
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Complete enzymatic digestion of double-stranded RNA to nucleosides enables accurate quantification of dsRNA†
Steven R.
Strezsak
 ab,
Penny J.
Beuning
a and
Nicholas J.
Skizim
*b
ab,
Penny J.
Beuning
a and
Nicholas J.
Skizim
*b
aDepartment of Chemistry & Chemical Biology, Northeastern University, Boston, MA 02115, USA
bGreenlight Biosciences, Medford, MA 02155, USA. E-mail: nskizim@greenlightbio.com; Tel: +1-617-279-0713
First published on 8th December 2020
Abstract
The rapid growth of research focusing on RNA, especially for RNA interference applications, has created a need for a robust method that can accurately determine the concentration of long dsRNA. As it is difficult to find a source for pure dsRNA reference material, the most common method for quantitation is using a reversed-phase HPLC method to determine purity, which is linked to a calibration curve prepared by measurements obtained using UV absorbance at 260 nm. In this study we developed a nucleic acid digestion method that can digest both double- and single-stranded RNA and DNA to nucleosides. A reversed-phase HPLC/UV method was used to separate and quantitate the monomeric nucleosides. Using this method, we were able to calculate the absorptivity coefficient (proxy for the extinction coefficient) for dsRNA to be 45.9 ± 0.52 μg mL−1/A260. This value agrees with the one report we were able to find but uses an orthogonal method. Moreover, this study allowed us to understand that sequence design can dramatically change the extinction coefficient of the molecule. For molecules with ssRNA overhangs, we observed a 5% reduction in the calculated extinction coefficient.
1. Introduction
One of the most popular methods to measure the concentration of nucleic acids is ultraviolet (UV) absorbance spectroscopy. With the development of nano spectrophotometers, as little at 2 μL of sample is adequate to determine the concentration and as little as 3 ng μL−1 can be detected within a matter of seconds.1 The concentration of nucleic acids is calculated using the Beer–Lambert law (A = ε × C × l), where A is absorbance, determined by the spectrophotometer, ε is the molar extinction coefficient of the measured sample, C is the concentration of nucleic acids in solution and l is the pathlength of the vessel in the spectrophotometer.2 While Beer–Lambert's law is straightforward, there are other factors that can complicate the analysis of nucleic acids, including conformational changes in the structure, base-stacking, and base pairing. These can cause a shift from the expected molar extinction coefficient relative to the simple sum of absorbance of individual nucleotides. This effect is referred to as either hypochromicity, which is a decrease in absorbance, or hyperchromicity, which is an increase in absorbance.Extinction coefficients for (ds)DNA and ssRNA are well characterized and mass concentration/A260 coefficients of 50 μg mL−1/A260 and 40 μg mL−1/A260 are commonly used for DNA and ssRNA, respectively.3 A study has calculated the mass concentration/A260 coefficient for ssDNA by 1H NMR to be between 37 and 38 μg mL−1/A260 although the most common value used is 33 μg mL−1/A260.4 A recent study determined the extinction coefficient for dsRNA to be between 46.18 and 47.29 μg mL−1/A260 by measuring the change in absorbance of samples upon thermal denaturation in the presence of DMSO.5 To our knowledge, this is the only published work that has reported a value for dsRNA and the goal of this research is to develop an orthogonal method that can be used to calculate the extinction coefficient for dsRNA.
Nucleic acid metabolism in nature includes both chemical reactions such as acid/base hydrolysis and enzymatic reactions including nucleases, phosphodiesterases and phosphatases.6,7 By digesting all of the intact nucleic acids to the monomeric nucleoside level, separation of analytes can be readily achieved using a reversed phase HPLC method.8 By quantifying the nucleosides of the digested product and calculating a molar extinction coefficient by summing the total amount of nucleosides, the effects of absorbance changes in solution as well as any base stacking and base pairing can be determined, and an accurate quantification of the dsRNA can be achieved. Additionally, high purity nucleoside standards are available, which is not the case for dsRNA material nor the nucleoside phosphates.9 Methods have been reported that have used either enzymatic or chemical digestion to yield monophosphorylated compounds, or with the addition of a phosphatase, nucleosides.10,11 HPLC separation of the nucleosides or nucleotides is then used to quantify the components in the mixture. One report described methods that use thermal hydrolysis to form 2′–3′ nucleotide monophosphate mixtures that would enable quantification of the total amount of RNA.12 We attempted to use this method to calculate the extinction coefficient for dsRNA, but observed differences in the extinction coefficients for the monomeric isomers, e.g. 2′-GMP vs. 3′-GMP. Without high purity standards for each of these isomers, which we were unable to source, quantification via this route was not possible.
Therefore, in this study we have developed a robust method to accurately determine the extinction coefficient of dsRNA using a combination of enzymatic digestion of nucleic acids to their monomeric nucleosides followed by an HPLC method to separate and quantitate the nucleosides using UV spectrophotometry. The dsRNA was first treated with RNase If, an RNase that has a preference to digest ssRNA overhangs and leave blunt-ended dsRNA products. Those products were then purified, yielding purely duplex dsRNA. Often dsRNA molecules have ssRNA regions that are not fully complexed, which can result from the specific DNA template design, for example, transcription initiation sequences at the 5′-mRNA end that are not complemented, or read-through from inefficient transcriptional termination. Removal of these ssRNA regions (often ssRNA overhangs at the 5′ or 3′ end of the molecule) from the test material was critical to obtain an accurate molar absorptivity coefficient for pure dsRNA. Notably, our enzymatic digestion method efficiently digests both RNA and DNA, and all nucleoside analytes can be resolved by our analytical HPLC method, enabling the analysis of mixtures of both RNA and DNA (Fig. 1).
 | ||
| Fig. 1 All dsRNA material used for analysis was treated with RNase If to remove ssRNA overhangs on the duplexed molecule. The samples were purified to remove digested nucleotides stemming from the ssRNA overhangs. A260 measurements were taken to determine the concentration of each sample prior to digestion. Samples were digested for 3 h and quantitated by a reversed-phase HPLC method. | ||
2. Materials and methods
2.1 Chemicals and materials
Triethylammonium acetate (1 M), acetonitrile, adenosine, ammonium acetate, cytidine, deoxyadenosine, deoxycytidine, deoxyguanosine, formic acid, guanosine, sodium chloride, thymidine, and uridine were all obtained from Sigma-Aldrich (MO, USA). RNase If, NEB Buffer 3, Cutsmart buffer, and alkaline phosphatase were obtained from New England Biolabs Inc. (MA, USA). RNase T1, ethanol, 10% sodium dodecyl sulfate, 1 M Tris–EDTA pH 7.0, 1 M Tris–EDTA pH 8.0, nuclease free water and ultrapure salmon sperm DNA were obtained from Thermo Fisher Scientific (NJ, USA). DNASep 4.6 × 50 mm HPLC columns were purchased from ADS Biotec (NE, USA) and Cosmosil 5C18-MS-II 4.6 × 150 mm columns were purchased from Nacalai (CA, USA). Oligonucleotides used in the study were from AgroRNA (Seoul, South Korea), Integrated DNA Technologies (IA, USA), and Trilink (San Diego, USA). Nucleic acids used in this study are shown in Table 1.| Nucleic acid | % GC content | Vendor |
|---|---|---|
| ssRNA (60 mer) | 63 | IDT |
| ssRNA (1929 mer) | 58 | Trilink |
| dsRNA (55 bp) | 53 | IDT |
| dsRNA (55 bp) | 49 | AgroRNA |
| dsRNA (200 bp) | 49 | AgroRNA |
| dsRNA (300 bp) | 40 | AgroRNA |
| dsRNA (300 bp) | 49 | AgroRNA |
| dsRNA (400 bp) | 48 | AgroRNA |
| dsRNA (425 bp) | 43 | AgroRNA |
| dsRNA (599 bp) | 53 | AgroRNA |
| ssDNA (594 bp) | 47 | IDT |
| ssDNA (594 bp) | 47 | IDT |
| ssDNA (545 bp) | 40 | IDT |
| ssDNA (574 bp) | 40 | IDT |
| dsDNA (2927 bp) | 46 | In-house |
| dsDNA (2963 bp) | 44 | In-house |
2.2 RNase If cleanup of purchased dsRNA
Samples were treated with RNase If and re-purified using a Qiaprep (Qiagen) solid phase extraction column. RNase If is a fusion protein of RNase I and maltose binding protein (MBP, New England Biolabs) and has been shown to exhibit preferential nuclease activity on ssRNA. Samples were warmed to 30 °C and 1 mL was transferred to a 1.5 mL Eppendorf tube. RNase If (100 μL of 50 U μL−1) and 100 μL NEB Buffer 3 (New England Biolabs) were added to each tube and heated at 37 °C for 1 h. Immediately after heating, samples were transferred to a 15 mL Falcon tube and 2.5 mL of a solution composed of 4% sodium dodecyl sulfate, 0.5 M NaCl and 5 mM EDTA. After mixing, 1 mL 5 M NaCl was added, vortexed for 15 s and centrifuged for 15 min at 4000 rpm at a temperature of 4 °C. A 3 mL aliquot of supernatant was transferred into 4 mL of solution containing 94% isopropanol with 6% guanidine HCl and mixed with a vortex for 15 s. A 700 μL aliquot of this mixture was added to ten different Qiaprep 2.0 spin columns, which were centrifuged to remove liquid at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 2 min. The columns were washed with 700 μL of 15 mM Tris–HCL, 1.5 mM disodium EDTA pH 7.0 in 80% ethanol. The columns were centrifuged at 10000 rpm for 2 min to remove liquid then dried by centrifugation for another minute at 10000 rpm. Nuclease-free water (100 μL) was added to each column and after 2 min, the samples were centrifuged at 10000 rpm to elute the dsRNA. Samples were pooled and stored at −80 °C until use. This method was adapted from the published RNASwift protocol.13
000 rpm for 2 min. The columns were washed with 700 μL of 15 mM Tris–HCL, 1.5 mM disodium EDTA pH 7.0 in 80% ethanol. The columns were centrifuged at 10000 rpm for 2 min to remove liquid then dried by centrifugation for another minute at 10000 rpm. Nuclease-free water (100 μL) was added to each column and after 2 min, the samples were centrifuged at 10000 rpm to elute the dsRNA. Samples were pooled and stored at −80 °C until use. This method was adapted from the published RNASwift protocol.13
2.3 Analysis of purified dsRNA and total RNA
RNA quantification was determined using half area UV-transparent plates (Thermo Catalog #675801) on a Synergy HTX multi-mode plate reader from Biotek (VT, USA). RNA concentrations were determined using absorbance at 260 nm. Additional analysis of the RNA was performed using ion-pair reverse-phase chromatography (IP-RP HPLC). Post-treatment/re-purified samples (2 μL) were analyzed by IP-RP-HPLC on an Agilent 1100 series HPLC using a DNASep column (50 mm × 4.6 mm I.D. ADS Biotec). Chromatograms were obtained using a UV detector at a wavelength of 260 nm. The chromatographic separation was performed using the following mobile phases: buffer A – 0.1 M triethylammonium acetate (TEAA) pH 7.0 (Sigma); buffer B – 0.1 M TEAA, pH 7.0 containing 25% acetonitrile (Sigma). RNA was analyzed using the following gradient starting at 20% buffer B to 52% in 0.5 min, followed by a hold at 52% buffer B to 2.25 min, then a linear gradient to 80% buffer B to 2.8 min followed by a hold at 80% buffer B to 4.25 min, followed by an immediate switch to 100% buffer B and held to 5 min, followed by an immediate return to 20% buffer B and held until 9 min at a flow rate of 0.75 mL min−1 at 50 °C.2.4 Enzymatic digestion to nucleosides
In a 200 μL working volume PCR strip tube, 40 μL of purified sample (6–40 μg nucleic acids) was added to 110 μL enzymatic digestion master mix containing 0.83 U μL−1 RNase A, 0.83 U μL−1 RNase T1, 0.83 U μL−1 phosphodiesterase I, 0.016 U μL−1 alkaline phosphatase and 0.083 mM 2′-O-methylcytidine. The samples were mixed by inversion and incubated at 37 °C for 3 h. This digestion protocol was optimized for an unpurified yeast RNA extract and was not subsequently optimized for purified nucleic acids.2.5 Reverse-phase high performance liquid chromatography of nucleosides
Digested samples were analyzed by reverse-phase chromatography on an Agilent 1100 series HPLC using a Cosmosil 5C18-MS-II column (50 mm × 4.6 mm I.D. Nacalai). Chromatograms were obtained using a UV detector at a wavelength of 260 nm. The chromatographic separation was performed using the following mobile phases: buffer A – 20 mM ammonium acetate with 0.1% formic acid; buffer B – methanol. Nucleosides were analyzed using the following gradient: starting at 2% buffer B to 27% over 8.5 min, followed by an immediate change to 100% buffer B and held until 9 min, followed by an immediate return to 2% B and held until 12 min at a flow rate of 0.75 mL min−1 at 35 °C. To normalize for any evaporation during hydrolysis or any variability in the HPLC injection, 2′-O-methylcytidine was added to the master mix and used as an internal standard for the quantitation of the individual nucleosides. Calibration curve slopes were generated by calculating the response factor for each analyte by dividing the area of the analyte by the area of the internal standard.2.6 Preparation of standard curves
Calibration curves were constructed in deionized water by preparing a stock solution containing adenosine, cytidine, guanosine, uridine each at 2.5 mM. The stock solution was serial diluted to form an additional six calibration levels (1.25, 0.625, 0.3125, 0.156, 0.078 and 0 mM). The purity of each standard was verified by HPLC and the concentration of the stock solution was adjusted to account for the measured purity. All purities were greater than 99% by HPLC.2.7 Method validation
The calibration curves are plots of the analyte area ratio (AAR) to the internal standard as a function of the analyte concentration (C). This gives the following equation: AAR = slope × C + intercept. The slope and the intercept are determined by the measured AAR and the nominal concentration of the analyte. The unknown concentrations are calculated from this equation for each analyte in the samples. All calculations were performed using Microsoft Excel.The precision of the method based on intra-day variability was determined by replicate analysis of two purchased ssRNA controls, a 60 nucleotide (nt) ssRNA and a 1929 nt ssRNA. The reproducibility was taken as the inter-day variability and was determined by replicate analysis of the controls in different days with two replicates being analyzed each day. The relative standard deviation values (RSD) were calculated from the ratios of the standard deviation (SD) to the mean and expressed as a percentage.
The accuracy of the method was determined by comparing practical amounts recovered from the control samples with actual values present in the samples (theoretical values). The literature accepted value of 40.0 ng μL−1/A260 was used for the ssRNA standards. The selectivity of the method was determined by examining the interference coming from the enzyme background.
The limit of quantification (LOQ) and limit of detection (LOD) were calculated from the standard error for the line of best fit of the calibration curve data collected during the validation. The STEYX function was used to calculate the standard error of Y given X for a least-squares linear regression line. LOD = 3 × STEYX value. Similarly, LOQ = 10 × STYEX value.
The specificity of the method was determined by titrating different amounts of RNA and DNA together to confirm a linear response proportional to the amount of RNA present in the sample.
3. Results and discussion
3.1 Method validation
The linearity of the nucleoside HPLC assay was evaluated using a six-point standard calibration curve with concentrations ranging from 0 to 2.5 mM. These standards were prepared in triplicate and evaluated over the course of three months. The slopes showed low inter-day variability (<4%) for all analytes and demonstrated good linearity between 0.0780–2.5 mM with a correlation coefficient (R2) of 0.999 or greater for all analytes.Three sources of nucleotides were used to determine the reaction time required to fully digest the samples to their nucleoside constituents. A mixture of ribonucleotide triphosphates; an RNA extract from Saccharomyces cerevisiae, labeled ‘RNA A’ (Worthington); and a chemically synthesized 60-mer oligonucleotide, labeled ‘RNA B’ (IDT) were used. We found that digestion was nearly complete at 100 min and was complete by 3 h (Fig. 2).
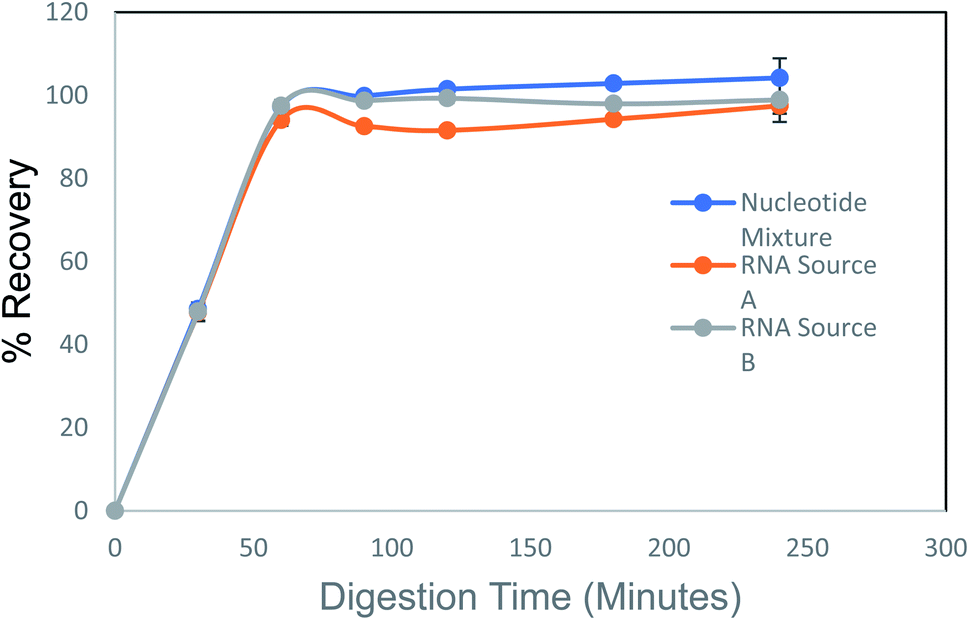 | ||
| Fig. 2 Evaluation of digestion time for complete recovery, three different nucleotide sources were digested through a time-course to determine optimal digestion time. At 3 h, all three samples reached 100% digestion. | ||
Evaluation of intra-day precision was determined by preparing 20 independent digestions of two different yeast sources and analyzing them using the nucleoside HPLC method on the same day. Recoveries were between 96 and 99% for both materials (Table 2).
| Sample name | Theoretical value (mM) | Calculated value (mM) | SD (mM) | RSD% | % recovery |
|---|---|---|---|---|---|
| a The theoretical value is calculated by using the vendor's purity claim and from the weight of the solutions that were prepared. Recoveries for both materials were greater than 96%. Relative standard deviations were less than 2.5% for 20 independent replicates. | |||||
| Source-A | 1.47 | 1.42 | 0.033 | 2.3 | 96.6 |
| Source-B | 1.18 | 1.17 | 0.01 | 0.9 | 98.8 |
Evaluation of inter-day precision was determined by preparing two independent digestions of three different RNA sources and running the nucleoside HPLC method over 20 d. All RSDs were less than 4% and shown in Table 3.
| Sample name | Average (mM) | Standard deviation | RSD (%) |
|---|---|---|---|
| Source-A | 1.4 | 0.05 | 3.42 |
| Source-B | 2.88 | 0.08 | 2.76 |
A titration of RNA and DNA mixtures was prepared in triplicate ranging from 0 to 100% of each analyte. The responses for both DNA and RNA were linear with R2 > 0.99 for both species (Fig. 3). A chromatogram of the analytical separation of a mixture of digested RNA and DNA is shown in Fig. 4.
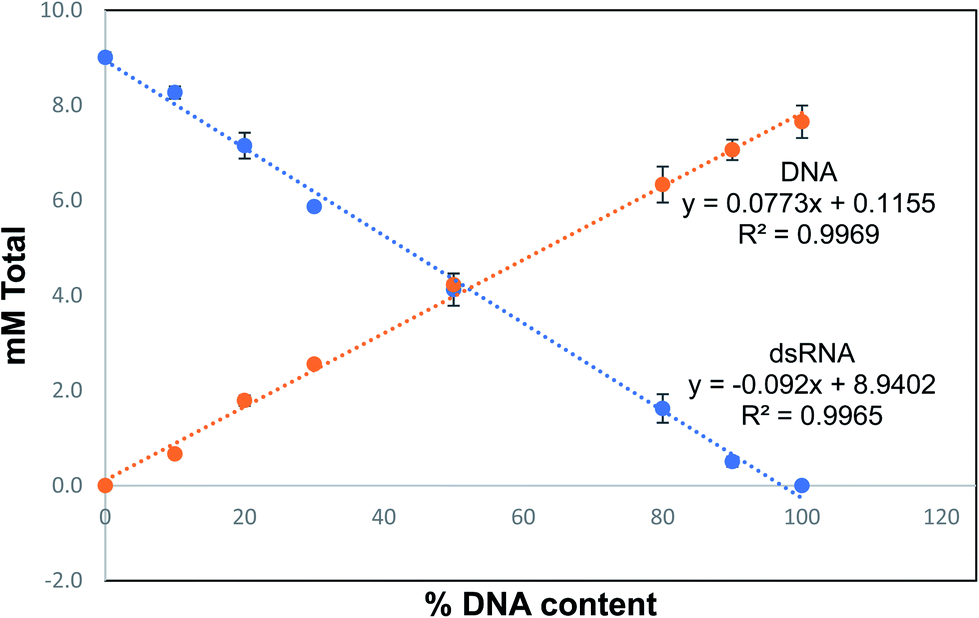 | ||
| Fig. 3 DNA/RNA titration. Mixtures of RNA and DNA were digested at varying amounts of each type to confirm that mixed nucleic acids could be digested together. There is a linear response to the amount of DNA that was added to the mixture demonstrating that mixed solutions could be digested together. | ||
 | ||
| Fig. 4 Reversed-phase separation of nucleosides and deoxynucleosides from a mixture of RNA and DNA. 2′-O-Methylcytidine is used as an internal standard and is spiked into the master mix prior to sample digestion. | ||
3.2 Equations and calculations to determine mass absorptivity coefficient (proxy for extinction coefficient)
Our approach initially assumes this coefficient to be 40.0, equivalent to that for ssRNA. We then scale up the coefficient based on the recovery observed by summation of the component nucleosides by HPLC assay, providing us the correct coefficient for dsRNA.Equations:
| Acorrected = Ana − Ablank | (1) |
| W260 = Acorrected × Y0 × dilution factor | (2) |
| RFx = Sx/SI | (3) |
| Cx = RFx/mx | (4) |
| CHPLC by HPLC = CdA + CA + CdC + CC + CdT + CdG + CG + CU | (5) |
| Q = CHPLC/W260 | (6) |
| Ycorrected = Y0 × Q | (7) |
 where * are known values for calibration curve, Cx = concentration of analyte (g L−1).
where * are known values for calibration curve, Cx = concentration of analyte (g L−1).
For each nucleic acid sample, 170 μL of sample was added to a half area plate, creating a path length of 1 cm. This was performed in triplicate and the average absorbance was subtracted from the average absorbance blank that ran on the plate (see eqn (1)). This value was multiplied by the dilution factor, if any, and by 40 (the widely accepted mass absorptivity coefficient for ssRNA) (eqn (2)). The concentration of nucleosides by HPLC were calculated from calibration curves containing all relevant compounds prepared along with the sample set. An internal standard 2′-O-methylcytidine was used for all compounds and response factors were calculated for all data (eqn (3)). Using the response factors, slopes were calculated using a linear model fit to calibration curves for each analyte (x). The concentrations for all analytes (x) were calculated by dividing the response factor by the analyte slope (eqn (4)). The concentration of nucleosides by HPLC was added up for each sample (eqn (5)) with g L−1 units and adjusting the mass by 61.811 (the addition of phosphate group per nucleoside minus the water loss from the formation of the phosphodiester bond). The corrected quotient (Q) for each sample was calculated by dividing the calculated concentration by HPLC by the calculated concentration by A260 in the spectrophotometer (eqn (6)). The extinction coefficient (mass absorptivity coefficient) was calculated by multiplying Q of the sample by the initial commonly accepted mass absorptivity coefficient Y0 (eqn (7)).
3.3 ssRNA overhang observations
Initial attempts to determine the extinction coefficient of the dsRNA led to values less than those previously reported.5 For these early experiments, we were not yet treating the RNA molecules with RNase If to remove ssRNA overhangs, and the DNA template from which this RNA was transcribed was designed to have ssRNA overhangs on the 3′ ends of the complex. These lower values were due to the effect of this un-complexed region on the bulk absorbance of the molecule. This molecule was digested by the RNase cocktail with or without a pretreatment by RNase If and the extinction coefficient increased from 42.9 to 45.8 μg mL−1/A260 with the addition of the RNase If step (Fig. 5). Furthermore, a blunt-ended RNA was chemically synthesized by IDT (different sequence) and digested without RNase If and the calculated extinction coefficient matched the higher value, supporting our theory that the ssRNA overhang lowers the overall calculated extinction coefficient for dsRNA. | ||
| Fig. 5 Calculated dsRNA extinction coefficient for RNase treated vs. untreated material. All dsRNA material used for analysis was treated with RNase If to remove ssRNA overhangs on the duplexed molecule. The samples were purified to remove any digested nucleotides from the ssRNA overhangs. A260 measurements were taken to determine the concentration of each sample prior to digestion. Samples were digested for 3 h and quantitated by a reversed-phase HPLC method. | ||
3.4 Quantification of dsRNA mass absorptivity coefficient (proxy for extinction coefficient)
Following the successful validation of the analytical method, we purchased dsRNA sequences of various lengths, treated them with RNase If to remove any ssRNA overhangs, and analyzed the resulting products for purity by HPLC. After cleanup, all sequences exhibited greater than 98.4% dsRNA as determined by HPLC. Chromatograms shown in Fig. S1–S8.† Those dsRNA samples were then digested with our enzyme cocktail in triplicate. For samples with sufficient volume, digestions were performed in triplicate and on multiple separate days (up to 3), yielding at least 6 replicates for all RNA the sequences. For ssDNA and dsDNA, we digested all sequences in triplicate for 1 day.To demonstrate acceptable sample recovery, we used the widely accepted mass absorptivity coefficient for ssRNA of 40.0 μg mL−1/A260 (with a pathlength of 1 cm). We used that coefficient along with the absorbance of a 1929 nt ssRNA control sequence at 260 nm to determine input RNA concentration. This ssRNA control was digested alongside the dsRNA products in each assay and its nucleoside constituents were used to determine RNA concentration by summing the monomers. We observed a recovery of 99.3 ± 0.9%. An additional ssRNA control (60-mer) was analyzed each day to verify assay performance and provide similar recovery information.
Using the equations in 3.2, we arrived at the mass-based absorptivity coefficients shown (Table 4).
| Size | Nature | GC% | Replicates | Average ε proxy μg mL−1/A260 | % A | % G | % C | % U or T | SD | CV (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Hairpins (stem-loop structures) exhibit some dsRNA nature (their stem) along with some ssRNA nature (their loop). This molecule would form a 55 bp dsRNA molecule if intermolecularly duplexed but would form a hairpin with a 25 bp stem if intramolecularly duplexed. Where possible, the samples were digested with three replicates over 3 days. The average calculated mass absorptivity coefficient for each sequence is shown as well as the variance observed for each sequence. | ||||||||||
| 60 | ssRNA | 63 | 9 | 39.7 | 18 | 32 | 32 | 18 | 0.37 | 0.9 |
| 1929 | ssRNA | 39 | 9 | 40.0 | 33 | 18 | 21 | 0 | 0.25 | 0.6 |
| 200 | dsRNA | 49 | 6 | 46.5 | 26 | 27 | 23 | 25 | 0.22 | 0.5 |
| 400 | dsRNA | 48 | 8 | 45.8 | 25 | 24 | 27 | 24 | 0.60 | 1.3 |
| 300 | dsRNA | 49 | 9 | 46.1 | 22 | 24 | 25 | 29 | 0.20 | 0.4 |
| 1000 | dsRNA | 48 | 7 | 46.7 | 27 | 25 | 23 | 25 | 0.67 | 1.4 |
| 300 | dsRNA | 45 | 6 | 45.8 | 33 | 24 | 21 | 22 | 1.34 | 2.9 |
| 425 | dsRNA | 43 | 6 | 45.7 | 32 | 26 | 17 | 26 | 0.93 | 2.0 |
| 599 | dsRNA | 53 | 7 | 45.1 | 26 | 23 | 30 | 21 | 0.49 | 1.1 |
| 55 | dsRNA | 49 | 6 | 45.6 | 22 | 22 | 27 | 29 | 0.44 | 1.0 |
| 55 (hairpina) | ss/dsRNAa | 53 | 8 | 42.1 | 25 | 20 | 33 | 9 | 0.36 | 0.9 |
| 594 | ssDNA | 47 | 3 | 37.3 | 27 | 23 | 24 | 26 | 0.19 | 1.4 |
| 594 | ssDNA | 47 | 3 | 37.2 | 27 | 24 | 23 | 26 | 0.17 | 1.3 |
| 574 | ssDNA | 40 | 3 | 38.4 | 32 | 24 | 16 | 28 | 0.24 | 1.7 |
| 574 | ssDNA | 40 | 3 | 37.2 | 32 | 25 | 15 | 28 | 0.12 | 0.9 |
| 2927 | dsDNA | 46 | 3 | 50.6 | 27 | 24 | 23 | 27 | 0.07 | 0.4 |
| 2963 | dsDNA | 44 | 3 | 49.3 | 28 | 23 | 22 | 28 | 0.08 | 0.5 |
We observed an extinction coefficient for the 55 bp dsRNA hairpin in between that of purely ssRNA and purely dsRNA. This observation is consistent with this molecule's propensity to form a hairpin with a 15 bp stem (ITS + ITS reverse complement) and a 25 nt loop; it would exhibit some ssRNA-like nature (the loop) and some dsRNA-like nature (the stem).
4. Conclusions
Ultraviolet spectroscopy is commonly used to quickly quantify nucleic acids in biology, but its use requires that an accurate extinction coefficient is known. The extinction coefficients for ssDNA, dsDNA, and ssRNA are indeed well characterized, but that for dsRNA is not.2,3,14,15 The absorptivity coefficients reported on vendor websites range between 40 and 50 μg mL−1/OD260. Here we demonstrate the absorptivity coefficient (proxy for the extinction coefficient) for dsRNA to be 45.9 ± 0.52 μg mL−1/A260. This value agrees well with the only other reported value for dsRNA at 46.52 μg mL−1/A260 (observed range in that study was 46.18 – 47.29 μg mL−1/A260).5 Neither oligonucleotide length (55–1000 bp) nor GC content (40–63%) showed a correlation with the extinction coefficient of the molecule. Graphs shown in Fig. S9 and S10.† To further build confidence in our assay, we digested ssDNA and dsDNA and calculated the absorptivity coefficients as 37.5 ± 0.59 and 50.0 ± 0.92 μg mL−1/A260 respectively. The coefficient of variation between replicates over different days was less than 3% for all molecules tested, demonstrating robustness of the assay presented here. Further, our methodology allows for mixtures of DNA and RNA to be digested simultaeously and provides separation of the ribonucleosides from the deoxyribonucleosides. This enables the method to be used to quantify DNA and RNA simultaneously in mixtures of the two, something that is impossible by UV spectroscopy alone or through RNA- or DNA-selective nucleic acid binding dyes, due to the significant amount of crossreactivity current dyes present.16 We also highlight that the structure of dsRNA molecules, specifically the presence of any ssRNA overhangs at the 5′ or 3′ ends of the molecule, or the propensity to form hairpins, will have significant impact on the overall absorbance of the product. Herein we utilized RNase If to remove these overhangs and enable the study, but researchers are encouraged to consider that dsRNA molecules from a given production process may have extinction coefficients somewhere between that of pure ssRNA and pure dsRNA if these features are present in their molecules.Conflicts of interest
There are no conflicts to declare.References
- K. Huikko, R. Kostiainen and T. Kotiaho, Introduction to micro-analytical systems: bioanalytical and pharmaceutical applications, Eur. J. Pharm. Sci., 2003, 20, 22, DOI:10.1016/S0928-0987(03)00147-7.
- M. Cavaluzzi and P. Borer, Revised UV extinction coefficients for nucleoside-5monophosphates and unpaired DNA and RNA, Nucleic Acids Res., 2004, 32(1), 9, DOI:10.1093/nar/gnh015.
- J. Sambrook and D. Russell, Fragmentation of DNA by sonication, Cold Spring Harb. Protoc., 2006 DOI:10.1101/pdb.prot4538.
- P. Yakovchuk, E. Protozanova and M. Kamenetskii, Base-stacking and base-pairing contributions into thermal stability of the DNA double helix, Nucleic Acids Res., 2006, 34(2), 11, DOI:10.1093/nar/gkj454.
- A. Nwokeoji, P. Kilby, D. Portwood and M. Dickman, Accurate Quantification of Nucleic Acids Using Hypochromicity Measurements in Conjunction with UV Spectrophotometry, Anal. Chem., 2017, 89, 8, DOI:10.1016/j.ab.2016.08.001.
- S. Linn and R. Roberts, Nucleases, 1982, 14, 243–274 Search PubMed.
- Y. Liu, Y. Zhang, P. Dong, R. An, C. Xue, Y. Ge, L. Wei and X. Liang, Digestion of Nucleic Acids Starts in the Stomach, Sci. Rep., 2015, 5, 11, DOI:10.1038/srep11936.
- J. Eadie, J. Mcbride, J. Efcavitch, L. Hoff and R. Cathcart, High performance liquid chromatography analysis of oligodeoxynucleotide base composition, Anal. Biochem., 1987, 165, 6, DOI:10.1016-0003-2697(87)90294-6.
- J. Liu, L. Ji, S. Wang, S. Chen, S. Guo and H. Wu, Characterisation of nucleosides and nucleobases in Mactra veneriformis by high performance liquid chromatography coupled with diode array detector-mass spectrometry (HPLC–DAD–MS), Food Chem., 2012, 135, 7, DOI:10.1016/j.foodchem.2012.05.019.
- G. Kallansrud and B. Ward, A comparison of measured and calculated single- and double-stranded oligodeoxynucleotide extinction coefficients, Anal. Biochem., 1995, 236, 134–138, DOI:10.1006/abio.1996.0141.
- A. Andrus and R. Kuimelis, Base composition analysis of nucleosides using HPLC, Curr. Protoc. Nucleic Acid Chem., 2001 DOI:10.1002/0471142700.nc1006s01.
- S. Wilson, D. Cohen, X. Wang and M. Hammond, A neutral pH thermal hydrolysis method for quantification of structured RNAs, RNA, 2014, 20(7), 1143–1160, DOI:10.1261/rna.045856.114.
- A. Nwokeoji, P. Kilby, D. Portwood and M. Dickman, RNASwift: a rapid, versatile RNA extraction method free from phenol and chloroform, Anal. Biochem., 2016, 512, 36–46, DOI:10.1021/acs.analchem.7b0400.
- R. Cantor, M. Warshaw and H. Shipiro, Oligonucleotide interactions. III. Circular dichroism studies of the conformation of deoxyoligonucleolides, Biopolymers, 1970, 9(9), 1059–1077, DOI:10.1002/bip.1970.360090909.
- M. Warshaw and I. Tinoco, Optical properties of sixteen dinucleoside phosphates, J. Mol. Biol., 1966, 20(1), 29–38, DOI:10.1016/0022-2836(66)90115-x.
- Y. Nakayama, H. Yamaguchi, N. Einaga and M. Esumi, Pitfalls of DNA Quantification Using DNA-Binding Fluorescent Dyes and Suggested Solutions, PLoS One, 2016, 11(3) DOI:10.1371/journal.pone.0150528.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ay01498b |
| This journal is © The Royal Society of Chemistry 2021 |