
A strategy based on isocratic and linear-gradient high-speed counter-current chromatography for the comprehensive separation of platycosides from Platycodi radix†
Jinqian
Yu‡
 a,
Xiangwei
Chang‡
bd,
Huasheng
Peng
b,
Xiao
Wang
a,
Jutao
Wang
b,
Daiyin
Peng
b and
Shuangying
Gui
*bcd
a,
Xiangwei
Chang‡
bd,
Huasheng
Peng
b,
Xiao
Wang
a,
Jutao
Wang
b,
Daiyin
Peng
b and
Shuangying
Gui
*bcd
aSchool of Pharmaceutical Sciences, Qilu University of Technology (Shandong Academy of Sciences), Jinan, 250014, China
bCollege of Pharmacy, Anhui University of Chinese Medicine, Hefei, 230012, China. E-mail: guishy0520@126.com
cAnhui Province Key Laboratory of Pharmaceutical Technology and Application, Anhui University of Chinese Medicine, Hefei, 230012, China
dInstitute of Pharmaceutics, Anhui Academy of Chinese Medicine, Hefei, 230012, China
First published on 7th January 2021
Abstract
Platycosides, the generally recognized main active constituents of Platycodi radix, have been studied extensively for their wide pharmacological activities. Herein, we have successfully developed an efficient method for the enrichment and comprehensive isolation of platycosides from Platycodi radix by MCI resin column chromatography (CC) and two different modes of high-speed counter-current chromatography (HSCCC). MCI resin CC was the preferable enrichment operation for platycosides from the 70%-ethanol extract of Platycodi radix and rendered target platycosides when eluted by 60% aqueous methanol solution. As for the separation, two different modes, including isocratic HSCCC and linear-gradient HSCCC, were applied together to separate the platycosides using a mixture of ethyl acetate, n-butanol and water coupled with evaporative light scattering detection, for the first time. Isocratic HSCCC was applied to separate crude platycosides from Platycodi radix using ethyl acetate–n-butanol–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2, v/v), yielding seven pure platycosides (compounds 1–6, 8) and two fractions of enriched mixtures of compounds 7, 9, 10, and 11. Linear-gradient HSCCC was employed to rapidly separate compounds 7, 9, 10, and 11 by constantly changing the proportions of ethyl acetate and n-butanol in the ethyl acetate–n-butanol–water solvent system. Finally, platycoside E (1), deapio-platycodin D3 (2), platycodin D3 (3), deapio-platycodin D2 (4), platycodin D2 (5), platycodin D (6), polygalacin D2 (7), polygalacin D (8), and three tautomers, namely 2′′-O-acetylplatycodin D (9) and 3′′-O-acetylplatycodin D (9′), 2′′-O-acetylpolygalacin D2 (10) and 3′′-O-acetylpolygalacin D2 (10′), and 2′′-O-acetylpolygalacin D (11) and 3′′-O-acetylpolygalacin D (11′), were obtained from 300 mg of crude platycosides from Platycodi radix.
:1:2, v/v), yielding seven pure platycosides (compounds 1–6, 8) and two fractions of enriched mixtures of compounds 7, 9, 10, and 11. Linear-gradient HSCCC was employed to rapidly separate compounds 7, 9, 10, and 11 by constantly changing the proportions of ethyl acetate and n-butanol in the ethyl acetate–n-butanol–water solvent system. Finally, platycoside E (1), deapio-platycodin D3 (2), platycodin D3 (3), deapio-platycodin D2 (4), platycodin D2 (5), platycodin D (6), polygalacin D2 (7), polygalacin D (8), and three tautomers, namely 2′′-O-acetylplatycodin D (9) and 3′′-O-acetylplatycodin D (9′), 2′′-O-acetylpolygalacin D2 (10) and 3′′-O-acetylpolygalacin D2 (10′), and 2′′-O-acetylpolygalacin D (11) and 3′′-O-acetylpolygalacin D (11′), were obtained from 300 mg of crude platycosides from Platycodi radix.
Introduction
Platycodon grandiflorum A. DC (Campanulaceae), the balloon flower, is a perennial flowering herb and widely distributed in China, South Korea, Japan, Mongolia and Russia. The roots derived from this plant have been documented as “Platycodi radix” in the Chinese Pharmacopoeia for the treatment of cough and phlegm, chest congestion, sore throat, and so on.1 Phytochemical reports on Platycodi radix have recorded the isolation of various platycosides, flavonoids, and fatty acid, with the first one being accepted as the main active constituent with broad pharmacological properties, including anti-inflammatory, anti-bacterial, anti-tumor, anti-oxidative and anti-hepatic injury roles.2–6 Platycosides belong to the family of pentacyclic triterpenoid saponins with aglycones assigned to polygalacic acid, platycodic acid and platycodigenic acid, and two carbohydrate side chains linked to C-3 and C-28. Nowadays, platycosides have been widely used as dietary supplements to cure respiratory diseases, especially pulmonary diseases, which make the isolation of platycosides from Platycodi radix more and more attractive not only for further pharmacological investigations but also for quality control. Currently, column chromatography and high-speed counter-current chromatography (HSCCC) have been used to separate platycosides from Platycodi radix. Kim et al.7,8 separated six major platycosides and four minor acetylated platycosides from Platycodi radix using a multi-step process including HSCCC and preparative reversed-phase high-performance liquid chromatography (RP-HPLC), which need high consumption of solvents and time.Consequently, an efficient separation method for the comprehensive separation of platycosides from Platycodi radix is urgently required. Nowadays, HSCCC has been accepted as a promising separation method due to its ability of liquid–liquid partitioning, which depends on the appropriate partition coefficients (KD) of the targets within the range of 0.5–2.0 in corresponding solvent systems.9–13 However, as for targets in complex extracts with a wide range of KD values, traditional isocratic HSCCC elution usually results in considerable time and solvent consumption. Hence, gradient HSCCC elution techniques, mainly including solvent gradient, salting-out gradient, and pH gradient,14,15 were developed to remedy the disadvantages of isocratic HSCCC mentioned above; among these, solvent-gradient HSCCC elution has become a prospective separation technique. Previous studies on solvent-gradient HSCCC have led to the frequent application of step-gradient and linear-gradient HSCCC elution modes for separation, and the latter one has been considered far more efficient and can operate as binary gradient HPLC using two gradient solvents as the mobile phase.15
Ethyl acetate, n-butanol and water are three representative solvents with low-to-high polarities, which are usually used as isocratic and linear-gradient solvents to separate the target components. As for isocratic HSCCC, the upper and lower phases of ethyl acetate–n-butanol–water are used as the stationary and mobile phases in the FWD (forward)-IN (head–end) mode, while in linear-gradient HSCCC, water saturated by ethyl acetate is used as the stationary phase, and ethyl acetate–n-butanol with changing ratios is used as the mobile phase in the FWD (forward)-OUT (end–head) mode.15 Herein, an efficient method combining isocratic HSCCC with linear-gradient HSCCC using the solvents ethyl acetate, n-butanol and water was successfully developed to separate platycoside E (1), deapio-platycodin D3 (2), platycodin D3 (3), deapio-platycodin D2 (4), platycodin D2 (5), platycodin D (6), polygalacin D2 (7), polygalacin D (8), and the three tautomers, namely 2′′-O-acetylplatycodin D (9) and 3′′-O-acetylplatycodin D (9′), 2′′-O-acetylpolygalacin D2 (10) and 3′′-O-acetylpolygalacin D2 (10′), and 2′′-O-acetylpolygalacin D (11) and 3′′-O-acetylpolygalacin D (11′) (Fig. 1), from Platycodi radix using evaporative light scattering detection (ELSD).
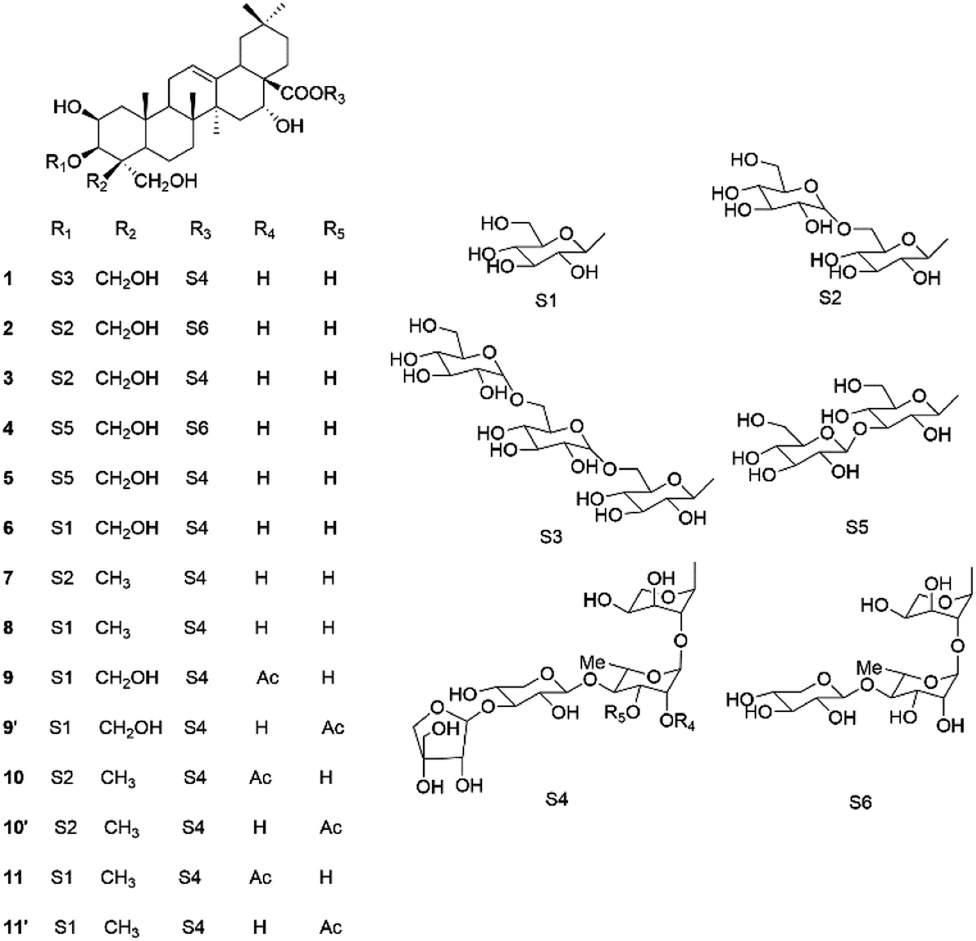 | ||
| Fig. 1 Chemical structures of the platycosides isolated from Platycodi radix. | ||
Experimental methods
Materials and reagents
Platycodi radix used in this study was obtained from the Inner Mongolia Traditional Chinese herbal medicine market. The title medicine was authenticated by Prof. Fengqin Zhou (Shandong University of Traditional Chinese Medicine) and its specimen (No. 20200711) was preserved at Anhui University of Chinese Medicine.Solvents used for the extraction and separation of platycosides, including ethanol, ethyl acetate, n-butanol, and acetonitrile, were all of analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Solvents used for HPLC and ESI-MS analysis were of analytical grade and purchased from Fisher Scientific (Tedia Company, USA). Water used for HSCCC, HPLC and ESI-MS was purified by an osmosis Milli-Q system (Millipore, Bedford, MA, USA). The MCI resin (CHP20P) was purchased from Beijing Update Biotechnology Co. Ltd (Mitsubishi Chemical Holdings, Japan).
Apparatus
A TBE-300A high-speed counter-current chromatography instrument (Tauto Biotechnique, Shanghai, China) was applied for isocratic HSCCC and linear-gradient HSCCC was equipped with a coil of multilayer polytetrafluoroethylene column (diameter of 1.6 mm and capacity of 300 mL) and a 20 mL manual sample loop. Two different pumps were used for these two different HSCCC elution modes: a TBP-5002 constant-flow pump (Tauto Biotechnique, Shanghai, China) for isocratic HSCCC separation and an LC3000 binary high-pressure gradient pump (Beijing Tong Heng Innovation Technology Co., Ltd, Beijing, China) for linear-gradient HSCCC separation. Other modules comprising of a SEDEX 80 ELSD with an N2000 workstation (Beijing Dikema Technology, Beijing, China) and a DC-0506 low-temperature bath at a constant temperature of 25 °C (Tauto Biotechnique, Shanghai, China) were equipped the same way in both isocratic HSCCC and linear-gradient HSCCC separation modes.As for the HPLC analysis of the crude platycosides and HSCCC elutions, an ACCHROM S6000 equipment and a Waters C18 column (250 mm × 4.6 mm, 5 μm) were used; the former was equipped with an ACCHROM S6000 solvent delivery unit, an ACCHROM S6000 Photodiode Array Detection and Alltech 3300 ELSD, an autosampler, and an ACCHROM S6000 column oven. As for the HRESI-MS analysis of the purified compounds, a Bruker Impact II mass spectrometer (Bruker Daltonic Inc., USA) was employed.
Preparation of crude platycosides
Particulate Platycodi radix (2 kg) was extracted under reflux conditions with 75% aqueous ethanol three times (2 h, 1 h, 1 h). The extracted solvents were combined together and then concentrated in a rotary evaporator to recover the ethanol solvent. After concentration, 500 g of crude extract was obtained. Subsequently, the crude ethanol extracts were subjected to MCI column chromatography and further eluted with water and 10% and 60% aqueous methanol to yield the crude platycosides. The combined platycosides were concentrated to remove the solvents and used for HSCCC separation.Measurement of partition coefficients
The KD values of the targets for isocratic and linear-gradient HSCCC separation were measured by HPLC. Solvent systems with different composition ratios of ethyl acetate, n-butanol and water (Tables 1 and 2) used for isocratic and linear-gradient HSCCC separation (each of 3 mL) were added into test tubes, followed by full equilibration. Approximately, 2 mg of sample was partitioned fully between equal volumes (each of 1 mL) of the upper and lower phases. After equilibration, equal volumes (300 μL) of each phase were transferred into separate tubes and dried by nitrogen to remove the effects of organic solvents. Then, 1 mL methanol was added to dissolve the obtained residues, which were further analyzed by HPLC to obtain the corresponding peak areas of the target compounds in the upper phase (Aupper) and lower phase (Alower). For isocratic HSCCC, the partition coefficient was defined as KD = Aupper/Alower. For linear-gradient HSCCC, the partition coefficient was defined as KCi = Alower/Aupper.| Solvent system | Ratio (v/v) | K D values of target compounds | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||
| n-Butanol–acetonitrile–water | 3:1:4 |
0.67 | 1.47 | 1.13 | 1.89 | 2.11 | 2.45 | 2.76 | 3.68 | 8.23 | 9.11 | 10.01 |
| Ethyl acetate–n-butanol–water | 2:1:3 |
0.09 | 0.23 | 0.15 | 0.63 | 0.32 | 0.41 | 4.24 | 1.97 | 1.34 | 1.46 | 3.52 |
| 1:1:2 |
0.68 | 1.05 | 0.92 | 2.54 | 1.15 | 1.78 | 8.47 | 4.37 | 3.97 | 4.11 | 7.51 | |
| 1:2:3 |
1.46 | 2.01 | 1.87 | 3.63 | 2.55 | 3.41 | 20.67 | 10.12 | 7.98 | 9.21 | 15.62 | |
| Compounds | Solvent system (ethyl acetate–n-butanol–water, v/v) | ln(KCi) = A (% wt) + B | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 100:0:100 |
90:10:100 |
80:20:100 |
70:30:100 |
60:40:100 |
50:50:100 |
A | B | R 2 | |
| 7 | 16.1 | 5.21 | 2.13 | 0.43 | 0.32 | 0.12 | −0.098 | 2.641 | 0.979 |
| 9 | 13.8 | 4.01 | 1.83 | 0.95 | 0.54 | 0.25 | −0.076 | 2.335 | 0.982 |
| 10 | 15.7 | 4.43 | 2.45 | 0.88 | 0.45 | 0.22 | −0.083 | 2.536 | 0.989 |
| 11 | 12.5 | 4.51 | 1.09 | 0.58 | 0.30 | 0.13 | −0.09 | 2.310 | 0.980 |
Preparation of two-phase solvent systems and sample solutions
For isocratic HSCCC separation, a solvent system of ethyl acetate–n-butanol–water (1:1:2, v/v) was prepared. The three different solvents were added into a separatory funnel to mix thoroughly and then separate fully, yielding the upper and lower phases. The two phases were employed as the stationary and mobile phases, respectively.
For linear-gradient HSCCC separation, ethyl acetate saturated with water and n-butanol saturated with water were used as the two mobile phases A and B, respectively, with the water-saturated ethyl acetate serving as the stationary phase. Firstly, 1000 mL ethyl acetate and 500 mL water were added into a separatory funnel to mix thoroughly and then separate fully, yielding the upper and lower phases, which were used as the mobile phase of A and the stationary phase, respectively. Then, 1000 mL n-butanol and 500 mL water were added into a separatory funnel to mix thoroughly and then separate fully, and the upper phase was used as the mobile phase of B.
The sample solutions for isocratic HSCCC separation were prepared by adding 0.3 g of crude platycosides into the mixed solvent with the lower and upper phases (5 mL of each phase). The samples for compounds 9 and 10, as well as the samples for compounds 7 and 11, were obtained three times by isocratic HSCCC separation. The sample solutions for linear-gradient HSCCC separation were prepared by adding 0.1 g of the enriched fraction into the mixed solvent made of mobile phase A and the stationary phase (5 mL of each phase).
The HSCCC separation procedure
For isocratic HSCCC separation, the solvent system of ethyl acetate–n-butanol–water (1:1:2, v/v) was applied. Before separation, TBE-300A was set to the FWD-IN mode. At first, the stationary phase was pumped into the HSCCC separation column at a flow rate of 20.0 mL min−1 until the solvent filled the column. Then, the mobile phase was pumped at a flow rate of 2.0 mL min−1 to achieve equilibrium with the stationary phase, and meanwhile, the column was rotated at a speed of 800 rpm. Finally, the prepared sample solutions were injected into the sample loop for further separation in the HSCCC column with a constant flow of the mobile phase.
For linear-gradient HSCCC separation, two solvents were used as two mobile phases, namely A and B. Before separation, TBE-300A was set to the FWD-OUT mode. At first, the stationary phase was pumped into the HSCCC separation column at a flow rate of 20.0 mL min−1 until the solvent filled the column. Then, mobile phase A was pumped at a flow rate of 2.0 mL min−1 to achieve equilibrium with the stationary phase, and meanwhile, the column was rotated at a speed of 800 rpm. Finally, the prepared sample solutions were injected into the sample loop for further separation in the HSCCC column by the set gradient solvents of the mobile phases A and B.
An evaporative light-scattering detector was used for both isocratic HSCCC and linear-gradient HSCCC to monitor the separation, and the HSCCC elutions were collected every 10 mL.
HPLC analysis and identification of the fractions
An ACCHROM S6000 equipment with an evaporative light-scattering detector and a Waters C18 column (250 mm × 4.6 mm, 5 μm) was used for the HPLC analysis of the crude platycosides and HSCCC fractions. The mobile phase was designated as acetonitrile (A) and water (B), with a gradient elution system of 0–4 min, 5–12% A; 4–6 min, 12–20.5% A; 6–25 min, 20.5–23% A; 25–32 min, 23–23.5% A; 32–45 min, 23.5–24% A; 45–55 min, 24–26% A; 55–65 min, 26–46% A. The flow rate was set as 1.0 mL min−1. The separated compounds were elucidated by comparing the HRESI-MS and NMR data with the literature.Results and discussion
Optimization of MCI resin column chromatography for the crude platycosides
It is well-known that Platycodi radix is rich in polysaccharides, which can be easily extracted together with platycosides using 70% ethanol. Hence, the removal of polysaccharides from the total extract is a key step to obtain the target platycosides. Herein, different resins and solvents were employed to investigate their effects on the enrichment of the platycosides. In this study, two kinds of macroporous resins (AB-8 and D101) using the elution solvent system of ethanol–water at different ratios (0:100, 10:90, 20:80, 30:70, 40:60, 50:50, 60:40, 70:30, 80:20, 95:5) and one pore resin (MCI CHP 20P) using the elution solvent system of methanol–water at different ratios (0:100, 10:90, 20:80, 30:70, 40:60, 50:50, 60:40, 70:30, 80:20, 90:10) were tested for the efficient enrichment of platycosides from the 70% ethanol extract (15 g). As indicated by the results, all these three resins could remove the polysaccharides and enrich the platycosides by column chromatography separation. Using the two kinds of macroporous resins (AB-8 and D101), the polysaccharide part was mainly obtained in water elutions and the platycoside part was enriched in the 80% and 90% ethanol–water elutions, with the other components that were polar than the platycosides eluted mainly in the 60% and 70% aqueous ethanol solutions. However, the platycosides enriched by these two macroporous resins (weights of platycosides enriched by AB-8 and D101 were 3.5 g and 4.3 g, respectively) were mixed with the minor polar components, enriching the content of the main platycoside (peak 6) to 13.2% and 9.5%, respectively. As for the pore resin MCI CHP 20P, the polysaccharide part was mainly obtained in the water and 10% ethanol–water elutions and the platycoside part was enriched in the 60% methanol aqueous solution, whereas the components with polarity larger than the platycosides gathered mainly in the 20%, 30%, 40% and 50% methanol aqueous solutions and the components with polarity lower than the platycosides gathered mainly in the 70%, 80% and 90% methanol aqueous solutions. This indicated that better enrichment of platycosides was achieved using the pore resin than the macroporous resins, which rendered the separation of platycosides from the other components with larger and smaller polarities. Overall, the results demonstrated MCI resin column chromatography as the preferable enrichment operation for platycosides from the 70% ethanol extract of Platycodi radix. Using water and 10% methanol aqueous solution to remove the polysaccharides and 60% methanol aqueous solution to obtain the platycosides (weighted 1.1 g) rendered the enriched content of the target platycoside (peak 6) at 45.5%.
Solvent system optimization for isocratic HSCCC
In traditional isocratic HSCCC elution, obtaining suitable KD values of the targets in the range of 0.5–2 in the chosen solvent system is a key step for successful separation (KD = Aupper/Alower). It is well-known that platycosides are of medium or high polarity, and medium or polar solvent systems are usually applied to separate this kind of saponins. As reported in the literature, solvent systems based on water, including chloroform–methanol–water–isopropanol, n-butanol–acetonitrile–water, n-butanol–water, n-hexane–n-butanol–water, ethyl acetate–methanol–water, have been broadly used for the separation of saponins. Since the crude platycosides were obtained by n-butanol extraction, solvent systems including n-butanol were applied to separate the crude platycosides, which can not only provide better solubility of the platycosides but also provide a wide range of polarity when coupled with different solvents. The solvent system of n-butanol–acetonitrile–water was tested first, in which compounds 1–7 showed better KD values since this solvent system exhibited high polarity. However, when n-butanol–acetonitrile–water (3:1:4, v/v) was applied for HSCCC to obtain the platycosides, no retention of the stationary phase was observed, which may be attributed to the nearly same density of the upper and lower phases of this solvent system (densities of the upper and lower phases were found to be 0.892 and 0.936, respectively). Subsequently, acetonitrile was withdrawn from this solvent system, resulting in a solvent system of n-butanol–water. However, when this solvent system was applied for the separation of crude platycosides, an emulsion appeared and existed for a long time. To eliminate the emulsion, different solvents were added to n-butanol–water. The results indicated that the crude platycosides exhibited better layering in ethyl acetate–n-butanol–water without emulsion formation. Then, various ratios of the ethyl acetate–n-butanol–water system were tested to obtain suitable KD values for the targets (Table 1). It was found that when the proportion of ethyl acetate–n-butanol–water was adjusted to 1:1:2, the best KD values, as well as better solubility of the crude platycosides, were obtained among the three different tested solvent systems of ethyl acetate–n-butanol–water. Finally, ethyl acetate–n-butanol–water (1:1:2, v/v) was applied for the separation of the crude platycosides, yielding seven pure compounds (1–6, 8), while compounds 9 and 10 co-eluted in a peak and compounds 7 and 11 drained together from the stationary phase at the end of the separation (Fig. 2A).
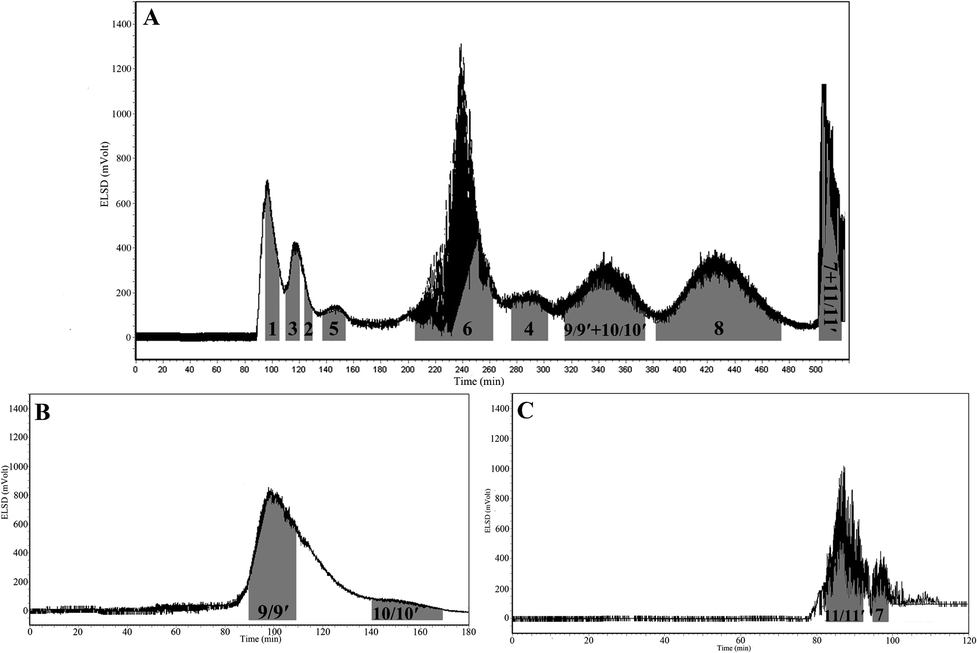 | ||
| Fig. 2 The high-speed counter-current chromatograms (HSCCC) of the separation of platycosides from Platycodi radix. Experimental conditions: (A) for isocratic HSCCC separation, the solvent system was ethyl acetate–n-butanol–water (1:1:2, v/v); for linear-gradient HSCCC, the upper organic phase was the stationary phase, and the lower aqueous phase was the mobile phase; (B) 0–40 min, 0–50% n-butanol; 40–120 min, 50–55% n-butanol; 120–180 min, 55–60% n-butanol for the separation of compounds 9, 9′, 10 and 10′, (C) 0–10 min, 0–50% n-butanol; 10–120 min, 50–70% n-butanol for the separation of compounds 7, 11 and 11′; ethyl acetate saturated with water and n-butanol saturated with water were used as the two mobile phases A and B, respectively, while water saturated with ethyl acetate acted as the stationary phase; revolution speed: 800 rpm; flow rate: 2.0 mL min−1; sample size: 300 mg; detection was with ELSD; retention of stationary phase: 53.3% in the first isocratic HSCCC separation, 56.4% in the linear-gradient HSCCC separation of 9, 9′, 10 and 10′, and 57.3% in the second dimension HSCCC separation of 7, 11 and 11′. | ||
Solvent system optimization for linear-gradient HSCCC
As for compounds 7, 9, 10 and 11, single isocratic HSCCC would not be enough to rapidly purify each of them. Linear-gradient HSCCC was chosen as the preferred technique not only for its efficient operation as binary gradient HPLC with two gradient solvents in the mobile phase, but also because it has employed ethyl acetate, n-butanol and water successfully as the linear-gradient solvents before. For linear-gradient HSCCC elution, the suitable KCi values (where i represents a logarithmic linear relation, KCi = Alower/Aupper) of the targets obtained in the chosen solvent system by the formula below. The constants A and B are key for successful separation and should exhibit a linear relationship with the changing content of n-butanol (% wt).15| ln(KCi) = A (% wt) + B |
In this case, the tested KCi values and calculated ln(KCi) values are shown in Table 2. The results indicated that the KCi values of compounds 7, 9, 10 and 11 increased with the decreasing content of n-butanol (% wt), which meant the increasing distribution of these four compounds in the upper organic phase. As for compounds 9 and 10, the tested KCi values in the solvent system of ethyl acetate–n-butanol–water (50:50:100, v/v) were a little higher than 0.2, indicating the main distribution of 9 and 10 in the organic phase. Based on the above results, the linear-gradient HSCCC system for compounds 9 and 10 was firstly set as 0–40 min, 0–50% n-butanol; 40–120 min, 50–60% n-butanol, which rendered most of 9 and 10 as a mixture within 40 min as the content of n-butanol increased from 55% to 60%, and only little of pure 9 and 10 were obtained. Subsequently, we lengthened the gradient range from 55% to 60% as: 0–40 min, 0–50% n-butanol; 40–120 min, 50–55% n-butanol; 120–180 min, 55–60% n-butanol. As shown in Fig. 2B, compounds 9 and 10 were eluted separately within 80 minutes as the content of n-butanol increased from 53% to 60%. As for compounds 7 and 11, the tested KCi values in the solvent system of ethyl acetate–n-butanol–water (50:50:100, v/v) were lower than 0.2, indicating the main distribution of 7 and 11 in the organic phase. Due to that, compounds 7 and 11 were drained together from the stationary phase composed of ethyl acetate–n-butanol–water (1:1:2, v/v) at the end of isocratic HSCCC separation. The linear-gradient HSCCC system for 7 and 11 was set as: 0–10 min, 0–50% n-butanol; 10–120 min, 50–70% n-butanol. As shown in Fig. 2C, compounds 7 and 11 were eluted separately within 25 minutes as the content of n-butanol increased from 62% to 66%.
After the optimization of the solvent systems for both isocratic HSCCC and linear-gradient HSCCC, the flow rate (1.0, 1.5, 2.0 mL min−1) and revolution speed (700, 800, 900 rpm) for HSCCC were also optimized. Finally, both isocratic HSCCC and linear-gradient HSCCC were carried out at a flow rate of 2.0 mL min−1 and a revolution speed of 800 rpm.
Purification of platycosides by isocratic HSCCC and linear-gradient HSCCC
300 mg of crude platycosides from Platycodi radix was successfully purified by two different elution modes, namely isocratic HSCCC and linear-gradient HSCCC, using the same solvent system composed of ethyl acetate–n-butanol–water.
Fig. 2A shows the isocratic HSCCC separation of crude platycosides using the ethyl acetate–n-butanol–water solvent system (1:1:2, v/v), yielding seven pure compounds (1–6, 8). While compounds 9 and 10 were co-eluted as a peak due to their closer KD values in the ethyl acetate–n-butanol–water system (1:1:2, v/v), compounds 7 and 11 drained together from the stationary phase at the end of the separation due to their relatively larger KD values in the employed solvent system. Therefore, another elution mode was applied in linear-gradient HSCCC separation for the separation of these four enriched compounds. As for compounds 9 and 10, the enriched mixture was subjected to a linear-gradient HSCCC system set as 0–40 min, 0–50% n-butanol; 40–120 min, 50–55% n-butanol; 120–180 min, 55–60% n-butanol, which eluted compounds 9 and 10 separately within 80 minutes when the content of n-butanol increased from 53% to 60% (Fig. 2B). As for compounds 7 and 11, the enriched mixture was subjected to a linear-gradient HSCCC system set as 0–10 min, 0–50% n-butanol; 10–120 min, 50–70% n-butanol, which could elute compounds 7 and 11 separately within 25 minutes when the content of n-butanol increased from 62% to 66% (Fig. 2C).
Finally, after both isocratic HSCCC and linear-gradient HSCCC modes of separation, 2.4 mg of platycoside E (1), 2.6 mg of deapio-platycodin D3 (2), 3.8 mg of platycodin D3 (3), 4.0 mg of deapio-platycodin D2 (4), 2.5 mg of platycodin D2 (5), 10.4 mg of platycodin D (6), 1.5 mg of polygalacin D2 (7), 7.2 mg of polygalacin D (8), 1.7 mg of 2′′-O-acetylplatycodin D (9) and 3′′-O-acetylplatycodin D (9′), 1.9 mg of 2′′-O-acetylpolygalacin D2 (10) and 3′′-O-acetylpolygalacin D2 (10′), and 1.4 mg of 2′′-O-acetylpolygalacin D (11) and 3′′-O-acetylpolygalacin D (11′) were obtained from 300 mg of crude platycosides from Platycodi radix, with purities of all components over 90.0%, as determined by HPLC (Fig. 3).
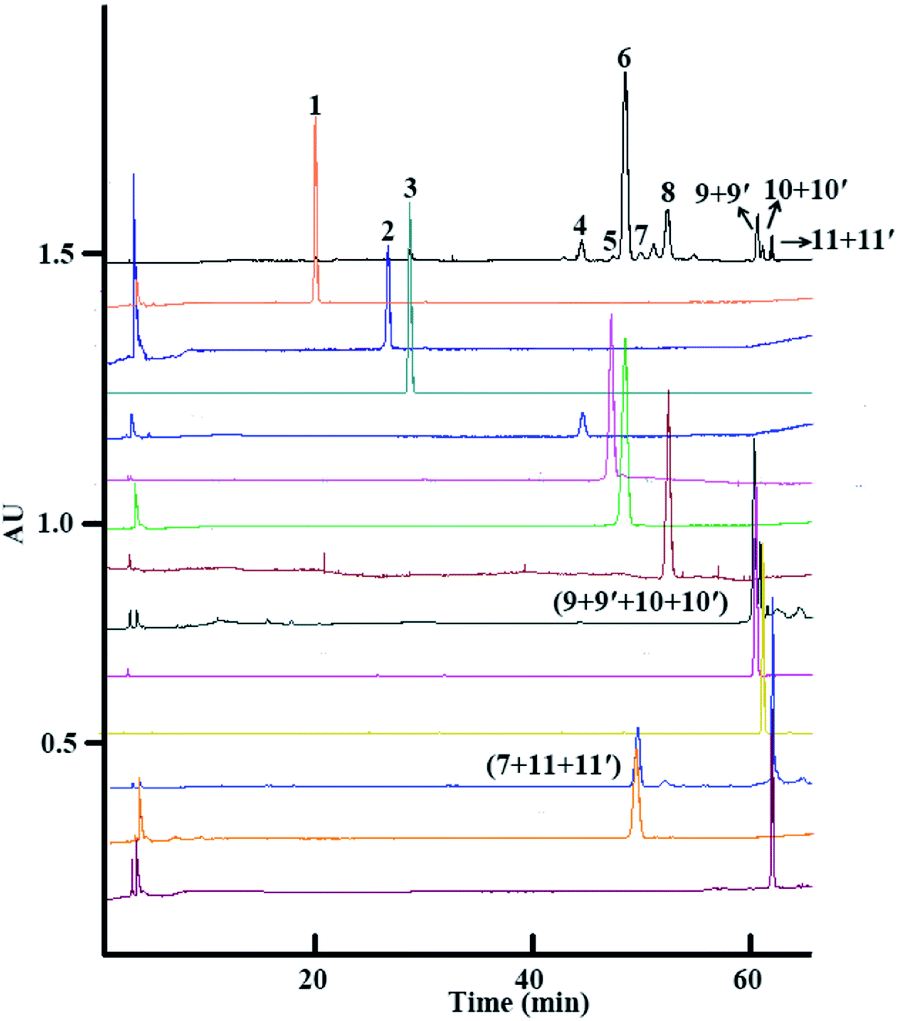 | ||
| Fig. 3 HPLC chromatograms of the crude platycosides and peak fractions from Platycodi radix. HPLC conditions: column: Waters C18 column (250 mm × 4.6 mm, 5 μm); mobile phase: acetonitrile (A)–water (B), 0–4 min, 5–12% A; 4–6 min, 12–20.5% A; 6–25 min, 20.5–23% A; 25–32 min, 23–23.5% A; 32–45 min, 23.5–24% A; 45–55 min, 24–26% A; 55–65 min, 26–46% A; column temperature: 30 °C; flow rate: 1.0 mL min−1; detection with ELSD; injection volume: 10 μL; peak 1: platycoside E, peak 2: deapio-platycodin D3, peak 3: platycodin D3, peak 4: deapio-platycodin D2, peak 5: platycodin D2, peak 6: platycodin D, peak 7: polygalacin D2, peak 8: polygalacin D, peak 9: 2′′-O-acetylplatycodin D (9) and 3′′-O-acetylplatycodin D (9′), peak 10: 2′′-O-acetylpolygalacin D2 (10) and 3′′-O-acetylpolygalacin D2 (10′), and peak 11: 2′′-O-acetylpolygalacin D (11) and 3′′-O-acetylpolygalacin D (11′). | ||
Structure identification of the isolated compounds
The isolated compounds 1–11 were initially elucidated by analyzing the HRESI-MS and HRESI-MS/MS data (see ESI†). Compound 1 had a molecular ion of [M + Cl]− at m/z 1583.8261, along with the MS/MS secondary ion fragments [M + Cl-api]− at m/z 1451.9676 and [M + Cl-api-xyl-rha-ara]− at m/z 1041.9704, distinctively leading to the structure of platycoside E. Compound 2 had a molecular ion of [M + Cl]− at m/z 1289.4697, along with the MS/MS secondary ion fragments of [M + Cl-xyl]− at m/z 1157.4357, [M + Cl-xyl-rha]− at m/z 1011.3885, and [M + Cl-xyl-rha-ara]− at m/z 879.1707, distinctively leading to deapio-platycodin D3. Compound 3 had a molecular ion of [M − H]− at m/z 1385.5179, along with the MS/MS secondary ion fragments of [M − H-api]− at m/z 1253.4862, [M − H-api-xyl]− at m/z 1121.0908, and [M − H-api-xyl-rha]− at m/z 975.7610, distinctively leading to the structure of platycodin D3. Compound 4 had a molecular ion of [M − H]− at m/z 1253.4912, as well as the MS/MS secondary ion fragments of [M − H-xyl]− at m/z 1121.4601, and [M − H-xyl-rha-ara]− at m/z 843.3782, which indicated that it was an isomer of compound 2. Compound 5 had a molecular ion of [M − H]− at m/z 1385.5074, as well as the MS/MS secondary ion fragment of [M − H-api]− at m/z 1253.4728, which indicated that it was an isomer of compound 3. Compound 6 had a molecular ion of [M − H]− at m/z 1223.4677, along with the MS/MS secondary ion fragments of [M − H-api]− at m/z 1091.4365 and [M − H-api-xyl-rha-ara]− at m/z 681.3621, distinctively leading to the structure of platycodin D. Compound 7 had a molecular ion of [M + Cl]− at m/z 1405.4869, along with the MS/MS secondary ion fragments of [M − H-api]− at m/z 1237.4451, [M − H-api-xyl]− at m/z 1105.4130, and [M − H-api-xyl-rha-ara]− at m/z 827.7734, which distinctively elucidated it as polygalacin D2. Compound 8 had a molecular ion of [M − H]− at m/z 1207.4783, along with the MS/MS secondary ion fragments of [M − H-api]− at m/z 1075.7404, [M − H-api-xyl-rha]− at m/z 797.2739, [M − H-api-xyl-rha-CO2]− at m/z 753.2581, and [M − H-api-xyl-rha-ara]− at m/z 665.0065, distinctively leading to the structure of polygalacin D. Compound 9 had a molecular ion of [M + Cl]− at m/z 1301.4472, along with the MS/MS secondary ion fragments of [M + Cl–COCH2]− at m/z 1259.4402, [M + Cl-api–COCH2]− at m/z 1127.4106, [M − H-api-xyl–COCH2]− at m/z 955.3774, and [M + Cl-api-xyl-rha-ara–COCH2]− at m/z 717.3011, which distinctively identified it as acetylated platycodin D. Compound 10 had a molecular ion of [M + Cl]− at m/z 1447.4901, along with the MS/MS secondary ion fragments of [M + Cl–COCH2]− at m/z 1405.4828, [M + Cl-api–COCH2]− at m/z 1273.4504, and [M − H-api-xyl-rha-ara-COCH2]− at m/z 831.3376, which distinctively elucidated it as acetylated polygalacin D2. Compound 11 had a molecular ion of [M + Cl]− at m/z 1285.4545, along with the MS/MS secondary ion fragments of [M + Cl–COCH2]− at m/z 1243.4522 [M + Cl–COCH2-api]− at m/z 1111.4171, [M + Cl–COCH2-api-xyl]− at m/z 979.3857, and [M + Cl–COCH2-api-xyl-rha]− at m/z 833.3395, which distinctively identified it as acetylated polygalacin D. Then, 1H and 13C NMR data (see ESI†) were employed to determine the structures of the isomers and the acetylated platycosides, as well as to verify the above-deducted structures. Interestingly, compound 9 was determined to exist as tautomers 2′′-O-acetylplatycodin D (9) and 3′′-O-acetylplatycodin D (9′) in a 1:1 ratio by the 13C NMR data (see ESI†) and by comparison with data in ref. 16 and 17, while compounds 10 and 11 were elucidated as tautomers: 2′′-O-acetylpolygalacin D2 (10) and 3′′-O-acetylpolygalacin D2 (10′) (see ESI†) in a 1:1 ratio and 2′′-O-acetylpolygalacin D (11) and 3′′-O-acetylpolygalacin D (11′) (see ESI†) a 1:1 ratio. Finally, these compounds were identified as platycoside E (1), deapio-platycodin D3 (2), platycodin D3 (3), deapio-platycodin D2 (4), platycodin D2 (5), platycodin D (6), polygalacin D2 (7), polygalacin D (8), tautomers 2′′-O-acetylplatycodin D (9) and 3′′-O-acetylplatycodin D (9′), tautomers 2′′-O-acetylpolygalacin D2 (10) and 3′′-O-acetylpolygalacin D2 (10′), and tautomers 2′′-O-acetylpolygalacin D (11) and 3′′-O-acetylpolygalacin D (11′).16,18–20
Conclusions
The roots of Platycodon grandiflorum A. DC, recorded as Platycodi radixin the Chinese Pharmacopoeia, are distributed widely around the world and are known for their edible and pharmacologic properties. As platycosides from the Platycodi radix are of medium or high polarity, MCI resin column chromatography was investigated and demonstrated as the preferable enrichment operation for platycosides from the 70% ethanol extracts of Platycodi radix, with the target platycosides eluted using 60% methanol aqueous solution. Then, two different elution modes, isocratic HSCCC and linear-gradient HSCCC, using the solvents ethyl acetate, n-butanol and water were firstly applied together to comprehensively separate the main platycosides, including platycoside E (1), deapio-platycodin D3 (2), platycodin D3 (3), deapio-platycodin D2 (4), platycodin D2 (5), platycodin D (6), polygalacin D2 (7), polygalacin D (8), tautomers 2′′-O-acetylplatycodin D (9) and 3′′-O-acetylplatycodin D (9′), tautomers 2′′-O-acetylpolygalacin D2 (10) and 3′′-O-acetylpolygalacin D2 (10′), and tautomers 2′′-O-acetylpolygalacin D (11) and 3′′-O-acetylpolygalacin D (11′), using evaporative light scattering detection. Linear-gradient HSCCC was employed to rapidly separate compounds 7, 9, 10, and 11, which were eluted out as mixtures at nearly the end of the isocratic HSCCC run. Linear-gradient HSCCC was equivalent to a binary gradient in HPLC, and the solvent system was varied by changing the proportions of the two mobile phases (ethyl acetate and n-butanol) to achieve ever-changing KCi values for the targets and successful separation. The results from this study demonstrate that this strategy of combined isocratic HSCCC and linear-gradient HSCCC was efficient and simple for the comprehensive separation of complex compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by National Natural Science Foundation of China (81873019).References
-
Committee of the Chinese Pharmacopia, The Chinese Pharmacopia, China Medical Science Press, Beijing, 2020, vol. 1, p. 289 Search PubMed
.
- Y. H. Choi, D. S. Yoo, M. R. Cha, C. W. Choi, Y. S. Kim, K. R. Lee and S. Y. Ryu, J. Nat. Prod., 2010, 73, 1863–1867 CrossRef CAS
- J. H. Choi, S. W. Jin, H. G. Kim, C. Y. Choi, H. S. Lee, S. Y. Ryu, Y. C. Chung, Y. J. Hwang, Y. J. Um, T. C. Jeong and H. G. Jeong, J. Agric. Food Chem., 2015, 63, 1468–1476 CrossRef CAS
- H. L. Kim, J. Park, H. Park, Y. Jung, D. H. Youn, J. W. Kang, M. Y. Jeong and J. Y. Um, J. Agric. Food Chem., 2015, 63, 7721–7730 CrossRef CAS
- S. H. Kang, T. H. Kim, K. C. Shin, Y. J. Ko and D. K. Oh, J. Agric. Food Chem., 2019, 67, 1470–1477 CrossRef CAS
- Z. Y. Zhang, M. C. Zhao, W. X. Zheng and Y. H. Liu, J. Biochem. Mol. Toxicol., 2017, e21934 CrossRef
- Y. W. Ha and Y. S. Kim, Phytochem. Anal., 2009, 20, 207–213 CrossRef CAS
- Y. W. Ha, M. Kang, Y. C. Na, Y. Park and Y. S. Kim, J. Sep. Sci., 2011, 34, 2559–2565 CrossRef
- Z. G. Jiang and Y. H. Yan, Anal. Methods, 2020, 12, 4674–4681 RSC
- Q. Liu, Y. L. Geng, X. Wang, J. Li and J. Q. Yu, Anal. Methods, 2019, 11, 4260–4266 RSC
- B. Li, L. Li, A. Q. Zhao, B. H. Han, Y. J. Fan, C. M. Liu and J. J. Liu, Anal. Methods, 2015, 7, 1321–1327 RSC
- A. Spórna-Kucab, N. Wróbel, A. Kumorkiewicz-Jamro and S. Wybraniec, J. Chromatogr. A, 2020, 1626, 461370 CrossRef
- Y. Yang, Y. X. Wang, W. Q. Zeng, J. Tian, X. Zhao, J. L. Han, D. Z. Huang and D. Y. Gu, J. Chromatogr. A, 2020, 1614, 460727 CrossRef CAS
- H. Ying, H. Y. Jiang, H. Liu, F. J. Chen and Q. Z. Du, J. Chromatogr. A, 2014, 1359, 117–123 CrossRef CAS
- X. Y. Song, K. Li, L. Cui, J. Q. Yu, I. Ali, H. Zhu, Q. Q. Wang, X. Wang and D. J. Wang, J. Chromatogr. A, 2020, 1615, 460719 CrossRef CAS
- H. Ishii, K. Tori, T. Tozyo and Y. Yoshimura, J. Chem. Soc., Perkin Trans. 1, 1984, 10, 661–668 RSC
- S. Asen and R. M. Horowitz, Phytochemistry, 1977, 16(1), 147–148 CrossRef CAS
- W. Li, L. Xiang, J. Zhang, Y. N. Zheng, L. K. Han and M. Saito, Chin. Chem. Lett., 2007, 18(3), 306–308 CrossRef CAS
- H. Ishii, K. Tori and T. Tozyo, Chem. Lett., 1978, 719–722 CrossRef CAS
- Z. D. He, C. F. Qiao, Q. B. Han, Y. Wang, W. C. Ye and H. X. Xu, Tetrahedron, 2005, 61(8), 2211–2215 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: NMR data and spectra of compounds 1–11. See DOI: 10.1039/d0ay02029j |
| ‡ These authors contribute equally to this work and joint first authors. |
| This journal is © The Royal Society of Chemistry 2021 |