
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct analysis of aromatic pollutants using a HPLC-FLD/DAD method for monitoring biodegradation processes†
Dilan Camille
Aydin
 *,
Julian
Zamudio Pineres
,
Fatma
Al-Manji
,
Huub
Rijnaarts
and
Tim
Grotenhuis
*,
Julian
Zamudio Pineres
,
Fatma
Al-Manji
,
Huub
Rijnaarts
and
Tim
Grotenhuis
Wageningen University & Research, P.O. Box 17, 6700 AA Wageningen, The Netherlands. E-mail: dilan.aydin@wur.nl
First published on 26th February 2021
Abstract
Industrial discharges resulting in contaminated groundwater is a global environmental problem. For such contaminated groundwater cases, bioremediation is a cost efficient and environmentally friendly approach. The determination and quantification of these pollutants has gained great importance and researchers are currently seeking to develop labor extensive, accurate and reliable methods for evaluating their biodegradation process. In this study, a HPLC method was developed and optimized for the quantification of 11 industrial pollutants studied as two different mixtures: benzene, toluene, ethylbenzene, o, m/p-xylene, indane, indene, and naphthalene (mixture A) and benzene, monochlorobenzene, 1,2-dichlorobenzene, and 1,4-dichlorobenzene (mixture B). The method uses two different detectors: fluorescence detection and diode array. The fluorescence detector was used for mixture A to achieve lower quantification limits and to quantify separately o-xylene and indene due to them showing similar wavelength behaviors. The limit of detection was found to be between 2 and 70 μg L−1 for mixture A and 290 μg L−1 for mixture B. The limit of quantitation was between 6 and 210 μg L−1 for mixture A and 980 μg L−1 for mixture B, respectively. The novel part of this study is that aqueous samples can be directly measured with one-step sample preparation and it comes with other advantages such as low volumes of sampling from batch bottles and also avoidance of high cost, relative to other analytical techniques. Therefore, this analytical method aims to facilitate the quantification of various aromatic hydrocarbons in laboratory batch samples and can be used as a routine monitoring tool for biological degradation processes of these 11 prevalent contaminants.
1 Introduction
Industrial wastes and accidental spills resulting in soil and groundwater contamination are becoming a global concern due to the toxicity and recalcitrance of many aromatic compounds.1 Common pollutants found in industrial or manufactured gas work sites are benzene, toluene, ethylbenzene, xylene (BTEX), polyaromatic hydrocarbons (PAHs) and various chlorinated aromatic hydrocarbons.2,3 These compounds are specified as hazardous, mostly classified as carcinogenic, and pose risks to human health and the environment.1,3 Therefore, current research efforts are focused on the application of efficient remediation strategies for these compounds.4 Among all remediation technologies available, biodegradation is the preferred approach due to being economical, energy efficient and environmental friendly.5 However, despite the wide range of biodegradation research studies reported, one of the main challenges is the lack of an adequate method for the analytical quantification of these pollutants. Developing a suitable analytical method is a prerequisite in order to obtain reliable and accurate data during measurements.6 Detailed information about different methods used for aromatic hydrocarbon determination and quantification was reviewed by Farhadian et al.5 and AlSalka et al.7 Each one of the discussed methods has its advantages and drawbacks, but the main focus on method selection should be based on the aim of the experiment and also other factors such as sample preparation, cost and the analysis time.8 In aromatic hydrocarbon bioremediation research, one needs a single method that is comprehensive for all relevant compounds present in the pollution mixture often encountered in remediation projects. The method is required to be accurate, fast, cost-efficient8 and thereby budget attractive and easy to handle in the laboratory and in situ pilot bioremediation technology development. For the measurements of monoaromatic compounds, a gas chromatography flame ionization detector (GC-FID), a GC-photo ionization detector (PID), GC-mass spectrometry (MS)9 and a GC-barrier ionization discharge detector (BID)10 are employed which are useful for detection in a low concentration range which is important to assess compliance with environmental thresholds (often in the μg L−1 range). Additionally, the GC technique and head-space (HS) sampling have been adopted by the Environmental Protection Agency (EPA) in many protocols. However these techniques come with disadvantages such as incompatibility with direct analyses in water samples and time consuming preparation steps (such as head-space extraction, purge and/or solid-phase microextraction) prior to analysis which may also require high cost equipment,11 and laborious methods to identify intermediate products formed during bioconversion.12 This renders these techniques less suitable to support lab and field technology development. The limitations of these GC-based methods can be overcome by using high performance liquid chromatography (HPLC), since it is more suited for direct aqueous sample analysis and can measure higher and wider ranges of concentrations for wide sets of aromatic pollutants, minimizing dilution or other pre-treatment steps.12In this study, such a HPLC based method was developed with focus on the quantification of two different mixtures of compounds, namely one composed of benzene, toluene, ethylbenzene, o, m/p-xylene, indene, indane, and naphthalene (which will be referred to as mixture A), and one with benzene, monochlorobenzene, 1,2-dichlorobenzene and 1,4-dichlorobenzene (referred to as mixture B) (Fig. 1). These contaminants were selected in view of their prominent presence in a former gas work site and a chemical industrial area, respectively. The method aimed for a fast and reliable quantification of all compounds listed in Fig. 1, by avoiding complex sample preparation steps. Additionally, this method was developed in order to facilitate the work of daily basis biodegradation experiments, where reliable results can be obtained for aqueous samples with low sampling volumes and by avoiding high use of organic solvents in the monitoring of biodegradation processes.
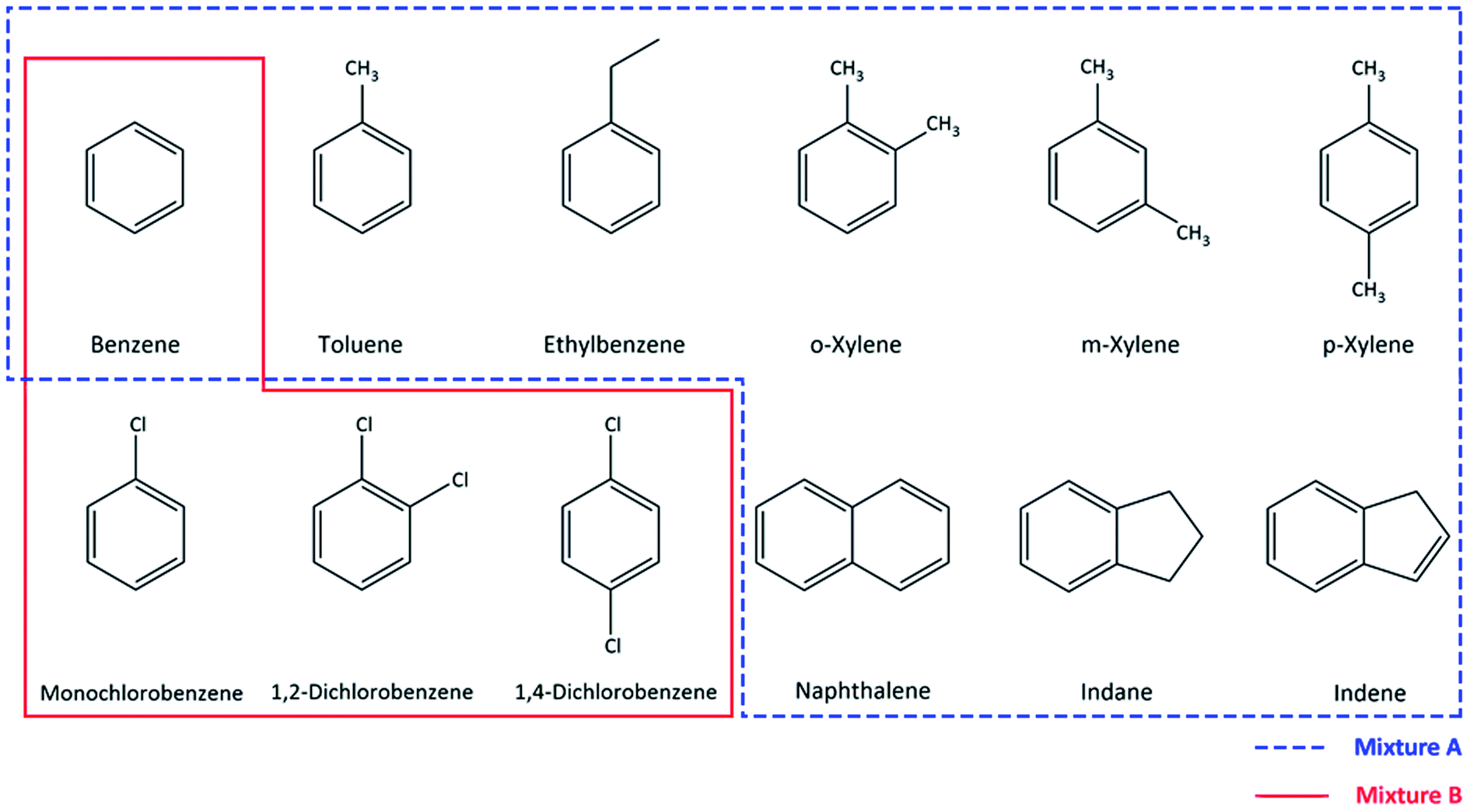 | ||
| Fig. 1 Chemical structure of the compounds used in this study. Mixture A (dashed line): benzene, toluene, ethylbenzene, o/m/p-xylene, naphthalene, indene and indane; mixture B (line): benzene, monochlorobenzene, 1,2-dichlorobenzene, and 1,4-dichlorobenzene. | ||
2 Experimental
The proposed method was developed and optimized by the use of HPLC with a fluorescence detector (FLD) and diode array detector (DAD) validated in terms of specificity, matrix effects, linearity, precision, recovery and limits of detection (LOD) and quantitation (LOQ).2.1 Chemicals
Chemicals used for standard solution and sample preparation are given in Table 1. ULC/MS grade absolute methanol was purchased from Biosolve (Netherlands).| Chemicals | Purity (%) | Supplier |
|---|---|---|
| Benzene | >99.7 | VWR Chemicals (USA) |
| Toluene | ≥99.9 | Merck KGaA (Germany) |
| Ethylbenzene | 99 | Alfa Aesar (Germany) |
| o-Xylene | 99 | Alfa Aesar (Germany) |
| m-Xylene | 99 | Alfa Aesar (Germany) |
| p-Xylene | 99 | Acros Organics (Czechia) |
| Indane | 95 | Alfa Aesar (Germany) |
| Indene | ≥99 | Sigma-Aldrich (Germany) |
| Naphthalene | 99 | Sigma-Aldrich (Germany) |
| Monochlorobenzene | 99 | VWR Chemicals (France) |
| 1,2-Dichlorobenzene | 99 | Acros Organics (Czechia) |
| 1,4-Dichlorobenzene | 99 | Alfa Aesar (Germany) |
2.2 Instrumentation
HPLC analyses were performed using a Thermo HPLC with an Ultimate 3000 RS Diode Array detector and Ultimate 3000 RS fluorescence detector (Thermo Scientific Dionex, USA). For the separation of each analyte, reversed phase chromatography was performed by using an Acclaim™ Phenyl-1 HPLC Column 150 × 4.6 mm, 3 μm (Thermo Scientific Dionex, USA). The mobile phase is an isocratic mixture of 60% methanol and 40% Milli-Q water.2.3 Standard calibration solutions
Stock solutions for mixture A and B were prepared at concentrations of 1 g L−1 and 10 g L−1 in 100 mL volumetric flasks, respectively. Each analyte was dissolved individually in methanol and was stored at 4 °C. Fresh working solutions were prepared daily by diluting the stock solutions to reach the desired concentration values in different matrices (Milli-Q water and groundwater for mixture A; Milli-Q water and medium for mixture B).2.4 Sample preservation
As the aromatics are volatile in water, tests demonstrated that all samples need to contain methanol as a co-solvent in order to limit evaporation losses during HPLC autosampler procedures. Samples prepared only in Milli-Q water showed incoherent concentrations while with methanol, the concentrations of the compounds were found to stay stable up to 12 hours (Table S1†). Further, methanol addition led to higher resolution and efficiency in chromatographic results. Therefore, methanol was added to standard solutions and to the samples before auto-sampled injection to the HPLC.The lowest methanol-to-sample ratio was determined for good reproducibility. In order to find optimal separation conditions and good stability in concentrations; different methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :sample ratios (0:100, 10:90, 25:75, 50:50 v/v) were analysed using mixture B (compounds in mixture B cover the same range of volatilization properties as for compounds in mixture A). For the analytical chromatographic method development, 25% of methanol was used in all samples unless otherwise specified.
:sample ratios (0:100, 10:90, 25:75, 50:50 v/v) were analysed using mixture B (compounds in mixture B cover the same range of volatilization properties as for compounds in mixture A). For the analytical chromatographic method development, 25% of methanol was used in all samples unless otherwise specified.
2.5 Method optimization
The operational parameters such as the sample injection volume, flow rate, mobile phase composition and column temperature were optimized.The injection volume varied between 10 and 80 μL and two different flow rates of 0.7 and 1.2 mL min−1 were tested as described by Taleuzzaman et al.13 Methanol was selected as an organic modifier in the mobile phase using the method developed by Amy Tan et al.12 and in consequence several methanol:water ratios were tested as eluents in HPLC. The column temperature was varied between 25 and 35 °C. All compounds of mixture A were measured with the HPLC-FLD, while analytes from mixture B were determined with the HPLC-DAD. The UV and excitation/emission wavelengths for each compound together with their retention times are presented in Table 2, and specific conditions of each detector are given in Table S2.† The run time is 27 min for mixture A and 24 min for mixture B.
| Compound | Detector: FLD | Detector: DAD | ||
|---|---|---|---|---|
| FLD (Exc/Em) | Retention time (min) | DAD (nm) | Retention time (min) | |
| a DAD; diode-array detector, FLD; fluorescence detector, Exc/Em; excitation/emission. | ||||
| Benzene | 210/260 | 9.0 | 210 | 7.76 |
| Toluene | 260/310 | 13.84 | ||
| Ethylbenzene | 210/260 | 16.30 | ||
| Indene | 280/330 | 17.10 | ||
| o-Xylene | 210/260 | 17.40 | ||
| m,p-Xylene | 260/310 | 18.10 | ||
| Indane | 260/310 | 22.10 | ||
| Naphthalene | 260/310 | 23.70 | ||
| Monochlorobenzene | 210 | 12.48 | ||
| 1,2-Dichlorobenzene | 210 | 19.83 | ||
| 1,4-Dichlorobenzene | 210 | 20.65 | ||
2.6 Specificity
Each compound was first measured individually to evaluate any possible interfering peaks when samples are prepared as mixtures. A fluorescence spectrum was generated by defining the spectral excitation and emission wavelength and the retention time for each compound was recorded. Samples prepared as in mixtures were run and peaks for each compound were compared with the spectra of the individual compound samples.Standard solutions for each compound individually at 1 g L−1 were prepared in methanol as the initial solution and measured at optimized parameters presented in Table 2. Blank samples and individual compounds were analysed in order to obtain a full UV spectrum between 210 and 340 nm. Then, mixture solutions of A and B (1 g L−1 and 10 g L−1, respectively) dissolved in methanol were tested and compared within the individual compound's results to confirm specificity.
2.7 Method development
This method was assessed in terms of matrix effects, linearity, intermediate precision, recovery and detection and quantitation limits as described by AlSalka et al.7 and Filho et al.14:given matrix (25:75 v/v) in order to reach final concentrations of 0.5, 1, 2.5, 5, 7.5 and 10 mg L−1.
For mixture B, the matrix effect was tested in samples with Milli-Q water and the medium as prepared according to Lindeboom et al.15 The same procedure as in mixture A was applied, where 10 g L−1 of mixture B stock solution was dissolved in 50 mL of methanol:given matrix (25:75 v/v) at five different concentrations (1, 2.5, 5, 7.5 and 12 mg L−1). Each peak for each compound was investigated for spectral information such as the retention time, peak height and area. The pair t-test was performed and p-values <0.05 were determined as an indicator of matrix effects.
| Intermediate precision (RSD%) = (SD/Cm) × 100 |
Recovery experiments were performed at 10 mg L−1 concentration for both mixtures A and B, in duplicate. The average percent recovery was calculated with the below equation:14
| Recovery (%) = (Cobs/Cr) × 100 |
SD is the standard deviation, Cm is the mean value for the replicates, Cobs refers to the concentration observed in the samples and Cr is the initial spiked concentration of the reference sample.
| LOD = 3 × (SD/S) |
| LOQ = 10 × (SD/S) |
3 Results and discussion
3.1 Sample preservation
Different methanol:sample ratios were tested with compounds of mixture B as shown in Table 3. As mentioned before, methanol was added to the samples in order to avoid evaporation of volatiles during HPLC autosampler procedures. According to Eurochem Guidelines,16 the acceptance criteria for a relative standard deviation correspond to 10% RSD response. Therefore, a ratio of 25% methanol was selected as the optimum due to the reason of showing minimum % RSD response values that are acceptable for all compounds. Additionally, 50% methanol was preferred due to the reasons of showing good reproducibility with the lowest LOD and LOQ possible for all the compounds, and also having lower methanol–sample ratios. The same methanol:sample ratio was applied to mixture A, since compounds in mixture A show a similar structure and similar physical/chemical properties to the compounds in mixture B.
| Methanol (%) | RSD (%) | |||
|---|---|---|---|---|
| Benzene | MCB | 1,2-DCB | 1,4-DCB | |
| a RSD; relative standard deviation, MCB; monochlorobenzene, DCB; dichlorobenzene. | ||||
| 0 | 12 | 3.0 | 4.9 | 16.5 |
| 10 | 5.9 | 5.7 | 10.5 | 17.0 |
| 25 | 6.5 | 3.7 | 5.2 | 7.2 |
| 50 | 1.7 | 5.3 | 2.1 | 8.9 |
3.2 Optimization
Optimal chromatographic conditions determined for this method are summarized in Table 4. Optimum conditions were determined according to better peak resolutions and smaller standard deviation outcomes.| Parameters | Mixture | |
|---|---|---|
| A | B | |
| Volume | 80 μL | 50 μL |
| Flow rate | 0.7 mL min−1 | 0.7 mL min−1 |
| Mobile phase (isocratic mode) | 60:40 (methanol:water) % |
60:40 (methanol:water) % |
| Column temperature | 35 °C | 35 °C |
| Run time | 27 min | 24 min |
The optimal range of injection volumes was determined to be 80 μL for mixture A and 50 μL for mixture B. Amy Tan et al.12 reported that high injection volumes may lead to the presence of some residual peaks at the end of the analysis. In our study, where the maximum injection volume was 80 μL, such a phenomenon was not encountered.
To determine the optimum conditions for compound separation, different flow rates were tested upon their effect on the duration of the analysis and the chromatographic resolution.12 For this study, 0.7 mL min−1 and 1.2 mL min−1 were tested. 0.7 mL min−1 showed higher efficiency and better analytical resolution.
3.3 Specificity
To evaluate any possible interfering peaks between compounds, samples prepared as individual compounds were compared to samples prepared as mixtures under optimal conditions given in Table 4. For mixture B, overlapping peaks were not recorded and all compounds were well separated with the use of a DAD (Fig. 2). For mixture A, all compounds could be quantified without interferences by the use of the FLD and the fluorescence properties of the compounds. Originally, o-xylene was detected with an excitation and emission wavelength of 210/260 nm with overlapping peaks between indene and o-xylene (Fig. 3a). Changing indene detection to 280/330 nm (Fig. 3b), differentiation and quantification of both compounds became possible.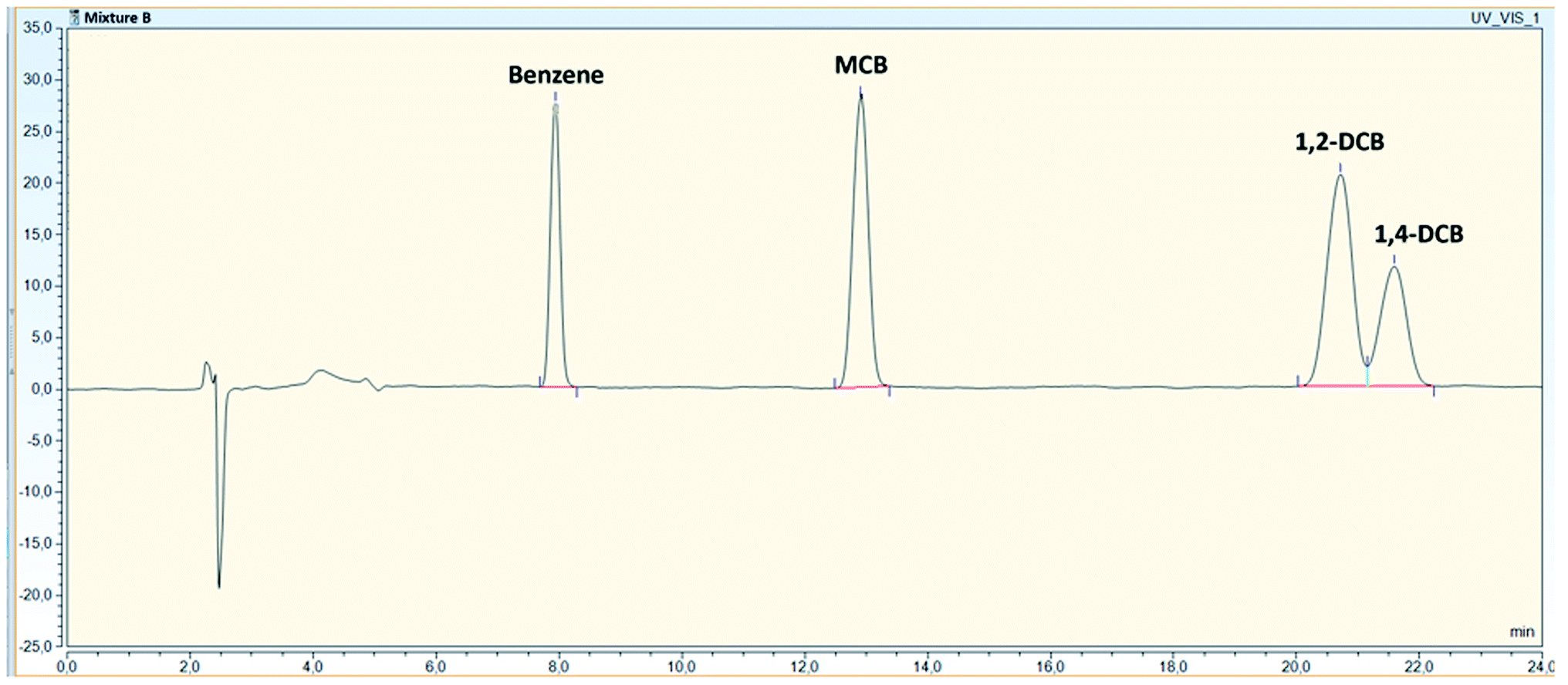 | ||
| Fig. 2 Chromatogram of all compounds in mixture B (12 mg L−1), identified with a HPLC-DAD at 210 nm. | ||
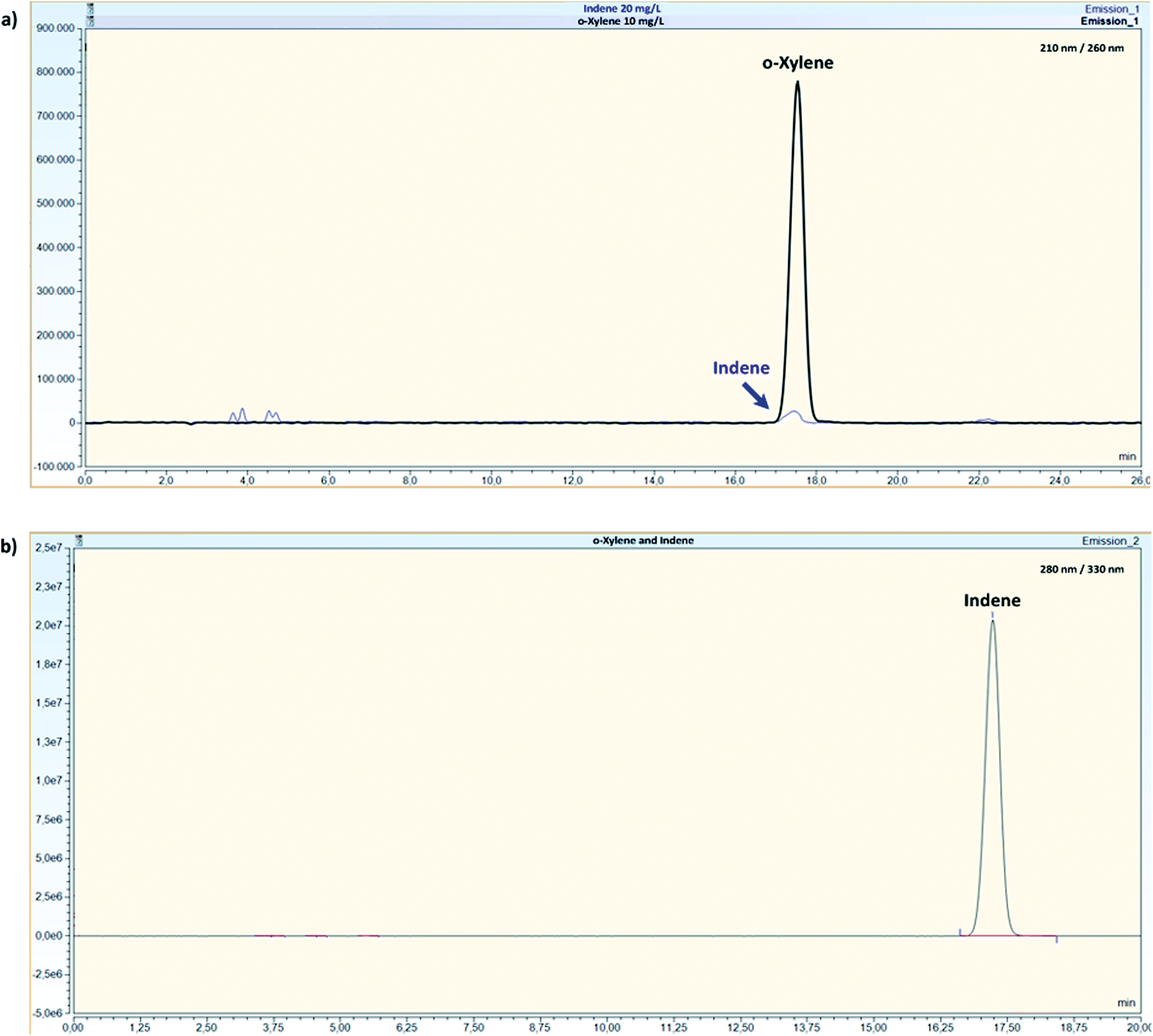 | ||
| Fig. 3 Chromatogram of o-xylene (10 mg L−1) detected at 210/260 nm. An interference with indene (20 mg L−1) can be seen as a small peak (a) and chromatogram of only indene at 280/330 nm (b) by the use of a HPLC-FLD. | ||
3.4 Method development
:groundwater showed RSD values between 0.25 and 14.39%. RSD values varied between 0.68 and 4.01% for compounds of mixture B, prepared in methanol:medium at different concentrations (Table S8†). The results were convenient because RSD values for all the compounds were below 20%, indicating a high level of precision as stated by Filho et al.14 and also meeting the US EPA quality control criteria.7 Recovery values for both mixtures were found to be between 82 and 110% as given in Table S9 and S10.† The obtained values were found to be within the acceptable recovery limits determined to be 80–120% according to US EPA quality control criteria.7
| Compound | LOD (μg L−1) | LOQ (μg L−1) |
|---|---|---|
| a LOD; limit of detection, LOQ; limit of quantitation. | ||
| Mixture A | ||
| Benzene | 70 | 210 |
| Toluene | 23 | 70 |
| Ethylbenzene | 36 | 110 |
| o-Xylene | 36 | 110 |
| p/m-Xylene | 5 | 15 |
| Indane | 12 | 36 |
| Indene | 2 | 6 |
| Naphthalene | 2 | 6 |
|
||
| Mixture B | ||
| Benzene | 290 | 980 |
| Monochlorobenzene | 290 | 980 |
| 1,2-Dichlorobenzene | 290 | 980 |
| 1,4-Dichlorobenzene | 290 | 980 |
The LOQ value was found to range between 6 μg L−1 (indene and naphthalene) and 210 μg L−1 (benzene) for mixture A and was 980 μg L−1 for all compounds in mixture B. These results were found to be convenient for researchers performing lab scale biodegradation monitoring experiments. When initial concentrations of 10 mg L−1 are applied, the proposed method shows a degradation of 90% (mixture B) to over 98% (mixture A) conversion of the studied compounds.
4 Discussion
4.1 Use of a phenyl column and FLD detector
This method was developed by following the study of Amy Tan et al.12 where they investigated BTEX and styrene with a HPLC-DAD with the use of a phenyl-1 column. According to the manufacturer's information, the selected column has an unique selectivity of aromatic compounds and higher chromatographic performance. The bonded phase displays greater π–π interactions than other phenyl phases and provides enhanced aromatic selectivity and higher hydrophobic retention. Therefore, we used this column as recommended by Amy Tan et al.12 contrary to the common use of C18 columns selected for highly volatile compounds. Unfortunately, the use of phenyl columns was not sufficient for the detection and quantification of all compounds in mixture A in one single run. This issue was solved by supplementing the method of Amy Tan et al.12 with the use of a FLD detector. This way, compounds showing peaks at similar retention times such as o-xylene and indene could be differentiated with the use of a fluorescence detector in different excitation/emission ranges. By the use of a HPLC-DAD method; benzene, monochlorobenzene, 1,2-dichlorobenzene and 1,4-dichlorobenzene were quantified in a single run. Using the same method with a FLD, BTEX, indane, indene and naphthalene could also be quantified in a single run.4.2 Sample preparation steps and low LOD concentrations
Farhadian et al.5 reported that HPLC is often not suitable for accurate separation of ethylbenzene from xylene isomers; however, in the study by AlSalka et al.7 they were able to quantify BTEX from environmental samples with the use of HPLC-DAD without any interference between the compounds of interest. Only, in the study of AlSalka et al., an additional step prior to injection to HPLC is needed where BTEX compounds are transferred from water to an organic solvent (acetonitrile) with the help of N2 gas. In that step, an amount of 75 mL of sample is needed for BTEX to be transferred to acetonitrile. Since they are dealing with environmental samples, this amount may not be considered as high. Solely, in laboratory spiked samples, minimizing the sample volume is extremely important especially if you need frequent sampling. This is why, this study focused on developing a method without a time-consuming preparation step where high sample volumes might be wasted. Keeping this in mind, the sample preparation step developed by AlSalka et al. allows reaching low LOD values that are crucial for environmental sampling, i.e. showing compliance to or exceeding legislative thresholds that are between 0.1 and 50 μg L−1 (BTEX).17Pascale et al.10 published a quantitative analytical GC-BID method for monitoring BTEX in low contaminated water samples and were able to determine BTEX in concentrations below legal limits (μg L−1). The disadvantage of this method is that again, high sample volumes are needed (15 mL) and it is only applicable for quantification at very low BTEX concentrations. When high concentrations are applied (higher than trace BTEX), chromatographic peaks of all analytes could interfere with the adjacent peaks or with the baseline. Their method can be a good option for quality control purposes but not ideal for laboratory spiked samples where high concentrations are studied and routine monitoring/sampling is needed.
4.3 Quantification of indane and indene
This study reports for the first time the direct quantification of indane and indene in water samples using HPLC even though these compounds are commonly found at high concentrations in groundwater of many industrial sites but are often not further studied for analytical or remediation purposes. Mundt and Hollender18 inspected indane and indene in groundwater samples and quantified these with the use of LC-DAD UV/-FLD/-MS but in their study, samples needed to be treated with solid phase extraction (SPE) materials prior to analysis. These authors also tested spiked non-contaminated groundwater samples, and found low LOD values, namely 0.4 μg L−1 for indene and 1.6 μg L−1 for indane, but recovery rates were found to be below 25%. In the study reported here, only one-step preparation (methanol addition) is needed before injection to HPLC, and recovery rates were between 82 and 110%, in the range of acceptable limits for recovery according to US EPA methods.4.4 HPLC method to facilitate routine lab-scale biodegradation experiments
Taking all results into account, the HPLC method described above is reliable, practical and fits for analysis of the contaminants commonly found in manufactured gas plant sites and chemical industrial areas. The tested method proved to be specific as all compounds could be identified and quantified at high accuracy applied in biological degradation experiments often in the range of 0.1 to 10 mg L−1. Benzene had a somewhat higher LOQ value of 210 μg L−1, which wasn't hampering the experiment for this study. Further, no laborious pre-treatment of samples is needed and sample volumes required for quantification is rather minimal (>375 μL). Proposed injection volumes within the method will allow a serial sampling of experimental batch bottles without significant changes of the total liquid volume in such batch experiments. Therefore, this method can facilitate the work of biodegradation researchers, as reliable results can be obtained with low sampling volumes and it is a relatively simple analytical method avoiding the use of organic solvents or complex extraction methods in the monitoring of in situ or laboratory based biodegradation processes.5 Conclusions
A single step analysis method for the quantification of 11 different (aromatic and chlorinated) compounds in aqueous samples was developed and validated by the use of a HPLC-FLD/DAD method. The optimum conditions for HPLC operation were determined to be a mobile phase of 60% methanol, with 50 μL (mixture B) and 80 μL (mixture A) injection volume, at a flow rate of 0.7 mL min−1 and a detection of 210 nm wavelength and the use of a FLD for the separation of indene and xylene isomers at different excitation and emission wavelength pairs. Under determined optimum conditions, chlorinated and non-chlorinated aromatic compounds could be quantified within 24 and 27 min, respectively. The method was also tested and confirmed in terms of intermediate precision and recovery, showing acceptable results fitting the quality control criteria of US EPA methods.To the best of our knowledge, this is the first time that indane and indene, together with BTEX and naphthalene were measured in aqueous samples with an HPLC method. Additionally, LOD and LOQ values were defined for all the compounds, within the detection range of 0.1 to 10 mg L−1, suitable for investigation of lab-scale biodegradation processes. The method proved to be simple and easy to use for daily basis biodegradation measurements.
Author contributions
Dilan C. Aydin: methodology, formal analysis, and writing-original draft. Julian Zamudio Pineres: formal analysis, methodology, and writing – review & editing. Fatma Al-Manji: formal analysis, methodology, and writing – review & editing. Huub Rijnaarts: supervision, and writing – review & editing. Tim Grotenhuis: supervision, and writing-review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful for the financing of this study by the Municipality of Utrecht, the Netherlands. This study is part of the project BestParc Utrecht, the Netherlands with the identification mark 4281188/170821/1002-gl.References
- P. S. Birak and C. T. Miller, J. Contam. Hydrol., 2009, 105, 81–98 CrossRef CAS PubMed.
- M. Mundt, K. Althoff, W. Dott and J. Hollender, Acta Hydrochim. Hydrobiol., 2003, 31, 204–212 CrossRef CAS.
- P. Jandera, J. Fischer and B. Prokes, Chromatographia, 2001, 54, 581–587 CrossRef CAS.
- R. Chakraborty and J. D. Coates, Appl. Microbiol. Biotechnol., 2004, 64, 437–446 CrossRef CAS PubMed.
- M. Farhadian, D. Duchez, C. Vachelard and C. Larroche, Water Res., 2008, 42, 1325–1341 CrossRef CAS.
- B. V. Kumar, V. K. Gaur, R. Kumar, S. Sharma and C. S. Akolkar, Adv. Appl. Sci. Res., 2014, 5, 201–209 Search PubMed.
- Y. AlSalka, F. Karabet and S. Hashem, Anal. Methods, 2010, 2, 1026–1035 RSC.
- T. M. S. Yamada, A. Dalva, C. R. Morais and A. A. Mozeto, J. Chromatogr. Sci., 2009, 47, 794–799 CAS.
- Y. Huang, J. Wei, J. Song, M. Chen and Y. Luo, Chemosphere, 2013, 92, 1010–1016 CrossRef CAS PubMed.
- R. Pascale, G. Bianco, S. Calace, S. Masi, I. M. Mancini, G. Mazzone and D. Caniani, J. Chromatogr. A, 2018, 1548, 10–18 CrossRef CAS PubMed.
- A. Sarafraz-Yazdi, A. H. Amiri and Z. Es'haghi, Chemosphere, 2008, 71, 671–676 CrossRef CAS PubMed.
- G.-Y. Amy Tan, C.-L. Chen, L. Zhao, Y. Mo, V. W. C. Chang and J.-Y. Wang, Anal. Methods, 2012, 4, 3545–3550 RSC.
- M. Taleuzzaman, S. Ali, S. J. Gilani, S. S. Imam and A. Hafeez, Austin J. Anal. Pharm. Chem., 2015, 2(6), 1056 Search PubMed.
- C. M. C. Filho, M. N. L. Neto, R. S. Teixeira, A. A. C. C. Pais and A. J. M. Valente, J. Liq. Chromatogr. Relat. Technol., 2017, 39, 837–846 CrossRef.
- R. E. Lindeboom, F. G. Fermoso, J. Weijma, K. Zagt and J. B. van Lier, Water Sci. Technol., 2011, 64, 647–653 CrossRef CAS PubMed.
- Eurachem Guide: the Fitness for Purpose of Analytical Methods – A Laboratory Guide to Method Validation and Related Topics, ed. B. Magnusson and U. Örnemark, 2nd edn 2014, ISBN 978-91-87461-59-0 Search PubMed.
- R. Sethi and A. Di Molfetta, in Groundwater Engineering: A Technical Approach to Hydrogeology, Contaminant Transport and Groundwater Remediation, Springer International Publishing, Cham, 2019, pp. 169–192 Search PubMed.
- M. Mundt and J. Hollender, J. Chromatogr. A, 2005, 1065, 211–218 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ay00083g |
| This journal is © The Royal Society of Chemistry 2021 |