
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid direct analysis of river water and machine learning assisted suspect screening of emerging contaminants in passive sampler extracts†
Alexandra K.
Richardson
 a,
Marcus
Chadha
b,
Helena
Rapp-Wright
ac,
Graham A.
Mills
d,
Gary R.
Fones
d,
Anthony
Gravell
e,
Stephen
Stürzenbaum
a,
David A.
Cowan
a,
David J.
Neep
f and
Leon P.
Barron
*ag
a,
Marcus
Chadha
b,
Helena
Rapp-Wright
ac,
Graham A.
Mills
d,
Gary R.
Fones
d,
Anthony
Gravell
e,
Stephen
Stürzenbaum
a,
David A.
Cowan
a,
David J.
Neep
f and
Leon P.
Barron
*ag
aDept. Analytical, Environmental & Forensic Sciences, School of Population Health & Environmental Sciences, Faculty of Life Sciences & Medicine, King's College London, 150 Stamford Street, London, SE1 9NH, UK
bAgilent Technologies UK Limited, 5500 Lakeside, Cheadle, SK8 3GR, UK
cSchool of Chemical Sciences, Dublin City University, Glasnevin, Dublin 9, Ireland
dFaculty of Science and Health, University of Portsmouth, White Swan Road, Portsmouth, PO1 2DT, UK
eNatural Resources Wales, Faraday Building, Swansea University, Singleton Campus, Swansea SA2 8PP, UK
fAgilent Technologies UK Limited, Church Stretton, Essex Road, SY6 6AX, UK
gEnvironmental Research Group, School of Public Health, Faculty of Medicine, Imperial College London, 80 Wood Lane, London W12 7TA, UK
First published on 30th December 2020
Abstract
A novel and rapid approach to characterise the occurrence of contaminants of emerging concern (CECs) in river water is presented using multi-residue targeted analysis and machine learning-assisted in silico suspect screening of passive sampler extracts. Passive samplers (Chemcatcher®) configured with hydrophilic–lipophilic balanced (HLB) sorbents were deployed in the Central London region of the tidal River Thames (UK) catchment in winter and summer campaigns in 2018 and 2019. Extracts were analysed by; (a) a rapid 5.5 min direct injection targeted liquid chromatography-tandem mass spectrometry (LC-MS/MS) method for 164 CECs and (b) a full-scan LC coupled to quadrupole time of flight mass spectrometry (QTOF-MS) method using data-independent acquisition over 15 min. From targeted analysis of grab water samples, a total of 33 pharmaceuticals, illicit drugs, drug metabolites, personal care products and pesticides (including several EU Watch-List chemicals) were identified, and mean concentrations determined at 40 ± 37 ng L−1. For targeted analysis of passive sampler extracts, 65 unique compounds were detected with differences observed between summer and winter campaigns. For suspect screening, 59 additional compounds were shortlisted based on mass spectral database matching, followed by machine learning-assisted retention time prediction. Many of these included additional pharmaceuticals and pesticides, but also new metabolites and industrial chemicals. The novelty in this approach lies in the convenience of using passive samplers together with machine learning-assisted chemical analysis methods for rapid, time-integrated catchment monitoring of CECs.
1. Introduction
Over 350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 chemicals and mixtures of chemicals are currently registered for commercial production and use globally.1 Consequently, chemical contamination of the aquatic environment via point and diffuse sources of pollution is increasingly evident. More recently, selected contaminants of emerging concern (CECs) have been identified such as pharmaceuticals, personal care products, pesticides, flame retardants, plastic-related chemicals, illicit drugs and a variety of other industrial chemicals in multiple environmental compartments.2–4 However, more comprehensive, flexible and rapid chemical characterisation approaches are required to improve understanding of the environmental risks such chemicals may pose.
000 chemicals and mixtures of chemicals are currently registered for commercial production and use globally.1 Consequently, chemical contamination of the aquatic environment via point and diffuse sources of pollution is increasingly evident. More recently, selected contaminants of emerging concern (CECs) have been identified such as pharmaceuticals, personal care products, pesticides, flame retardants, plastic-related chemicals, illicit drugs and a variety of other industrial chemicals in multiple environmental compartments.2–4 However, more comprehensive, flexible and rapid chemical characterisation approaches are required to improve understanding of the environmental risks such chemicals may pose.
The majority of studies characterising CEC occurrence in aquatic media have focussed on the use of grab or composite sampling. Whilst these methods enable near real-time monitoring of CECs, they require time and labour-intensive monitoring campaigns using repeated sampling to capture the breadth of CEC occurrence and their fluctuation. As an alternative, passive sampling enables time-weighted average occurrence characterisation over extended periods. Analyte accumulation in the sorbent can also improve the analytical performance through enhanced sensitivity using appropriately selective chemistries. However, passive samplers often fail to capture pulsed sources of CECs. Several different passive sampling approaches and formats exist, for CECs, mixed-mode sorbents within metal or plastic housings configured with porous membranes are popular, including polar organic chemical integrative samplers (POCIS) or Chemcatcher® devices.
With the advent and increased availability of full-scan high-resolution accurate mass spectrometry (HRMS), the potential for simultaneous targeted, untargeted and suspect screening of environmental samples for larger numbers of CECs in all environmental compartments has been realised.5,6 For the latter in particular, suspect screening with HRMS now offers the ability to retrospectively interrogate acquired full-scan sample data to potentially identify additional compounds post hoc. However, by comparison with its application to water (e.g., either directly or following solid-phase extraction),5,7 few reports of untargeted or suspect screening of passive sampler extracts exist for CECs. Soulier and colleagues analysed POCIS extracts for CECs and demonstrated occurrence of ∼30 industrial chemicals, pesticides, pharmaceuticals and personal care products across two selected sites in France using database matching by retention time and HRMS.6 The likelihood of larger numbers of contaminants being present in passive sampler extracts is high, as demonstrated by the complexity of the untargeted data analysis subsequently performed by these authors. Rimayi et al. recently used Chemcatcher® samplers for suspect screening of CECs in South Africa, revealing the occurrence of >200 compounds including general medicines and psychotropic compounds in wastewater impacted river catchments, of which ∼180 were detected for the first time.7 Again, suspect matching was performed using large databases incorporating accurate mass ±5 ppm, isotopic fit and ±0.5 min retention time thresholds. However, in many cases, retention data is either not available for such large numbers of compounds in databases, or the analytical methods used do not match the chromatographic datasets, rendering them unusable for matching. For unknowns, HRMS allows the collection of full-scan data at high sensitivity, mass accuracy and resolution8–10 enabling in silico tentative identification to be performed in many cases, either by exact mass matching or through comparison with accurate-mass databases.10,11 Current mass spectral libraries are extensive, containing reference data for thousands of compounds, thus allowing for a single sample to be screened and deliver a list of potentially matching contaminants in a relatively short period of time. However, in many cases, identification of suspects using HRMS libraries still requires a reference chromatographic retention time for comparison. Obtaining reference standards for suspect compounds can be costly and these are not always commercially available and particularly for metabolites or transformation products.12 In these circumstances, predictive retention time models have recently shown to be useful tools to raise assurance much further for shortlisted suspect contaminants identified using HRMS spectral libraries and where retention data does not exist or is not usable. For example, previous work in our laboratory showed the application of retention time prediction reduced the number of suspects shortlisted in untreated wastewater by one third, allowing prioritisation for reference standard purchase.13 In this way, the combination of a suitably accurate matched predicted retention time and appropriate MS criteria could arguably elevate a lower level match to Level 2(a) “probable structure” classification according to the widely adopted framework proposed by Schymanski et al.14
Multiple methods for in silico prediction of liquid chromatography retention times have been published in the literature, ranging from simple logP based models12,15,16 to complex multivariate quantitative structure–retention relationships (QSRR) models.17,18 More recently, machine learning-based QSRR methods have emerged for retention time prediction including support vector machines,19 tree-based learners,20 and artificial neural networks (ANNs).8,13,21–23 We have extensively evaluated the latter and even demonstrated good generalisability across multiple reversed-phase LC methods, instruments and sample types for >1100 unique compounds.21 Recently, we applied this approach to identify retrospectively 37 additional CECs in influent and effluent wastewaters in London in LC-HRMS data.13 Given that the River Thames is subject to regular wastewater impact from CECs arising from combined sewer overflows (CSOs),24 the potential combination of passive sampling and machine-learning assisted high-resolution suspect screening analysis could present a powerful new method for CEC characterisation, including the ability to utilise HRMS databases more fully where LC methods do not match or where analyte retention data is lacking. With the constant development and improving performance of analytical tools and methods especially for large numbers of compounds, better prediction of gradient retention time is now possible.
The aim of this work was to improve understanding of the occurrence of CECs using passive samplers deployed in the River Thames (UK) using both targeted LC-MS/MS analysis and machine learning-assisted in silico LC-HRMS suspect screening. To achieve this, the objectives were: (a) to perform differential targeted analysis of river water and passive sampler extracts using a rapid, direct injection LC-MS/MS method;25 (b) to develop and apply an ANN-based model for multi-analyte retention prediction in a gradient reversed-phase LC method and (c) application of the developed LC-HRMS suspect screening workflow to the occurrence of new and additional CECs in two river monitoring campaigns in winter and summer in 2018/19. This new approach is likely to improve the value of passive sampler extract data as a more rapid in silico shortlisting step for new or additional CECs.
2. Methods
2.1 Materials and reagents
All reagents were of analytical grade or purer. Acetonitrile (MeCN) and methanol (MeOH) were from Sigma-Aldrich (Gillingham, Dorset, UK). LC-MS grade formic acid, ammonium formate and hydrochloric acid (37% v/v, HCl) were purchased from Millipore (Millipore, Bedford, USA), Agilent Technologies UK Ltd. (Santa Clara, CA, USA), and Sigma-Aldrich (Steinheim, Germany), respectively. Ultra-pure water was obtained from an 18.2 Ω cm Millipore Milli-Q water purification system. A mix of n = 164 analytical standards and n = 34 deuterated internal standards (SIL-IS) (purity ≥97%) were used for targeted analysis including confirmatory identification and quantification (full details, including all sources, can be found in the ESI S1†).2.2 Passive sampler preparation, deployment and extraction procedures
Chemcatcher® housings were obtained from AT Engineering (Tadley, UK) and were cleaned as per Castle et al.26 Briefly, Supor poly(ether sulfone) (PES) 0.2 mm membranes (Pall Europe, Portsmouth, UK) were cut to size (52 mm) using a wad punch. Membranes were soaked for 24 h in MeOH to eliminate manufacturing residues.27 Post soak, the membranes were washed using fresh MeOH and then soaked for an additional 24 h in water. Hydrophilic–lipophilic balanced (HLB) sorbent disks (47 mm diameter) were purchased from Biotage (Uppsala, Sweden) and Affinisep (Val de Reuil, France). HLB disks were conditioned with MeOH (50 mL) and water (50 mL) before assembly. Chemcatcher® samplers were prepared by placing the HLB disk onto the sampler body and overlaying with a PES membrane before screwing the retaining ring in place. Prior to deployment, assembled samplers were stored in ultrapure water.Chemcatcher® samplers were deployed on two occasions in the River Thames UK at two proximal sites located in Central London. This region of the river is tidal, brackish and CEC concentrations at both sites were previously found not to be statistically different over a weeklong grab sampling period.24 This sampling area is also close to several CSO vents, which discharge untreated wastewater into the Thames with a frequency of roughly once a week, especially during times of heavy rainfall. During both deployments, Chemcatcher® samplers were fastened via drilled pilot holes and cable ties to 34 × 15 cm solid plastic boards. These were then affixed to pontoons and submerged at a relatively consistent 1 m depth underwater using a 3 kg dive weight. The first campaign (winter, 21st December 2018–6th January 2019) was performed at the London Fire Brigade (LFB) Lambeth River Fire Station pontoon (51°29′35.1′′N; 0°07′19.9′′W) using four Chemcatcher® devices. This site allowed secure access away from the shore and over the holiday period to deploy and collect samplers, as needed. The second campaign (summer, 27th August 2019–9th September 2019) was located ∼2 km downriver at the Transport for London (TFL) Blackfriars Pier (51°30′38.5′′N; 0°06′00.6′′W) again allowing access to a Central London region of the catchment and three devices were deployed. A field blank was exposed during both deployments and retrieval and analysed using LC-MS/MS (as for deployed samplers using LC-HRMS). After the deployment periods, the Chemcatcher® housing was disassembled, the PES membranes discarded and the HLB disks were removed and air-dried overnight at room temperature alongside the field blank to account for contamination before storage at −20 °C in the dark until analysis. HLB disks (samples and field blanks) were eluted using 40 mL of MeOH at ambient temperature under vacuum. The use of successive elution steps with solvents of different pH was not considered here to minimise complexity, but could be used to potentially increase the number of compounds eluted from the sorbent. Extracts were dried using a Genevac centrifugal rotary evaporator (SP Scientific, Ipswich, UK) at 40 °C for 2 h. Prior to instrumental analysis samples were reconstituted in 1 mL of MeOH. The full procedure is described in Taylor et al.28
2.3 River water sampling, preparation and CEC quantification procedures
Grab samples (500 mL) were collected in pre-rinsed Nalgene® bottles (Sigma-Aldrich) at the start of each passive sampler deployment and were transported to the laboratory, acidified (to pH 2 with HCl) and frozen until analysis. Water samples were prepared for direct injection LC-MS/MS analysis as described by Ng et al.25 In brief, river water samples (10 mL sub-sample, n = 3) were first centrifuged at 2000 rpm for 10 min. Aliquots (900 μL) of supernatant were spiked with 100 μL of SIL-IS (prepared in MeOH) to give a final concentration of 500 ng L−1. For quantification, background-corrected external matrix-matched calibration was performed using 900 μL of pooled river water from each campaign spiked with constant 100 μL volumes, again containing each analytical standard and SIL-IS (at 500 ng L−1 used only for quality control in LC-MS/MS) to yield final concentrations over the range 10–2000 ng L−1. Following this, samples were then filtered directly into deactivated HPLC vials (Agilent A-Line) using BD Plastipak™ syringes (Fisher Scientific UK Ltd., Loughborough, UK) coupled to 0.2 μm Teflon membrane filters.2.4 Instrumentation
For suspect screening, the analysis was performed using an Agilent 1290 (Infinity II) LC system coupled to a 6546 LC/Q-TOF mass spectrometer. Analytical separations were performed on an Agilent Zorbax Eclipse Plus C18, 2.1 × 100 mm, 1.8 μm column. A 15 min binary gradient of 0.1% formic acid and 5 mM ammonium formate (mobile phase A – MPA) to 0.1% formic acid and 5 mM ammonium formate in MeOH (mobile phase B – MPB). The elution gradient consisted of 100% MPA from 0 to 1 min, followed by a linear increase to 100% MPB from 1 to 12 min. The re-equilibration time was 3 min at 100% MPA. The column was maintained at 40 °C with a constant flow rate of 0.4 mL min−1 and an injection volume of 6 μL. All passive sampler extracts were run separately in both positive and negative mode with the same mobile phases over a scan range of m/z 50–1000. The data were acquired at 10 GHz giving a resolution range of 30000–60000 full width at half maximum (FWHM) over the measured mass range and the scan rate was 3 Hz. Sheath and drying gas settings were both 12 L min−1, with the temperatures at 350 °C and 250 °C, respectively. All data were acquired using data-independent acquisition (DIA) using alternating collision energies of 0 eV and 20 eV to collect alternating mass spectra for all ions with and without fragmentation, respectively. All data were acquired in centroid mode and processed using Agilent MassHunter software.
Targeted direct injection LC-MS/MS analysis of water samples and extracts from the passive samplers was performed using a Nexera X2 LC system coupled to an LCMS-8060 (Shimadzu Corp., Kyoto, Japan) fitted with an electrospray ionisation source. Rapid separations (Fig. S1†) were performed on a 5 × 3.0 mm, 2.7 μm Raptor biphenyl guard column (Restek, Pennsylvania, USA). The LC method comprised a binary gradient of 0.1% v/v aqueous formic acid (mobile phase C – MPC) and 0.1% v/v formic acid in 50:50 MeOH:MeCN (mobile phase D – MPD). The elution profile consisted of 10% MPD for 0.2 min, 10–60% MPD from 0.2 to 3.0 min, and 100% MPD to 4.0 min. The re-equilibration time was 1.5 min at 10% MPD. The column was kept at ambient temperature with a flow rate of 0.5 mL min−1 and an injection volume of 10 μL. Where possible, two transitions for each analyte were monitored using multiple reaction monitoring (MRM) with the dwell time varying between 1 to 20 ms depending on the analyte (Table S1†). The threshold for a retention time match to a reference standard was set to 0.2 min. Further method details can be found in Ng et al.25 For river water samples, these guard columns were replaced after every 3000 injections, approximately. Qualitative and quantitative method performance for detected compounds in water samples were assessed in accordance with the tripartite guidelines published by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH).29
2.5 Procedures for suspect screening
For LC-QTOF-MS/MS data, retrospective suspect screening was performed on all passive sampler extracts using three commercial Agilent mass spectral libraries (Forensic Toxicology, Pesticides and Water Screening), each containing curated MS/MS spectra for matching to shortlisted suspects (each also acquired using collision energies of 0 and 20 eV). All samples were screened through each database using the ‘find-by-formula’ match criteria, where MS features are selected for against the mass spectral databases. Initially, a broad data screen of all samples was performed based on the [M + H]+ or the [M − H]− ion accurate m/z and to within 5 ppm tolerance without a co-eluting fragment ion. The weighting of the scoring criteria was 100 for the mass accuracy, and 10 for each of isotope spacing (the distance between the ions of the isotope pattern) and abundance (observed height compared to the theoretical). The minimum threshold for MassHunter software to return an identification was set to a 25% match, incorporating these weightings. Subsequent curation and shortlisting of this original search was implemented by further selecting the [M + Na]+, [M + NH4]+ and [M + HCOO]− adducts with a co-elution of at least 75% overlap to the [M + H]+ or the [M − H]− ion. In terms of match criteria, the weightings for isotope abundance and spacing were increased to 60 and 50, respectively. Similarly, the overall threshold for reporting an identification was then increased to a match of 90%. All extracted ion chromatogram (EIC) peaks for compounds were retained, including those with more than one peak. Assigning the correct EIC peak would be done through retention modelling.Following this, and to refine this initial shortlist of compounds further, ANN-based retention time prediction was employed.21 Measured retention data used to train the model were generated from LC-QTOF-MS/MS measurements for a mix of 239 pesticide standards (see S2 for details†) injected in triplicate commercially available through Agilent Technologies UK Ltd. Simplified molecular-input line-entry specifications (SMILES) from Pub-Chem were used to generate data on each compound for 16 molecular descriptors. These descriptors were selected based on a combination of curated descriptors relevant to reversed-phase liquid chromatography mechanisms, correlation with tR and genetic feature selection. In addition, and based on 239 compounds used for model development, the ratio of cases to inputs far exceeded the 5:1 ratio threshold proposed by Topliss and Costello.30 Full details can be found in Mollerup et al.,31 Munro et al.24 and Miller et al.22 Dragon version 7.0 (Kode Chemoinformatics srl, Pisa, Italy) was used to generate data for hydrophilic factor (Hy), unsaturation index (Ui), Ghose–Crippen and Moriguchi logP (AlogP, MlogP), number of benzene-like rings (nBnz), number of oxygen and carbon atoms (nC, nO), number of double and triple bonds (nDB, nTB) and number of 4–9 membered rings (nR04–nR09). For logD (mobile phase pH = 3.0), data were generated using Percepta PhysChem Profiler (ACD Laboratories, Ontario, Canada). See Table S2† for all molecular descriptor data and selection of such descriptors was based on previous work. The data were used as inputs to train a three-layer multilayer perceptron (3MLP) using Trajan v6.0 (Trajan Software Ltd., Lincolnshire, UK) with a 16-4-1 architecture (optimised) and with retention time as the output. The training of models was performed in two phases. In Phase 1, the dataset was split into 70:15:15 (training:verification:test) and, using random sampling, the most appropriate neural network type selected from linear models, probabilistic neural networks (PNNs), generalised regression neural networks (GRNNs), radial basis functions (RBFs), and 3MLP and four-layer multilayer perceptron (4MLPs). Thousands of models were built and evaluated over several separate 10 min training phases and the performance of the best 50 summarised in each case. The best model type was then selected based on the lowest and most consistent error returned and across each set. In Phase 2, the architecture of the best model was further optimised. The dataset was partitioned into 70:30 and bootstrap sampling applied and in ten replicated rounds of training of 5 min intervals each. The best multilayer perceptron model used conjugated gradient descent and backwards propagation to optimise performance32,33 (in this case, a 3MLP with a 16-4-1 architecture).
3. Results & discussion
3.1 Rapid targeted CEC occurrence in water using direct injection LC-MS/MS
Abiotic river conditions including pH, temperature, dissolved oxygen, ammonium concentration and flow as well as rainfall data are given in the ESI (S4).† Importantly, no sewer overflows were reported before either campaign. Following targeted analysis of water samples across both campaigns, a total of 33 unique CEC compounds were detected (20% of the total number included in the method, see Table 1). These were broadly classified as pharmaceuticals and personal care products (PPCPs), pesticides, controlled drugs, industrial chemicals and drug metabolites (Fig. 1). Within this, 18 contaminants were confirmed in winter river water samples and of these, 12 were quantifiable (a compound was considered quantifiable if the calculated concentration was greater than the LLOQ presented in Table 1). In summer, 33 compounds were confirmed in water samples and 19 were quantifiable. Eleven compounds identified in the water samples (amitriptyline, benzoylecgonine, carbamazepine, cocaine, diclofenac, ketamine, propranolol, sulfapyridine, temazepam, tramadol and trimethoprim) were consistent with previous studies monitoring the Central London region of the Thames.24,34 Four of these contaminants (carbamazepine, diclofenac, propranolol and trimethoprim) were also consistent with past reports of surrounding catchment regions of the Thames.35–37 However, the reported concentrations were higher in comparison to this work, between 17 and 140 ng L−1, and this is perhaps unsurprising given the temporal variability associated with grab sampling.24 Three contaminants (acetamiprid, diclofenac and imidacloprid) were/are listed on the European WFD Watch Lists,38 with the neonicotinoids clothianidin and imidacloprid in particular now banned for use in the European Union.39 In several cases, controlled substances were also detected in water including abused drugs (e.g. benzodiazepines and ketamine) and pesticides (e.g., fenuron, present in all samples at 37 ± 6 ng L−1), which was also consistent with previous reports of UK river waters.40 The average concentrations of all quantified compounds were 33 ± 23 ng L−1 and 44 ± 44 ng L−1 for winter and summer water samples, respectively. In both campaigns' tramadol was, in most cases, the compound with the highest concentration at an average of 164 ± 65 ng L−1. Overall, this direct-injection analytical method performed better in this river water matrix than previous assessments in wastewater. Mean ± standard deviation for limits of detection (LOD) and lower limits of quantification (LLOQ) across all compounds detected were 5 ± 3 ng L−1 and 14 ± 10 ng L−1, respectively. This represented a significant improvement (p < 0.05) over the LLOQs determined in wastewater previously (43 ng L−1, on average)25 and was most likely due to lower sample complexity.25 The performance of this method was compared to other direct-injection methods for surface waters found in the literature (Table 1) though it was challenging to find methods with significant analyte commonality. A method by Boix et al.41 included nine compounds in common with LLOQs ranging from 0.2–37.5 ng L−1, and was more sensitive on the whole by comparison. However, this method employed a ten-fold larger injection volume (100 μL). In comparison to the Hermes et al.42 method, two compounds are comparable in terms of performance (tramadol and trimethoprim) and six exhibited higher sensitivity than our method (carbamazepine, diclofenac, imidacloprid, lidocaine, terbutryn, venlafaxine). This method also employsed a larger injection volume (80 μL). It is unclear how such large injection volumes impact analytical performance across batch analyses of large numbers of samples over extended periods. Martínez Bueno et al.43 used the same injection volume (10 μL) with six common compounds, the LLOQ reported in this study were significantly higher for three compounds (amphetamine, MDMA and nicotine) by 8–17 fold. For the remaining three, the LLOQs were comparable. In addition to sensitivity at the lower limits of range, the linearity of the method used herein was excellent for most compounds and over three orders of magnitude. This performance was likely aided by the removal of added imprecision from advanced sample preparation steps such as solid-phase extraction. Lastly, the speed of this targeted LC-MS/MS method potentially enables both grab and time-integrated sampling to be performed simultaneously on a much larger catchment scale if necessary (with approximately 260 injections possible in a single 24 h period).| Analyte | Linearity | LODb (ng L−1) | LLOQc (ng L−1) | Other DI LC-MS/MS methods LLOQs (ng L−1) | Matrix effectsd | Winter | Summer | ||
|---|---|---|---|---|---|---|---|---|---|
| Min, max [CEC] (ng L−1) | Frequencye | Min, max [CEC] (ng L−1) | Frequencye | ||||||
| a Represents n ≥ 5 calibrants measured in river water matrix and all tested over the range 10–2000 ng L−1. b LOD determined using 3 × standard deviation of the regression line divided by the slope. c LLOQ determined as 3.3 × LOD. d Represents the mean of n = 6 replicate measures of the percentage of background-subtracted responses measured for a 1000 ng L−1 spiked Thames river water sample compared to a standard at the same concentration (negative values represent suppression and vice versa). e Frequency represents the number of passive sampler extracts where occurrence was confirmed for that compound. f Hermes et al. (2018) LLOQ in surface waters.42 g Boix et al. (2015) LLOQ in surface waters.41 h Martínez Bueno et al. (2011) LLOQ in surface waters;43 — not detected. | |||||||||
| 4-Fluoromethcathinone (4-FMC) | 0.992 | 4 | 13 | — | −18 | — | — | 18 | 1/6 |
| Acetamiprid | 0.998 | 4 | 11 | — | +6 | — | — | 17, 33 | 4/6 |
| Amitriptyline | 0.991 | 4 | 11 | — | −35 | 12 | 1/3 | <LLOQ | 1/6 |
| Amphetamine | 0.994 | 4 | 12 | 6.3g, 200.0h | −14 | 25 | 1/3 | 19, 41 | 6/6 |
| Azoxystrobin | 0.997 | 4 | 11 | — | −5 | — | — | <LLOQ | 4/6 |
| Benzoylecgonine | 0.998 | 4 | 12 | 0.1g, 13.0h | +1 | <LLOQ | 3/3 | <LLOQ | 3/6 |
| Bisoprolol | 0.997 | 4 | 12 | — | +14 | — | — | <LLOQ | 3/6 |
| Carbamazepine | 0.960 | 4 | 13 | 1.0f, 0.2g | +12 | 24, 33 | 3/3 | 77, 117 | 6/6 |
| Citalopram | 0.987 | 5 | 14 | 10.0f | +26 | <LLOQ | 3/3 | <LLOQ, 14 | 6/6 |
| Clopidogrel | 0.998 | 4 | 11 | 0.5f | +2 | — | — | <LLOQ | 1/6 |
| Clozapine | 0.998 | 4 | 11 | — | +59 | — | — | <LLOQ | 5/6 |
| Cocaine | 0.997 | 4 | 11 | 1.0g, 10.0h | −4 | — | — | <LLOQ | 4/6 |
| Cyclouron | 0.985 | 4 | 12 | +2 | — | — | 50 | 1/6 | |
| Diclofenac | 0.987 | 4 | 12 | 2.0f, 6.8g | +2 | — | — | 24, 31 | 2/6 |
| Fenuron | 0.987 | 4 | 12 | — | −6 | 33, 43 | 3/3 | 27, 46 | 6/6 |
| Imidacloprid | 0.927 | 8 | 24 | 15.0f | +12 | — | — | 26, 30 | 2/6 |
| Ketamine | 0.995 | 4 | 11 | 25.0h | +4 | <LLOQ, 13 | 2/3 | 21, 31 | 6/6 |
| Lidocaine | 0.999 | 4 | 11 | 2.0f | +1 | 15, 19 | 3/3 | 31, 51 | 6/6 |
| MDMA | 0.996 | 4 | 12 | 0.5g, 100.0h | +3 | — | — | <LLOQ | 5/6 |
| Memantine | 0.992 | 4 | 13 | — | +9 | — | — | <LLOQ | 2/6 |
| Methamphetamine | 0.994 | 4 | 13 | — | +2 | — | — | <LLOQ | 3/6 |
| Nicotine | 0.987 | 5 | 14 | 200.0h | −3 | 32 | 1/3 | 17 | 1/6 |
| Oxazepam | 0.902 | 7 | 22 | — | +8 | 41, 58 | 3/3 | 52, 75 | 6/6 |
| Propamocarb | 0.995 | 4 | 11 | — | −2 | <LLOQ | 3/3 | <LLOQ, 12 | 4/6 |
| Propranolol | 0.992 | 4 | 12 | — | +19 | <LLOQ | 1/3 | 16 | 1/6 |
| Pyracarbolid | 0.992 | 3 | 9 | — | −8 | — | — | 12 | 1/6 |
| Salicylic acid | 0.995 | 3 | 10 | 37.5g | +128 | 64 | 1/3 | 44, 78 | 4/6 |
| Sulfapyridine | 0.991 | 4 | 13 | — | +3 | <LLOQ, 17 | 3/3 | <LLOQ | 1/6 |
| Temazepam | 0.985 | 3 | 10 | — | +2 | <LLOQ | 1/3 | 10, 20 | 6/6 |
| Terbutryn | 0.996 | 4 | 11 | 1.0f | 0 | — | 0/3 | <LLOQ | 6/6 |
| Tramadol | 0.990 | 4 | 11 | 15.0f | 0 | 78, 93 | 3/3 | 169, 251 | 6/6 |
| Trimethoprim | 0.998 | 4 | 11 | 10.0f, 1.8g | −4 | <LLOQ | 2/3 | <LLOQ, 13 | 5/6 |
| Venlafaxine | 0.997 | 4 | 11 | 2.0f, 0.2g | +1 | 19, 20 | 3/3 | 37, 75 | 6/6 |
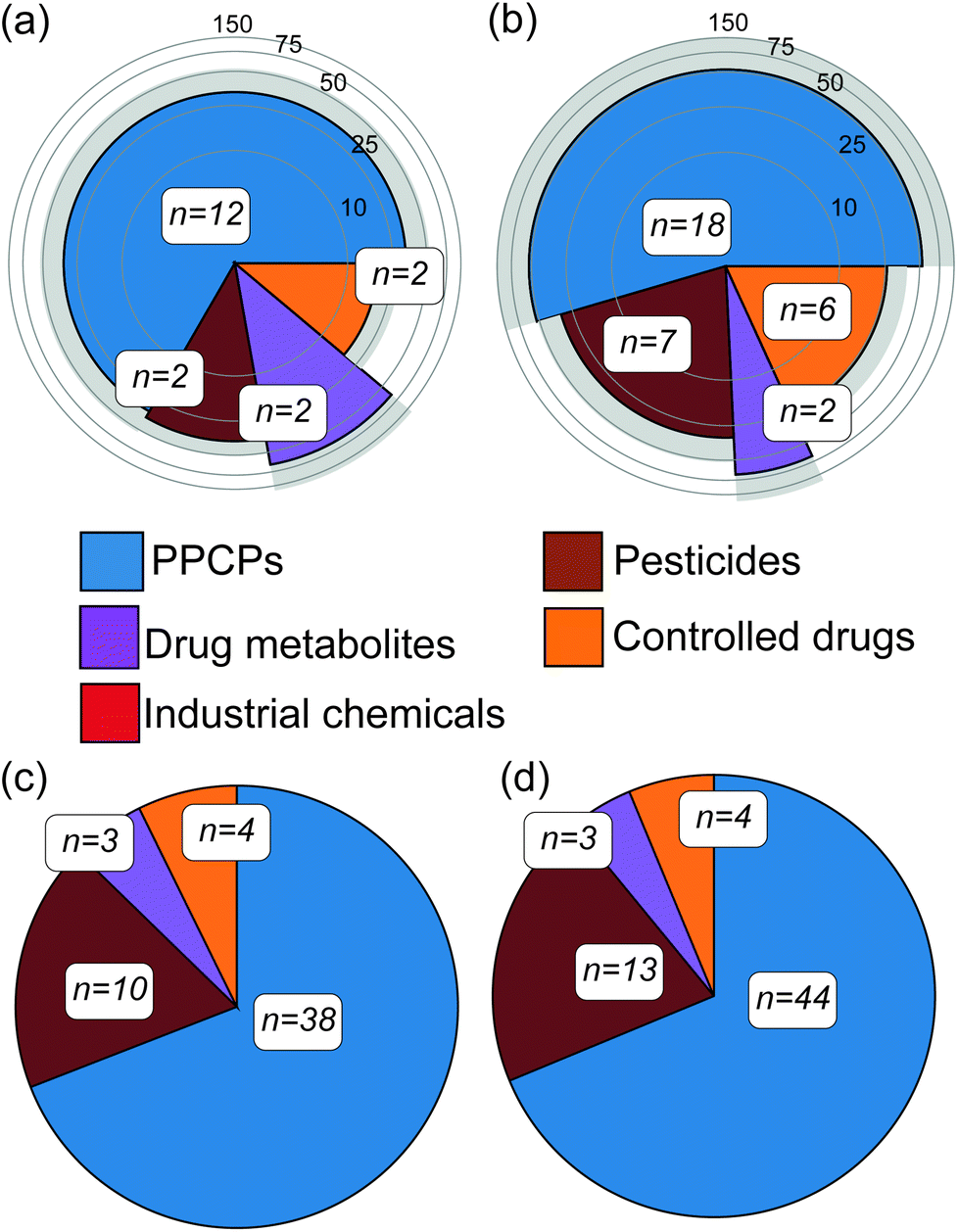 | ||
| Fig. 1 (Top) Mean concentration (ng L−1) of compound classes detected directly in river water using rapid targeted LC-MS/MS analysis for winter (a) and summer (b) samples (standard deviation indicated by the grey rings). Concentrations represented by concentric rings on logarithmic scales for clarity. (Bottom) Proportions of each chemical class detected in passive sampler extracts in winter (c) and summer (d), again using targeted analysis (qualitative only). n = number of unique compounds within each class. | ||
3.2 Rapid targeted LC-MS/MS analysis of passive sampler extracts
Combination of this rapid analytical method with passive sampling offered a new approach to catchment chemical contaminant profiling for potentially large numbers of devices and water samples. Unsurprisingly, and likely due to the enhanced detection capability offered by using an HLB-sorbent in Chemcatcher® samplers, a larger number of CECs was sequestered by passive samplers during both deployments (i.e., n = 65 unique compounds across both seasonal deployments, see Fig. S4 for details†). Quantification was not performed for CECs in passive sampler extracts. Extracts of field blanks contained traces of six to eight compounds, but peak intensity was <104 in all cases and negligible in comparison to measured signals in deployed samplers in the river. An additional 43 and 38 compounds were identified in winter and summer passive sampler extracts, respectively, that were not present in their corresponding water samples, most likely due to insufficient method sensitivity. The LLOQ and LOD of the passive samplers were not assessed as no quantification was performed using matrix-matched calibrants. With respect to detection, a signal-to-noise ratio greater than 3:1 was checked manually in all cases and to ensure that all detected features represented a chromatographic peak.
In addition to those EU Watch List compounds detected in water samples, the macrolide antibiotics (azithromycin and clarithromycin), neonicotinoid pesticides (clothianidin and thiacloprid) and the triazine herbicide (ametryn) were identified in passive sampler extracts in across campaigns.38 Seven additional compounds were also identified (i.e., diazepam, fluoxetine, metoprolol, nortriptyline, sulfamethazine, sulfamethoxazole and warfarin) and this occurrence was consistent with previous studies of the Thames and its surrounding catchments over the range of 5–305 ng L−1 (median: 50 ng L−1).24,34–37 Conversely, 17 and 8 compounds were not detected in the winter and summer Chemcatcher® extracts, respectively, that were present in water samples (18 unique compounds in total). Unfortunately, given the time-integrated averaging nature of passive sampling, pulse introduction of contaminants are missed, which could partly explain this. However, there were no reported sewer overflow events during either deployment (S3 for more details†).
The range of logD values for all compounds sequestered onto these HLB sorbents during both campaigns was −1.16 to 6.09 at the mean river pH and similar to previous works.44–49 The logD of all 18 compounds unique to water samples covered a range of −0.3 to 4.21. Despite methanol being a recognised solvent for passive sampler sorbent elution,28 incomplete elution or ion suppression for some compounds may have occurred. However, a stronger solvent is likely to elute more heavily retained matrix components and successive elution using different solvents or at different pH was considered excessive for practical application. For matrix effects, LC-MS signals stability was relatively low for most analytes following direct measurement of river water samples, despite their brackish nature (Table 1).25 Therefore, despite this limitation for passive sampler extracts, the combination of both direct injection and passive sampler methods was still considered to be very useful for rapid targeted monitoring of river catchments for a relatively large number of CECs.
3.3 In silico suspect screening of passive sampler extract with LC-QTOF-MS/MS
Passive sampler extracts were subjected to suspect screening using machine LC-QTOF-MS and subsequent data mining. Direct injection LC-QTOF-MS suspect screening of the water samples was not considered but could provide added information for comparison with passive sampler data where the method is suitably sensitive.The first step of the suspect screening workflow involved comparing passive sampler extract data to the Agilent MS databases (forensic toxicology database = 9002 compounds; pesticide database = 1684 compounds; and water screening database = 1451 compounds). This resulted in an initial shortlist of 8485 unique possible compounds in extracts. When these data were further curated using the methods described in 2.5, this was reduced to 237 unique compounds identified across all passive sampler extracts (149 in winter and 157 in summer). Within this set, multiple matches were returned for 95 compounds. The scale of this occurrence data not only demonstrates the advantages of using HLB-type passive samplers for time-integrated catchment occurrence characterisation but also that the scale of data generated would make routine monitoring impractical. Thus, to prioritise rapidly potential compounds present and increase confidence in compound identity, machine learning was employed to predict retention time as a further data curation process to reduce the number of candidates to a practicable number for risk management purposes. Of course, candidate shortlists are all dependent on the database selected. Larger databases such as the US EPA CompTox Chemicals Dashboard would have returned more suspect candidates. Nevertheless, the use of the vendor-supplied database in the first instance was taken as a starting point to demonstrate the proof of concept. As sensitivity was expected to be poorer for CECs than that of the targeted method, suspect screening of directly injected water samples on the LC-QTOF/MS was not performed. However, this could prove beneficial for wider xenobiotic exposure characterisation in future work as technology advances.
The optimised model (a 16-4-1 3MLP) for the prediction of retention time showed excellent correlation and agreement across training, verification and blind test data (coefficient of determination, R2 = 0.885, 0.871 and 0.874, respectively, (Fig. S5(a)†)). The mean average error (MAE) across all cases in the training, verification and blind test sets were 26, 26 and 29 s, respectively (Fig. S5(b)†). The applicability domain of the prediction model was defined by investigating the molecular descriptors used to generate the prediction modes using principal component analysis (PCA) in Python (Fig. S6(a)†). Following mass spectral database suspect shortlisting, the model was applied to all 237 compounds tentatively identified in the passive sampler extracts (Table S3†). For all compounds, the retention time difference (ΔtR) between the measured (tR) and predicted (tPR) retention times were calculated. Compounds with ΔtR outside the 75th percentile of model error (52 s) were discarded, as previously proposed by our group.13 Predictions may have been improved if a more diverse set of training case examples were used including other classes of chemicals. Furthermore, ab initio molecular descriptor selection for this specific method was considered, which may have also been similarly successful. However, these descriptors were previously found to generalise well across several reversed-phase LC-based methods and was the preferred option.21
This process resulted in a shortlist of 59 (n = 43 in winter and n = 37 in summer) compounds across all passive sampler extracts with ΔtR data within this threshold (Fig. 2 and Table S4†). The majority of compounds clustered well within a 95% confidence interval of PCA data for molecular descriptors used to define the applicability domain (Fig. S6(b)†). A range of classes was tentatively identified including flame retardants, PPCPs, controlled drugs, pesticides, industrial chemicals and metabolites. Of all 59 compounds detected, 21 were common to both winter and summer. The largest class of compounds detected in common overall were PPCPs. Eight compounds were present in all sampler extracts in each campaign and of these, two were present in all samplers from both campaigns, i.e., O-desmethylvenlafaxine (a metabolite of the antidepressant, venlafaxine) and tri-(2-chloroisopropyl)phosphate (TCPP, a flame retardant). Others were only prevalent in the winter campaign, including 4-hydroxyphenyl-pyruvic acid (an intermediate metabolite of phenylalanine), butylacetanilide (insect repellent), aniline (industrial synthetic precursor) and dicamba (a broad-spectrum herbicide). Unique to summer were amisulpride (an antiemetic and antipsychotic) and dilaurylthiodipropionate (an antioxidant prevalent in food and cosmetics). Importantly, nine shortlisted compounds could not be found in the literature for river water (Table 2).
 | ||
| Fig. 2 (Top) Frequency of detection of compounds in replicate passive sampler extracts (blue = winter; n = 4, green = summer; n = 3 samplers) identified using in silico suspect screening. (Bottom) Differences in compound occurrence between campaigns and overall proportion based on chemical classification. n = number of unique compounds within each class. | ||
| Compound | Primary use(s) | CAS | Measured m/z + isotope match | ppm | Qualifier fragment(s) | Measured tR (min) | ΔtPRa (min) | Current Schymanski framework level (now all raised to 2(a) with tR prediction or higher with reference standard confirmation) |
|---|---|---|---|---|---|---|---|---|
| a Error in retention time prediction. b [M + H]+ adduct. c [M − H]− adduct. d ‘Probable structure’ based on precursor ion + at least one product ion with and only one database match from MassBank. e ‘Unequivocal molecular formula’ based on precursor ion + isotope pattern match to the library. f ‘Tentative candidate(s)’ based on precursor ion + product ions. | ||||||||
| 2,4-Dinitro-o-cresol (DNOC) | Herbicide | 534-52-1 | 198.0235b | −1.17 | 180.0177 | 7.00 | −0.65 | 3f |
| 8-Hydroxy-efavirenz | Antiretroviral pharmaceutical metabolite | 205754-32-1 | 330.0159c | −2.77 | 257.9963, 246.0139, 286.0252, 250.0485 | 8.69 | −0.56 | 2(a)d |
| 9-Octadecenamide | Manufacturing lubricant | 301-02-0 | 282.2790b | 0.47 | 247.242, 97.1012, 83.0855, 135.1168, 265.2526 | 11.34 | 0.79 | 2(a)d |
| Benhepazone | Nonsteroidal anti-inflammatory | 363-13-3 | 237.1022b | 0.34 | — | 7.23 | −0.09 | 4e |
| Benzhydryl cyanide | Pharmaceutical precursor | 86-29-3 | 194.0972b | −3.89 | — | 7.23 | −0.46 | 4e |
| Dilaurylthiodipropionate | Antioxidant | 123-28-4 | 515.4123b | 1.18 | 143.0161, 329.2145, 115.0212, 161.0267, 89.0056 | 11.28 | −0.12 | 2(a)d |
| Furegrelate | Cardiovascular pharmaceutical | 85666-24-6 | 271.1080b | −1.00 | 210.0913 | 6.03 | 0.75 | 3f |
| Methylthiouracil | Antithyroid agent | 56-04-2 | 143.0275b | −1.03 | 84.0444 | 3.33 | 0.39 | 2(a)d |
| Proguanil | Malaria prevention | 500-92-5 | 254.1169b | −0.75 | 170.0480, 153.0214, 102.1026, 128.0262 | 6.72 | 0.03 | 2(a)d |
Among those tentatively identified were a few interesting cases to illustrate the performance of the new in silico suspect screening workflow. Firstly, an active metabolite of lidocaine (3-hydroxylidocaine, 3-HL) was shortlisted in passive sampler extracts. A clear precursor ion was detected at m/z 251.1762 [M + H]+ (Fig. 3(a)). Based on this ion alone, four chromatographic peaks were detected. Application of the predictive retention time model isolated a single chromatographic peak within a 19 s error which also corresponded to the presence of its qualifier fragment at m/z 89.0964 ([CH2N(CH2CH3)2]+).50 This, therefore, allowed a 2(a) identification according to the Schymanski et al. framework. 3-HL is formed in humans from cytochrome P450 enzymes 1A2 and 3A4 but has not been reported in river water before, but it is unsurprising given that lidocaine itself was detected in the targeted analysis of river water in both campaigns. Lidocaine is widely used as a local anaesthetic in both animals and humans and is available on prescription and as an over-the-counter medication to treat teething pain in children, skin burns/irritations, poisonous stings/bites and haemorrhoids. Lidocaine is also regularly used as an adulterant in illicit street drugs, such as cocaine.51 A second novel metabolite tentatively identified using in silico suspect screening was 8-hydroxyefavirenz, the primary metabolite of the antiretroviral, efavirenz,52 used to treat HIV-1 infection in the UK. A matching [M − H]− isotope abundance, several fragment ions and predicted retention time were all detected (Fig. 3(b)). To our knowledge, this is the first reported environmental occurrence of this metabolite in river water. In human liver microsomal studies, CYP2B6 was shown to play a major role in efavirenz clearance via 8-hydroxylation (∼77% (ref. 53)). Globally, reports of efavirenz occurrence are limited,54 but recently, concentrations as high as 37.6 μg L−1 have been measured in wastewater effluent in South Africa despite high sorption potential via sludge treatment. Lastly, tris(1-chloro-2-propyl)phosphate (TCPP) (Fig. 3(c)) was identified and is an organophosphate flame retardant that has multiple applications, including electronics and in furniture manufacture. In these applications TCPP is typically used in a film coating format rather than chemically bonded to the material, thus is prone to release into the environment.55 TCPP has been previously reported at ng L−1 concentrations in seawater and is known to cause detrimental effects in multiple animal taxa.55,56 In zebrafish, the lethal concentration (LC50) 96 h post fertilisation of TCPP was observed to be 3.7 mg L−1.57 Exposure to TCPP has resulted in decreases in neurobehavioral responses in fish, invertebrate and rodent species58–60 as well as endocrine disruption, and developmental and reproductive toxicity.56 When human cells have been exposed to TCPP through in vitro experiments, studies report inhibition in cell viability, growth rate, protein synthesis and cell cycle arrest.56 As such, TCPP is classified as a high hazard by the US EPA.61 Again the [M + H]+ ion was detected at m/z 327.0081 along with two fragments at m/z 98.9842 ([H4PO4]+) and m/z 174.9921 ([C3H6ClO4PH3]+). No other compound was shortlisted that corresponded to mass spectral data alone, but retention prediction was again accurate to within 13 s of the detected peak in the extract. Previous work focussing on evaluation of retention time prediction models for suspect screening in wastewater showed a success rate of between 83–73%.24
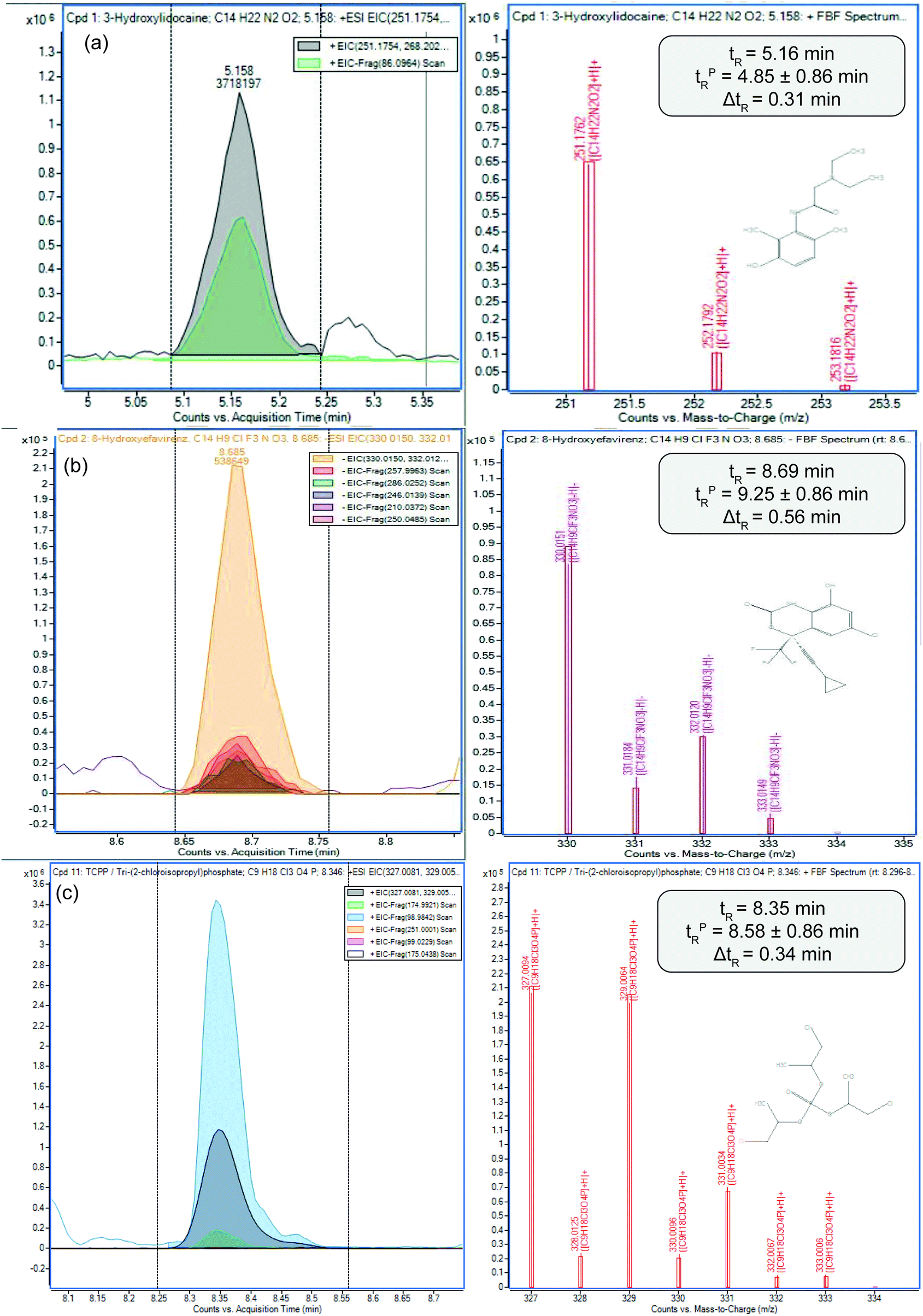 | ||
| Fig. 3 Extracted ion chromatograms of (a) 3-hydroxylidocaine, (b) 8-hydroxyefavirenz and (c) TCPP in the passive sampler extracts. (Left) Extracted ion chromatograms of the [M + H]+ (a and c) and [M − H]− (b) ion and relevant fragments measured by DIA. Right: denotes the isotopic fit of the [M + H]+ (a and c) and [M − H]− (b) and matching predicted retention times. | ||
Of all compounds tentatively identified using suspect screening, 15 more were confirmed using curated database entries which included retention time data. Of these, one compound was confirmed using database retention times within the Agilent Forensic database (phenytoin). Passive sampler extracts were also analysed on a separate LC-QTOF-MS method which held curated database LC retention time and accurate MS data for 14 more compounds and their presence was confirmed in all cases (see S4 for method details†). These included amisulpride, atenolol, bicalutamide, celiprolol, disopyramide, erythromycin, flecainide, irbesartan, O-desmethylvenlafaxine, practolol, proguanil, sotalol, sulpiride, tapentadol. Therefore, with respect to the Schymanski et al. identification framework,14 the compounds initially shortlisted using the Agilent HRMS databases were mostly classified between Level 4 (unequivocal molecular formula) and Level 2(a) (probable structure), depending on the presence of unique fragment ions. With the addition of predicted and curated library retention time data, we propose that matching compounds which had only one positive library spectrum match could be elevated to Level 2(a). However, to elevate compounds to Level 1 (confirmed structure), confirmation with an analytical standard is still required. That being said, the workflow presented above rapidly and efficiently aided compound occurrence confirmation workflows in environmental samples. Furthermore, according to the manufacturer, the LC-MS/MS instrument used for targeted analysis is capable of monitoring 555 transitions simultaneously and there is sufficient scope to add these and several more compounds to the targeted analytical method if required, including multiple transitions for each (to this point, 292 transitions were monitored including two for each compound and at least one for each SIL-IS). Even where the number of transitions to be monitored exceeds this threshold, the speed of the LC-MS/MS method leaves scope for the incorporation of multiple rapid injections of the same small sample using different target analyte sets, each of several hundred CECs. Using passive sampling together with both targeted analysis and machine learning-assisted suspect screening, therefore, offers a new, flexible and rapid capability for near time-integrated catchment monitoring of CECs and potentially at large scale.
4. Conclusion
A new methodology that successfully integrated new, flexible and more rapid approaches for CEC identification and monitoring in river water was demonstrated in two separate river monitoring studies. In particular, the potential for targeted direct LC-MS/MS analysis of river water for 164 CECs was possible for the subsequent detection of 33 compounds at low-mid ng L−1 concentrations using small volume injection in 5.5 min. To boost sensitivity even further and to perform time-integrated catchment monitoring, passive sampling was also successfully used with this new rapid targeted extract analysis method together with an in silico LC-QTOF-MS/MS suspect screening workflow to detect 65 CECs and subsequently shortlist an additional 59 compounds across both campaigns, respectively, including new compounds and metabolites. Specifically, the inclusion of retention time prediction reduced the number of suspects by roughly two thirds in comparison to the use of HRMS database searching alone, offering a new approach to rapidly prioritise reference standard acquisition for confirmation. This new workflow offers a new capability to perform near real-time catchment monitoring and/or triage impacted sites for potential in-depth time-integrated monitoring of river sites impacted by CECs.Author contributions
Alexandra K. Richardson: conceptualisation, methodology, validation, formal analysis, investigation, data curation, writing – original draft, writing – review & editing. Marcus Chadha: resources, data curation, investigation, writing – original draft, writing – review & editing. Helena Rapp Wright: investigation, validation. Graham A. Mills: resources, writing – review & editing. Gary R. Fones: resources, writing – review & editing. Anthony Gravell: investigation, data curation, writing – review & editing. Stephen Stürzenbaum: writing – review & editing, supervision. David A. Cowan: writing – review & editing, supervision. David J. Neep: resources, writing – review & editing, supervision. Leon P. Barron: conceptualisation, resources, writing – original draft, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Biotechnology and Biological Sciences Research Council under the London Interdisciplinary Doctoral Training Programme (LIDO) programme (BB/M009513/1) with Agilent Technologies UK Limited as iCASE partners. Thanks are also extended to Keng Tiong Ng, James Fielding and Adam Taylor for their assistance and to the London Fire Brigade, Transport for London and London Port Authority for granting access to the field sampling sites in this work.References
- Z. Wang, G. W. Walker, D. C. G. Muir and K. Nagatani-Yoshida, Environ. Sci. Technol., 2020, 54, 2575–2584 CrossRef CAS.
- J. L. Wilkinson, P. S. Hooda, J. Swinden, J. Barker and S. Barton, Environ. Pollut., 2018, 234, 864–875 CrossRef CAS.
- A. Zenker, M. R. Cicero, F. Prestinaci, P. Bottoni and M. Carere, J. Environ. Manage., 2014, 133, 378–387 CrossRef CAS.
- T. aus der Beek, F. A. Weber, A. Bergmann, S. Hickmann, I. Ebert, A. Hein and A. Küster, Environ. Toxicol. Chem., 2016, 35, 823–835 CrossRef CAS.
- P. Gago-Ferrero, A. A. Bletsou, D. E. Damalas, R. Aalizadeh, N. A. Alygizakis, H. P. Singer, J. Hollender and N. S. Thomaidis, J. Hazard. Mater., 2020, 387, 121712 CrossRef CAS.
- C. Soulier, C. Coureau and A. Togola, Sci. Total Environ., 2016, 563–564, 845–854 CrossRef CAS.
- C. Rimayi, L. Chimuka, A. Gravell, G. R. Fones and G. A. Mills, Environ. Monit. Assess., 2019, 191, 388 CrossRef.
- R. Bade, L. Bijlsma, T. H. Miller, L. P. Barron, J. V. Sancho and F. Hernández, Sci. Total Environ., 2015, 538, 934–941 CrossRef CAS.
- A. Agüera, M. J. Martínez Bueno and A. R. Fernández-Alba, Environ. Sci. Pollut. Res., 2013, 20, 3496–3515 CrossRef.
- M. J. Gómez, M. M. Gómez-Ramos, O. Malato, M. Mezcua and A. R. Férnandez-Alba, J. Chromatogr. A, 2010, 1217, 7038–7054 CrossRef.
- M. Ibáñez, J. V. Sancho, F. Hernández, D. McMillan and R. Rao, TrAC, Trends Anal. Chem., 2008, 27, 481–489 CrossRef.
- R. Bade, L. Bijlsma, J. V. Sancho and F. Hernández, Talanta, 2015, 139, 143–149 CrossRef CAS.
- K. Munro, T. H. Miller, C. P. B. Martins, A. M. Edge, D. A. Cowan and L. P. Barron, J. Chromatogr. A, 2015, 1396, 34–44 CrossRef CAS.
- E. L. Schymanski, J. Jeon, R. Gulde, K. Fenner, M. Ruff, H. P. Singer and J. Hollender, Environ. Sci. Technol., 2014, 48, 2097–2098 CrossRef CAS.
- J. Nurmi, J. Pellinen and A. L. Rantalainen, J. Mass Spectrom., 2012, 47, 303–312 CrossRef CAS.
- S. Kern, K. Fenner, H. P. Singer, R. P. Schwarzenbach and J. Hollender, Environ. Sci. Technol., 2009, 43, 7039–7046 CrossRef CAS.
- K. Héberger, J. Chromatogr. A, 2007, 1158, 273–305 CrossRef.
- C. Giaginis and A. Tsantili-Kakoulidou, Chromatographia, 2013, 76, 211–226 CrossRef CAS.
- X. Li, F. Luan, H. Si, Z. Hu and M. Liu, Toxicol. Lett., 2007, 175, 136–144 CrossRef CAS.
- M. Cao, K. Fraser, J. Huege, T. Featonby, S. Rasmussen and C. Jones, Metabolomics, 2015, 11, 696–706 CrossRef CAS.
- L. P. Barron and G. L. McEneff, Talanta, 2016, 147, 261–270 CrossRef CAS.
- T. H. Miller, A. Musenga, D. A. Cowan and L. P. Barron, Anal. Chem., 2013, 85, 10330–10337 CrossRef CAS.
- A. G. Fragkaki, E. Farmaki, N. Thomaidis, A. Tsantili-Kakoulidou, Y. S. Angelis, M. Koupparis and C. Georgakopoulos, J. Chromatogr. A, 2012, 1256, 232–239 CrossRef CAS.
- K. Munro, C. P. B. Martins, M. Loewenthal, S. Comber, D. A. Cowan, L. Pereira and L. P. Barron, Sci. Total Environ., 2019, 657, 1099–1111 CrossRef CAS.
- K. T. Ng, H. Rapp-Wright, M. Egli, A. Hartmann, J. C. Steele, J. E. Sosa-Hernández, E. M. Melchor-Martínez, M. Jacobs, B. White, F. Regan, R. Parra-Saldivar, L. Couchman, R. U. Halden and L. P. Barron, J. Hazard. Mater., 2020, 398, 1229933 Search PubMed.
- G. D. Castle, G. A. Mills, A. Bakir, A. Gravell, M. Schumacher, I. Townsend, L. Jones, R. Greenwood, S. Knott and G. R. Fones, Talanta, 2018, 179, 57–63 CrossRef CAS.
- R. Guibal, S. Lissalde, A. Charriau and G. Guibaud, Talanta, 2015, 144, 1316–1323 CrossRef CAS.
- A. C. Taylor, G. R. Fones and G. A. Mills, Trends Environ. Anal. Chem., 2020, 27, e00096 CrossRef CAS.
- ICH Expert Working Group, International Council for Harmonisation of Technical, Requirements for Pharmaceuticals for Human Use (ICH), 2005, pp. 1–7 Search PubMed.
- J. G. Topliss and R. J. Costello, J. Med. Chem., 1972, 15, 1066–1068 CrossRef CAS.
- C. B. Mollerup, M. Mardal, P. W. Dalsgaard, K. Linnet and L. P. Barron, J. Chromatogr. A, 2018, 1542, 82–88 CrossRef CAS.
- M. W. Gardner and S. R. Dorling, Atmos. Environ., 1998, 32, 2627–2636 CrossRef CAS.
- C. A. Murthy and J. Pittman, Inf. Sci., 1998, 112, 137–150 CrossRef.
- D. White, D. J. Lapworth, W. Civil and P. Williams, Environ. Pollut., 2019, 249, 257–266 CrossRef CAS.
- N. Nakada, S. Hanamoto, M. D. Jürgens, A. C. Johnson, M. J. Bowes and H. Tanaka, Sci. Total Environ., 2017, 575, 1100–1108 CrossRef CAS.
- S. Letsinger, P. Kay, S. Rodríguez-Mozaz, M. Villagrassa, D. Barceló and J. M. Rotchell, Sci. Total Environ., 2019, 678, 74–84 CrossRef CAS.
- K. V. Thomas and M. J. Hilton, Mar. Pollut. Bull., 2004, 49, 436–444 CrossRef CAS.
- European Commission, Off. J. Eur. Union, 2013, 56, 1–17 Search PubMed.
- European Commission, Off. J. Eur. Union, 2011, 153, 1–186 Search PubMed.
- T. H. Miller, K. T. Ng, S. T. Bury, S. E. Bury, N. R. Bury and L. P. Barron, Environ. Int., 2019, 129, 595–606 CrossRef CAS.
- C. Boix, M. Ibáñez, J. V. Sancho, J. Rambla, J. L. Aranda, S. Ballester and F. Hernández, Talanta, 2015, 131, 719–727 CrossRef CAS.
- N. Hermes, K. S. Jewell, A. Wick and T. A. Ternes, J. Chromatogr. A, 2018, 1531, 64–73 CrossRef CAS.
- M. J. Martínez Bueno, S. Uclés, M. D. Hernando and A. R. Fernández-Alba, Talanta, 2011, 85, 157–166 CrossRef.
- B. Petrie, A. Gravell, G. A. Mills, J. Youdan, R. Barden and B. Kasprzyk-Hordern, Environ. Sci. Technol., 2016, 50, 9469–9478 CrossRef CAS.
- R. Guibal, S. Lissalde and G. Guibaud, Environ. Toxicol. Chem., 2020, 39, 1186–1195 CrossRef CAS.
- J. A. Baz-Lomba, C. Harman, M. Reid and K. V. Thomas, Water Res., 2017, 121, 221–230 CrossRef CAS.
- K. M. Stroski, J. K. Challis and C. S. Wong, Anal. Chim. Acta, 2018, 1018, 45–53 CrossRef CAS.
- H. Li, P. A. Helm, G. Paterson and C. D. Metcalfe, Chemosphere, 2011, 83, 271–280 CrossRef CAS.
- Y. Li, C. Yang, D. Zha, L. Wang, G. Lu, Q. Sun and D. Wu, Sci. Total Environ., 2018, 610–611, 1356–1363 CrossRef CAS.
- J. D. Harkins, G. D. Mundy, W. E. Woods, A. Lehner, W. Karpiesiuk, W. A. Rees, L. Dirikolu, S. Bass, W. G. Carter, J. Boyles and T. Tobin, J. Vet. Pharmacol. Ther., 1998, 21, 462–476 CrossRef CAS.
- F. O. Chagas, M. C. Hespanhol, L. H. M. Da Silva, P. R. Patrício, A. O. Maldaner, T. S. Soares, A. S. B. Castro and P. A. Marinho, Analyst, 2020, 145, 6562–6571 RSC.
- M. Brandmann, U. Nehls and R. Dringen, Neurochem. Res., 2013, 38, 2524–2534 CrossRef CAS.
- E. T. Ogburn, D. R. Jones, A. R. Masters, C. Xu, Y. Guo and Z. Desta, Drug Metab. Dispos., 2010, 38, 1218–1229 CrossRef CAS.
- T. T. Mosekiemang, M. A. Stander and A. de Villiers, Chemosphere, 2019, 220, 983–992 CrossRef CAS.
- C. Ji, Z. Lu, L. Xu, F. Li, M. Cong, X. Shan and H. Wu, Sci. Total Environ., 2020, 724, 138307 CrossRef CAS.
- C. Wang, H. Chen, H. Li, J. Yu, X. Wang and Y. Liu, Environ. Int., 2020, 143 Search PubMed.
- S. P. McGee, E. M. Cooper, H. M. Stapleton and D. C. Volz, Environ. Health Perspect., 2012, 120, 1585–1591 CrossRef CAS.
- L. V. Dishaw, D. L. Hunter, B. Padnos, S. Padilla and H. M. Stapleton, Toxicol. Sci., 2014, 142, 445–454 CrossRef CAS.
- C. Wang, J. An, Y. Bai, H. Li, H. Chen, D. Ou and Y. Liu, Environ. Pollut., 2019, 246, 904–913 CrossRef CAS.
- V. C. Moser, P. M. Phillips, J. M. Hedge and K. L. McDaniel, Neurotoxicol. Teratol., 2015, 52, 236–247 CrossRef CAS.
- K. Baker, R. Baker, W. Baur, J. Bulmahn, E. Greenwood, T. Hitchcock, J. Jacobs, N. Logue, F. Mentzer, E. Mona, C. Pramas, S. K. Reynolds, F. W. Schneider, M. A. Stackpole, L. Stevens, J. L. Sutter, R. Beisner, J. Fares, W. Mahy, B. Sola, F. Solhan, B. Wootten, L. Bonner, D. Team, L. Bonner and J. Compton, U.S. EPA Design for the Environment, 2014, 4121233–4121235.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ay02013c |
| This journal is © The Royal Society of Chemistry 2021 |