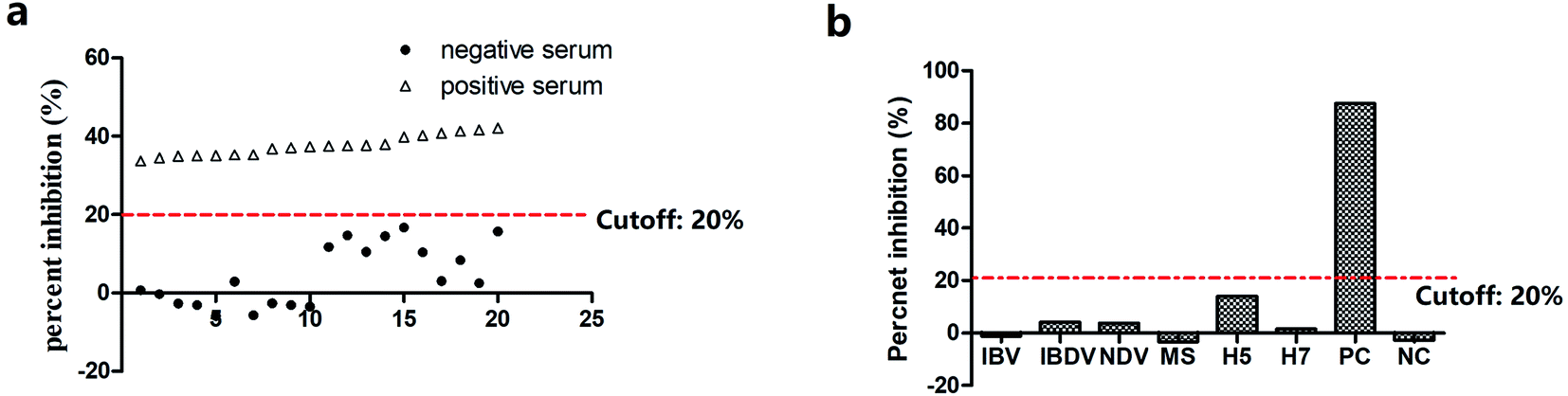 | ||
| Fig. 4 ib-ELAA for detection of antibodies against M. gallisepticum. (a) Determination of the cutoff value of percent inhibition. (b) Binding inhibition of aptamer Apt-236 by various positive sera against pathogens IBV, IBDV, NDV, M. synoviae, H5, H7. | ||
Detection of sera antibodies in chickens immunized with M. gallisepticum using ib-ELAA
To compare different assays for M. gallisepticum antibody detection, 33 sera samples from three chickens immunized with inactivated M. gallisepticum BJ44T strain were collected from 6–192 days. Four assays were used to detect M. gallisepticum antibodies, including ib-ELAA, ELISA, IDEXX, and HI, of which the cutoff was 20% (PI), 0.28 (OD450 nm), 0.5 (S/P), and 1:4 (HI titer), respectively. As shown in Fig. 5, the growth and decline of the specific antibodies in the immunized chicken determined by ib-ELAA were consistent with other three widely used assays. Approximately after 25 days of being immunized, antibodies could be detected by all four assays and lasted up to 192 days, with the antibody titer reaching the peak at 50–75 days.
 | ||
| Fig. 5 M. gallisepticum antibody detection of the sera samples from 3 immunized chickens. Sera samples were collected from 3 chickens at 6, 16, 24, 31, 46, 61, 79, 102, 132, 164, and 192 days after immunization. One point represents the average value of the three chickens. | ||
In addition, 230 clinical chicken sera samples randomly collected from five different provinces in China were also used to validate the effectiveness of ib-ELAA for the detection of M. gallisepticum antibodies. All the samples were detected by ib-ELAA and IDEXX i-ELISA in parallel and the results are listed in Table 4. The positive coincidence rate was 93.10% (135/145), and the negative coincidence rate is 89.41% (76/85), with a total coincidence rate of 91.74% (211/230). The kappa value was the intrinsic efficacy index among the reagents which was used to assess the degree of agreement between two tests; agreement between different strategies was interpreted as the kappa value, and the kappa value of more than 0.75 was considered as substantial agreement.37 In this study, the kappa value between ib-ELAA and the commercial ELISA assay (IDEXX i-ELISA) was 0.82 as analyzed using SPSS v19.0, indicating a high agreement.
| ib-ELAA results | IDEXX i-ELISA results | Total | |
|---|---|---|---|
| Positive | Negative | ||
| Positive | 135 | 9 | 144 |
| Negative | 10 | 76 | 86 |
| Total | 145 | 85 | 230 |
As reported by many researchers, competitive/blocking ELISAs have been confirmed to be significantly sensitive and specific, showing even a higher sensitivity than i-ELISA.38–42 Here Aptamer Apt-236, instead of monoclonal antibody, was used to develop a blocking format M. gallisepticum antibody detection assay, which is chemically, animal-free produced, can be easily modified without affecting their affinity, and is used and restored in a variety of conditions.30
Conclusion
An indirect blocking ELAA was successfully used for M. gallisepticum antibody detection, using a ssDNA aptamer Apt-236 that can specifically bind to PvpA protein of M. gallisepticum. The antibody results from hundreds of sera samples detected by this ib-ELAA are well consistent with ELISA, HI and the commercial ELISA assay indicating that ib-ELAA developed here was a useful method for the antibody surveillance of M. gallisepticum and helpful to gain a better understanding of the ecology and epidemiology of M. gallisepticum.Author contributions
Ping Fu and Feng Wang: investigation, formal analysis, and writing-original draft; Feng Wang and Yunke Zhang: formal analysis, and aptamer SELEX and ib-ELAA experiments; Xilan Qiao, Xinbo Yan: sample collection and data curation; Yuewei Zhang and Wenyan Zhou: writing-reviewing and editing; Wenxue Wu: supervision, funding acquisition, and project administration.All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the National Key R&D Program of China (2016YFD0500800).References
- O. Buyuktanir, T. Yildirim, C. Yakicier, O. Genc and N. Yurdusev, Vet. Microbiol., 2008, 129, 139–149 Search PubMed.
- K. M. Sulyok, Z. Kreizinger, K. Beko, B. Forro, S. Marton, K. Banyai, S. Catania, C. Ellis, J. Bradbury, O. M. Olaogun, A. B. Kovacs, T. Cserep and M. Gyuranecz, J. Clin. Microbiol., 2019, 57, e01084-18 Search PubMed.
- S. Sato, I. Nonomura, F. Shimizu, S. Shoya and T. Horiuchi, Natl. Inst. Anim. Health Q., 1970, 10, 58–65 Search PubMed.
- X. Xiao, D. H. Zhao, X. Yang, W. Shi, H. Deng, J. Ma, S. Zhang and Y. H. Liu, J. Vet. Pharmacol. Ther., 2014, 37, 99–102 Search PubMed.
- B. Yuan, M. Zou, Y. Zhao, K. Zhang, Y. Sun and X. Peng, Int. J. Mol. Sci., 2018, 19, 2172 Search PubMed.
- M. Z. Ali, M. M. Rahman and S. Sultana, Vet. World, 2015, 8, 9–14 Search PubMed.
- S. H. Kleven, C. J. Morrow and K. G. Whithear, Avian Dis., 1988, 32, 731–741 Search PubMed.
- S. Kahya, S. Temelli, A. Eyigor and K. T. Carli, Vet. Microbiol., 2010, 144, 319–324 Search PubMed.
- M. Garcia, M. W. Jackwood, S. Levisohn and S. H. Kleven, Avian Dis., 1995, 39, 606–616 Search PubMed.
- X. Lin, S. Li, B. Zhang, H. Yang, K. Zhang and H. Huang, Anal. Methods, 2020, 12, 5496–5502 Search PubMed.
- L. S. Rotherham, C. Maserumule, K. Dheda, J. Theron and M. Khati, PLoS One, 2012, 7, e46862 Search PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 Search PubMed.
- D. W. Drolet, L. Moon-McDermott and T. S. Romig, Nat. Biotechnol., 1996, 14, 1021–1025 Search PubMed.
- P. Fu, Z. Sun, Z. Yu, Y. Zhang, J. Shen, H. Zhang, W. Xu, F. Jiang, H. Chen and W. Wu, Anal. Chem., 2014, 86, 1701–1709 Search PubMed.
- S. Y. Toh, M. Citartan, S. C. B. Gopinath and T. H. Tang, Biosens. Bioelectron., 2015, 64, 392–403 Search PubMed.
- D. Kamashev, J. Oberto, M. Serebryakova, A. Gorbachev, Y. Zhukova, S. Levitskii, A. K. Mazur and V. Govorun, Biochemistry, 2011, 50, 8692–8702 Search PubMed.
- P. Fu, Z. Sun, Y. Zhang, Z. Yu, H. Zhang, D. Su, F. Jiang and W. Wu, BMC Vet. Res., 2014, 10, 42 Search PubMed.
- J. Xu, D. Teng, F. Jiang, Y. Zhang, S. A. El-Ashram, H. Wang, Z. Sun, J. He, J. Shen, W. Wu and J. Li, Appl. Microbiol. Biotechnol., 2015, 99, 1859–1871 Search PubMed.
- J. L. Yunlei Zhou, F. Wei, X. Zhang and H. Jiang, Chin. J. Prev. Vet. Med., 2011, 33, 713–717 Search PubMed.
- S. Boguslavsky, D. Menaker, I. Lysnyansky, T. Liu, S. Levisohn, R. Rosengarten, M. Garcia and D. Yogev, Infect. Immun., 2000, 68, 3956–3964 Search PubMed.
- D. Yogev, D. Menaker, K. Strutzberg, S. Levisohn, H. Kirchhoff, K. H. Hinz and R. Rosengarten, Infect. Immun., 1994, 62, 4962–4968 Search PubMed.
- A. Rhodes, A. Deakin, J. Spaull, B. Coomber, A. Aitken, P. Life and S. Rees, J. Biol. Chem., 2000, 275, 28555–28561 Search PubMed.
- C. Marimuthu, T. H. Tang, J. Tominaga, S. C. Tan and S. C. Gopinath, Analyst, 2012, 137, 1307–1315 Search PubMed.
- N. Kamatkar, M. Levy and J. M. Hebert, Mol Ther Nucleic Acids, 2019, 17, 530–539 Search PubMed.
- J. Wang, Q. Wang, Y. Luo, T. Gao, Y. Zhao and R. Pei, Talanta, 2019, 204, 424–430 Search PubMed.
- K. Pleiko, L. Saulite, V. Parfejevs, K. Miculis, E. Vjaters and U. Riekstina, Sci. Rep., 2019, 9, 8142 Search PubMed.
- M. Berezovski, R. Nutiu, Y. Li and S. N. Krylov, Anal. Chem., 2003, 75, 1382–1386 Search PubMed.
- M. Zuker, Nucleic Acids Res., 2003, 31, 3406–3415 Search PubMed.
- J. G. Bruno, M. P. Carrillo, A. M. Richarte, T. Phillips, C. Andrews and J. S. Lee, BMC Res. Notes, 2012, 5, 633 Search PubMed.
- E. Baldrich, J. L. Acero, G. Reekmans, W. Laureyn and C. K. O'Sullivan, Anal. Chem., 2005, 77, 4774–4784 Search PubMed.
- R. Stoltenburg, P. Krafcikova, V. Viglasky and B. Strehlitz, Sci. Rep., 2016, 6, 33812 Search PubMed.
- S. Y. Toh, M. Citartan, S. C. Gopinath and T. H. Tang, Biosens. Bioelectron., 2015, 64, 392–403 Search PubMed.
- H. Park and I. R. Paeng, Anal. Chim. Acta, 2011, 685, 65–73 Search PubMed.
- S. Wang, J. Liu, W. Yong, Q. Chen, L. Zhang, Y. Dong, H. Su and T. Tan, Talanta, 2015, 131, 562–569 Search PubMed.
- S. Brehm, S. Hoch and J. Hoch, Biochem. Biophys. Res. Commun., 1975, 63, 24–31 Search PubMed.
- K. A. Webster, M. Giles and C. Dawson, Vet. Parasitol., 1997, 68, 155–164 Search PubMed.
- S. V. Khadse, G. Bajaj, P. Vibhakar, P. Nainani, R. Ahuja and G. Deep, J. Clin. Diagn. Res., 2016, 10, BC12–14 Search PubMed.
- N. H. Ferrin, Y. Fang, C. R. Johnson, M. P. Murtaugh, D. D. Polson, M. Torremorell, M. L. Gramer and E. A. Nelson, Clin. Diagn. Lab. Immunol., 2004, 11, 503–514 Search PubMed.
- A. Ghadersohi, Z. Fayazi and R. G. Hirst, Vet. Immunol. Immunopathol., 2005, 104, 183–193 Search PubMed.
- J. Hirota and S. Shimizu, J. Virol. Methods, 2013, 188, 132–138 Search PubMed.
- J. J. Reddington, G. M. Reddington and N. J. MacLachlan, J. Vet. Diagn. Invest., 1991, 3, 144–147 Search PubMed.
- N. Sharma, A. Hotta, Y. Yamamoto, O. Fujita, A. Uda, S. Morikawa, A. Yamada and K. Tanabayashi, Clin. Vaccine Immunol., 2013, 20, 9–16 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1, top 400 sequences of the second and fifth round by high-throughput sequencing. Fig. S1, binding inhibition of aptamer Apt-236 in an indirect ELAA format by various positive sera against pathogens IBV, IBDV, NDV, M. synoviae, H5, H7, H9, and five negative sera against M. gallisepticum (N1–N5). See DOI: 10.1039/d1ay00515d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |