
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple, rapid and low-cost spectrophotometric method for irinotecan quantification in human plasma and in pharmaceutical dosage forms†
Georgia Eleni
Tsotsou
 *,
Panagiota
Gkotzamani‡
,
Victoria
Petro‡
,
Ariadne
Argyropoulou
and
Petros
Karkalousos
*,
Panagiota
Gkotzamani‡
,
Victoria
Petro‡
,
Ariadne
Argyropoulou
and
Petros
Karkalousos
Laboratory of Chemistry, Biochemistry and Cosmetology, Department of Biomedical Sciences, University of West Attica, Egaleo 122 43, Greece. E-mail: gtsotsou@uniwa.gr
First published on 8th December 2020
Abstract
Irinotecan is an anticancer drug for which significant benefits from personalised dosing are expected. Quick procedures are therefore essential for monitoring irinotecan in treated patients. The objective of this work was to develop and validate a rapid and simple visible spectrophotometric method for quantitative determination of irinotecan in pharmaceutical dosage forms and to further investigate its usefulness for irinotecan analysis in plasma. Based on the shift of the irinotecan 355/368 nm-peak at very low pH (0.2) to 400 nm, we established a linear relationship between absorbance at 400 nm and irinotecan concentration in dilutions of an irinotecan solution for injection (R2 ≥ 0.999) and in plasma containing irinotecan (R2 ≥ 0.995). Background absorbance correction at 455 nm was essential to minimise background interference, solely in plasma samples. We fully validated the assay for quality control of the irinotecan solution in the injection dosage form: the standard curve was linear over the concentration range of 0.90 to at least 37.00 μg ml−1. The CV% on all quality control levels was determined to be ≤5.81% for repeatability and ≤6.62% for intermediate precision. Recovery was between 96.5% and 101.9%. Upon comparison with the LC/UV method, we demonstrated very good agreement and acceptable bias between the two methods (slope 0.973, y-intercept 0.0064). Similarly, the technical parameters of the assay in plasma satisfied international guidelines for method validation: the useful analytical range was determined to be between 0.93 and at least 10.00 μg ml−1. This is suitable for quantifying irinotecan in the plasma of treated patients, in the upper region of its therapeutic window, to decide whether dose adjustment is required. Repeatability and intermediate precision (CV%) were within 4.49% and 9.91%, respectively. Recovery was between 96.3% and 103.8%. There was a lack of significant interference by mild hemolysis or by icterus. Irinotecan extraction efficiency from plasma was within 77.9–68.5%. Our results indicated that the proposed method allows quantitative determination of irinotecan plasma levels with acceptable analytical characteristics. The advantages of the proposed method in both matrices, in terms of specificity, rapidity, simplicity, environmental impact and cost effectiveness, are discussed.
Introduction
Irinotecan (7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxy-camptothecin or CPT-11) is a semi-synthetic antineoplastic prodrug agent and is a water-soluble analogue of the natural product camptothecin. Irinotecan is converted in vivo by carboxylesterase to its active metabolite SN-38. SN-38 and irinotecan bind to the mammalian topoisomerase I–DNA complex, preventing re-annealing, causing double strand DNA breaks and thus leading to cell death. SN-38 is 100–1000 times more potent than irinotecan. SN-38 peak serum levels achieved in humans are, however, 100 to 75-fold lower than the corresponding irinotecan concentrations, depending on therapeutic regimen specificities.1–4 Large interindividual pharmacokinetic variability has been elucidated for irinotecan, which in combination with a narrow therapeutic window and dose-dependent adverse effects, are mandating individualised patient treatment.5Many HPLC methods for quantitative determination of irinotecan and its metabolites in biological fluids or cell lysates and for analysis of irinotecan dosage forms have been reported in the literature. They were coupled with UV detection for analysis of irinotecan dosage forms, or mainly with MS or fluorescence detection for irinotecan analysis in biological fluids.3,6–9 A method based on solid phase extraction coupled with differential pulse voltammetry has recently been proposed for irinotecan quantification in plasma for personalised dose optimisation purposes.10 Additionally, the fluorescence characteristics of irinotecan have been exploited for its direct determination in biological fluids and pharmaceuticals.11–14 In recent publications, fluorescent imprinted nanopolymers were designed, specifically detecting irinotecan in deproteinised plasma at clinically relevant concentrations,11 while the distinct pH-dependent fluorescence properties of irinotecan and SN-38 have been used to selectively quantify each compound at clinical concentrations.14
A review of irinotecan absorbance-based assays yielded two publications proposing its direct quantitative determination in pharmaceutical dosage forms. The first assay relies on irinotecan absorption at 247 nm.15 The second method is based on the formation of ion pair complexes of the drug with two acidic dyes, bromocresol green and bromophenol blue, in acidic buffer solutions, followed by their extraction in chloroform, before measuring the absorbance of the organic layer.16 No absorbance-based method for irinotecan analysis in plasma has been proposed.
Two pKa values have been experimentally determined for irinotecan. The pKa value for the piperidino group has been reported in the range of 8.7–8.93 (ref. 17 and 18) (pKa2), while that for the quinoline functionality is at 2.08 (ref. 18) (pKa1). Irinotecan can exist in a neutral form (Fig. 1, structure C) or mono-cationic (Fig. 1, structure B) or di-cationic (Fig. 1, structure A) form, whose relative abundance depends on the medium's pH.19 Aqueous solutions of irinotecan in the neutral or mono-cationic form display characteristic spectra with absorption maxima at around 355 and 368 nm (double peak). Protonation of the quinoline moiety, at the N1 atom, at a pH as low as 1.11 generates di-cationic irinotecan species (Fig. 1, structure A), with a λmax of 400 nm, at equilibrium with the mono-cationic species (Fig. 1, structure B), the latter prevailing at pH 2.61.19 Similarly, a red shift of the absorption bands of CPT (pKa = 1.14) and 10-CPT (pKa = 1.8) by 35–40 nm has been observed as a result of protonation of the quinoline moiety.20 Upon further significant lowering of the pH to 0.2 where, based on a pKa2 of 2.08, irinotecan should practically exist exclusively in its diprotonated form, we observed a total shift of the 355/368 nm double peak to 400 nm. In this work we demonstrate the linear relationship between absorbance at 400 nm at pH 0.2 and irinotecan concentration in aqueous solutions of pharmaceutical dosage forms and in treated plasma samples containing irinotecan. We also investigate the analytical parameters of the proposed method for irinotecan quantification in the two matrices.
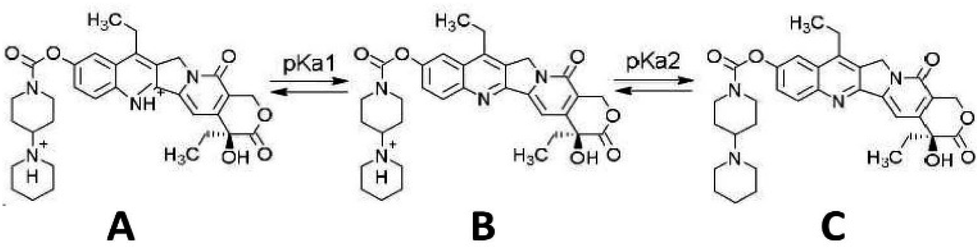 | ||
| Fig. 1 Structure of the irinotecan molecule in the dicationic (A), monocationic (B) and neutral (C) form, with respect to pKa values. | ||
Results and discussion
Spectrophotometric method development
The partial shift of the camptothecin broad absorption peak at 355/368 nm to higher wavelengths, upon pH change towards strongly acidic values, has been described in the literature.19,20 Upon further significant lowering of the pH to about 0.2 we observed light green colouration of the solution with a full shift of the 355/368 nm peak to 400 nm (Fig. 2A and B). No additional peaks in the visible region were observed. The irinotecan molar extinction coefficient (ε) plot vs. wavelength at different pH values where the drug is present in the monocationic (pH 4.9) or the dicationic form (pH 0.2), or as a mixture of the two forms (pH 2.0 and 1.25) is displayed in Fig. 2A. We determined two isosbestic points, at 375 and 305 nm, in accordance with the literature.19 The molar extinction coefficient of irinotecan at 400 nm in water and at pH 0.2 (i.e. ε of the dicationic form) was calculated to be 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 823 M−1 cm−1. We evaluated whether irinotecan absorbance at 400 nm at very low pH values can be applied to the quantification of irinotecan, in plasma or in pharmaceutical dosage forms, by investigating the correlation between the absorption of the resulting green product and the concentration of irinotecan in treated plasma or in treated aqueous dilutions of pharmaceutical dosage forms. In all plasma experiments, plasma was prepared from blood and spiked with the drug before processing (HCl/NaCl/centrifugation), as in the Experimental section.
823 M−1 cm−1. We evaluated whether irinotecan absorbance at 400 nm at very low pH values can be applied to the quantification of irinotecan, in plasma or in pharmaceutical dosage forms, by investigating the correlation between the absorption of the resulting green product and the concentration of irinotecan in treated plasma or in treated aqueous dilutions of pharmaceutical dosage forms. In all plasma experiments, plasma was prepared from blood and spiked with the drug before processing (HCl/NaCl/centrifugation), as in the Experimental section.
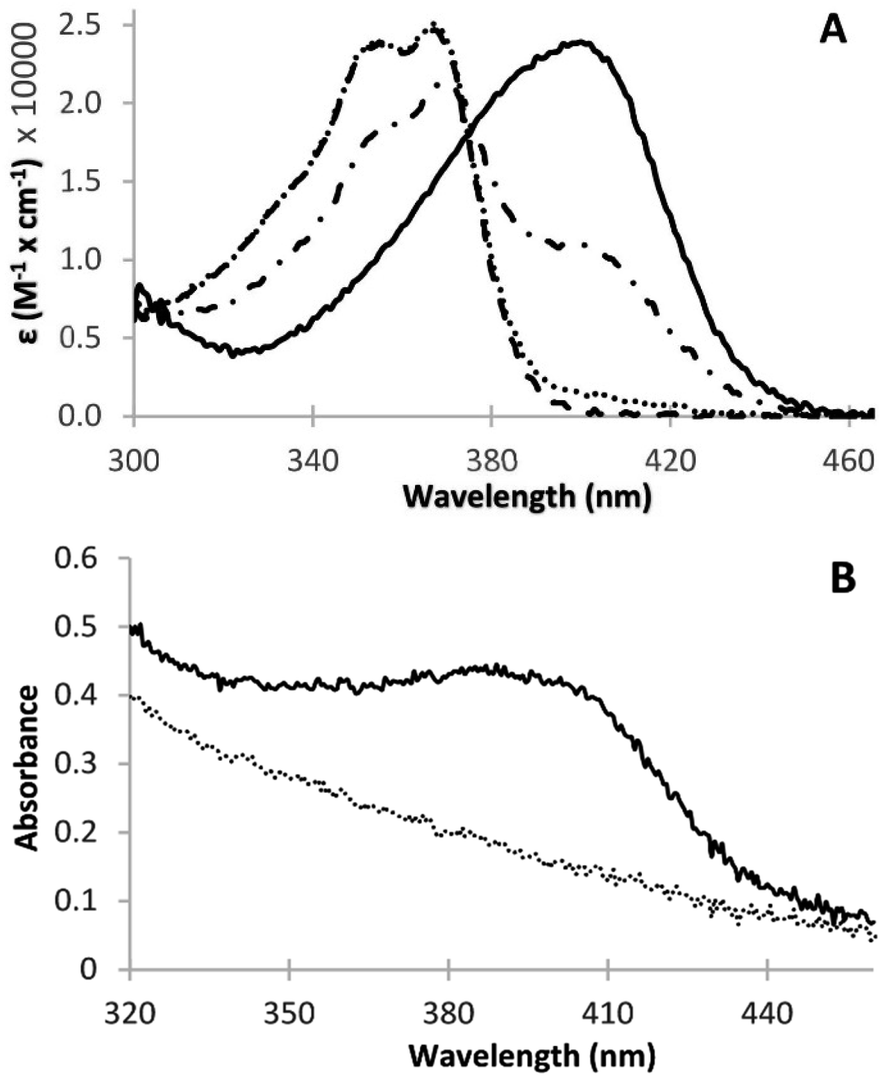 | ||
| Fig. 2 (A) Extinction coefficient vs. wavelength plot for irinotecan at different pH values (aqueous solution containing 20.0 μg ml−1 irinotecan). Dashed line, pH 4.9; dotted line, pH 2.0; dotted-dashed line, pH 1.25; solid line, pH 0.2. (B) Vis absorbance spectra of HCl/NaCl-treated plasma at pH 0.2, where an extra peak at 400 nm is observed in plasma containing irinotecan (solid line, 10.0 μg ml−1) when compared to the blank plasma matrix (dotted line). | ||
Deproteination prior to measurement was deemed necessary to remove interfering proteins upon irinotecan determination in plasma. We found that pretreating irinotecan in plasma with acetonitrile (1 part plasma plus 2 parts acetonitrile), as commonly used in the state-of-the art, with subsequent acidification was sufficient for deproteination. However, a pre-treatment procedure based simply on plasma acidification in the presence of sodium chloride was preferred, since it introduced lower sample dilution. The addition of sodium chloride was imperative to retain sample clarity for a few hours. In its absence, fast turbidity formation was apparent in plasma samples processed solely by acidification.
In the case of irinotecan analysis in plasma, absorbance was measured simultaneously at two wavelengths, a primary (A1: 400 nm) and a secondary (A2:455 nm) wavelength, to compensate for the effects of turbidity (light scattering) developing under the conditions of the assay. Standard curves were based on the (A1–A2) difference, as described in detail in the Experimental section of this article.
The plots of absorbance of the coloured product versus irinotecan concentration in plasma or aqueous solutions demonstrated a strong positive, linear correlation (Fig. 3A and B). They therefore validated the absorption of the coloured product as a measure of irinotecan concentration. The residual sum of squares was very low (in the range of 10−4 to 10−5) for both fittings, indicating a very tight fit of the linear model to the experimental data.
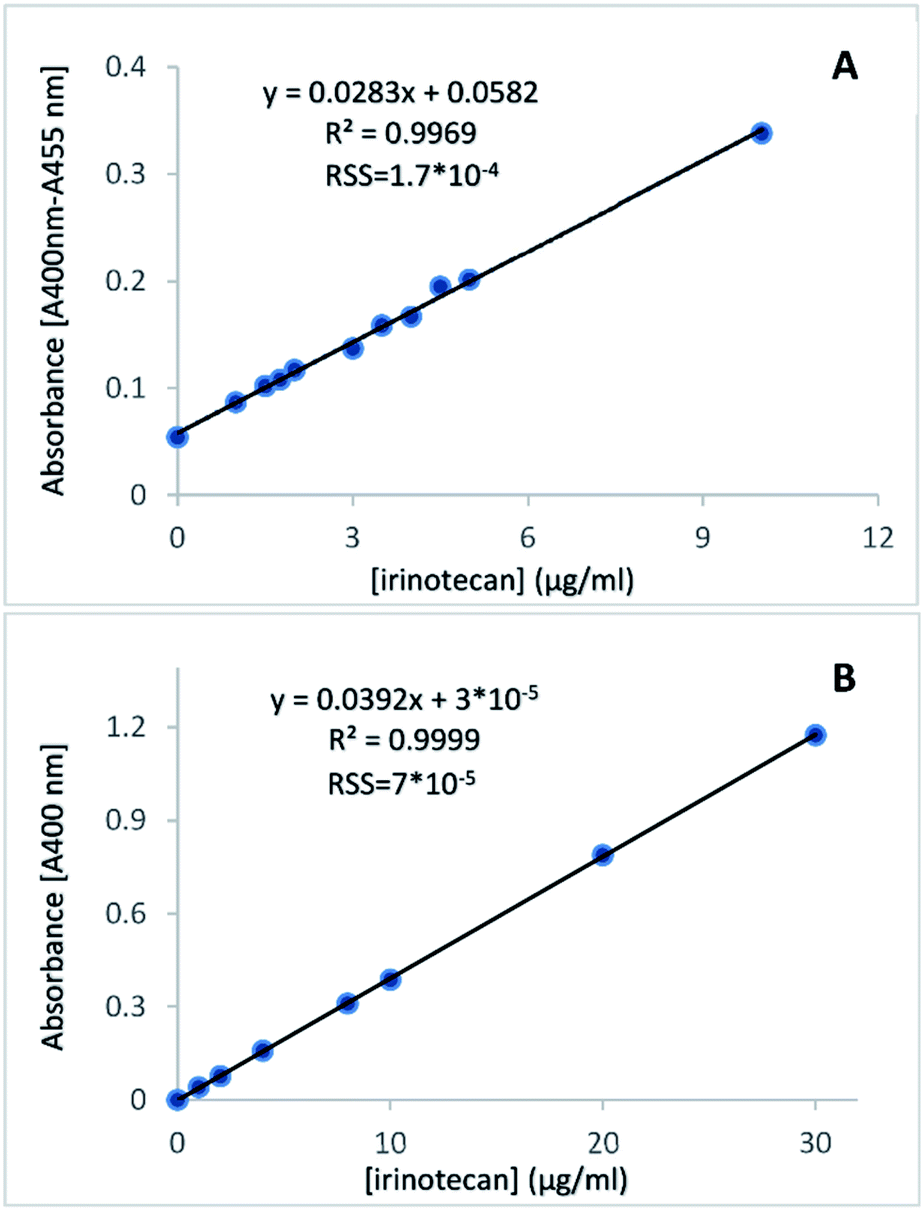 | ||
| Fig. 3 Calibration plots of irinotecan in HCl/NaCl-treated plasma (A) and in HCl-treated aqueous solution (B), where the absorbance difference (A400 nm − A455 nm) or the absorbance at 400 nm is plotted against irinotecan concentration in standards. The correlation coefficient (R2), equation of linear fitting and Residual Sum of Squares (RSS) are given. | ||
Spectrophotometric method validation
After linearity was established, the new spectrophotometric method was validated as per the ICH21 in terms of the useful analytical range, specificity, accuracy, precision (repeatability and intermediate precision), limit of detection and limit of quantitation. In particular, accuracy was determined by bias calculation upon standard concentration back-calculation, recovery studies and method comparison studies. Separate validation studies were performed for the assay in plasma and the assay in aqueous solutions of pharmaceutical dosage forms.Useful analytical range, precision, LOD and LOQ
The useful analytical range, precision, LOD and LOQ of the proposed method are listed in Table 1, where relevant validation characteristics were established across the specified range of the analytical procedure (pharmaceutical dosage form matrix) or across the upper region of the irinotecan therapeutic window (plasma matrix).| Aqueous dilution of irinotecan solution for injection | Irinotecan in plasma | ||||
|---|---|---|---|---|---|
| Intermediate precision | Intermediate precision | ||||
| a n = number of replicates, CV = coefficient of variation, C.I. = confidence interval. | |||||
| Nominal [irinotecan], μg ml−1 | CV% (n) | C.I. (95.4%) | Nominal [irinotecan], μg ml−1 | CV% (n) | C.I. (95.4%) |
| 1.80 | 3.66 (n = 8) | 1.68–1.95 | 0.90 | 5.57 (n = 7) | 0.77–0.96 |
| 2.00 | 6.62 (n = 8) | 1.82–2.38 | 1.00 | 9.91 (n = 6) | 1.00–1.49 |
| 5.00 | 2.64 (n = 8) | 4.73–5.26 | 3.50 | 4.03 (n = 6) | 3.12–3.66 |
| 12.00 | 3.16 (n = 6) | 10.75–12.20 | 5.00 | 1.25 (n = 6) | 4.89–5.15 |
| 24.00 | 4.21 (n = 6) | 21.62–25.60 | |||
| Repeatability | Repeatability | ||||
|---|---|---|---|---|---|
| Nominal [irinotecan], μg ml−1 | CV% (n) | C.I. (95.4%) | Nominal [irinotecan], μg ml−1 | CV% (n) | C.I. (95.4%) |
| 1.80 | 5.81 (n = 10) | 1.44–1.82 | 0.90 | 3.42 (n = 7) | 0.81–0.93 |
| 2.00 | 5.30 (n = 9) | 1.70–2.10 | 1.00 | 4.49 (n = 7) | 0.91–1.09 |
| 5.00 | 1.46 (n = 10) | 4.74–5.03 | 3.50 | 0.99 (n = 8) | 3.42–3.56 |
| 12.00 | 0.57 (n = 10) | 11.61–11.87 | 5.00 | 0.44 (n = 8) | 5.07–5.16 |
| 36.36 | 1.25 (n = 10) | 32.55–34.22 | 7.00 | 0.37 (n = 7) | 6.77–6.87 |
| Linearity | Linearity | ||
|---|---|---|---|
| R 2 (coefficient of linearity) | ≥0.999 (n = 13) | R 2 (coefficient of linearity) | ≥ 0.995 (n = 10) |
| Range, μg ml−1 | 0.90 to at least 37.00 | Range, μg ml−1 | 0.93 to at least 10.00 |
| LoD, μg ml−1 | 0.30 | LoD, μg ml−1 | 0.31 |
| LoQ, μg ml−1 | 0.90 | LoQ, μg ml−1 | 0.93 |
| Recovery | Recovery | ||
|---|---|---|---|
| Nominal [irinotecan], μg ml−1 | Recovery (%) | Nominal [irinotecan], μg ml−1 | Recovery (%) |
| 2.50 | 96.5 | 1.08 | 103.8 |
| 5.00 | 98.4 | 2.75 | 96.3 |
| 7.50 | 101.9 | 4.50 | 99.6 |
| 11.00 | 99.9 | 5.25 | 103.3 |
| 15.00 | 100.2 | ||
| 17.00 | 100.1 | ||
| 24.00 | 98.1 | ||
Calibration curves prepared on different days showed good linearity and acceptable results of the back-calculated concentrations over the validated range of 0.90–37.00 μg ml−1 for aqueous dilutions of irinotecan and 0.93–10.00 μg ml−1 for irinotecan determination in plasma. Pearson's coefficient of determination R2 was ≥0.995 for all replicates in both matrices. For all standards employed in all replicate calibration curves within the validation experiments, the standard concentration was back-calculated within ±7.6% and ±10.8% of their nominal concentration (±14.5% and ±19.6% at the LOQ) in aqueous dilutions of irinotecan and in plasma, respectively, indicating acceptable assay accuracy (bias(%)). Precision, expressed as CV%, on all quality control levels was determined to be ≤5.81% and ≤4.49% for repeatability as well as ≤6.62% and ≤9.91% for intermediate precision in the aqueous and plasma matrix, respectively. Thus, repeatability and intermediate precision of the method proposed herein were both acceptable across the useful analytical range in both matrices. The very comparable technical characteristics (i.e. performance) of the assay in aqueous solutions of the irinotecan dosage form and in irinotecan-spiked human plasma are a good indication of the specificity of the assay in plasma.
The LOD and LOQ were determined to be 0.30 and 0.90 μg ml−1, respectively, and the useful analytical range of the spectrophotometric assay was quite sufficient for irinotecan quantitative determination in pharmaceutical dosage forms.
The expected irinotecan concentration range in patient plasma after standard dose administration and subsequent elimination is approximately 15 ng ml−1 to 4 μg ml−1.1–4 The useful analytical range of the method proposed herein in plasma is 0.93 to at least 10.00 μg ml−1, covering only the upper region of irinotecan clinical concentrations and the therapeutic window. The irinotecan total AUC is widely considered an essential pharmacological parameter for calculation of body clearance of the drug and correlates to the SN-38 AUC and antitumor activity.22,23 Pharmacokinetic modelling studies have demonstrated that the irinotecan total AUC can be predicted adequately using three timed plasma samples, within the first 5.5 h or 6 h after drug infusion.2,24 Within this time range, irinotecan levels are typically above or at the LOQ of the assay proposed herein in the plasma matrix.2,24 Additionally, quantifying irinotecan in the plasma of treated patients within 0.5–3.0 h from drug infusion, when irinotecan levels are in the upper region of the therapeutic window, is considered sufficient to decide whether a subsequent dose adjustment is required.10 Our assay in plasma may thus be useful for the purposes of personalised dose adjustment or for calculating the irinotecan AUC based on PK models.
Stabilities
We found that HCl-treated aqueous dilutions of the irinotecan solution for injection at pH 0.2 remained stable at RT in the dark, for at least two weeks. Irinotecan in HCl/NaCl-treated plasma remained stable at RT for at least 3 hours and in a −20 °C freezer for a period of at least 2 weeks. A mild turbidity, apparent to the eye, was developed after storage of HCl/NaCl-treated plasma for several hours at RT. Quantitative determination of irinotecan in mildly turbid samples was sufficiently accurate and precise when the standards used for calibration were stored under the same conditions, having also developed a mild turbidity. In concludion, the stability of the treated samples in both matrices was quite sufficient for analytical purposes.Interferents' effect
The effect of interferents (hemolysis and icterus) on analyte recovery upon irinotecan determination in plasma was investigated. This effect was not expected to be significant since the plasma blank contribution to absorbance is taken into account in the proposed assay protocol, through the usage of a second wavelength (455 nm). We did not investigate the effect of lipemia since high-speed centrifugation (10000 × g for 15 min), incorporated in our protocol, is appropriate for lipemia removal in plasma samples.25 The fitted linear data in the calibration plots obtained for each interferent and the baseline sample were described by the following equations:| Icteric serum: A(400–455 nm) = 0.0450 [irinotecan, μg ml−1] − 0.0077, R2 = 0.999. |
| Mildly-hemolysed plasma: A(400–455 nm) = 0.0385 [irinotecan, μg ml−1] + 0.0022, R2 = 0.999. |
| Non-icteric, non-hemolysed plasma: A(400–455 nm) = 0.0419 [irinotecan, μg ml−1] − 0.0040, R2 = 0.999. |
In all cases there was a lower than 10% difference in the slope of the calibration curves in the presence of the interferents relative to their absence, indicating a non-significant effect of likely interferents on the proposed assay. Moreover, the calculated change (bias) in the analyte concentration for each irinotecan concentration investigated (Table 2) was found to be lower than ±10%, indicating an acceptable bias.
| [Irinotecan], μg ml−1 | Icteric Samples | Hemolysates |
|---|---|---|
| Bias% | Bias% | |
| 2.13 | +1.22 | +6.56 |
| 4.24 | +2.59 | −2.71 |
| 6.35 | +4.60 | −6.77 |
| 8.44 | +5.57 | −8.7 |
| 10.52 | +6.37 | −7.95 |
| 12.59 | +7.23 | −7.36 |
| 14.65 | 7.24 | −5.41 |
No interference of the excipients used in the solution for injection under test, namely lactic acid, sorbitol and potassium hydroxide, was noted in either matrix. Other common excipients used in the pharmacopeia were not expected to interfere, due to their loss of solubility at the extreme pH of the assay or when judging based on the presumed mechanism of colour change.
Extraction efficiency
It is reported that irinotecan remains bound in the blood mainly to plasma proteins (47%) and erythrocytes (33%).26 The very good linear relationship we observed between the nominal standard irinotecan concentration in plasma and measured absorbance indicates a constant irinotecan loss when applying the proposed assay in plasma. Indeed, when we evaluated irinotecan extraction efficiency from plasma by the HCl/NaCl method, using duplicates at nine QC concentrations, we found it to be in the range of 77.9 (at LOQ) – 68.5% (at 10 μg ml−1 irinotecan). In all cases, duplicate extraction recoveries did not differ by more than 6.5%. Literature studies have confirmed similar extraction recoveries from plasma, irrespective of the irinotecan concentration: published recoveries were 66–70% upon deproteinization with three volumes of 0.1% acetic acid in methanol relative to the plasma sample volume or with one volume of acetonitrile relative to the plasma sample volume.3,10,27Comparison of the spectrophotometric method with the HPLC method
In order to further evaluate the analytical performance of the proposed method in aqueous dilutions of pharmaceutical dosage forms against a standard method, a scatter plot was constructed after analysing aqueous dilutions of the irinotecan solution for injection by the proposed method in parallel with a validated literature method, based on LC/UV. Correlation analysis demonstrated a linear relationship between the two series of measurements (Fig. 4) with each of the methods – LC/UV and the proposed photometric method – (R2 = 0.995), in aqueous dilutions of a pharmaceutical dosage form. The slope was determined to be close to 1 (0.973), while the y-intercept was 0.0064, demonstrating very good agreement and acceptable bias between the two methods. The residual sum of squares was low (6.7), indicating a good linear relationship between the concentrations determined by the two methods.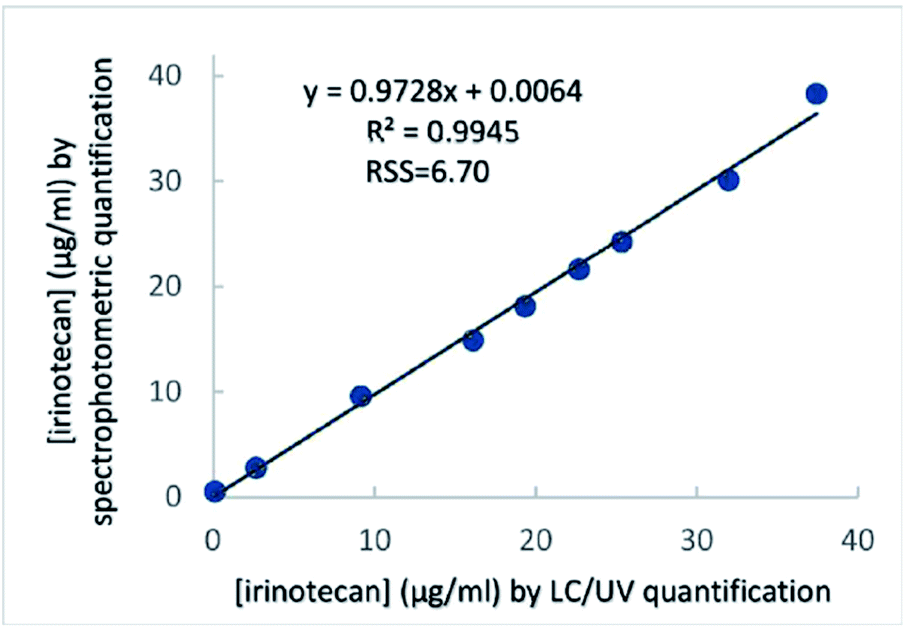 | ||
| Fig. 4 Correlation analysis between two series of measurements performed by LC/UV and the proposed photometric assay. The concentrations of nine aqueous QC irinotecan samples were determined by each method and were plotted one against the other. | ||
Our results above have validated the proposed spectrophotometric method for determining the irinotecan concentration in the plasma, within the upper region of its therapeutic window. They have also validated the same method for determining the irinotecan concentration in a pharmaceutical dosage form. It is worth considering that the overall procedure in plasma requires, for the full monitoring of a patient (i.e. measurement of 3–5 samples, including plasma pre-treatment and irinotecan quantification using a standard curve), about 35 min. The overall procedure for analysis of aqueous dilutions of irinotecan requires only a couple of minutes per sample, after standard curve construction.
When compared to the literature spectrophotometric methods for irinotecan determination in aqueous samples, the proposed method is superior in being more selective since the increase in absorbance at 400 nm requires the presence of specific structural characteristics (quinolinium moiety). On the other hand, since camptothecin-based molecules share the required structural feature, the assay is most likely applicable to all CPT derivatives and their quinolinium-bearing metabolites SN-38 and SN-38G (SN-38 glucuronide).
The proposed assay is also more specific in comparison to the alternative assay based on ion pair formation with acidic dyes,16 since the capacity for ion pair formation is a common property of molecules carrying a positive charge at acidic pH. Finally, detection at 400 nm is much more specific than that at 247 nm, employed by an alternative literature method.15 To our knowledge, no absorbance-based method for irinotecan analysis in plasma has been proposed.
Since SN-38 also bears the quinolinium moiety, it is safe to presume that SN-38 and irinotecan would be simultaneously detected by the proposed method. Studies have demonstrated that SN-38 levels are at least 75-fold lower than the corresponding irinotecan concentrations, within the 0.5–3.0 h interval after drug infusion.1–4 As a result, concomitant determination of SN-38 and irinotecan by the method proposed herein is not expected to significantly affect irinotecan determination accuracy and precision within the recommended sampling interval.
The new method's simplicity and velocity, requirements for very basic laboratory equipment (vis spectrophotometer and centrifuge) and common reagents (HCl, NaCl) mean that it can be more widely used in basically equipped laboratories. Last, but not least, the assay involves no use of organic solvents, rendering it a greener alternative than HPLC and ion-pair formation-based methods.
Experimental
Chemicals and reagents
The irinotecan Pharmaceutical Dosage Form used was IRITEC Solution for Infusion (irinotecan hydrochloride, trihydrate 40 mg/2 ml (Vianex Pharmaceutical Company, Attica, Greece)), containing lactic acid, sorbitol (Ε420), hydrochloric acid and potassium hydroxide as excipients. Acetonitrile and water used in the LC/UV experiments were HPLC Grade (Chem lab NV, Zedelgem, Belgium). Sodium hydroxide, sodium chloride, phosphoric acid, glacial acetic acid and potassium permanganate were all ACS reagent grade from JT Baker. Hydrochloric acid, fuming 37%, for analysis was from Merck, while sodium laureth sulphate (SLES) was from Goldschmidt AG (Essen, Germany). Ascorbic acid, ferrozine and iron standard were supplied in the form of a ferrozine method-based iron determination kit by Biosis (Attica, Greece). Standard aqueous buffer solutions from Fisher Scientific were used to calibrate the pH meter. The plasma used in all experiments was pooled plasma collected from healthy volunteers not undergoing any medical treatment.Apparatus
HPLC experiments were performed using an Ultimate 3000 High Performance Liquid Chromatography System from Dionex Corporation (Sunnyvale, CA, USA), equipped with a PDA 3000 detector also from Dionex Corporation. RP-HPLC analysis was performed using an ACE 5 column (110 Å, C18, 5 μm, 250 × 4.6 mm) from Hichrom (Leicestershire, UK). Photometric measurements were conducted in an Ultrospec 2100 pro UV/vis photometer (Amersham Biosciences). The spectrophotometer was controlled using Datrys Lite software (Biochrom Ltd, Cambridge, UK). pH measurements were performed on a Consort C860 model pH meter (Consort bvba, Belgium), with a Consort combination glass electrode.Preparation of standards and quality control (QC) samples
Stock solutions of irinotecan were freshly prepared in water at concentrations of 100 and 200 μg ml−1, prior to usage. They were then used to prepare fresh irinotecan standards and quality control (QC) solutions by diluting an appropriate amount of the stock solution with water and adding 50 μL of each dilution to 1 ml of the respective blank matrix (water or blank plasma). For blank plasma preparation whole blood was collected into commercially available EDTA-treated tubes. Cells were removed from pooled blood collections after centrifugation for 10 min at 2000 × g. The resulting supernatant (blank plasma) was apportioned into 5 ml aliquots and stored at −20 °C. On the day of the experiment, blank plasma was thawed and spiked with the appropriate dilution of irinotecan standards or QCs, as above. After preparation in the relevant matrix, standards and QCs were processed as described below.Sample processing for irinotecan quantification
Plasma standards or plasma QC samples were pretreated by the addition of HCl to a final concentration of 1.30 M, in the presence of NaCl at 2.7% w/w, to ensure more prolonged clarity of the solution. Salt was added in the form of powder to minimize dilution. The mixture was incubated at RT for 10 min. This was followed by centrifugation at 15000 × g at RT for 10 min (or for 15 min in lipemic samples) and the supernatant was removed for subsequent use.
Aqueous standards or aqueous quality control solutions of irinotecan were simply pretreated by the addition of HCl to a final concentration of 1.30 M.
Spectrophotometric quantification of irinotecan
Upon spectrophotometric determination of irinotecan in pharmaceutical dosage forms, monochromatic analysis was applied at 400 nm, 5–10 min after sample acidification.Upon spectrophotometric determination of irinotecan in HCl/NaCl-treated plasma, bichromatic analysis was applied after sample centrifugation to eliminate the contribution of endogenous substances to absorption at 400 nm. 455 nm was chosen as the second wavelength, where only the interfering components of the HCl-treated plasma contribute to absorbance, and not irinotecan.
Spectrophotometric method validation
The proposed photometric method was validated as per the ICH21 in terms of the linearity, useful analytical range, specificity, accuracy, precision (repeatability and intermediate precision), limit of detection and limit of quantitation. Distinct validation studies were performed for the assay in plasma and the assay in aqueous solutions. The number of QC replicate measurements are mentioned in the Results section. For the method concerning pharmaceutical dosage form quantification, validation characteristics were established across the specified useful analytical range of the procedure. Validation parameters for the method in plasma were defined across the clinically relevant range (LOQ to around 7 μg ml−1). Accuracy, in particular, was determined by Method comparison studies (pharmaceutical dosage form matrix) and by Bias calculation upon standard concentration back-calculation as well as recovery studies (in both matrices). Further details on method validation procedures are provided in the ESI.†Method comparison studies
For method comparison studies, measurements in aqueous irinotecan QC samples were performed by the proposed spectrophotometric method in parallel to the HPLC/UV method developed herein. The HPLC separation method applied was based on closely related reversed-phase literature HPLC methods proposed for the analysis of pharmaceutical preparations of irinotecan or the analysis of irinotecan in plasma in its lactone form (pH around 3.0).9 We performed analysis on an ACE 5 column (110 Å, C18, 5 μm, 250 × 4.6 mm, Hichrom Ltd, Leicestershire, UK). The mobile phase consisted of acetonitrile: 20 mM phosphoric acid/sodium phosphate buffer (pH 3.2) at a relative volume ratio of 28:72. Analysis was isocratic with a total analysis time of 10 min. The flow rate was set to 1.0 ml min−1 and the column temperature to 29 °C and analysis was performed at 220 nm using a PDA detector. The injection volume was 20 μl. All standards and samples of irinotecan were prepared in 20 mM phosphoric acid/sodium phosphate buffer (pH 3.2). We performed triplicate measurements of nine concentrations of pharmaceutical dosage forms, covering the entire specified range of the analytical procedure, in order to minimize random variation effects.
Irinotecan extraction efficiency
Percentage irinotecan extraction efficiency from plasma was determined using duplicates of QC samples at nine concentration levels within the 1.0–5.0 μg ml−1 range. The entire pre-treatment protocol (i.e. pH lowering to 0.2, centrifugation and absorbance measurement) was applied to the plasma QC samples. Extraction efficiency was calculated as the ratio of the (A400–A455 nm) absorbance difference of the analyte in blank plasma spiked before treatment over the (A400–A455 nm) absorbance difference of the analyte spiked at the same concentration into the pre-treated blank plasma matrix.Verification of the applied LC/UV method
Verification of the applied LC/UV method was as per the ICH guidelines,21 as described for the spectrophotometric assay, above. The linearity, range, specificity, accuracy, precision (repeatability and intermediate precision), limit of detection and limit of quantitation were defined. Where relevant, validation characteristics were established across the specified range of the analytical procedure.Conclusions
The technical characteristics of the proposed spectrophotometric method (linearity, useful analytical range, precision, accuracy, specificity) are quite adequate for irinotecan quantification in pharmaceutical dosage forms. Acceptable bias, as well as lack of interference, were also demonstrated for the new method. We have moreover provided evidence of a strong positive correlation between our method and HPLC/UV analysis. The method validated herein is thus proposed as a simpler alternative to established HPLC methods for the quantitative determination of irinotecan in pharmaceutical dosage forms.Being an anticancer drug for which significant benefits from personalised dosing are expected, quick procedures are essential for monitoring irinotecan in treated patients. We have demonstrated that our spectrophotometric method can be employed after simple plasma pre-treatment for irinotecan quantification even in plasma. The technical characteristics of this modified assay (linearity, useful analytical range, precision, and accuracy) were determined to be appropriate for quantifying irinotecan within the upper region of its therapeutic window in plasma and could thus be useful for the purposes of personalised dose adjustment or for calculating the irinotecan AUC based on PK models.
HPLC methods are characterized by higher analysis cost due to the expensive equipment and consumables involved, lengthy analysis time and specialized personnel required. As a result of the use of organic solvents upon analysis and/or upon plasma pre-treatment, the environmental impact of chromatographic methods is greater. The same holds for one of the literature spectrophotometric methods based on the formation of ion-pair complexes. When further compared to the existing photometric methods, the proposed method is also superior in being more selective. This is since, unlike literature methods, absorbance is measured in the visible part of the electromagnetic spectrum, where interferences are less likely. Moreover, the increase in absorbance at 400 nm requires the presence of specific structural characteristics of the irinotecan molecule, rendering the assay specific. This new proposed method can be more widely used since it only requires very common laboratory reagents, a simple vis spectrophotometer and no organic solvents.
In conclusion, the proposed method is superior in terms of specificity, rapidity, simplicity, environmental impact and cost effectiveness and has appropriate technical characteristics to be used in routine analysis of irinotecan in bulk manufacture and in pharmaceutical formulations and upon monitoring of irinotecan levels in plasma within the upper region of its therapeutic window.
We are currently investigating the concentration of irinotecan in biological fluids (e.g. by binding to DNA-columns or by solid-phase extraction), before assaying. Such pre-treatment may lower the assay LOQ in plasma and expand its application towards characterising the pharmacokinetic attributes of novel irinotecan delivery systems/formulations.
Conflicts of interest
There are no conflicts of interest to declare.Abbreviations
| CPT-11 | Irinotecan |
| CPT | Camptothecin |
| 10-CPT | 10-Hydroxycamptothecin |
| SLES | Sodium laureth sulphate |
| RSS | Residual sum of squares |
| AUC | Area under the plasma concentration versus time curve |
| CV | Coefficient of variation |
| C.I. | Confidence interval |
Acknowledgements
We would like to acknowledge Prof. George-Albert Karikas, Emeritus Professor of Biochemistry-Clinical Chemistry, University of West Attica, Greece, for assistance with the provision of irinotecan solution for injection.References
- G. G. Chabot, Clinical Pharmacokinetics of Irinotecan, Clin. Pharmacokinet., 1997, 33, 245 CrossRef CAS.
- R. H. J. Mathijssen, R. J. van Alphen, M. J. A. de Jonge, J. Verweij, P. de Bruijn, W. J. Loos, K. Nooter, L. Vernillet, G. Stoter and A. Sparreboom, Sparse-data set analysis for irinotecan and SN-38 pharmacokinetics in cancer patients co-treated with cisplatin, Anti-Cancer Drug., 1999, 10, 9 CrossRef CAS.
- E. Marangon, B. Posocco, E. Mazzega and G. Toffoli, Development and Validation of a High-Performance Liquid Chromatography–Tandem Mass Spectrometry Method for the Simultaneous Determination of Irinotecan and Its Main Metabolites in Human Plasma and Its Application in a Clinical Pharmacokinetic Study, PLoS ONE, 2015, 10, e0118194 CrossRef.
- M. Czejka, B. Gruenberger, A. Kiss, A. Farkouh and J. Shueller, Pharmacokinetics of Irinotecan in Combination with Biweekly Cetuximab in Patients with Advanced Colorectal Cancer, Anticancer Res., 2010, 30, 2355 CAS.
- F. M. de Man, A. K. L. Goey, R. H. N. van Schaik, R. H. J. Mathijssen and S. Bins, Individualization of Irinotecan Treatment: A Review of Pharmacokinetics, Pharmacodynamics, and Pharmacogenetics, Clin. Pharmacokinet., 2018, 57, 1229 CrossRef CAS.
- Z. P. Hu, X. X. Yang, X. Chen, E. Chan, W. Duan and S. F. Zhou, Simultaneous determination of irinotecan (CPT-11) and SN-38 in tissue culture media and cancer cells by high performance liquid chromatography: application to cellular metabolism and accumulation studies, J. Chromatogr. B Analyt. Technol. Biomed Life Sci., 2007, 850, 575 CrossRef CAS.
- C. Atasilp, P. Chansriwong, E. Sirachainan, T. Reungwetwattana, A. Puangpetch, S. Prommas, S. Sirilerttrakul, B. Rerkarmnuaychoke, S. Wongwaisayawan and C. Sukasem, Determination of irinotecan, SN-38 and SN-38 glucuronide using HPLC/MS/MS: Application in a clinical pharmacokinetic and personalized medicine in colorectal cancer patients, J. Clin. Lab. Anal., 2018, 32, e22217 CrossRef.
- R. K. Konda, B. R. Chandu and K. B. C. Sekhar, An improved HPLC-UV method for the estimation of Irinotecan in bulk and tablet dosage, Int. J. Pharm. Biomed. Res., 2011, 2, 188 CAS.
- R. Mullangi, P. Ahlawat and N. R. Srinivas, Irinotecan and its active metabolite, SN-38: review of bioanalytical methods and recent update from clinical pharmacology perspectives, Biomed. Chromatogr., 2010, 24, 104 CrossRef CAS.
- G. Bonazza, S. Tartaggia, G. Toffoli, F. Polo and D. Salvatore, A fast method for the detection of irinotecan in plasma samples by combining solid phase extraction and differential pulse voltammetry, Anal. Bioanal. Chem., 2020, 412, 1585 CrossRef CAS.
- M. Tommasini, E. Pellizzoni, V. Iacuzzi, E. Marangon, P. Posocco, C. Forzato, P. Bertoncin, G. Toffoli, M. Resmini and F. Berti, Fluorescent Imprinted Nanoparticles for the Effective Monitoring of Irinotecan in Human Plasma, Nanomaterials, 2020, 10, 1707 CrossRef CAS.
- M. I. Rodríguez Cáceres, I. Durán-Merás, N. E. Ornelas Soto, P. L. López de Alba and L. López Martínez, Spectrofluorimetric determination of irinotecan in the presence of oxidant agents and metal ions, Talanta, 2008, 74, 1484 CrossRef.
- H. Y. Fu, H. L. Wu, J. F. N. Yong, J. Y. Hong, Y. Z. Ru and Q. Yu, Highly sensitive fluorescence quantification of irinotecan in biological fluids with the aid of second-order advantage, Chin. Chem. Lett., 2010, 21, 1482 CrossRef CAS.
- L. A. Serrano, Y. Yang, E. Salvati, F. Stellacci, S. Krol and S. Guldin, pH-Mediated molecular differentiation for fluorimetric quantification of chemotherapeutic drugs in human plasma, Chem. Commun., 2018, 54, 1485 RSC.
- S. Sharma and M. C. Sharma, Development and validation of Spectrophotometric method and TLC Densitometric Determination of Irinotecan HCl in pharmaceutical dosage forms, Arab. J. Chem., 2016, 9, S1368 CrossRef CAS.
- V. M. Balaram and J. V. Rao, Spectrophotometric Determination of Irinotecan in Pharmaceutical Formulations, Asian J. Chem., 2008, 20, 4205 CAS.
- N. Karadas, S. Sanli, B. Akmese, B. Dogan-Topal, A. Can and S. A. Ozkan, Analytical application of polymethylene blue-multiwalled carbon nanotubes modified glassy carbon electrode on anticancer drug irinotecan and determination of its ionization constant value, Talanta, 2013, 115, 911 CrossRef CAS.
- M. I. Rodríguez Cáceres, I. Durán-Merás, N. E. Ornelas Soto, P. L. López de Alba and L. López Martínez, Spectrofluorimetric determination of irinotecan in the presence of oxidant agents and metal ions, Talanta, 2008, 74, 1484 CrossRef.
- M. R. Nunzio, Y. Douhal, J. A. Organero and A. Douhal, Structural and photodynamic properties of the anti-cancer drug irinotecan in aqueous solutions of different pHs, Phys. Chem. Chem. Phys., 2018, 20, 14182 RSC.
- K. M. Solntsev, E. N. Sullivan, L. M. Tolbert, S. Ashkenazi, P. Leiderman and D. Huppert, Excited-state proton transfer reactions of 10-Hydroxycamptothecin, J. Am. Chem. Soc., 2004, 126, 12701 CrossRef CAS.
- European Medicines Agency, ICH Q2 (R1) Validation of Analytical Procedures: Text and Methodology-step 5, Published 01/06/ 1995 Search PubMed.
- G. G. Chabot, D. Abigerges, G. Catimel, S. Culine, M. de Forni, J.-M. Extra, M. Mahjoubi, P. Herait, J.-R. Armand, R. Bugat, M. Clavel and M. E. Marty, Population pharmacokinetics and pharmacodynamics of irinotecan (CPT-11) and active metabolite SN-38 during phase I trials, Ann. Oncol., 1995, 6, 141 CrossRef CAS.
- M. de Forni, R. Bugat, G. G. Chabot, S. Culine, J.-M. Extra, A. Gouyette, I. Madelaine, M. E. Marty and A. Mathieu-Boué, Phase I and Pharmacokinetic Study of the Camptothecin Derivative Irinotecan, Administered on a Weekly Schedule in Cancer Patients, Cancer Res., 1994, 54, 4347 CAS.
- G. G. Chabot, Limited sampling models for simultaneous estimation of the pharmacokinetics of irinotecan and its active metabolite SN-38, Cancer Chemother. Pharmacol., 1995, 36, 463 CrossRef CAS.
- M. J. Castro-Castro, B. Candás-Estébanez, M. Esteban-Salán, P. Calmarza, T. Arrobas-Velilla, C. Romero-Román, M. Pocoví-Mieras and J. A. Aguilar-Doreste, Removing Lipemia in Serum/Plasma Samples: A Multicenter Study, Ann. Lab. Med., 2018, 38, 518 CrossRef CAS.
- O. Combes, J. Barré, J. Duché, L. Vernillet, Y. Archimbaud, M. P. Marietta, J. P. Tillement and S. Urien, Vitro Binding and Partitioning of Irinotecan (CPT-11) and its Metabolite, SN-38, in Human Blood, Invest. New Drugs, 2000, 18, 1 CrossRef CAS.
- S. Ragot, P. Marquet, F. Lachâtre, A. Rousseau, E. Lacassie, J. M. Gaulier, J. L. Dupuy and G. Lachâtre, Sensitive determination of irinotecan (CPT-11) and its active metabolite SN-38 in human serum using liquid chromatography–electrospray mass spectrometry, J. Chromatogr. B Biomed. Sci. Appl., 1999, 736, 175 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the methodology of assay validation and results regarding HPLC method verification are reported. See DOI: 10.1039/d0ay02201b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |