
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron oxide xerogels for improved water quality monitoring of arsenic(III) in resource-limited environments via solid-phase extraction, preservation, storage, transportation, and analysis of trace contaminants (SEPSTAT)†
Michael S.
Bono
Jr.
 *abc,
Emily B.
Hanhauser
ab,
Chintan
Vaishnav
bd,
A. John
Hart
a and
Rohit
Karnik
*a
*abc,
Emily B.
Hanhauser
ab,
Chintan
Vaishnav
bd,
A. John
Hart
a and
Rohit
Karnik
*a
aDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: msb294@cornell.edu; karnik@mit.edu
bTata Center for Technology and Design, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cDepartment of Biomedical Engineering, Boston University, Boston, MA 02215, USA
dSloan School of Management, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
First published on 20th April 2021
Abstract
Arsenic is a widespread trace groundwater contaminant that presents a range of health risks and has an acceptable level of only 10 μg L−1 in drinking water. However, in many countries arsenic quantification in water is limited to centralized laboratories because it requires the use of elemental analysis techniques with high capital cost. As a result, routine water samples are frequently not tested for trace contaminants such as arsenic. In order to facilitate improved arsenic monitoring, we present the use of iron oxide xerogels for adsorption of arsenic(III) from water samples at neutral pH, dry storage for over 120 days, and desorption of stored arsenic at elevated pH. Iron oxide xerogels offer high surface area (340 m2 g−1) and an As(III) adsorption capacity of 165 mg g−1. Using an extraction solution of 100 mM sodium hydroxide and 1 mM sodium phosphate, As(III) is reliably eluted from iron oxide xerogels for initial As(III) concentrations from 10 μg L−1 to 1000 μg L−1, with a calculated detection limit of less than 4 μg L−1 and less than 17% difference in recovered As(III) between test solutions with low and high interfering ion concentrations. By demonstrating the ability for iron oxide xerogels to reliably adsorb, store, and release arsenic, we enable the development of protocols for solid-phase extraction, preservation, storage, transportation, and analysis of trace contaminants (SEPSTAT), where arsenic would be adsorbed from water samples onto xerogel-based sorbents and shipped to centralized laboratories for recovery and quantification.
1 Introduction
Arsenic is a trace water contaminant occurring naturally in many groundwater sources and affecting the water supply for approximately 150 million people worldwide, with chronic effects including a range of cancers, skin lesions, nerve damage, and cardiovascular disease.1 Because of these health risks, the World Health Organization has designated a permissible limit of 10 μg L−1 for arsenic in drinking water, with a higher provisional limit of 50 μg L−1 for resource-limited nations with high arsenic occurrence such as India and Bangladesh. However, in arsenic-stricken regions such as the Bengal Basin, groundwater arsenic levels can reach as high as 3200 μg L−1.2 Arsenic levels in groundwater can vary widely in space, even between wells 100 m apart,3 necessitating source-level testing for effective water quality management via source selection or remediation.Arsenic monitoring is a formidable challenge using current testing technology. Existing arsenic field test kits generally operate using the Gutzeit reaction, wherein aqueous arsenic is reduced to toxic arsine gas before reaction with mercuric bromide to effect a yellow color change.4 These tests are not sufficiently accurate for reliable source labeling, with false negatives as high as 68% and false positives as high as 35%.5 As a result, accurate arsenic quantification requires the use of elemental analysis techniques such as inductively coupled plasma atomic emission spectroscopy (ICP-AES), inductively coupled plasma mass spectrometry (ICP-MS), and atomic adsorption spectroscopy (AAS).6,7 Elemental analysis can detect arsenic at μg L−1 levels in minutes.4 However, the instrumentation cost for these methods—ranging from 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 USD for flame AAS systems to as much as 200000 USD for ICP-MS systems4—and the need for highly trained laboratory technicians limit their use to centralized testing locations. For example, in India, elemental analysis instrumentation is available at the state level, but is frequently not available at the local laboratories where routine water samples are analyzed.
000 USD for flame AAS systems to as much as 200000 USD for ICP-MS systems4—and the need for highly trained laboratory technicians limit their use to centralized testing locations. For example, in India, elemental analysis instrumentation is available at the state level, but is frequently not available at the local laboratories where routine water samples are analyzed.
Current arsenic monitoring practices require the use of bulky liquid samples which are difficult to transport to the laboratories where they can be accurately analyzed. In India, arsenic testing protocols require 35–100 mL of liquid per sample for analysis,8–10 collected as part of large (250–2000 mL) water samples,9,11,12 with sample acidification or refrigeration recommended for analyte preservation during transportation.11,12 These samples' volume and recommended preservation measures make them difficult to transport to centralized laboratories from both the local laboratories where samples are received and the rural water sources where samples are collected, particularly when considering the cumulative volume of samples collected from all monitored sources. Transportation of water samples is one of the major costs of water testing,13 and this expense is exacerbated for monitoring of trace contaminants in rural environments. As a result, routine water samples are frequently not tested for arsenic.
These challenges for arsenic and other difficult-to-analyze trace contaminants such as heavy metals and pesticides have motivated our development of a new dry sampling paradigm where trace contaminants are adsorbed onto compact solid sorbents, which are then dried and transported to laboratories having appropriate analytical instrumentation for quantification14 (Fig. 1). This sampling paradigm builds upon the success of dried blood spot testing, which has facilitated widespread testing of newborn blood samples for phenylalanine and other small molecules;15,16 the wide range of existing and emerging solid-phase extraction materials;17–19 and sampling techniques such as stir bar sorptive extraction,20 stir cake sorptive extraction,21 and stir-disc solid-phase extraction22 which offer the potential for compact sampling devices amenable to shipping. We recently demonstrated the use of commercial ion-exchange resins to adsorb, store, and release heavy metals in order to enable this new sampling paradigm of Solid-phase Extraction, Preservation, Storage, Transportation, and Analysis of Trace contaminants (SEPSTAT),14 and now extend this paradigm to arsenic sampling.
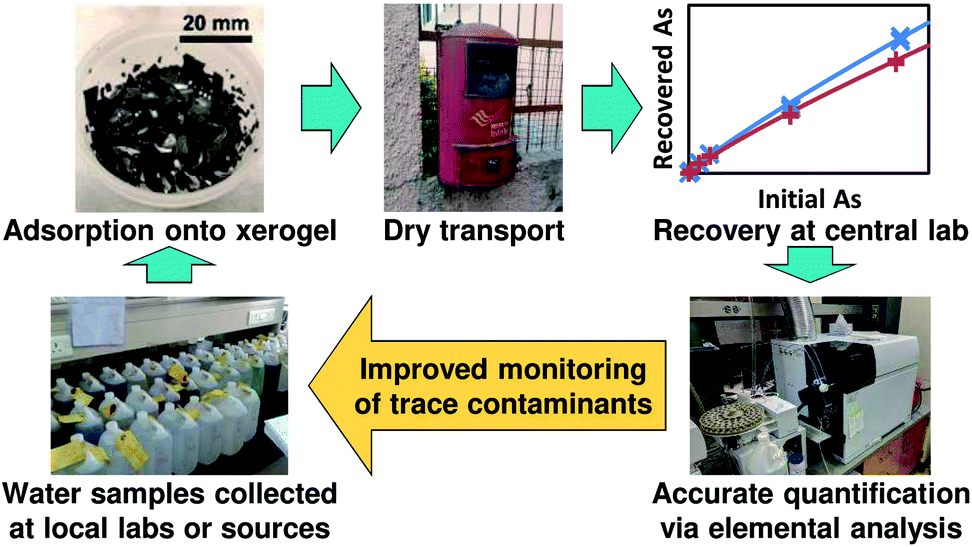 | ||
| Fig. 1 Intended SEPSTAT measurement paradigm for monitoring of arsenic and other trace water contaminants. | ||
Iron oxide is a promising material for solid-phase extraction of arsenic. Iron-oxide-based sorbents have been widely demonstrated for arsenic removal,23–26 offering the potential for capacities as high as 171 mg g−1 (ref. 26) and adsorption in the presence of other common interfering ions.23,25,27,28 Jia et al.19 used magnetic iron oxide nanoparticles for solid-phase extraction of arsenic from complex fish tissue samples before dissolving the nanoparticles in acid for arsenic quantitation, demonstrating that iron oxide is capable of quantitatively extracting arsenic from complex matrices. However, to date, quantitative arsenic recovery via desorption from iron oxide has not yet been demonstrated, creating the opportunity to develop protocols to reliably elute adsorbed arsenic from iron oxide sorbents. One particularly promising elution mechanism is the widely demonstrated pH dependence of arsenic adsorption onto iron oxide, with high arsenic capacity at neutral pH and decreased arsenic capacity at basic pH.23,25 High-pH sodium hydroxide solutions have been used for regeneration of iron-oxide based arsenic sorbents,29,30 suggesting the potential for arsenic quantitation via adsorption at neutral pH and recovery under basic conditions.
Our intended use case of large-scale sampling and transport in resource-limited settings motivates the selection of low-cost, compact iron oxide sorbents with high arsenic capacity. High arsenic capacity can be achieved through the use of high-surface area iron oxide nanomaterials.30 One of the most promising such nanomaterials are iron oxide xerogels, which are inexpensive, easily fabricated via sol–gel chemistry, and offer high surface area and low pore volume.31 This is attractive for widespread use in resource-limited settings, where transportation to centralized laboratory facilities necessitates the use of low-cost and compact sorbents.
Groundwater arsenic contamination predominantly occurs as inorganic oxyanions of arsenic(III), also referred to as arsenite, or arsenic(V), also referred to as arsenate.2 For our initial demonstration of arsenic sampling, we focused our investigation on arsenite due to its greater toxicity than arsenate.32–34 Arsenite is prevalent in reducing environments such as low-oxygen groundwater;2,35 constitutes the majority of groundwater arsenic content in locations as diverse as West Bengal, India24 and Michigan, USA;36 and persists in drinking water after groundwater is brought to the surface for storage and eventual consumption, as aeration and aerobic storage do not oxidize arsenite.37
In this work, we present the use of iron oxide xerogels to reliably adsorb, store, and release arsenic(III) from water samples in order to enable a new arsenic sampling paradigm of Solid-phase Extraction, Preservation, Storage, Transportation, and Analysis of Trace contaminants (SEPSTAT). We show that iron oxide xerogels reliably adsorb aqueous arsenic for compact dry storage and recovery at high pH. Furthermore, in order to ensure reliable sampling of arsenic from real-world groundwater samples, we investigated arsenic adsorption in the presence of interfering ions at concentrations representative of groundwater sources, followed by elution and quantification, in order to develop an optimized recovery protocol to enable robust arsenic sampling in diverse sample matrices. This investigation resulted in a solid-phase extraction protocol for reliable sampling of aqueous arsenic which will enable the development of methods and devices for improved arsenic monitoring.
2 Experimental section
2.1 Materials
Sodium arsenite was selected as an arsenic(III) source for this investigation due to its previous use in investigation of arsenic adsorption by iron oxides.23 Other reagents and materials were selected to minimize the potential for inadvertent arsenic concentration, and are listed in the ESI (Section S1†).2.2 Xerogel fabrication and characterization
Iron oxide xerogels were fabricated using an epoxide-assisted sol–gel synthesis method adapted from Gash et al.31 using iron chloride hexahydrate (FeCl3·6H2O) as an iron precursor, with five ethanol solvent exchanges conducted after gelation in order to remove residual organic impurities.38 The detailed xerogel fabrication process is provided in the ESI (Section S2†).We characterized the xerogels using nitrogen adsorption porosimetry, optical microscopy, and scanning electron microscopy. For nitrogen adsorption porosimetry, xerogel samples were dried for 3 days at 130 °C and ambient pressure, prior to degassing and subsequent analysis in a Micromeritics ASAP 2020 Accelerated Surface Area and Porosimetry system. At least 100 mg xerogel was placed in a glass sample tube and capped with a porous frit (both Micromeritics) before degassing with an evacuation phase consisting of a 200 °C target temperature (10 °C min−1 ramp) and 5 μm Hg vacuum setpoint, followed by a 240 minute heating phase at 150 °C. After degassing, the sample was weighed before measuring nitrogen adsorption and desorption isotherms at 77.350 K with an equilibration time of 5 s at each point. Surface area was calculated via Brunauer–Emmett–Teller (BET) analysis of eight points along the adsorption isotherm, with partial pressures ranging from 0.065 to 0.199, and pore volume and pore size distribution was calculated via Barrett–Joyner–Halenda (BJH) analysis of the adsorption isotherm. Optical microscopy was conducted using a Nikon Eclipse TE2000-U inverted microscope. Scanning electron microscopy was conducted using a JEOL JSM-6010LA microscope after sputtering samples with gold using a SC7640 sputter coater (Quorum Technologies).
2.3 Arsenic quantification
We quantified sample arsenic concentration via inductively coupled plasma atomic emission spectroscopy (ICP-AES), also known as inductively coupled plasma optical emission spectroscopy (ICP-OES), and inductively coupled plasma mass spectrometry (ICP-MS), with ICP-AES used for measuring adsorption capacity and ICP-MS used for all other arsenic quantification unless otherwise noted. ICP-AES measurements were conducted on an Agilent 5100 ICP-OES using a 3% nitric acid rinse solution and argon snout purge. Arsenic was quantified by averaging the radial view signals at 188.980 nm and 193.696 nm. ICP-MS measurements were conducted on an Agilent 7900 ICP-MS using a rinse solution consisting of 2% nitric acid + 2% hydrochloric acid, a 10 μg L−1 indium internal standard in all standards and samples, and helium collision mode (4.5 mL min−1) in order to minimize isobaric interferences from polyatomic ions.39Samples were analyzed after dilution and acidification, where acid contents of 2% and 2.67% nitric acid were used for samples with originally neutral pH and originally basic pH (pH ≥12), respectively. Samples were diluted by a factor of at least 3, with greater dilution factors used as needed in order to ensure that the total dissolved solids (TDS) content was less than the generally recommended maximum value of 2000 mg L−1 for ICP-MS samples. Standards for adsorption solutions were matrix-matched, after accounting for the dilution factor, to contain the starting concentration of all solution components except for the elements which were monitored: arsenic, phosphorus, and silicon. Standards for extraction solutions were matrix-matched to all components present at concentrations greater than 100 mg L−1 in the diluted recovery samples.
2.4 Adsorption and recovery measurements
For xerogel adsorption and recovery measurements, xerogels were pulverized to minimize variation in properties within the dried xerogels, with the pulverized xerogel fragments exhibiting a lognormal size distribution (diameter mean ± s.d. 109 ± 93 μm, see ESI Section S5† for additional information). For each adsorption/recovery sample, we weighed out approximately 25 mg pulverized xerogel into a 15 mL polypropylene centrifuge tube (VWR International) before the addition of 7.5 mL adsorption solution for 3.33 g L−1 xerogel loading. For adsorption measurements, sample tubes were vortexed in order to evenly disperse the xerogel within the adsorption solution and placed on their sides for shaking at 100 rpm on a benchtop orbital shaker (MaxQ 2000, Barnstead). Arsenic adsorption capacity was measured after 87 hours of adsorption, for an adsorption solution consisting of sodium arsenite solution diluted to an initial concentration of 1214 mg L−1 As with no added interfering ions. An adsorption period of 24 hours was used for all other adsorption measurements.At the end of adsorption, we centrifuged the samples for 6 minutes at 2000g, removed the supernatant, and allowed the xerogel to dry at ambient conditions in the loosely capped upright centrifuge tubes used for adsorption, which were sealed after the xerogel was observed to have dried completely. Similarly, for recovery measurements we added 7.5 mL of the desired extraction solution to each sample tube, vortexed the tubes to redisperse the xerogel, placed the tubes on their sides for 24 hours of shaking at 100 rpm, and centrifuged for 6 minutes at 2000g before removing samples of the recovery solution to quantify the eluted arsenic. Initial samples of each adsorption and extraction solution were analyzed in order to properly control for initial arsenic concentration, with additional control samples processed and analyzed as appropriate to quantify leaching of residual arsenic present in xerogels after fabrication and confirm negligible adsorption or desorption of arsenic from the polypropylene sample tubes. Safety note: dermal arsenic exposure can pose a health risk40 which can be exacerbated for acidic or basic arsenic solutions due to pH-related skin damage. For high arsenic concentrations, and when there is a significant risk of skin contact due to splashing or spillage, the use of gloves made of butyl rubber or similar materials is recommended.
Arsenic sampling in the presence of interfering ions was evaluated by adsorbing arsenic from adsorption test solutions intended to span the full range of potential interferent concentrations: deionized water (DI Water), Low Mix, Medium Mix, and High Mix, with compositions as shown in Table 1. For this investigation, we focused on interference from inorganic anions previously observed to affect arsenic adsorption onto iron oxides: chloride, nitrate, sulfate, bicarbonate, silicate, and phosphate.23,25,27,28 Low Mix and Medium Mix were formulated as representative compositions of groundwater sources with low and high salt contents, respectively, whereas High Mix was formulated based on the maximum expected concentration of each individual interfering ion. Adsorption test solutions were prepared from stock solutions of their corresponding sodium salts. Medium Mix and High Mix solutions were pH-adjusted to pH = 7 via addition of 1 M hydrochloric acid. For our screening of arsenic adsorption and recovery across different recovery protocols, we used initial arsenic concentrations at a reference level of 85 ± 3 μg L−1, selected based on previous measurements of the concentration range where interference is most noticeable,27 in matrices consisting of DI Water, Medium Mix, and High Mix in order to span the maximum potential range of interferent concentrations.
| Anion species | Regulated levels8 | Typical concentrations | Test solution mixes | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Acceptable | Permissible | Assam41 | Agra42 | Sindh43 | BGW46 | Low | Medium | High | |
| Chloride, Cl | 250 | 1000 | 39(111) | 350(2705) | 431(900) | — | 125 | 250 | 1000 |
| Nitrate, NO3 | 45 | 45 | 0.11(0.72) | — | 22(97) | — | 1 | 45 | 100 |
| Sulfate, SO4 | 200 | 400 | 21(83) | — | 877(1120) | 7.5(15) | 100 | 200 | 1200 |
| Bicarbonate, HCO3 | N/A | N/A | 70(119) | — | 508(782) | 488(500) | 125 | 500 | 800 |
| Silicate, as SiO2 | N/A | N/A | — | 10(21) | — | 41(64) | 10 | 50 | 65 |
| Phosphate, PO4 | N/A | N/A | 0.16(1.27) | 0.03(0.67) | 0.79(5.00) | 4.5(9.2) | 1 | 5 | 10 |
| TDS | 500 | 2000 | 118(349) | — | 763(3350) | — | 549 | 1566 | 4807 |
In order to optimize our recovery protocol, arsenic recovery was measured for several extraction solutions, selected based on previous observations of conditions where arsenic adsorption was decreased for iron oxides.23,25,28 100 mM sodium hydroxide (NaOH) was used to investigate recovery under highly basic conditions (pH = 13) without any other interfering ions, as well as with the addition of 1 mM silicate (Na2SiO3) and 1 mM phosphate (Na2HPO4) based on previous studies finding that the presence of silicate and phosphate result in a substantial decrease in arsenic adsorption.25,28 In addition, a slightly acidic phosphate solution (30 mM NaH2PO4, pH = 4) was investigated based on observations that this was the pH where arsenic adsorption was most impeded by the presence of phosphate.23 Based on these results, additional formulations of NaOH and phosphate were investigated: 10 mM NaOH, 20 mM NaOH + 2 mM Na3PO4, 20 mM NaOH + 30 mM Na3PO4, 200 mM NaOH + 2 mM Na3PO4, and 200 mM NaOH + 30 mM Na3PO4.
2.5 Evaluation of SEPSTAT method performance
An extraction solution of 100 mM NaOH + 1 mM Na3PO4 (pH = 13) was used for the optimized recovery protocol and evaluated for performance in adsorption and recovery of arsenic(III) over the concentration range found in drinking water. Low Mix and High Mix solutions were prepared as described in Table 1 for nominal arsenic concentrations of 0, 10, 50, 100, 500, and 1000 μg L−1 As, with exact measured arsenic concentrations of {0.01, 10.74, 52.30, 105.11, 512.60, and 1058.76} μg L−1 As for Low Mix solutions and {0.03, 11.25, 52.53, 107.47, 508.95, and 1037.31} μg L−1 As for High Mix solutions. Each solution was adsorbed onto xerogel samples (N = 4) at a xerogel loading of 3.33 g L−1 over 24 hours as described above, and xerogel samples were stored at ambient conditions for 124–125 days before elution over 24 hours in 100 mM NaOH + 1 mM Na3PO4 at the same xerogel loading. The detection limit for each sample matrix was calculated as a modified version of the method detection limit (MDL),47 with calculation details described in the ESI (Section S6†).3 Results and discussion
3.1 Material characterization and adsorption capacity
Our characterization results suggest that xerogels are well-suited to dry sampling and transport, resulting in high arsenic adsorption capacity. The fabricated iron oxide xerogels were glassy in appearance, with a porous structural morphology visible under scanning electron microscopy (Fig. 2). BET analysis yielded a surface area measurement of 340 m2 g−1, and BJH pore volume analysis (Fig. 2d) yielded a pore volume of 0.29 cm3 g−1 and an average pore diameter of 3.0 nm. This combination of high surface area and low pore volume maximizes the ratio of sorbent surface area to overall sorbent volume, making xerogels an excellent geometry for compact dry storage of adsorbed trace contaminants such as arsenic. Arsenic adsorption capacity measurements confirmed our hypothesis that xerogels would have a high arsenic adsorption capacity, with a capacity of 165 mg As per g xerogel measured at an equilibrium aqueous arsenic concentration of 692 mg L−1 (calculation described in ESI Section S3†). Furthermore, preliminary measurements confirmed our hypothesis of pH-dependent adsorption, with high, consistent adsorption at neutral pH and decreased adsorption at high pH (Fig. S1†).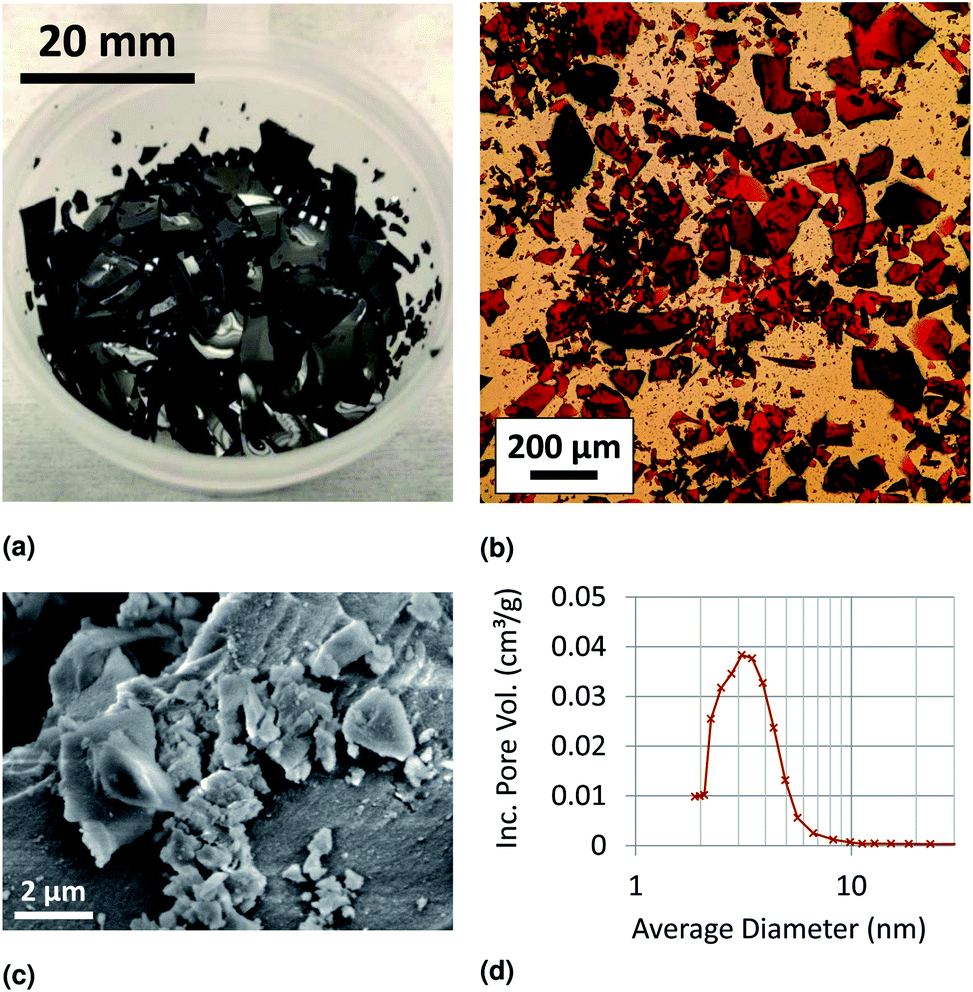 | ||
| Fig. 2 (a) Photograph of iron oxide xerogels, (b) optical microscopy image of pulverized iron oxide xerogel, (c) scanning electron microscopy (SEM) image of iron oxide xerogel, and (d) xerogel pore size distribution according to Barrett–Joyner–Halenda (BJH) analysis of a nitrogen adsorption isotherm. | ||
3.2 Adsorption and recovery characterization
In our investigation of arsenic adsorption and elution, we characterized iron oxide xerogels' ability to adsorb and release arsenic from a range of sample matrices designed to replicate groundwater samples in arsenic-afflicted areas. As part of this investigation, we screened a range of extraction solutions in order to identify an optimal recovery protocol to yield consistent arsenic recovery for accurate quantitation of initial sample arsenic content.Our results confirmed our initial hypothesis that high-pH extraction solutions would facilitate recovery of adsorbed arsenic from iron oxide xerogels. Preliminary experiments showed that arsenic could be recovered after adsorption at neutral pH using a simple extraction solution of 100 mM sodium hydroxide (NaOH), with no observed effects due to variations in initial solution pH and storage time (Fig. S2†).
In order to ensure reliable arsenic sampling in real-world water samples, we measured adsorption and recovery for arsenic in adsorption test solutions formulated based on typical concentrations of expected groundwater interferents (Table 1). For the scope of our interference investigation, we focused on inorganic anions known to affect arsenic adsorption onto iron oxides, consistent with the scope of previous interference studies conducted as part of the development of arsenic removal technology.28,46 Previous research has observed decreased arsenic adsorption onto iron oxides in the presence of chloride, nitrate, sulfate, bicarbonate, phosphate, and silicate,23,25,27,28 so we formulated three test solutions, denoted as Low Mix, Medium Mix, and High Mix, based on regulated levels of these anions (when applicable)8 and observed concentrations in four arsenic-affected areas: Assam, India;41 Agra district, Uttar Pradesh, India;42 Khairpur district, Sindh, Pakistan;43 and a national survey of Bangladesh groundwater (BGW).44–46 Low Mix is intended to replicate the typical composition of groundwater sources with lower salt content, Medium Mix is intended to replicate the typical concentration of groundwater sources with higher salt content, and High Mix is intended to represent the worst-case scenario for each individual interfering ion.
When arsenic adsorption and recovery were measured in the presence of interfering anions (Fig. 3), arsenic adsorption was high regardless of interfering ion content. However, our preliminary tests (Fig. S3†) showed that recovery via 100 mM NaOH was substantially reduced for arsenic adsorbed in the presence of interfering ions, with low recovery efficiency ηrec, defined as the fraction of arsenic in the initial sample which was eluted after adsorption:
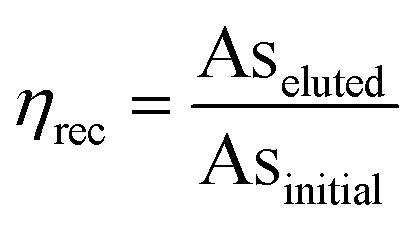 | (1) |
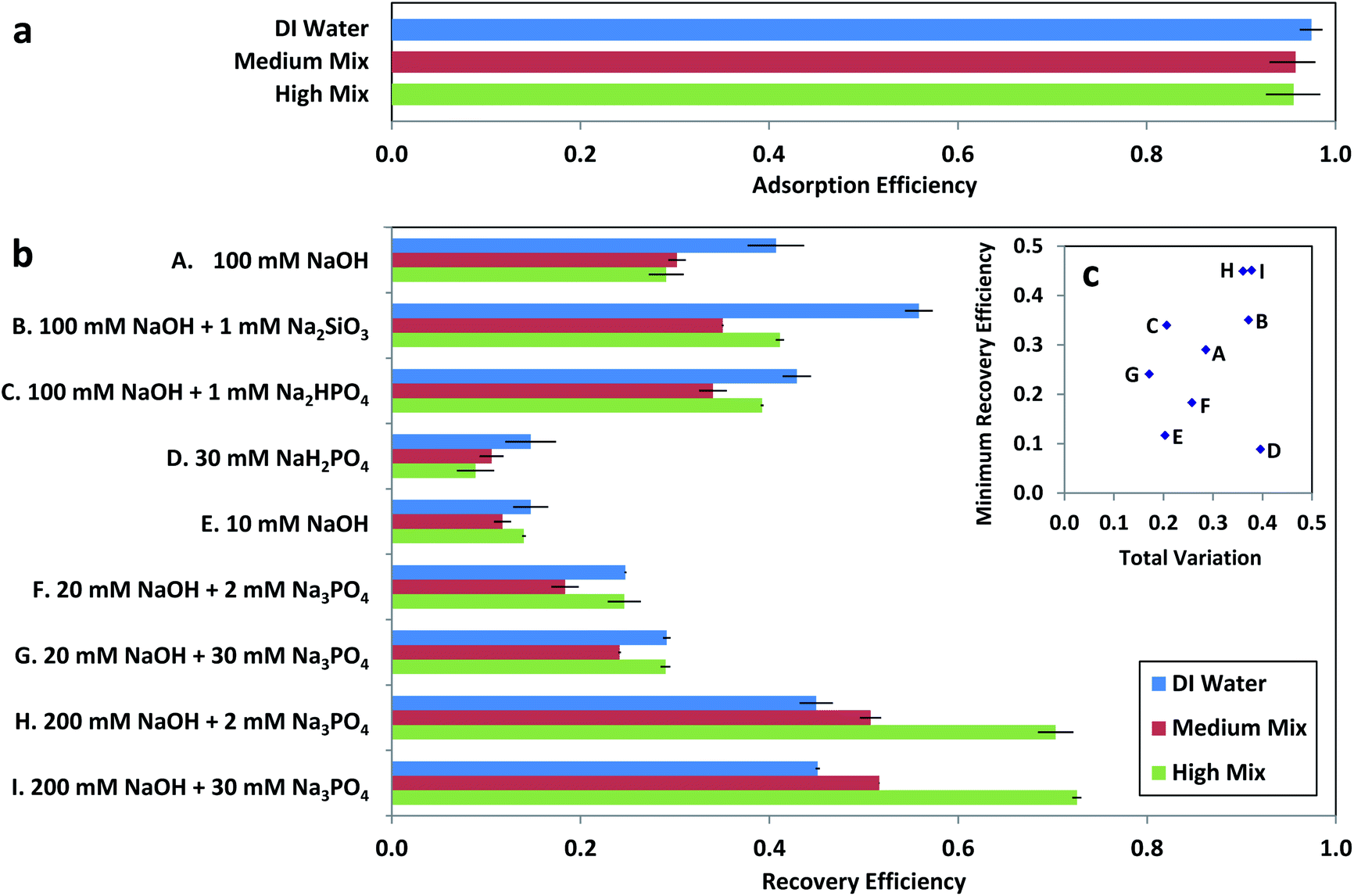 | ||
| Fig. 3 Arsenic adsorption and recovery for test solutions with 85 ± 3 μg L−1 arsenic in adsorption matrices of deionized (DI) water, Medium Mix, and High Mix, where Medium and High Mixes are adsorption test solutions as defined in Table 1. (a) Arsenic adsorption efficiency as a function of test solution. Error bars are the measured range (N = 10). (b) Arsenic recovery efficiency as a function of extraction solution. Error bars are the measured range for each extraction solution (N = 2). (c) Minimum recovery efficiency and total variation for each extraction protocol, where total variation is defined as the difference between the highest and lowest recovery efficiency measured for each extraction solution normalized by the highest recovery efficiency (eqn (4)). | ||
The observed consistency in adsorption but variability in recovery suggests the presence of adsorption sites with a range of pH dependency, such that the presence of additional ions during adsorption results in a displacement of arsenic ions to adsorption sites which maintained greater affinity at increased pH. This behavior of consistent adsorption and variable recovery was observed even for xerogel loadings as high as 13.33 g L−1 (Fig. S3†), necessitating further development of the extraction solution used for arsenic elution.
In order to ensure reliable recovery regardless of the presence of interfering ions during adsorption, we systematically investigated the use of extraction solutions containing phosphate and silicate (Fig. 3), as these ions have been previously shown to substantially decrease arsenic adsorption.23,25,28 For evaluation of potential extraction solutions, we used each extraction solution's mean recovery efficiencies ηrec for the three test solutions investigated to calculate the extraction solution's minimum recovery efficiency (ηrec,min), maximum recovery efficiency (ηrec,max), and total variation (σtot) as defined below:
| ηrec,min = min{ηrec(DI Water), ηrec(Medium Mix), ηrec(High Mix)} | (2) |
| ηrec,max = max{ηrec(DI Water), ηrec(Medium Mix), ηrec(High Mix)} | (3) |
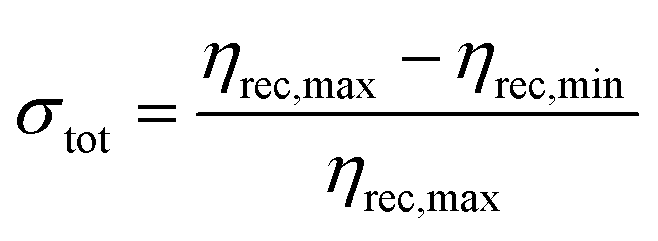 | (4) |
For our intended application of quantitative arsenic sampling, the ideal extraction solution would minimize variability for samples from different sample matrices while still offering an acceptable minimum recovery efficiency for the worst-performing sample matrix. As a result, our most important selection criterion among different extraction solutions was minimizing total variation σtot, with maximization of minimum recovery efficiency ηrec,min used as a secondary selection criterion among extraction solutions with comparable total variation.
Among the first three potential improved extraction solutions that we investigated (solutions B, C, and D in Fig. 3), acidic phosphate (30 mM NaH2PO4, pH = 4) yielded substantially less recovery than the original 100 mM NaOH (solution A in Fig. 3), whereas recovery in NaOH with the addition of silicate (100 mM NaOH + 1 mM Na2SiO3) resulted in an increase in both minimum recovery efficiency and total variation between adsorption test solutions. However, recovery in NaOH with the addition of phosphate (100 mM NaOH + 1 mM Na2HPO4) resulted in an increase in minimum recovery efficiency accompanied by a decrease in total variation between adsorption test solutions. Additional combinations of NaOH and phosphate were investigated (solutions E, F, G, H, and I in Fig. 3), with the lowest total variation observed for 20 mM NaOH + 30 mM Na3PO4 and the greatest minimum recovery efficiencies observed for 200 mM NaOH + 2 mM Na3PO4 and 200 mM NaOH + 30 mM Na3PO4. As 100 mM NaOH + 1 mM Na2HPO4 exhibited a comparable total variation to the minimal total variation exhibited by 20 mM NaOH + 30 mM Na3PO4 (σtot = 0.21 vs. 0.17) along with substantially higher minimum recovery efficiency (ηrec,min = 0.34 vs. 0.24), it offered the best combination of low total variation and high minimum recovery efficiency (Fig. 3c), with the phosphate species changed to 1 mM Na3PO4 for the optimized recovery protocol in order to minimize protonation of the NaOH.
These results are consistent with adsorption sites with a distribution of adsorption efficiencies, in agreement with the isotherms measured in our preliminary experiments (Fig. S1†) and suggesting a smaller number of sites with a greater decrease in affinity at elevated pH but limited selectivity between arsenic(III) and interfering ions, as well as a larger number of sites with a higher selectivity for arsenic adsorption but a lower decrease in affinity at elevated pH. The presence of interfering ions during adsorption would then displace arsenic adsorption to sites with greater selectivity against other ions but lower pH dependence, resulting in decreased recovery at elevated pH which can be enhanced through the addition of phosphate. Phosphate appears to assist in desorption from these sites with lower pH dependence, presumably because it is capable of competing with arsenic for adsorption at these sites at elevated pH, similar to previous usage of higher concentrations (100–1000 mM) of chloride ions for improved regeneration of iron–oxide-based arsenic sorbents.29
3.3 SEPSTAT method performance
Our optimized extraction solution of 100 mM NaOH + 1 mM Na3PO4 resulted in reliable arsenic recovery for initial arsenic(III) concentrations from 10 μg L−1 to 1000 μg L−1 after as long as 125 days' dry storage, the longest period tested. For each interferent level (Low Mix and High Mix, as defined in Table 1), recovered and initial arsenic exhibited a consistent power-law dependence over 10 μg L−1 to 1000 μg L−1 As, with r2 = 0.9968 for Low Mix samples and r2 = 0.9979 for High Mix samples (Fig. 4, results tabulated in Tables S3 and S4†). The relative standard deviation in recovered arsenic at each concentration level (excluding the 0 μg L−1 As level) was <5% for Low Mix samples and <7.5% for High Mix samples, with an average value of 3.1% over all arsenic and interferent levels.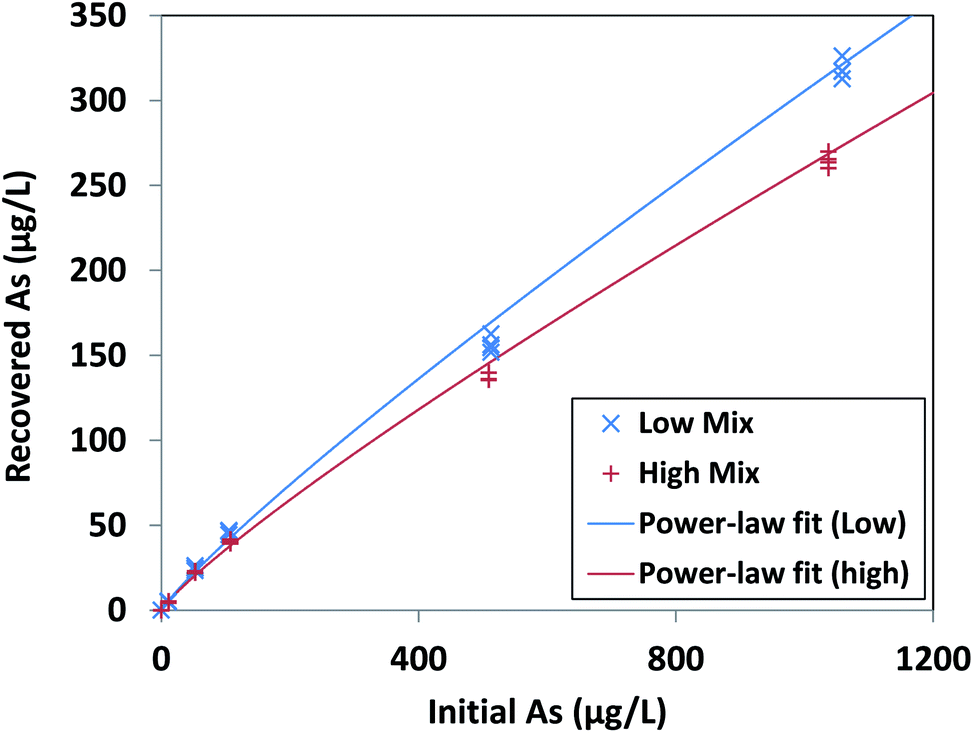 | ||
| Fig. 4 Recovered arsenic concentration as a function of initial arsenic concentration for arsenic adsorbed from Low Mix and High Mix test solutions, as defined in Table 1, recovered from iron oxide xerogels after dry storage for 124–125 days and extraction in the optimized extraction solution of 100 mM sodium hydroxide and 1 mM tribasic sodium phosphate. Power-law fits are [Recovered As] = 0.69 × [Initial As]0.88 for Low Mix samples (r2 = 0.9968) and [Recovered As] = 0.68 × [Initial As]0.86 for High Mix samples (r2 = 0.9979). Data points are individual replicates (N = 4 for each initial solution composition). | ||
Recovery efficiencies were consistent with the observed power-law dependence, ranging from 30.1% to 47.6% for Low Mix samples and 25.5% to 44.5% for High Mix samples, with the greatest recovery efficiencies at the lowest (10 μg L−1) concentration level. The root mean square error (RMSE) between the measured recovery efficiencies (excluding those at the 0 μg L−1 As level) and those estimated by the fitted power-law relationships was only 3.8% for the Low Mix samples and 3.1% for the High Mix samples. The fitted power-law relationships from our performance evaluation could also accurately estimate the measured recovery efficiencies from our initial screening experiment for extraction with 100 mM sodium hydroxide + 1 mM sodium phosphate, which utilized a different xerogel fabrication batch and much shorter storage time (approximately 24 hours). We found an RMSE of 2.7% between the measured and estimated recovery efficiencies for the High Mix samples from our screening experiment, as well as an RMSE of only 2.8% between the measured recovery efficiencies for the DI Water samples from our screening experiment and the estimated recovery efficiency for a Low Mix sample with the same initial arsenic concentration. The observed consistent recovery relationship allows for reliable calculation of initial arsenic concentration from measured concentration of eluted arsenic after dry storage, enabling the implementation of dry sampling methods for arsenic monitoring.
Moreover, there was less than 17% difference in recovered arsenic between Low Mix and High Mix samples at all concentration levels, demonstrating the robustness of the sodium hydroxide and phosphate extraction solution. We were able to easily detect arsenic at the 10 μg L−1 initial concentration level, with calculated detection limits of 1.8 μg L−1 initial As for samples in a Low Mix matrix and 3.7 μg L−1 initial As for samples in a High Mix matrix. Less than 100 ng L−1 As was recovered from xerogels after adsorption from solutions with no added arsenic (i.e., at the 0 μg L−1 initial concentration level), demonstrating that any residual arsenic present in the xerogels after fabrication has minimal effect on the measured recovered arsenic.
These results demonstrate the suitability of iron oxide xerogels for arsenic sampling via the SEPSTAT paradigm, paving the way for development of xerogel-based arsenic sampling devices similar to the devices incorporating commercial ion-exchange resins that we previously developed for sampling of heavy metals.14 Iron oxide xerogels are easily fabricated via one-pot synthesis followed by solvent exchanges for impurity removal, with a bulk reagent cost of approximately 1 USD per gram (see Table S5†); therefore, we do not anticipate barriers to scaling the synthesis to commercially-relevant quantities of inexpensive single-use xerogel sorbents. Xerogels can be incorporated into organic polymer composites48 or coated onto materials such as fabric,49 offering multiple potential manufacturing strategies for compact, easily shippable devices for arsenic sampling. Once developed, these devices could be used to adsorb arsenic from drinking water samples collected at local labs, then shipped in the mail to centralized labs where the arsenic would be recovered and quantified via existing elemental analysis techniques such as ICP-AES or ICP-MS (Fig. 1). The demonstration of a reliable relationship between initial and recovered arsenic across different sample matrices (Fig. 4) enables the implementation of adsorption and recovery protocols where the initial sample arsenic concentration could be accurately determined using the measured arsenic concentration after recovery.
4 Conclusion
We present the use of iron oxide xerogels to reliably adsorb, store, and release arsenic(III), enabling monitoring of arsenic in drinking water using standard laboratory instruments with vastly reduced need for transportation of bulky liquid water samples. This work extends existing usage of solid-phase extraction to solid-phase storage of arsenic in order to facilitate improved monitoring of trace water contaminants in resource-limited settings.One key area for future work is extension of the demonstrated sampling technique from arsenic(III) to arsenic(V), whether by demonstrating adsorption and recovery protocols that are robust across both arsenic species or by converting all sample arsenic to one species during the sampling process. Sample arsenic(V) could potentially be reduced to arsenic(III) before adsorption by pretreating samples with prereductants such as L-cysteine, which has been demonstrated to reduce arsenic(V) to arsenic(III) for analysis without the need for strongly acidic environments required by other prereductants such as potassium iodide.50,51 Alternatively, species conversion could be implemented during adsorption by incorporating additional materials capable of facilitating arsenic oxidation or reduction, such as manganese oxides28 or zerovalent iron,52 respectively, into iron oxide xerogel-based sorbents. Additional areas for future work include investigation of adsorption and recovery kinetics in order to fully develop protocols for use during water monitoring;14 investigation of adsorption and recovery for sample matrices incorporating organic matter;27,53–55 development of compact devices for adsorption, transportation, and recovery;14 and stakeholder engagement in order to better understand use cases and design priorities for implementation of the SEPSTAT paradigm.56
Author contributions
Conceptualization: MSB, EBH, CV, AJH, RK. Funding acquisition: CV, AJH, RK. Methodology: MSB, EBH, RK. Investigation: MSB. Formal analysis: MSB. Software: MSB, EBH. Project administration: MSB, EBH, RK. Supervision: MSB, CV, AJH, RK. Visualization: MSB, RK. Writing – original draft: MSB, RK. Writing – review & editing: MSB, EBH, CV, AJH, RK.Conflicts of interest
EH, RK, and CV are involved with a startup company to commercialize the described technology. The authors are co-inventors on two patent applications related to the technology described in this paper that may be licensed to the company.57,58 The described collaboration and patent applications do not alter our adherence to the Royal Society of Chemistry Code of Conduct.Acknowledgements
This research was supported primarily by the Tata Center for Technology and Design at MIT, which was funded with generous support from the Tata Trusts, as well as by the Abdul Latif Jameel Water and Food Security Lab (J-WAFS) at MIT, which was funded with generous support from Mohammed Abdul Latif Jameel. The authors thank Xiaoyuan (Charlene) Ren, Sydney Beasley, Ramprasad Venkatesha, and Prof. James Wescoat for assistance with planning and conducting stakeholder engagement in India. In addition, the authors thank Shashank Deshpande (Maharashtra Groundwater Surveys Development Agency); Malini Shankar and S. C. Kollur (both Maharashtra Pollution Control Board); Karthik Seshan (Arghyam); Puneet Srivastava (WaterAid); and Prof. Manish Kumar (BIT-Mesra) for helpful meetings regarding water quality management in India. This work made use of facilities at the MIT Center for Environmental Health Sciences, which was supported by a core center grant P30-ES002109 from the National Institute of Environmental Health Sciences, National Institutes of Health; the MRSEC Shared Experimental Facilities at MIT, supported by the National Science Foundation under award number DMR-14-19807; and the Institute for Soldier Nanotechnologies at MIT, supported in part by the US Army Research Office. The authors thank Koli Taghizadeh, Stephen Slocum, Maharaja Kumar, Timothy McClure, Amy Tatem-Bannister, and William DiNatale for training and assistance with the analytical instrumentation used in this work, as well as Jay Patel, Susanna Kahn, and members of the Microfluidics and Nanofluidics Laboratory for helpful discussions.References
- P. Ravenscroft, H. Brammer and K. Richards, Arsenic Pollution: A Global Synthesis, John Wiley & Sons, Hoboken, New Jersey, USA, 2009, vol. 28 Search PubMed.
- P. Smedley and D. Kinniburgh, Appl. Geochem., 2002, 17, 517–568 CrossRef CAS.
- C. Davis, in Arsenic Exposure and Health Effects V: Proceedings of the fifth International Conference on Arsenic Exposure and Health Effects, ed. W. R. Chappel, C. O. Abernathy, R. I. Calderon and D. J. Thomas, Elsevier, Amsterdam, Netherlands, 2003, pp. 421–437 Search PubMed.
- N. Yogarajah and S. S. H. Tsai, Environ. Sci.: Water Res. Technol., 2015, 1, 426–447 RSC.
- M. M. Rahman, D. Mukherjee, M. K. Sengupta, U. K. Chowdhury, D. Lodh, C. R. Chanda, S. Roy, M. Selim, Q. Quamruzzaman, A. H. Milton, S. M. Shahidullah, M. T. Rahman and D. Chakraborti, Environ. Sci. Technol., 2002, 36, 5385–5394 CrossRef CAS PubMed.
- L. H. Keith, Compilation of EPA's Sampling and Analysis Methods, Lewis Publishers, New York City, New York, USA, 2nd edn, 1996 Search PubMed.
- R. E. Wagner, Guide to Environmental Analytical Methods, Genium Publishing Corporation, Amsterdam, New York, USA, 4th edn, 1998 Search PubMed.
- Bureau of Indian Standards, IS 10500: Drinking water — Specification, 2012 Search PubMed.
- Bureau of Indian Standards, IS 3025 (Part 2): Methods of Sampling and Test (Physical and Chemical) for Water and Waste Water Part 2 Determination of 33 Elements by Inductively Coupled Plasma Atomic Emission Spectroscopy, 2004 Search PubMed.
- Bureau of Indian Standards, IS 3025 (Part 37): Methods of Sampling and Test (Physical and Chemical) for Water and Waste Water: Part 37 Arsenic (First Revision), 1988 (reaffirmed 2003) Search PubMed.
- Ministry of Drinking, Water and Sanitation, Uniform Drinking Water Quality Monitoring Protocol, Government of India, 2013 Search PubMed.
- Bureau of Indian Standards, IS 3025 (Part 1): Methods of Sampling and Test (Physical and Chemical) for Water and Waste Water: Part 1 Sampling (First Revision), 1987 (reaffirmed 2003) Search PubMed.
- J. Crocker and J. Bartram, Int. J. Environ. Res. Public Health, 2014, 11, 7333–7346 CrossRef PubMed.
- E. Hanhauser, M. S. Bono Jr, C. Vaishnav, A. J. Hart and R. Karnik, Environ. Sci. Technol., 2020, 54, 2646–2657 CrossRef CAS PubMed.
- R. Guthrie and A. Susi, Pediatrics, 1963, 32, 338–343 CAS.
- W. Li and F. L. S. Tse, Biomed. Chromatogr., 2010, 24, 49–65 CrossRef CAS PubMed.
- L. Zhang, D. Ishi, K. Shitou, Y. Morita and A. Isozaki, Talanta, 2005, 68, 336–342 CrossRef CAS PubMed.
- X. Jiang, K. Huang, D. Deng, H. Xia, X. Hou and C. Zheng, Trends Anal. Chem., 2012, 39, 38–59 CrossRef CAS.
- Y. Jia, H. Yu, L. Wu, X. Hou, L. Yang and C. Zheng, Anal. Chem., 2015, 87, 5866–5871 CrossRef CAS PubMed.
- P. Popp, C. Bauer and L. Wennrich, Anal. Chim. Acta, 2001, 436, 1–9 CrossRef CAS.
- Y. Zhang, M. Mei, T. Ouyang and X. Huang, Talanta, 2016, 161, 377–383 CrossRef CAS PubMed.
- P. Tomai, A. Martinelli, S. Morosetti, R. Curini, S. Fanali and A. Gentili, Anal. Chem., 2018, 90, 6827–6834 CrossRef CAS PubMed.
- S. Dixit and J. G. Hering, Environ. Sci. Technol., 2003, 37, 4182–4189 CrossRef CAS PubMed.
- S. Sarkar, L. M. Blaney, A. Gupta, D. Ghosh and A. K. SenGupta, Environ. Sci. Technol., 2008, 42, 4268–4273 CrossRef CAS PubMed.
- E. Arifin, J. Cha and J.-K. Lee, Bull. Korean Chem. Soc., 2013, 34, 2358–2366 CrossRef CAS.
- I. Polowczyk, P. Cyganowski, J. Ulatowska, W. Sawiński and A. Bastrzyk, Water, Air, Soil Pollut., 2018, 229, 203 CrossRef PubMed.
- M. Kanematsu, T. M. Young, K. Fukushi, D. A. Sverjensky, P. G. Green and J. L. Darby, Environ. Sci. Technol., 2011, 45, 561–568 CrossRef CAS PubMed.
- S. Kong, Y. Wang, H. Zhan, S. Yuan, M. Yu and M. Liu, Water Environ. Res., 2014, 86, 147–155 CrossRef CAS PubMed.
- B. K. Chaudhary and J. Farrell, Environ. Eng. Sci., 2015, 32, 353–360 CrossRef CAS PubMed.
- W. Zhang, C. Liu, L. Wang, T. Zheng, G. Ren, J. Li, J. Ma, G. Zhang, H. Song, Z. Zhang and Z. Li, Colloids Surf., A, 2019, 561, 364–372 CrossRef CAS.
- A. E. Gash, T. M. Tillotson, J. H. Satcher Jr, J. F. Poco, L. W. Hrubesh and R. L. Simpson, Chem. Mater., 2001, 13, 999–1007 CrossRef CAS.
- W. R. Byron, G. W. Bierbower, J. B. Brouwer and W. H. Hansen, Toxicol. Appl. Pharmacol., 1967, 10, 132–147 CrossRef CAS PubMed.
- T. Maitani, N. Saito, M. Abe, S. Uchiyama and Y. Saito, Toxicol. Lett., 1987, 39, 63–70 CrossRef CAS PubMed.
- N. E. Korte and Q. Fernando, Crit. Rev. Environ. Control, 1991, 21, 1–39 CrossRef CAS.
- D. Mohan and C. U. Pittman Jr, J. Hazard. Mater., 2007, 142, 1–53 CrossRef CAS PubMed.
- M.-J. Kim, J. Nriagu and S. Haack, Environ. Pollut., 2002, 120, 379–390 CrossRef CAS PubMed.
- J. D. Lowry and S. B. Lowry, Oxidation of Arsenic(III) by Aeration and Storage, U.S. Environmental Protection Agency EPA/600/R-01/102, 2002 Search PubMed.
- S. J. Juhl, N. J. Dunn, M. K. Carroll, A. M. Anderson, B. A. Bruno, J. E. Madero and M. S. Bono Jr., J. Non-Cryst. Solids, 2015, 426, 141–149 CrossRef CAS.
- N. Yamada, Spectrochim. Acta, Part B, 2015, 110, 31–44 CrossRef CAS.
- S. Ouypornkochagorn and J. Feldmann, Environ. Sci. Technol., 2010, 44, 3972–3978 CrossRef CAS PubMed.
- N. J. Khound, P. Phukon and K. G. Bhattacharyya, Int. J. Appl. Sci. Eng. Res., 2012, 1, 512–521 CrossRef CAS.
- M. K. Gupta, V. Singh, P. Rajwanshi, M. Agarwal, K. Rai, S. Srivastava, R. Shrivastav and S. Dass, Environ. Monit. Assess., 1999, 59, 275–285 CrossRef CAS.
- J. A. Baig, T. G. Kazi, A. Q. Shah, G. A. Kandhro, H. I. Afridi, M. B. Arain, M. K. Jamali and N. Jalbani, Ecotoxicol. Environ. Saf., 2010, 73, 914–923 CrossRef PubMed.
- D. G. Kinniburgh and P. L. Smedley, Arsenic contamination of groundwater in Bangladesh, British Geological Survey; Bangladesh Department for Public Health Engineering, 2001 Search PubMed.
- L. C. Roberts, S. J. Hug, T. Ruettimann, M. M. Billah, A. W. Khan and M. T. Rahman, Environ. Sci. Technol., 2004, 38, 307–315 CrossRef CAS PubMed.
- M. C. Ciardelli, H. Xu and N. Sahai, Water Res., 2008, 42, 615–624 CrossRef CAS PubMed.
- T. Martin, C. Brockhoff and J. Creed, Method 200.7, Revision 4.4: Determination of Metals and Trace Elements in Water and Wastes by Inductively Coupled Plasma-Atomic Emission Spectrometry, U.S. Environmental Protection Agency, 1994 Search PubMed.
- E.-J. Lee, S.-H. Teng, T.-S. Jang, P. Wang, S.-W. Yook, H.-E. Kim and Y.-H. Koh, Acta Biomater., 2010, 6, 3557–3565 CrossRef CAS PubMed.
- F. Li, Y. Xing and X. Ding, Surf. Coat. Technol., 2008, 202, 4721–4727 CrossRef CAS.
- H. Chen, I. D. Brindle and X. C. Le, Anal. Chem., 1992, 64, 667–672 CrossRef CAS.
- P. K. Dasgupta, H. Huang, G. Zhang and G. P. Cobb, Talanta, 2002, 58, 153–164 CrossRef CAS PubMed.
- F. Sun, K. A. Osseo-Asare, Y. Chen and B. A. Dempsey, J. Hazard. Mater., 2011, 196, 311–317 CrossRef CAS PubMed.
- M. S. Mak, P. Rao and I. M. Lo, Environ. Pollut., 2011, 159, 377–382 CrossRef CAS PubMed.
- M. Lawson, D. A. Polya, A. J. Boyce, C. Bryant and C. J. Ballentine, Geochim. Cosmochim. Acta, 2016, 178, 160–177 CrossRef CAS.
- A.-R. Schittich, U. J. Wënsch, H. V. Kulkarni, M. Battistel, H. Bregnhøj, C. A. Stedmon and U. S. McKnight, Environ. Sci. Technol., 2018, 52, 13027–13036 CrossRef CAS PubMed.
- M. S. Bono Jr, S. Beasley, E. Hanhauser, A. J. Hart, R. Karnik and C. Vaishnav, PLoS One, 2020, 15, e0228140 CrossRef PubMed.
- E. B. Hanhauser, M. S. Bono, X. Ren, C. Vaishnav, A. J. Hart and R. N. Karnik, System and Method for Preservation, Transport, and Analysis of Water Samples, US PCT Patent, Application No. PCT/US2017/029614, Filed 2017 Search PubMed.
- I. Sen, K. S. Harsha, E. B. Hanhauser, R. N. Karnik, A. J. Hart and M. Bono, A Vessel and a Method for Purifying Water and Monitoring Quality of Water, Indian Patent Application No. 202011024458, Filed 2020 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full list of reagents and materials; detailed description of xerogel fabrication process; isotherm and adsorption capacity calculation methodology; methods and results for preliminary experiments; particle size analysis for pulverized xerogels; detection limit calculation methodology; tabulated method performance evaluation measurements; and xerogel bill of materials and cost calculation. See DOI: 10.1039/d0ay02365e |
| This journal is © The Royal Society of Chemistry 2021 |