
Characterization of ovarian cancer-derived extracellular vesicles by surface-enhanced Raman spectroscopy†
Nina M.
Ćulum
a,
Tyler T.
Cooper
 b,
Gilles A.
Lajoie
b,
Thamara
Dayarathna
c,
Stephen H.
Pasternak
c,
Jiahui
Liu
d,
Yangxin
Fu
d,
Lynne-Marie
Postovit
de and
François
Lagugné-Labarthet
*a
b,
Gilles A.
Lajoie
b,
Thamara
Dayarathna
c,
Stephen H.
Pasternak
c,
Jiahui
Liu
d,
Yangxin
Fu
d,
Lynne-Marie
Postovit
de and
François
Lagugné-Labarthet
*a
aUniversity of Western Ontario (Western University), Department of Chemistry, 1151 Richmond St., London, Ontario N6A 5B7, Canada. E-mail: flagugne@uwo.ca
bUniversity of Western Ontario (Western University), Department of Biochemistry, 1151 Richmond St., London, Ontario N6A 5B7, Canada
cUniversity of Western Ontario (Western University), Robarts Research Institute, 1151 Richmond St., London, Ontario N6A 5B7, Canada
dUniversity of Alberta, Department of Oncology, 116 St. & 85 Ave., Edmonton, Alberta T6G 2R3, Canada
eQueen's University, Department of Biomedical & Molecular Sciences, 99 University Ave., Kingston, Ontario K2L 3N6, Canada
First published on 21st October 2021
Abstract
Ovarian cancer is the most lethal gynecological malignancy, owing to the fact that most cases are diagnosed at a late stage. To improve prognosis and reduce mortality, we must develop methods for the early diagnosis of ovarian cancer. A step towards early and non-invasive cancer diagnosis is through the utilization of extracellular vesicles (EVs), which are nanoscale, membrane-bound vesicles that contain proteins and genetic material reflective of their parent cell. Thus, EVs secreted by cancer cells can be thought of as cancer biomarkers. In this paper, we present gold nanohole arrays for the capture of ovarian cancer (OvCa)-derived EVs and their characterization by surface-enhanced Raman spectroscopy (SERS). For the first time, we have characterized EVs isolated from two established OvCa cell lines (OV-90, OVCAR3), two primary OvCa cell lines (EOC6, EOC18), and one human immortalized ovarian surface epithelial cell line (hIOSE) by SERS. We subsequently determined their main compositional differences by principal component analysis and were able to discriminate the groups by a logistic regression-based machine learning method with ∼99% accuracy, sensitivity, and specificity. The results presented here are a great step towards quick, facile, and non-invasive cancer diagnosis.
1. Introduction
In 2021, ovarian cancer is estimated to be the fifth most lethal cancer in the United States and the most lethal gynaecological malignancy with 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 770 projected deaths.1 The 5-year survival rate for women diagnosed with ovarian cancer is 47%.2,3 However, ovarian cancer usually presents at a late stage when the 5-year relative survival rate is 27%, and few cases are diagnosed when the tumour is localized with a 5-year relative survival rate of 92%.4 The high mortality rate of ovarian cancer is due to the diagnostic delay arising from the lack of early disease warning signs as well as the lack of early ovarian cancer screening options.3 There is a need for the development of non-invasive approaches for the diagnosis of ovarian cancer, particularly at the early stage, which can be achieved by the characterization of extracellular vesicles (EVs) and the analysis of their biomarkers.
770 projected deaths.1 The 5-year survival rate for women diagnosed with ovarian cancer is 47%.2,3 However, ovarian cancer usually presents at a late stage when the 5-year relative survival rate is 27%, and few cases are diagnosed when the tumour is localized with a 5-year relative survival rate of 92%.4 The high mortality rate of ovarian cancer is due to the diagnostic delay arising from the lack of early disease warning signs as well as the lack of early ovarian cancer screening options.3 There is a need for the development of non-invasive approaches for the diagnosis of ovarian cancer, particularly at the early stage, which can be achieved by the characterization of extracellular vesicles (EVs) and the analysis of their biomarkers.
EVs are a complex group of membrane-bound vesicles secreted by all cells and found in bodily fluids.5,6 EVs are generally classified into three groups (exosomes, microvesicles, and apoptotic bodies) based on their size and mechanism of formation.6 Exosomes, which have a diameter of 30–150 nm, are generated within endosomal compartments and released by the fusion of multivesicular bodies with the plasma membrane.7,8 Exosomes are enriched in tetraspanins (e.g., CD9, CD63, CD81) or membrane proteins that are involved in exosome release and sorting of cargo molecules.9 Microvesicles and apoptotic bodies, with diameters of 100–1000 nm and 1–5 μm, respectively, both bud directly from the plasma membrane, but the latter is released by cells undergoing apoptosis.7,10 Microvesicles also include a subtype of EVs called oncosomes, which are EVs released by cancer cells. The presence of larger oncosomes (1–10 μm) has been implicated in driving metastatic spread through integrin signalling regulation.11 EVs, particularly exosomes and microvesicles, play an important role in intercellular communication via the transfer of proteins and RNA.10,12 Furthermore, EVs are involved in physio-pathological activities including cancer progression by mediating crosstalk between tumour and stromal cells.9,13 Cancer cells have additionally been associated with an increase in EV production in comparison to normal cells, which could be related to the specific conditions of the tumour microenvironment.9 Since EVs carry complex biological information from their parent cells, they are particularly interesting in cancer research as they can be exploited as cancer biomarkers.14,15
Surface-enhanced Raman spectroscopy (SERS) is a promising technique for EV detection and characterization.16 Conventional Raman spectroscopy has also been explored in EV characterization and cancer diagnosis,17–19 but it is often difficult to obtain well-resolved spectra of biological samples due to weak Raman scattering and signal intensity, as well as high background fluorescence.20 SERS can be used to determine the same chemical information as in conventional Raman spectroscopy, but it is advantageous in that single molecule detection is possible, background fluorescence can be quenched, and signal intensity can see enhancement by 103–1010.21 For the SERS enhancement effect to be observed, the laser wavelength must be at or near the localized surface plasmon resonance (LSPR) of the chosen SERS substrate. The LSPR of a SERS substrate or probe is determined by the size and shape of nanoscale roughness features on the metal surface.21,22 When the LSPR is excited, large areas of electromagnetic enhancement are generated at the nanoscale metallic features, and the Raman signal intensity of an analyte confined to these regions is greatly enhanced.22 Typical SERS substrates and probes for EV analysis are composed of gold or silver since their LSPRs are located around common laser wavelengths used in Raman spectroscopy (i.e., 532 nm, 632 nm, and 785 nm).20 These substrates also usually consist of nanoparticles,23–36 arrays of nanopillars or nanorods,37–43 or porous structures or nanoholes44–46 that are sometimes further functionalized for specific EV capture.35,41,43,46–48 SERS analysis of cancer-derived EVs has been greatly explored, particularly in breast,36,38,40,41,43,48,49 lung,26,28,34,39,42,46,50 and pancreatic cancer.27,31,35 Some studies have explored the SERS characterization of ovarian cancer-derived (OvCa) EVs,23,25,44,47 but analysis has generally been less thorough compared to the other cancer-derived EVs listed.
Early work in SERS characterization of OvCa EVs dates back to 2014, in which simple gold nanoparticles (NPs) were utilized to analyse EVs from cell line A2780 grown in normoxic (normal O2) and hypoxic (1% O2) conditions.23 However, with principal component analysis (PCA) followed by discriminant function analysis, the two EV types could be only be differentiated with 57.1% sensitivity and 53.8% specificity, and accuracy was not reported. One year later, a silver film-coated plasmonic nanobowl substrate fabricated by soft lithography on flexible polydimethylsiloxane was proposed for SERS analysis of EVs isolated from the SKOV-3 cell line.44 The authors did not evaluate the diagnostic potential of the substrate since the purpose of the study was to compare EV purity from different isolation methods. In 2017, a more selective approach using a thiolated peptide ligand for the capture of EVs from SKOV-3 cells was reported.25 The ligand was also bound to silver NPs by a thiol-metallic bond for SERS analysis. While the authors were able to demonstrate the targeted detection of OvCa EVs, they did not perform statistical analysis to evaluate the diagnostic potential of the probe, and so accuracy, sensitivity, and specificity were not reported. Most recently in 2020, a simple plasmonic scaffold consisting of a cysteamine-treated microscale biosilicate substrate embedded with silver NPs was introduced for SERS analysis of EVs.47 The authors investigated OvCa EVs derived from SKOV-3 cells as well as from patients with ovarian and endometrial cancer. With PCA followed by linear discriminant analysis, cancer could be diagnosed with 99.4% accuracy, 100% sensitivity, and 99.2% specificity. However, the authors warned that these numbers must be interpreted with caution given the small sample size used in analysis that could lead to biased results.
As evidenced by these four studies, most OvCa EVs analysed by SERS are derived from the SKOV-3 cell line. For this work, we sought to characterize EVs derived from OVCAR3 and OV-90 cells, which are well-established model systems for epithelial ovarian adenocarcinomas. Given that most (90%) malignant ovarian tumours are epithelial in origin, and of these cancers, 70% present as high-grade serous and <5% present as low-grade serous,4 we also chose to characterize EVs from high-grade serous (EOC6) and low-grade serous (EOC18) primary cell lines. These four cell lines were compared to EVs derived from a non-malignant human immortalized ovarian surface epithelial (hIOSE) cell line as a control. As a SERS substrate, we again employed gold nanohole arrays fabricated by electron-beam lithography (EBL) that we have extensively reported and characterized in past work.45,51 Herein, we report SERS spectra of the five aforementioned cell lines and were able to discriminate their spectral signals by PCA and logistic regression with extremely high accuracy, sensitivity, and specificity of approximately 99% each.
2. Materials and methods
2.1. Cell culture
OV-90 (ATCC® CRL-11732) and NIH:OVCAR3 (ATCC® HTB-161) were obtained from the ATCC. Human immortalized surface epithelial cells hIOSE (OSE364) were obtained from the Canadian Ovarian Tissue Bank at the BC Cancer Agency, kindly provided by Dr David Huntsman. Primary cell lines EOC6 and EOC18 were isolated from the ascites of patients with high-grade and low-grade serous ovarian cancer, respectively. All cell lines, except OVCAR3, were maintained in M199 + MCDB105 supplemented with 5–15% fetal bovine serum (FBS). NIH:OVCAR3 cells were cultured in RPMI-1640 supplemented with 20% FBS and 5 μg mL−1 insulin. Media was exchanged with serum-free media for 20–30 hours to generate conditioned media (CM) for EV purification. All work involving the use of patient samples (cell lines, plasma, and ascites) was approved by the Health Research Ethics Board of the Alberta Cancer Committee. Informed consent was obtained for any experimentation with human subjects.2.2. Extracellular vesicle (EV) isolation
OvCa CM samples were first centrifuged at 200–300g at 4 °C to pellet cells. Supernatants (except CM) were diluted 1:10 in phosphate-buffered saline (PBS) and centrifuged at 3000g for 20 minutes at 4 °C to remove cell debris. To remove large membrane fragments, supernatants were spun at 10000g for an additional 20 minutes at 4 °C. Lastly, supernatants were ultracentrifuged at 120000–140000g (SW-28 rotor) for 2 hours at 4 °C to pellet EVs on an Optima™ L-100 XP ultracentrifuge (Beckman Coulter). The supernatant was removed and EVs were resuspended in 100–300 μL of PBS and stored at −80 °C until further use.
2.3. EV protein extraction and digestion
To prepare CM for proteomic analysis, ∼30 μg of EVs were lyophilized to dryness and reconstituted in 8 M urea, 50 mM ammonium bicarbonate (ABC), 10 mM dithiothreitol (DTT), and 2% SDS lysis buffer. Proteins were sonicated at 10 × 0.5 s pulses (Level 1) with a probe sonicator (Fisher Scientific, Waltham, MA), reduced in 10 mM DTT for 30 minutes at room temperature (RT), alkylated in 100 mM iodoacetamide for 30 minutes at RT in the dark, and precipitated in chloroform/methanol.52 On-pellet in-solution protein digestion was performed in 100 μL of 50 mM ABC (pH 8) by adding 1/50 Trypsin/Lys-C (Promega) to digest EV proteins. EV proteins were incubated at 37 °C overnight (∼20 h) in a ThermoMixer C (Eppendorf) at 900 rpm before acidifying to pH 3–4 with 10% formic acid (FA). Salts and detergents were removed from peptide samples using C18 stagetips made in-house. Briefly, 10 layers were stacked into 200 μL pipette tips and rinsed with ice-cold methanol. Stagetips were conditioned with solution A (80/20/0.1%; acetonitrile (ACN)/water/trifluoroacetic acid (TFA)), followed by solution B (5/95/0.1%; ACN/water/TFA) prior to loading ∼20 μg of peptides resuspended in solution B. Duplicate washes were performed with solution B prior to elution of peptides using solution C (80/20/0.1%; ACN/water/FA) and final elution using a 50/50 mixture of ACN/0.1% FA. Peptides were centrifuged at 45 °C under vacuum and resuspended in 0.1% FA prior to quantification by the bicinchonic acid assay (BCA) and injection into the mass spectrometer.2.4. Ultraperformance liquid chromatography coupled to tandem mass spectrometry (UPLC-MS/MS)
Peptides were analysed using an ACQUITY UPLC M-class system (Waters) connected to a Q Exactive™ Plus mass spectrometer (Thermo Scientific) using a nonlinear gradient. Buffer A consisted of water/0.1% FA and buffer B consisted of ACN/0.1% FA. Peptides (∼1 μg estimated by BCA) were initially loaded onto an ACQUITY UPLC M-Class Symmetry C18 Trap Column (100 Å, 5 μm, 180 μm × 20 mm) and trapped for 5 minutes at a flow rate of 5 μL min−1 at 99% A/1% B. Peptides were separated on an ACQUITY UPLC M-Class Peptide BEH C18 Column (130 Å, 1.7 μm, 75 μm × 250 mm) operating at a flow rate of 300 nL min−1 at 35 °C using a non-linear gradient consisting of 1–10% B, 10–20% B, 20–30% B, 30–40% B, 40–50% B, 60–70% B, and 80–90% B for 10 minutes intervals before cyclic washing between 5–95% B, equalling 140 minutes gradient total. Settings for data acquisition on the Q Exactive™ Plus are outlined in Table S1.†2.5. UPLC-MS/MS data analysis
MS raw files were searched with de novo peptide sequencing software PEAKS (version 10.5) using the Human Uniprot database (reviewed only, updated November 2020). Missed cleavages were set to 5 and I = L. Cysteine carbamidomethylation was set as a fixed modification. Oxidation (M), N-terminal acetylation (protein), and deamidation (NQ) were set as variable modifications (maximum number of modifications per peptide = 7) and all other settings were left as default. Precursor mass deviation was left at 20 ppm and 4.5 ppm for first and main search, respectively. Fragment mass deviation was left at 20 ppm. Protein and peptide false discovery rate was set to 0.01 (1%) and the decoy database was set to revert. Proteomic data analysis, including PCA for Fig. 1E, and visualization were performed in a Python 3+ environment (Perseus software). The proteins identified by PCA were ranked within the software by their suitability to their respective PC scores. Comparison of the proteomes identified within each sample to Vesiclepedia databases was performed using the open-source FunRich (version 3.1.3) software.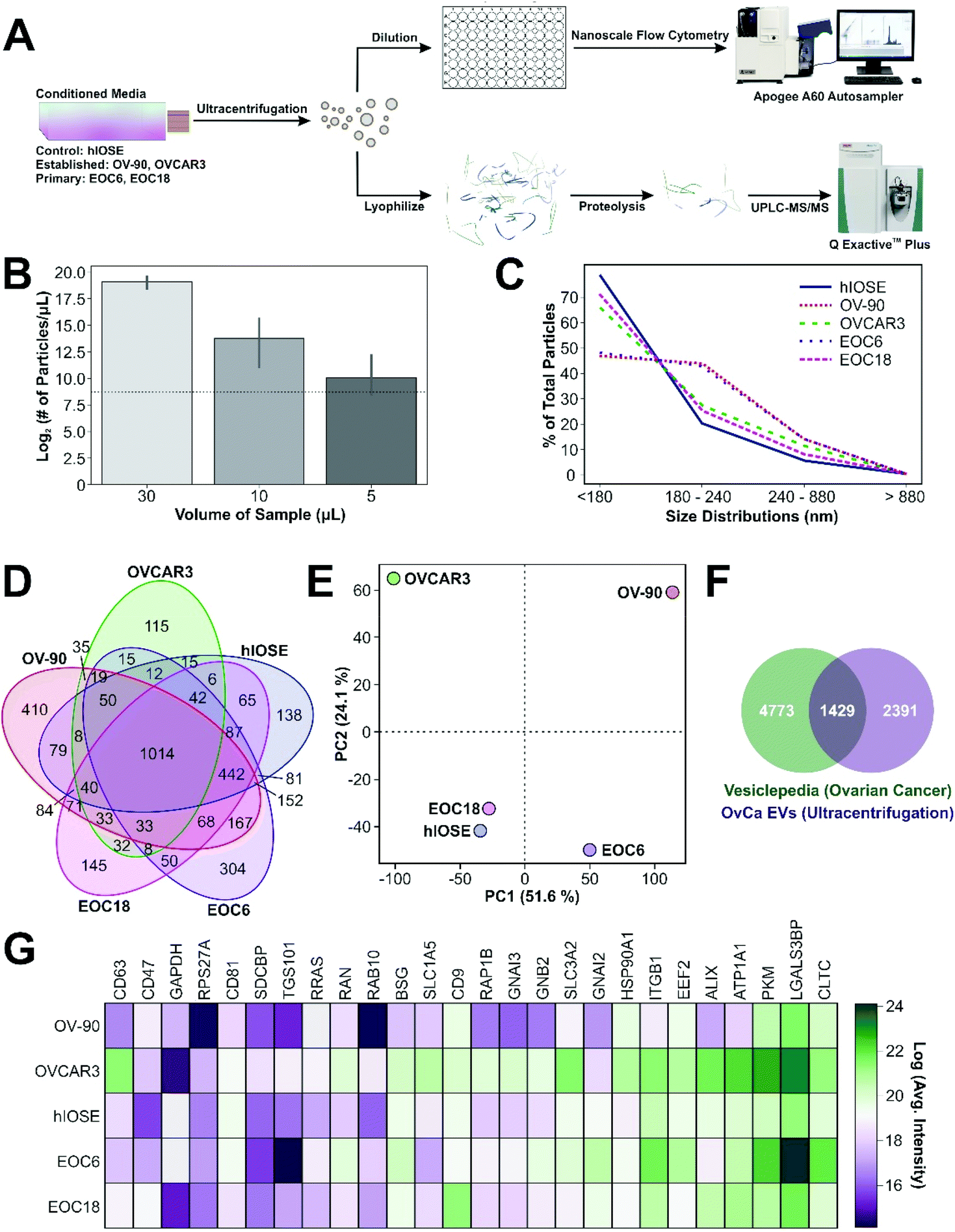 | ||
| Fig. 1 (A) Schematic illustration of nFC and UPLC-MS/MS analysis of OvCa. (B) nFC results revealed a near-linear decrease in the number of particles as the volume of sample analysed diminished. The horizontal dashed line indicates the mean particle number when an equal volume of background media (PBS) was analysed. (C) Size distribution of EVs. (D) Venn diagram highlighting the distinct proteomics cargo contained within EVs from various OvCa cell lines, leading to distinct proteomic “fingerprints” as demonstrated by (E) PCA. (F) Overlap of OvCa EV proteomes with Vesiclepedia database filtered for OvCa. (G) Heat map indicating that classical EV markers (e.g., CD9/81/63) were confidently identified in all OvCa EV samples. | ||
2.6. Nanoscale flow cytometry (nFC)
OvCa EVs were analysed for the number of microparticles per mL and EV size distribution by nFC. Serial injections (2, 5, 10, or 20 mL) of each concentrate were diluted to 300 mL with 0.22 mm-filtered PBS within low-attachment 96-well plates at RT. EVs were enumerated in duplicate on the Apogee A60 nanoscale flow cytometer with autosampler, capable of EV resolution between 150 and 1000 nm.53 130 μL of diluted CM was injected and analysed at 1.5 μL min−1 for 1 minute. The size of secreted microparticles was estimated using silica beads ranging 110–1300 nm using properties of large-angle light scatter and small-angle light scatter. Silica beads provide a refractive index (k = 1.42) that is closer to cells (k = 1.35–1.39) than commonly used polystyrene beads (k = 1.59).2.7. Electron-beam lithography (EBL)
Nanohole arrays were fabricated by EBL following the protocol established by Kaufman et al.45 A negative-tone resist (ma-N 2405, MicroChem) was spin-coated onto reactive O2-cleaned (Trion Technology) glass coverslips at 3000 rpm for 45 seconds (corresponding to thickness of ∼500 ± 50 nm) and baked at 90 °C for 90 seconds. AquaSAVE™ conductive polymer (Sigma-Aldrich) was subsequently spin coated at 1000 rpm for 45 seconds and baked at 90 °C for 45 seconds. EBL was performed using a LEO 1530 (Zeiss) scanning electron microscope (SEM) with a 30.0 kV EHT voltage, 10.0 μm aperture, and 25.0–30.0 pA current. Square and triangular arrays were written at sizes of 0.5 and 1.0 μm with 1.0 μm spacing between holes using ELPHY Quantum software (Raith Fabrication). All array patches measured 50 × 50 μm2. Following beam exposure, samples were soaked in DI water to remove the conductive layer and samples were developed in MF-319 (MicroChem) for 40–45 seconds. Samples were soaked in DI water and air dried to avoid collapsing the nanopillars. Samples were subjected to an O2 plasma descum process (Trion Technology) for 60 seconds to remove residual resist surrounding the nanopillars. A 3 nm adhesion layer of titanium and 30 nm layer of gold was deposited onto the samples by electron beam evaporation (Angstrom Engineering). For lift-off, samples were exposed to Remover-PG (MicroChem) heated to 80 °C for 2 hours. Remover-PG was removed from the samples by soaking in a 1:3 solution of methyl isobutyl ketone (MIBK) and isopropanol (IPA). Samples were rinsed with IPA and dried under N2. For final cleaning, samples were bathed in Nano-Strip® (Cyantek) heated to 80 °C for 30 minutes, then DI water for 15 minutes, and dried under N2. Finally, samples were subjected to O2 plasma (Trion Technology) for 5 minutes to remove any remaining resist from the holes.
2.8. Surface-enhanced Raman spectroscopy (SERS)
Concentrated EV samples were diluted 1:20 with Milli-Q water. 10–20 μL of dilute EV samples were drop-cast onto nanohole arrays and subsequently removed using cohesive properties allowed by a Kimwipe absorbent paper (Kimberly-Clark Inc.). The edge of the absorbent paper was placed on the corner of the solution droplet, allowing for solution removal via capillary action. EV solutions were allowed to dry for 15–30 minutes prior to SERS measurements. EVs were located in nanoholes by SERS mapping, and spectra were extracted from these maps. SERS spectra were acquired with an XploRA™ PLUS spectrometer (Horiba Scientific) using a 785 nm excitation laser source, 600 grooves per mm grating, 100 × objective (N.A. = 0.9), and 100 μm pinhole. Laser power was set to 5 mW with an acquisition time of 4 seconds per spectrum.
2.9. Principal component analysis (PCA) and machine learning
PCA of SERS spectra was completed with Orange software (version 3.28.1). All SERS spectra were standardized to μ = 0, σ2 = 1 prior to PCA, and outliers were removed by the Local Outlier Factor method with contamination set to 10%. Retained spectra were analysed by PCA and the first 25 principal components (PCs) were selected to explain 97.2% of variance among spectra. Machine learning was subsequently done by the same Orange software. The first 25 PCs were used as input data in a logistic regression-based machine learning algorithm. Models were trained and tested using 5-fold cross validation.3. Results and discussion
3.1. Validation of EV size and proteomic cargo
Size quantification of EVs by nFC and proteomic analysis by UPLC-MS/MS was conducted to validate the enrichment of EVs by ultracentrifugation (UC) from CM generated by four OvCa cell lines and one normal control cell line (Fig. 1A). Amongst several methods available for EV purification,7,54–56 this study considered UC as an optimal balance between EV yield and purity. UC provides a heterogenous mixture of EVs which includes both microvesicles and exosomes. Therefore, the use of the term EV throughout this study is in accordance with the International Society for Extracellular Vesicles.57 In support of previous studies,53 we demonstrate a near-linear detection of particles as the volume of sample analysed was diminished, indicating acceptable EV purity (Fig. 1B). The size distribution of EVs was verified by nFC (Fig. S1†) and estimated by small-angle light scatter measurements. The majority of EVs detected were estimated to be less than 240 nm in diameter (Fig. 1C), which is consistent with the size of microvesicles and exosomes.58 However, the resolution of exosomes less than 100 nm in diameter from background noise was unattainable based on the properties of the cytometer used in this study, but subsequent proteomic analysis was able to verify the presence of common EV markers in each OvCa sample.UPLC-MS/MS is a powerful tool to detect and quantify proteomic cargo within EVs (Fig. S2†). In total, we identified over 3000 proteins in EVs generated by all five cell lines. However, unique proteomic cargo was detected among each. Specifically, 1014 peptides were shared by EVs from all five cell lines, whereas 138, 410, 115, 304, and 145 peptides were unique to hIOSE, OV-90, OVCAR3, EOC6, and EOC18, respectively (Fig. 1D). Distinct proteomic fingerprints were identified by PCA, where PC1 and PC2 scores correspond to tetraspanin-8 (TSPAN8) and histone H2A type 1 (HIST1H2A), respectively (Fig. 1E). EVs isolated from OVCAR3, OV-90, and EOC6 each demonstrate a distinguishing proteome compared to EVs isolated from hIOSE and EOC18. A high PC1 score indicates the presence of TSPAN8 in EVs isolated from OV-90 and EOC6, while a high PC2 score indicates the presence of HIST1H2A in EVs isolated from OVCAR-3 and OV-90. EVs isolated from hIOSE and EOC18 are both associated with low PC1 and PC2 scores, indicating a low presence of TSPAN8 and HIST1H2A in these EVs. Furthermore, EVs from hIOSE and EOC18 are clustered closely to one another on the score plot, indicating a high similarity between normal EVs and low-grade OvCa EVs, as expected. OvCa proteomes identified here were compared with those available in the Vesiclepedia database filtered for OvCa cell lines (Fig. 1F). Of 6202 proteins with OvCa EV proteomes in this database, 1429 were shared with EV proteomes identified within our samples. Classical EV markers (e.g., CD9/81/63) were also confidently detected in EVs isolated from all five cell lines in addition to 22 core exosome proteins recently identified by UPLC-MS/MS (Fig. 1G).59 Collectively, our nFC and proteomic data support UC as a suitable method for the enrichment of EVs for downstream SERS analysis.
3.2. SERS characterization of EVs
Gold nanohole arrays of different shapes (squares, triangles) and sizes (500 nm, 1000 nm) were fabricated by EBL as plasmonic substrates for EV capture and SERS analysis. The protocol for the fabrication of nanohole arrays has been well established and documented by our laboratory.45,51 The EBL process is illustrated in Fig. 2A. Briefly explained, EBL is a technique that allows for the fabrication of custom features on a substrate covered with a photoresist, and with some setups achieving 10 nm resolution.60 For nanohole arrays, a negative-tone resist is used, which is an electron-sensitive polymer that undergoes bond formations in areas exposed to the electron beam.61 After development, unexposed regions of the resist are removed, leaving nanopillars on the substrate surface. Metals (i.e., gold) are then deposited to allow for the propagation of surface plasmons for SERS sensing. Finally, after lift-off, these nanopillars are removed, leaving in place the desired nanoholes (Fig. S3†).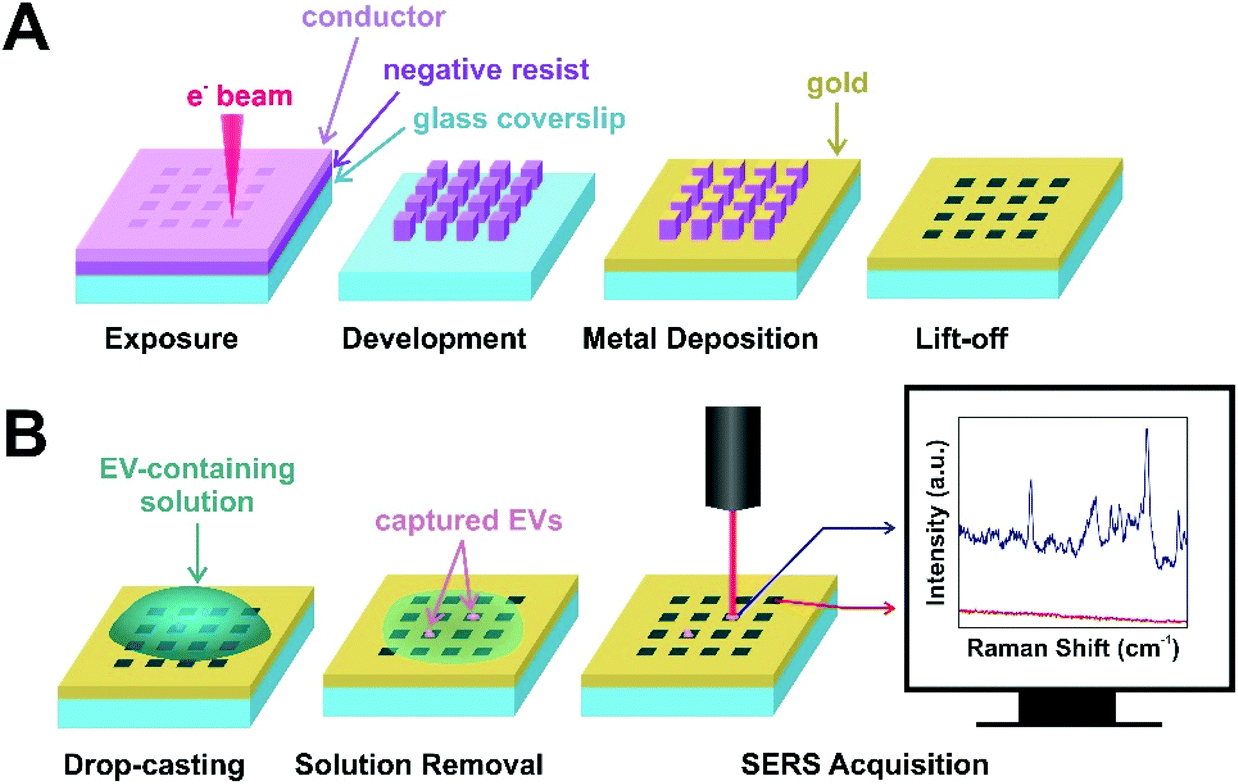 | ||
| Fig. 2 (A) Schematic illustration of the fabrication of nanohole arrays by EBL and (B) subsequent sample preparation and SERS acquisition. | ||
The benefits of these nanohole array platforms are two-fold: the nanoscale size of the holes allows for capture of smaller EVs (i.e., exosomes and microvesicles), and the plasmonic nature of the substrate allows for SERS sensing. Prior to SERS, EVs are drop-casted on the substrate surface, and the solution is removed by placing the corner of an absorbent wipe on the edge of the droplet. This capillary flow induces the EVs in solution to fall into these holes or “traps” and the solution is dried before SERS analysis. The mechanics of this capture have been previously demonstrated with polystyrene beads by Kaufman et al.45 Furthermore, we have previously demonstrated that these gold substrates display absorbance bands (i.e., LSPRs) around 650–690 nm and 750–780 nm.51 When the incident laser wavelength matches the LSPR, surface plasmons are excited to collective oscillation, producing large areas of electromagnetic radiation at the nanoholes. Therefore, when EVs are confined in these holes, we expect to observe a signal that is greatly enhanced compared to traditional Raman spectroscopy. For acquisition of SERS spectra, mapping experiments are necessary to locate trapped EVs, since the EVs are smaller in diameter than can be viewed optically. Areas on the maps with higher intensity are presumed to be EVs, while areas with low intensity and no discernible peaks are identified as the background. The process of sample preparation for SERS and the SERS acquisition process is illustrated in Fig. 2B.
EVs isolated from the hIOSE cell line serve as a control group (Fig. 3A). The majority of peaks found in hIOSE EVs are attributed to proteins and amino acids, which include the peaks at 755 cm−1 (tryptophan), 818 cm−1 (C–C stretching in collagen), 935 cm−1 (proline, valine, protein backbone), 1029 cm−1 (phenylalanine), 1303 cm−1 (collagen, amide III), and 1545 cm−1 (amide II).62,63 Several nucleic acid peaks are located at 724 cm−1 (adenine), 787 cm−1 (cytosine, uracil, thymine), 1185 cm−1 (cytosine, guanine, adenine), 1356 cm−1 (guanine), and 1482 cm−1 (guanine, adenine).62,63 Lipid and carbohydrate peaks can be found at 1254 cm−1 and 865 cm−1, respectively.62,63 A complete list of SERS peaks of hIOSE EVs are summarized in Table S2.†
 | ||
| Fig. 3 Average SERS spectra of (A) ovarian epithelial cell line-derived EVs (control), (B) established OvCa cell line-derived EVs, and (C) primary OvCa cell line-derived EVs. Average SERS spectra of hIOSE, OV-90, OVCAR3, EOC6, and EOC18 are comprised of 207, 123, 106, 166, and 156 spectra, respectively. | ||
EVs from two established OvCa cell lines, OV-90 and OVCAR3, serve as cancer models (Fig. 3B). SERS spectra of EVs from both OV-90 and OVCAR3 are dominated by protein and amino acid peaks. For OV-90 EVs, these peaks are located at 755 cm−1 (tryptophan), 908 cm−1 (tyrosine), 1008 cm−1 (phenylalanine), 1036 cm−1 (phenylalanine), 1151 cm−1 (C–N stretch), 1274 cm−1 (amide III), 1335 cm−1 (collagen, amide III), and 1533 cm−1 (amide II).62,63 For OVCAR3 EVs, the protein and amino acid peaks are found at 741 cm−1 (tryptophan), 818 cm−1 (collagen), 935 cm−1 (proline, valine, protein backbone), 956 cm−1 (CH3 stretching), 1055 cm−1 (C–O, C–N stretching), 1176 cm−1 (tyrosine), 1197 cm−1 (tryptophan), 1226 cm−1 (amide III), and 1584 cm−1 (phenylalanine).62,63 Second most abundant for OV-90 and OVCAR3 EVs are nucleic acid peaks. For OV-90 EVs, these nucleic acid peaks are located at 675 cm−1 (guanine), 782 cm−1 (thymine, cytosine, uracil), 822 cm−1 (phosphodiester), 1186 cm−1 (cytosine, guanine, adenine), and 1201 cm−1 (nucleic acids and phosphates).62,63 For OVCAR3 EVs, the nucleic acid peaks are located at 675 cm−1 (guanine), 724 cm−1 (adenine), 1176 cm−1 (cytosine, guanine), 1376 cm−1 (adenine), and 1483 cm−1 (guanine, adenine).62,63 Carbohydrate peaks in the OV-90 SERS spectrum are found at 943 cm−1, 1118 cm−1, and 1370 cm−1, and in the OVCAR3 SERS spectrum are found at 848 cm−1, 929 cm−1, and 994 cm−1.62,63 Lastly, OV-90 lipid peaks are located at 714 cm−1, 719 cm−1, and 1467 cm−1, while an OVCAR3 lipid peak is found at 1299 cm−1.62,63 The complete list of spectral peaks for OV-90 and OVCAR3 EVs are summarized in Tables S3 and S4,† respectively.
EVs from two primary OvCa cell lines, EOC6 and EOC18, were also probed as models of high-grade and low-grade serous cancer, respectively (Fig. 3C). Like the EVs from the three aforementioned cell lines, the SERS spectra of EOC6 and EOC18 EVs are dominated by protein and amino acid peaks. For EOC6 EVs, these peaks are located at 639 cm−1 (tyrosine), 939 cm−1 (C–C skeletal stretching), 1003 cm−1 (phenylalanine), 1159 cm−1 (C–C/C–N stretching), 1162 cm−1 (tyrosine), 1225 cm−1 (amide III), 1265 cm−1 (collagen, phenylalanine), 1439 cm−1 (collagen), and 1558 cm−1 (tryptophan, tyrosine, amide II).62,63 For EOC18 EVs, protein and amino acid peaks are found at 756 cm−1 (tryptophan), 852 cm−1 (proline, hydroxyproline, tyrosine), 935 cm−1 (proline, valine, protein backbone), 987 cm−1 (phenylalanine), 1032 cm−1 (phenylalanine, proline), 1209 cm−1 (tryptophan, phenylalanine), 1248 cm−1 (amide III), 1338 cm−1 (amide III), and 1362 cm−1 (tryptophan).62,63 However, much fewer nucleic acid peaks are located compared to OV-90 and OVCAR3 EVs, which are found at 797 cm−1 (uracil) for EOC6 EVs and 677 cm−1 (guanine) and 1577 cm−1 (guanine, adenine) for EOC18 EVs.62,63 Lipid peaks for EOC6 EVs are located at 736 cm−1, 775 cm−1, and 1367 cm−1, and in EOC18 EVs are found at 1166 cm−1, 1300 cm−1, 1466 cm−1.62,63 Lastly, carbohydrate peaks of EOC6 EVs are present at 845 cm−1 and 1023 cm−1, while one carbohydrate peak at 1425 cm−1 is present in the SERS spectra of the EOC18 EVs.62,63 The complete list of spectral peaks of the EOC6 and EOC18 EVs are summarized in Tables S5 and S6†, respectively.
We would like to note that these spectral assignments have been made tentatively based on our expectations for EVs to show protein, nucleic acid, and lipid SERS signals. Since EVs are molecularly heterogeneous, it is difficult to precisely validate the origin of these chemical moiety fingerprints as there is a great amount of spectral overlap among protein, nucleic acid, and lipid peaks in the fingerprint region.64,65 Furthermore, these moieties can be detected not only from EVs themselves, but from CM and other contaminants. To alleviate these difficulties, researchers often use dimensionality reduction tools such as PCA to determine key differences and patterns among groups of samples.21
3.3. Spectral analysis by PCA
Often, the first two PCs are used to compare different classes of samples as they contain most of the information of the original data set. However, sometimes the first two PCs alone are not enough to differentiate classes of samples,66 as demonstrated with our data, since PCA is an unsupervised technique. The goal of PCA is to maximize variance in a dataset, which is achieved while ignoring class labels. In our case, the PC18 and PC25 scores best separated the EV types, although they account for a very small percentage of the original data set (Fig. 4A–C). These PCs were determined by the informative projections feature in Orange software, which ranks attribute pairs by classification accuracy. PC18 best differentiates the normal group from the cancer groups, where the latter tends to be associated with more negative PC18 scores (Fig. 4D). PC25 interestingly best separates the cancer groups in terms of the severity of cancer, where higher-grade cancers (e.g., EOC6, OVCAR3, OV-90) tend to be associated with more positive PC25 scores.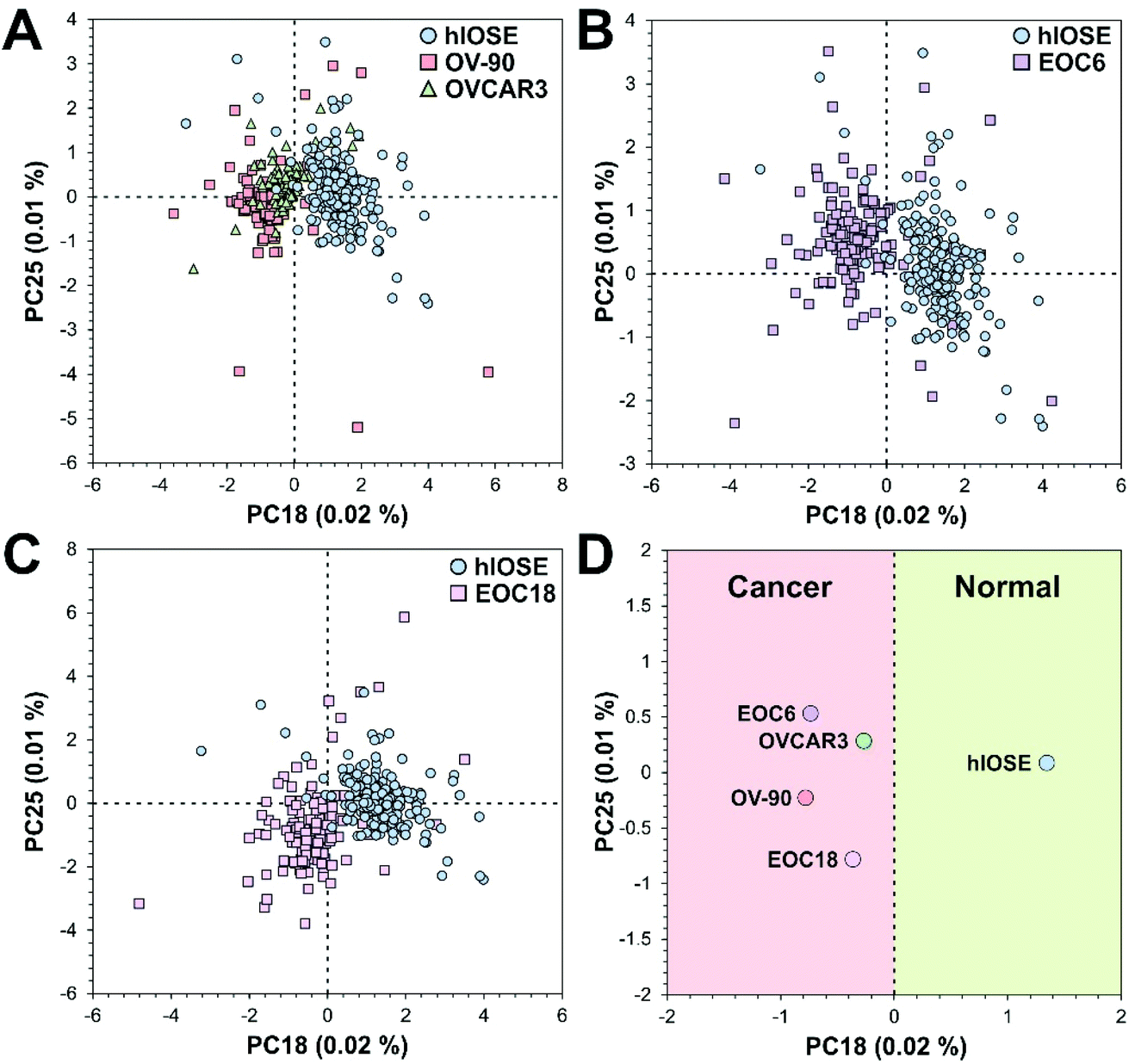 | ||
| Fig. 4 PC score plots comparing (A) normal (hIOSE) vs. cancer (OV-90, OVCAR3) EVs, (B) normal vs. high-grade cancer (EOC6) EVs, (C) normal vs. low-grade cancer (EOC18) EVs, and (D) all groups to highlight separation along PC18. In (A)–(C), each point corresponds to one SERS spectrum while the points in (D) correspond to the centroids of each group in (A)–(C). | ||
PC loading spectra of PC18 and PC25 scores are compared to the average spectrum of each EV type presented in Fig. 3 to interpret which spectral peaks are responsible for the most variance in the data set (Fig. 5). PC18 is best described by protein assignments at 728 cm−1 (tryptophan) and 1237 cm−1 (amide III), nucleic acid assignments at 782 cm−1 (thymine, cytosine, uracil), 1180 cm−1 (cytosine, guanine), and 1483 cm−1 (guanine, adenine), and a carbohydrate peak at 942 cm−1 (polysaccharides) (Table 1 and Fig. 5A). The more positive PC18 peaks at 782 cm−1, 1181 cm−1, and 1483 cm−1 tend to be more associated with the normal EVs than the OvCa EVs (i.e., all three peaks are found in the hIOSE spectrum but not in each of the OvCa spectra). Interestingly, these three peaks are all nucleic acid in origin. PC25 is best described by protein assignments at 743 cm−1 (tryptophan), 832 cm−1 (tyrosine), 973 cm−1 (CH3, CCH vibrations), and 1170 cm−1 (tyrosine), a carbohydrate peak at 940 cm−1 (polysaccharides), and a lipid peak at 1060 cm−1 (ceramide) (Table 2 and Fig. 5B). More positive PC25 peaks located at 940 cm−1, 1060 cm−1, and 1170 cm−1 tend to be associated more with the high-grade cancer EVs (EOC6) than the low-grade cancer EVs (EOC18). While PCA is a valuable tool to determine how spectra differ from one another, the PC scores determined can also serve as classifiers in machine learning algorithms to better differentiate classes of data.
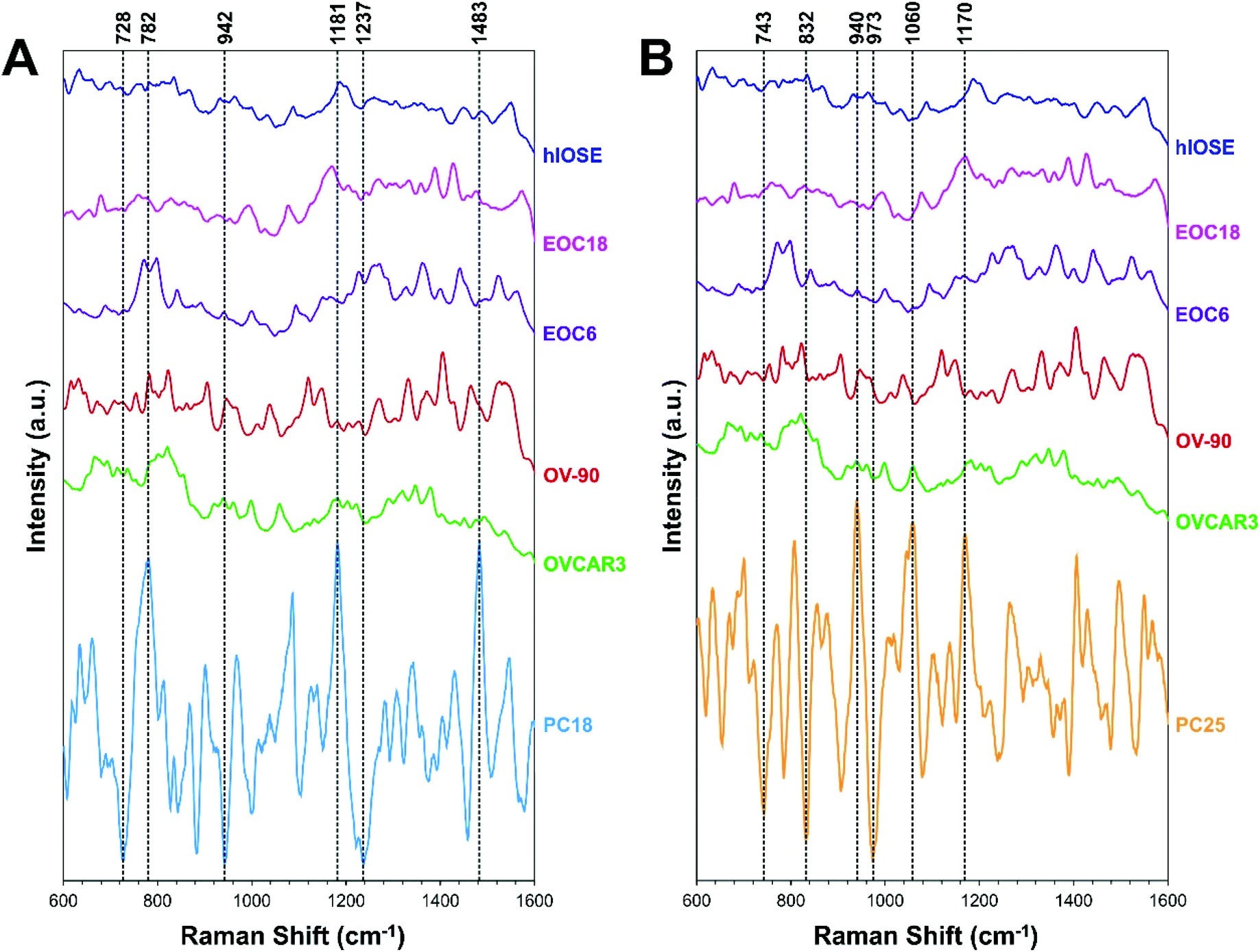 | ||
| Fig. 5 Average SERS spectra of EVs derived from each cell type (also shown in Fig. 3) compared with (A) PC18 and (B) PC25 loading spectra. Key peaks (i.e., spectral peaks that best describe each PC) are highlighted with vertical dashed lines. | ||
| Raman shift (cm−1) | Presumed origin | Ref. peak (cm−1) |
|---|---|---|
| 728 | Ring breathing of tryptophan | 728 (ref. 62) |
| 782 | Thymine, cytosine, uracil ring breathing modes | 782 (ref. 62 and 63) |
| 942 | Skeletal modes of polysaccharides | 941 (ref. 62 and 63) |
| 1181 | Cytosine, guanine | 1180 (ref. 62 and 63) |
| 1237 | Amide III, CH2 wagging vibrations from glycine | 1237 (ref. 62 and 63) |
| 1483 | Guanine, adenine ring breathing modes | 1485 (ref. 62 and 63) |
| Raman shift (cm−1) | Presumed origin | Ref. peak (cm−1) |
|---|---|---|
| 743 | DNA, tryptophan | 742 (ref. 62) |
| 832 | Asymmetric O–P–O stretching, tyrosine | 731 (ref. 62 and 63) |
| 940 | Skeletal modes of polysaccharides | 941 (ref. 62 and 63) |
| 973 | CH3, CCH vibrations in proteins | 973 (ref. 62 and 63) |
| 1060 | C–C in-plane bending, C–N stretching, ceramide | 1061 (ref. 62) |
| 1170 | C–H in-plane bending mode of tyrosine | 1170 (ref. 62 and 63) |
3.4. Machine learning for ovarian cancer diagnosis
A logistic regression-based machine learning algorithm was used to classify normal and OvCa EVs based on the first 25 PCs calculated. Several algorithms were tested and compared based on the value of the area under the curve (AUC) of the receiver operating characteristic (ROC) curves (Fig. S4†). Logistic regression was chosen for the high AUC obtained as well as extremely high accuracy, precision, and recall, although some algorithms like support vector machine worked almost as well (Table S7†). The algorithm was trained and tested with 5-fold cross validation to reduce potential overfitting.With the logistic regression machine learning method, we have monitored how classification accuracy increases with an increasing number of PCs retained (Table S8†). The first 5 PCs (91.5% total explained variance), 15 PCs (95.5% total explained variance), and 20 PCs (96.5% total explained variance) were investigated as classifiers for the algorithm (Fig. S5D†). As shown in the traditional score plot, PC1 and PC2 do not well-differentiate the classes of spectral data (Fig. S5A†). Informative projections calculated by Orange software indicated that from the first 5 PCs, PC2 and PC3 provide the best classification accuracy, but substantial overlap is still observed among the data clusters (Fig. S5B†). The resulting accuracy, recall, and precision achieved by the algorithm is therefore poor, and corresponds to 46.3%, 50.6%, and 46.3%, respectively. When the first 15 PCs are included as classifiers for the algorithm, accuracy, sensitivity, and specificity increases marginally to 69.0%, 69.6%, and 69.0%, respectively. For the first 15 PCs, PC6 and PC9 are able to better differentiate the EOC6 and EOC18 EVs from the hIOSE EVs compared to when the 5 PCs are used as classifiers, but substantial overlap remains among OV-90, OVCAR3, and hIOSE EVs (Fig. S5C†).
The importance of including lower rank PCs such as PC18 and PC25 is demonstrated by including the first 20 and 25 PCs as machine learning classifiers. When the first 20 PCs are included, the informative projection indicates that PC6 and PC18 best differentiate OvCa EVs from normal EVs (Fig. S5D†). As aforementioned, PC18 contains information that best classifies the cancer-derived EVs. Therefore, accuracy, sensitivity, and specificity increase to 94.6%, 94.8%, and 94.6%, respectively. PC25, as aforementioned, contains spectral information that best differentiates the high-grade OvCa EVs from the low-grade OvCa EVs. Thus, when the first 25 PCs, which contain 97.2% total explained variance, are included as machine learning classifiers, 98.6% accuracy, sensitivity, and specificity are achieved.
We further wanted to determine the algorithm's capability of discriminating (1) normal (hIOSE) EVs from established OvCa cell line (OV-90, OVCAR3) EVs (Fig. 6A), (2) normal EVs from high-grade OvCa EVs (Fig. 6B), (3) normal EVs from low-grade OvCa (EOC18) EVs (Fig. 6C), and (4) low-grade OvCa EVs from high-grade OvCa EVs (Fig. 6D). Heat maps shown in Fig. 6 were created to visualize the machine learning output scores of each sample. The output scores given represent the probability of a test sample belonging to a cancerous group. For Fig. 6A–C, an output score of 1.0 corresponds to a cancerous sample and is shown in red on the heat maps, while an output score of 0 corresponds to a normal sample and is shown in yellow on the heat maps. A vast majority of OvCa EVs were successfully classified as cancerous, as indicated by the red and dark orange bars in Fig. 6A–C. In Fig. 6D, an output score of 1.0 corresponds to a high-grade cancer sample and is shown in red on the heat maps, while an output score of 0 corresponds to a low-grade cancer sample and is shown in yellow on the heat maps. Similarly, a majority of the EOC6 EVs were successfully classified as high-grade, also indicated by the red and dark orange bars in Fig. 6D.
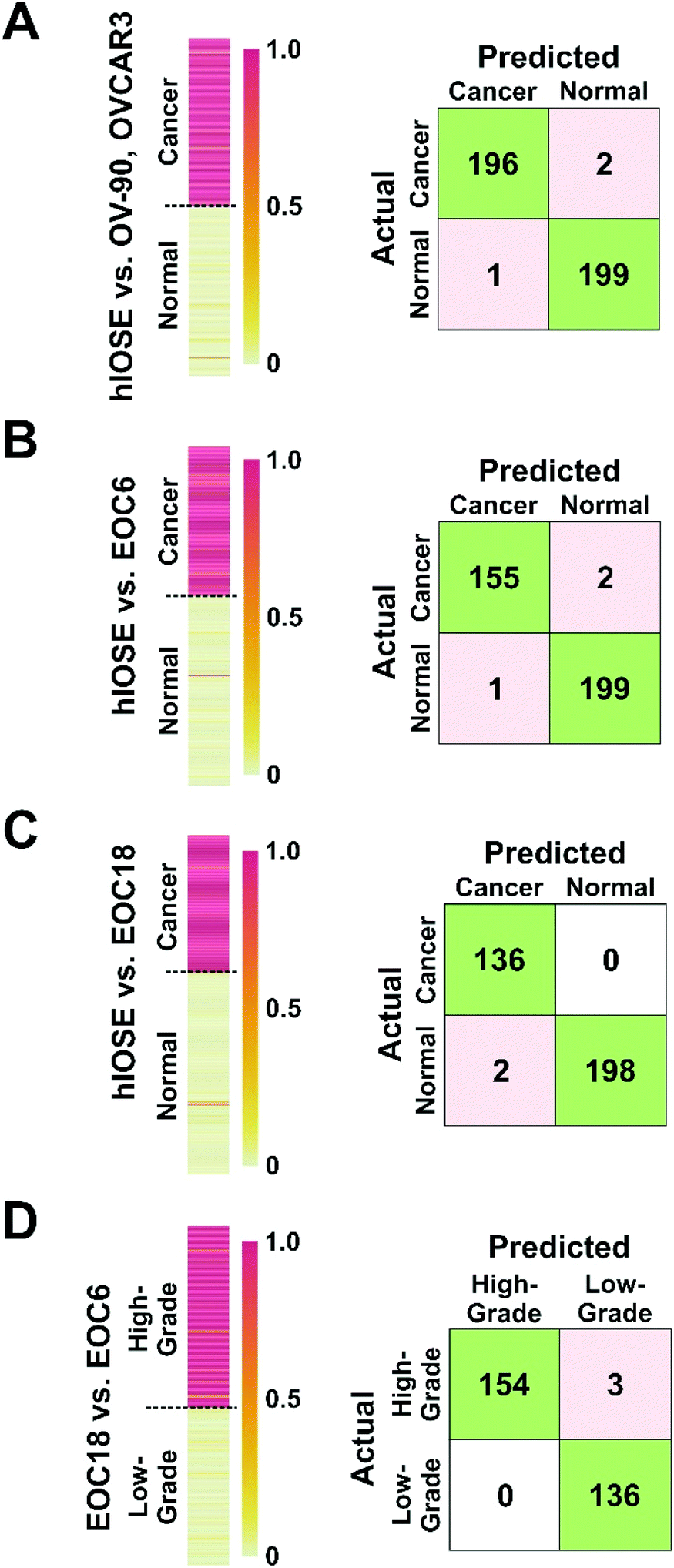 | ||
| Fig. 6 Heat maps (left) to visualize output scores of (A) normal vs. established cancer cell line EVs, (B) normal vs. high-grade cancer EVs, (C) normal vs. low-grade cancer EVs, and (D) low-grade vs. high-grade cancer EVs, with corresponding confusion matrices (right). In the heat maps, each horizontal bar corresponds to one SERS spectrum. Groups labelled on the left indicate the origin of the sample, while the colour of the bar indicates the algorithm's prediction. In (A)–(C), a red or dark orange colour (output score >0.5) corresponds to a cancer prediction and a yellow or light orange colour (output score <0.5) corresponds to a normal prediction. In (D), a red or dark orange colour (output score >0.5) corresponds to a high-grade cancer prediction and a yellow or light orange colour (output score <0.5) corresponds to a low-grade cancer prediction. | ||
EVs isolated from hIOSE were compared to EVs isolated from OV-90, OVCAR3, and EOC6 to determine the platform's capability of correctly diagnosing OvCa. When the established cell line EVs were used as training sets for the cancer group, 196 out of 198 OvCa EV samples were correctly identified as cancerous, while 199 out of 200 normal EV samples were correctly identified as non-cancerous (Fig. 6A), corresponding to 99.2% classification accuracy, 99.0% sensitivity, and 99.5% specificity. When the EOC6 EVs were used as a training set for a cancer group, 155 out of 157 OvCa samples were correctly identified as cancerous, while 199 out of 200 normal EVs were again correctly classified as non-cancerous (Fig. 6B). The resulting classification accuracy was identical to the normal vs. established OvCa cell line model at 99.2%. The sensitivity dipped slightly to 98.7%, but the specificity remained the same at 99.5%.
EVs isolated from hIOSE were compared to EVs isolated from EOC18 to determine the platform's capability of correctly diagnosing low-grade OvCa, which can be more of a challenge compared to high-grade OvCa since low-grade OvCa cells tend to resemble normal cells more closely. With the EOC18 EVs as a training set for the cancer group, all 136 OvCa samples were correctly identified as cancerous, while 198 out of 200 normal EVs were correctly classified as non-cancerous (Fig. 6C). The 99.4% accuracy achieved here is comparable to the previous two models, suggesting we can successfully classify both low-grade and high-grade OvCa samples with extremely high accuracy. Furthermore, we were also able to achieve high sensitivity and specificity at 100% and 99.0%, respectively.
EVs isolated from EOC6 and EOC18 were compared to determine the platform's capability of predicting whether an OvCa sample is high-grade or low-grade, since the grade of OvCa can help predict the prognosis of the disease as well as how the cancer may respond to treatment. 154 out of 157 EOC6 samples were correctly identified as high-grade, while all 136 EOC18 samples were correctly identified as low-grade, corresponding to 99.0% accuracy, 98.1% sensitivity, and a 100% specificity (Fig. 6D).
The accuracies, sensitivities, and specificities achieved with each of the four models compared in Fig. 6 is summarized in Table 3. The high accuracies presented here indicate that this platform and methodology is not only promising in terms of ovarian cancer diagnosis, but also promising in determining the grade of disease. Since EVs are found in bodily fluids such as urine, plasma, and saliva, this approach has a strong potential for non-invasive ovarian cancer diagnosis.
| Model | Accuracy | Sensitivity | Specificity |
|---|---|---|---|
| hIOSE vs. OV-90, OVCAR-3 | 99.2% | 99.0% | 99.5% |
| hIOSE vs. EOC6 | 99.2% | 98.7% | 99.5% |
| hIOSE vs. EOC18 | 99.4% | 100% | 99.0% |
| EOC6 vs. EOC18 | 99.0% | 98.1% | 100% |
4. Conclusions
In conclusion, we have presented plasmonic gold nanohole arrays for the capture of single EVs which we subsequently characterized by SERS. To our knowledge, this is the first time EVs from these five cell lines (OV-90, OVCAR3, EOC6, EOC18, and hIOSE) have been characterized by SERS. We then were able to determine their main compositional differences by PCA. We found that normal EVs could generally be differentiated from OvCa EVs by the presence of peaks at 782 cm−1 (thymine, cytosine, uracil ring breathing modes), 1181 cm−1 (cytosine, guanine), and 1483 cm−1 (guanine, adenine ring breathing modes). Interestingly, high-grade cancer could also be predicted based on the presence of peaks at 940 cm−1 (polysaccharides), 1060 cm−1 (C–C in-plane bending, C–N stretching, ceramide), and 1170 cm−1 (tyrosine).The PC scores calculated were then used as classifiers in a logistic regression-based machine learning algorithm, which was able to differentiate normal EVs from the established OvCa cell line EVs with 99.2% accuracy, 99.0% sensitivity, and 99.5% specificity. Normal EVs could also be discriminated from the high-grade primary cell line EVs with 99.2% accuracy, 98.7% sensitivity, and 99.5% specificity. Interestingly, we were also able to differentiate the normal EVs from the low-grade primary cell line EVs with 99.4% accuracy, 100% sensitivity, and 99.0% specificity. Lastly, the low-grade and high-grade primary cell line EVs could also be discriminated with 99.0% accuracy, 98.1% sensitivity, and 100% specificity. These results are similar to those achieved most recently by Rojalin et al.,47 but it is worth noting that gold-based SERS substrates generally offer higher stability in air compared to silver-based substrates and would perhaps be more suitable in a clinical setting.
The results presented in this paper are a great step towards early, non-invasive, facile, and rapid ovarian cancer diagnosis. However, it must be noted that at this stage, the results are proof-of-concept, and should be interpreted with caution. Until clinical samples are obtained from ovarian cancer patients and healthy individuals, we cannot determine precisely how the model will perform with new test data. It should also be noted that without a method for specific EV capture (e.g., by antibodies), EVs must first be isolated from complex patient biofluids prior to SERS analysis using this platform. Future work in this study will involve challenging these SERS platforms with EVs from real ovarian cancer patient samples as well as from healthy individuals to determine the feasibility of using this methodology in a clinical setting.
Author contributions
Nina M. Ćulum: conceptualization; data curation; formal analysis; investigation; methodology; visualization; writing (original draft, review & editing). Tyler T. Cooper: conceptualization; data curation; formal analysis; investigation; methodology; visualization; writing (original draft, review & editing). Gilles A. Lajoie: resources; supervision. Thamara Dayarathna: investigation. Stephen H. Pasternak: resources; supervision. Jiahui Liu: resources. Yangxin Fu: resources. Lynne-Marie Postovit: conceptualization; supervision. François Lagugné-Labarthet: conceptualization; resources; writing (review & editing); supervision.Ethics statement
All work involving the use of patient samples (i.e., EOC6 and EOC18) was approved by the Health Research Ethics Board of Alberta-Cancer Committee (#25132).Conflicts of interest
The authors declare no known competing financial or non-financial interests.Acknowledgements
The authors would like to gratefully thank the Nanofabrication Facility at the University of Western Ontario (Western University) for their assistance with the preparation of the platforms by electron-beam lithography. The authors would also like to thank Dr David Huntsman at the BC Cancer Agency for providing the human immortalized surface epithelial cells. This research was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada (DG RGPIN-2020-06676).References
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, CA Cancer J. Clin., 2021, 71, 7–33 CrossRef PubMed.
- L. C. Peres, K. L. Cushing-Haugen, M. Köbel, H. R. Harris and A. Berchuck, et al., J. Natl. Cancer Inst., 2019, 111, 60–68 CrossRef PubMed.
- S. Lheureux, M. Braunstein and A. M. Oza, CA Cancer J. Clin., 2019, 69, 280–304 Search PubMed.
- B. M. Reid, J. B. Permuth and T. A. Sellers, Cancer Biol. Med., 2017, 14, 9–32 CAS.
- C.-Y. Chiang and C. Chen, J. Biomed. Sci., 2019, 26, 9 CrossRef PubMed.
- C. P. R. Xavier, H. R. Caires, M. A. G. Barbosa, R. Bergantim and J. E. Guimarães, et al., Cells, 2020, 9, 1141 CrossRef CAS PubMed.
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 373–383 CrossRef CAS PubMed.
- L. M. Doyle and M. Z. Wang, Cells, 2019, 8, 727 CrossRef CAS PubMed.
- A. Lucidi, D. Buca, C. Ronsini, S. Tinari and G. Bologna, et al., Int. J. Mol. Sci., 2020, 21, 8762 CrossRef CAS PubMed.
- E. M. Veziroglu and G. I. Mias, Front. Genet., 2020, 11, 700 CrossRef CAS PubMed.
- C. Ciardiello, A. Leone, P. Lanuti, M. S. Roca and T. Moccia, et al., J. Exp. Clin. Cancer Res., 2019, 38, 317 CrossRef PubMed.
- L. Margolis and Y. Sadovsky, PLoS Biol., 2019, 17, e3000363 CrossRef CAS PubMed.
- T. An, S. Qin, Y. Xu, Y. Tang and Y. Huang, et al., J. Extracell. Vesicles, 2015, 4, 27522 CrossRef PubMed.
- Y. Fujita, Y. Yoshioka and T. Ochiya, Cancer Sci., 2016, 107, 385–390 CrossRef CAS PubMed.
- T. Kinoshita, K. W. Yip, T. Spence and F. F. Liu, J. Hum. Genet., 2017, 62, 67–74 CrossRef CAS PubMed.
- T. Rojalin, B. Phong, H. J. Koster and R. P. Carney, Front. Chem., 2019, 7, 279 CrossRef CAS PubMed.
- P. Beekman, A. Enciso-Martinez, H. S. Rho, S. P. Pujari and A. Lenferink, et al., Lab Chip, 2019, 19, 2526–2536 RSC.
- H. Zhang, A. C. Silva, W. Zhang, H. Rutigliano and A. Zhou, PLoS One, 2020, 15, e0235214 CrossRef CAS PubMed.
- O. J. Old, L. M. Fullwood, R. Scott, G. R. Lloyd and L. M. Almond, et al., Anal. Methods, 2014, 6, 3901–3917 RSC.
- U. K. Sur, Resonance, 2010, 15, 154–164 CrossRef CAS.
- H. J. Butler, L. Ashton, B. Bird, G. Cinque and K. Curtis, et al., Nat. Protoc., 2016, 11, 664–687 CrossRef CAS PubMed.
- C. L. Haynes, A. D. McFarland and R. P. Van Duyne, Anal. Chem., 2005, 77, 338 A–346 A CrossRef CAS.
- L. T. Kerr, L. Gubbins, K. W. Gorzel, S. Sharma, M. Kell, et al., presented in part at the Proc. SPIE 9129, 2014.
- S. Stremersch, M. Marro, B.-E. Pinchasik, P. Baatsen and A. Hendrix, et al., Small, 2016, 12, 3292–3301 CrossRef CAS PubMed.
- C. Lee, R. Carney, K. Lam and J. W. Chan, J. Raman Spectrosc., 2017, 48, 1771–1776 CrossRef CAS.
- J. Park, M. Hwang, B. Choi, H. Jeong and J.-h. Jung, et al., Anal. Chem., 2017, 89, 6695–6701 CrossRef CAS PubMed.
- T.-D. Li, R. Zhang, H. Chen, Z.-P. Huang and X. Ye, et al., Chem. Sci., 2018, 9, 5372–5382 RSC.
- H. Shin, H. Jeong, J. Park, S. Hong and Y. Choi, ACS Sens., 2018, 3, 2637–2643 CrossRef CAS PubMed.
- Y.-F. Tian, C.-F. Ning, F. He, B.-C. Yin and B.-C. Ye, Analyst, 2018, 143, 4915–4922 RSC.
- Z. Wang, S. Zong, Y. Wang, N. Li and L. Li, et al., Nanoscale, 2018, 10, 9053–9062 RSC.
- J. Carmicheal, C. Hayashi, X. Huang, L. Liu and Y. Lu, et al., Nanomedicine, 2019, 16, 88–96 CrossRef CAS PubMed.
- J. C. Fraire, S. Stremersch, D. Bouckaert, T. Monteyne and T. De Beer, et al., ACS Appl. Mater. Interfaces, 2019, 11, 39424–39435 CrossRef CAS PubMed.
- D. Chalapathi, S. Padmanabhan, R. Manjithaya and C. Narayana, J. Phys. Chem. B, 2020, 124, 10952–10960 CrossRef CAS PubMed.
- H. Shin, S. Oh, S. Hong, M. Kang and D. Kang, et al., ACS Nano, 2020, 14, 5435–5444 CrossRef CAS PubMed.
- S. Jiang, Q. Li, C. Wang, Y. Pang and Z. Sun, et al., ACS Sens., 2021, 6, 852–862 CrossRef CAS PubMed.
- G. Li, N. Zhu, J. Zhou, K. Kang and X. Zhou, et al., J. Mater. Chem. B, 2021, 9, 2709–2716 RSC.
- L. Tirinato, F. Gentile, D. Di Mascolo, M. L. Coluccio and G. Das, et al., Microelectron. Eng., 2012, 97, 337–340 CrossRef CAS.
- S. Zong, L. Wang, C. Chen, J. Lu and D. Zhu, et al., Anal. Methods, 2016, 8, 5001–5008 RSC.
- K. Sivashanmugan, W.-L. Huang, C.-H. Lin, J.-D. Liao and C.-C. Lin, et al., J. Taiwan Inst. Chem. Eng., 2017, 80, 149–155 CrossRef CAS.
- E. A. Kwizera, R. O'Connor, V. Vinduska, M. Williams and E. R. Butch, et al., Theranostics, 2018, 8, 2722–2738 CrossRef CAS PubMed.
- J. U. Lee, W. H. Kim, H. S. Lee, K. H. Park and S. J. Sim, Small, 2019, 15, 1804968 CrossRef PubMed.
- Z. Yan, S. Dutta, Z. Liu, X. Yu and N. Mesgarzadeh, et al., ACS Sens., 2019, 4, 488–497 CrossRef CAS PubMed.
- N. Kim, M. R. Thomas, M. S. Bergholt, I. J. Pence and H. Seong, et al., Nat. Commun., 2020, 11, 207 CrossRef CAS PubMed.
- C. Lee, R. P. Carney, S. Hazari, Z. J. Smith and A. Knudson, et al., Nanoscale, 2015, 7, 9290–9297 RSC.
- L. Kaufman, T. Cooper, G. Wallace, D. Hawke, D. Betts, et al., presented in part at the Proc. SPIE 10894, 2019.
- S. Dong, Y. Wang, Z. Liu, W. Zhang and K. Yi, et al., ACS Appl. Mater. Interfaces, 2020, 12, 5136–5146 CrossRef CAS PubMed.
- T. Rojalin, H. J. Koster, J. Liu, R. R. Mizenko and D. Tran, et al., ACS Sens., 2020, 5, 2820–2833 CrossRef CAS PubMed.
- T. Kang, J. Zhu, X. Luo, W. Jia and P. Wu, et al., Anal. Chem., 2021, 93, 2519–2526 CrossRef CAS PubMed.
- N. Ferreira, A. Marques, H. Águas, H. Bandarenka and R. Martins, et al., ACS Sens., 2019, 4, 2073–2083 CrossRef CAS PubMed.
- M. Avella-Oliver, R. Puchades, S. Wachsmann-Hogiu and A. Maquieira, Sens. Actuators, B, 2017, 252, 657–662 CrossRef CAS.
- N. M. Ćulum, T. T. Cooper, G. I. Bell, D. A. Hess and F. Lagugné-Labarthet, Anal. Bioanal. Chem., 2021, 413, 5013–5024 CrossRef PubMed.
- D. Wessel and U. I. Flügge, Anal. Biochem., 1984, 138, 141–143 CrossRef CAS PubMed.
- J. Gomes, F. Lucien, T. T. Cooper, Y. Kim and K. C. Williams, et al., Thromb. Haemostasis, 2018, 118, 1612–1624 CrossRef PubMed.
- F. Royo, C. Théry, J. M. Falcón-Pérez, R. Nieuwland and K. W. Witwer, Cells, 2020, 9, 1955 CrossRef CAS PubMed.
- A. Bobrie, M. Colombo, S. Krumeich, G. Raposo and C. Théry, J. Extracell. Vesicles, 2012, 1, 18397 CrossRef CAS PubMed.
- T. T. Cooper, S. E. Sherman, G. I. Bell, T. Dayarathna and D. M. McRae, et al., Stem Cells Dev., 2021, 30, 247–264 CrossRef CAS PubMed.
- C. Théry, K. W. Witwer, E. Aikawa, M. J. Alcaraz and J. D. Anderson, et al., J. Extracell. Vesicles, 2018, 7, 1535750 CrossRef PubMed.
- D. S. Choi, D. K. Kim, Y. K. Kim and Y. S. Gho, Mass Spectrom. Rev., 2015, 34, 474–490 CrossRef CAS PubMed.
- F. G. Kugeratski, K. Hodge, S. Lilla, K. M. McAndrews and X. Zhou, et al., Nat. Cell Biol., 2021, 23, 631–641 CrossRef CAS PubMed.
- C. Vieu, F. Carcenac, A. Pépin, Y. Chen and M. Mejias, et al., Appl. Surf. Sci., 2000, 164, 111–117 CrossRef CAS.
- A. S. Gangnaik, Y. M. Georgiev and J. D. Holmes, Chem. Mater., 2017, 29, 1898–1917 CrossRef CAS.
- A. C. S. Talari, Z. Movasaghi, S. Rehman and I. U. Rehman, Appl. Spectrosc. Rev., 2015, 50, 46–111 CrossRef CAS.
- I. U. Rehman, Z. Movasaghi and S. Rehman, in Vibrational Spectroscopy for Tissue Analysis, CRC Press, Boca Raton, 2012, vol. 1, pp. 213–294 Search PubMed.
- N. Kuhar, S. Sil, T. Verma and S. Umapathy, RSC Adv., 2018, 8, 25888–25908 RSC.
- E. Willms, C. Cabañas, I. Mäger, M. J. A. Wood and P. Vader, Front. Immunol., 2018, 9, 738 CrossRef PubMed.
- M. Bukva, G. Dobra, J. Gomez-Perez, K. Koos and M. Harmati, et al., Cancers, 2021, 13, 1407 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an01586a |
| This journal is © The Royal Society of Chemistry 2021 |