
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssessment of shifted excitation Raman difference spectroscopy in highly fluorescent biological samples†
Florian
Korinth
ab,
Tanveer Ahmed
Shaik
 a,
Jürgen
Popp
ac and
Christoph
Krafft
*a
a,
Jürgen
Popp
ac and
Christoph
Krafft
*a
aLeibniz Institute of Photonic Technology and Member of Leibniz Research Alliance “Health Technologies”, 07745 Jena, Germany. E-mail: christoph.krafft@leibniz-ipht.de
bLeibniz Institute for Astrophysics Potsdam and Member of Leibniz Research Alliance “Health Technologies”, 14482 Potsdam, Germany
cInstitute of Physical Chemistry and Abbe Center of Photonics, Friedrich Schiller University, 07743 Jena, Germany
First published on 27th October 2021
Abstract
Shifted excitation Raman difference spectroscopy (SERDS) can be used as an instrumental baseline correction technique to retrieve Raman bands in highly fluorescent samples. Genipin (GE) cross-linked equine pericardium (EP) was used as a model system since a blue pigment is formed upon cross-linking, which results in a strong fluorescent background in the Raman spectra. EP was cross-linked with 0.25% GE solution for 0.5 h, 2 h, 4 h, 6 h, 12 h, and 24 h, and compared with corresponding untreated EP. Raman spectra were collected with three different excitation wavelengths. For the assessment of the SERDS technique, the preprocessed SERDS spectra of two excitation wavelengths (784 nm–786 nm) were compared with the mathematical baseline-corrected Raman spectra at 785 nm excitation using extended multiplicative signal correction, rubberband, the sensitive nonlinear iterative peak and polynomial fitting algorithms. Whereas each baseline correction gave poor quality spectra beyond 6 h GE crosslinking with wave-like artefacts, the SERDS technique resulted in difference spectra, that gave superior reconstructed spectra with clear collagen and resonance enhanced GE pigment bands with lower standard deviation. Key for this progress was an advanced difference optimization approach that is described here. Furthermore, the results of the SERDS technique were independent of the intensity calibration because the system transfer response was compensated by calculating the difference spectrum. We conclude that this SERDS strategy can be transferred to Raman studies on biological and non-biological samples with a strong fluorescence background at 785 nm and also shorter excitation wavelengths which benefit from more intense scattering intensities and higher quantum efficiencies of CCD detectors.
Introduction
A frequent problem in Raman spectroscopy of pristine biological samples is that high auto-fluorescence is simultaneously excited masking weaker Raman bands and making spectral interpretation of measured data challenging.1–4 A good example of a strong fluorescent biological sample is equine pericardium (EP) after its cross-linking with Genipin (GE).5 The pericardium of animals, which consists primarily of type I collagen and elastin,6 is regularly used for tissue engineering as a scaffold for vascular grafts, patches, aortic valves, and wound healing procedures. To strengthen the pericardium structure and enhance its durability, cross-linking protocols have been developed. The cross-linking with GE is less cytotoxic compared to glutaraldehyde (GA)7 and results in a strong tissue scaffold without the risk of calcification which is the main cause of graft failure of GA crosslinked scaffolds. The reaction of GE with lysine or hydroxy-lysine residues of collagen results in the formation of cross-links and leads to the formation of a pigment,8–11 which turns the sample from white to a deep blue color leading to very strong background contributions in the Raman spectra.Several approaches exist to correct spectra with high spectral backgrounds, which can be broadly classified into instrumental and computational methods.12 The instrumental methods can be separated into time-gated,13,14 wavelength and frequency modulated15–18 methods. Examples for computational baseline correction methods are the sensitive nonlinear iterative peak (SNIP) algorithm,19,20 multiplicative signal correction (MSC),21 extended multiplicative signal correction (EMSC),22,23 polynomial fitting,24 rubberband method25,26etc.
The instrumental method of shifted excitation Raman difference spectroscopy (SERDS)27 can be regarded as a wavelength modulated method, which has already been employed for biological samples.28–39 In SERDS, two Raman spectra are acquired at the same spatial position with two slightly shifted excitation wavelengths. The shift in excitation wavelength should be chosen according to the full width at half maximum (FWHM) of the expected Raman bands and is limited by the transmission of the bandpass filter for laser clean-up in the setup. The shift in excitation wavelength, which yielded the best results for biological samples36 and the given Raman setup, was 2 nm near 785 nm corresponding to 32 cm−1. The Raman bands follow this shift in excitation, whereas the auto-fluorescence and other wavelength-independent background contributions (such as ambient light) remain unaltered. Thus, the difference spectrum resulting from the subtraction of the two Raman spectra (SERDS spectrum) is ideally without fluorescence (background-free) and shows a characteristic difference profile for the Raman bands, which approximately corresponds to the 1st derivative of the Raman spectrum. For a better interpretation of the background free data, a reconstruction of a Raman spectrum is usually performed on the SERDS spectrum.40–44
In this study, we aimed to explore the limits of the SERDS method and other mathematical baseline correction algorithms by treating EP for increasing GE cross-linking durations which leads to Raman spectra with increasing fluorescence backgrounds. The fluorescence background ranges from a very subtle background for untreated EP to a very strong fluorescence at longer GE cross-linking durations. The Raman data at excitation wavelengths 784 and 786 nm were processed by the SERDS approach. For comparison, Raman spectra at 785 nm excitation of these samples were also background corrected using different computational correction algorithms like EMSC, polynomial, rubberband, and SNIP. We have already compared the results of the SERDS method with the mathematical baseline correction method EMSC for moderate background.36 However, this comparison did not include very strong auto-fluorescing samples in the practical part.
Methods
Decellularized equine pericardium (EP) was obtained from Auto Tissue (Berlin, Germany). The tissue was equally divided into 24 small pieces with approximately 2.5 mm × 2.5 mm dimensions each. EP was incubated with 0.25% GE prepared in phosphate-buffered solution (PBS) at 37 °C for 0.5 h, 2 h, 4 h, 6 h, 12 h, and 24 h (n = 3 per time point). The time points were selected to observe GE cross-linking EP for shorter durations (0.5 h, 2 h, and 4 h) and longer durations (6 h, 12 h, and 24 h). An untreated EP at each of the time points remained in PBS which served as a control (n = 1 per time point). All cross-linked tissues were rinsed at room temperature with PBS. The samples were kept in PBS during the entire spectroscopic measurements.All Raman spectra were acquired using a Shamrock (SR-303i) spectrometer (grating: 300 grooves per mm blazed at 860 nm, slit width 160 μm) in combination with an open electrode CCD detector (DV420A-OE) which was thermoelectrically cooled to −60 °C (all from Andor, Belfast, UK). The tunable excitation light source consisted of diode laser with an operating range of 765 nm–795 nm, a laser controller and an amplifier BoosTa pro (all from Toptica, Gräfelfing, Germany), and was coupled into a microscope cage system (Thorlabs, Newton, NJ, USA). The laser light was focused through a 60×/NA 1.0 water immersion objective (Nikon, Tokyo, Japan). For each sample 9 spectra per excitation wavelength were acquired in a raster scan fashion using a motorized stage (Optiscan, Prior, Rockland, MA, USA) with a step size of 20 μm to offset possible inhomogeneities. At each spatial position of the Raman array, three spectra with consecutive excitation wavelengths (784 nm/85 mW, 785 nm/150 mW, and 786 nm/85 mW) were collected with an exposure time of 3 s, 10 accumulations, and a dwell time of 0.5 s between each accumulation. The instrument control and Raman spectra acquisition were performed using an in-house developed LabVIEW (National Instruments, Austin, TX, USA) routine. For a schematic of the SERDS setup see Fig. S1 in the ESI.†
All measured spectral data was processed using the statistical analysis software R45 and the following R packages: hyperSpec,46 cbmodels,47 Ramancal,48 pracma,49 and MALDIquant.50 Preprocessing of all measured Raman spectra encompassed correcting cosmic spikes, calibrating the wavelength axis using the spectra of 4-Acetaminophenol collected at λex = 785 nm, and calibrating the intensity axis by calculating the system response function with the measured and known reference spectrum of a white-light source calibration lamp (Raman Calibration Accessory—HCA, Kaiser Optical Systems, Inc., USA). An overview of the spectral processing steps to generate the results in Fig. 1 is shown in Fig. S2 in the ESI.†
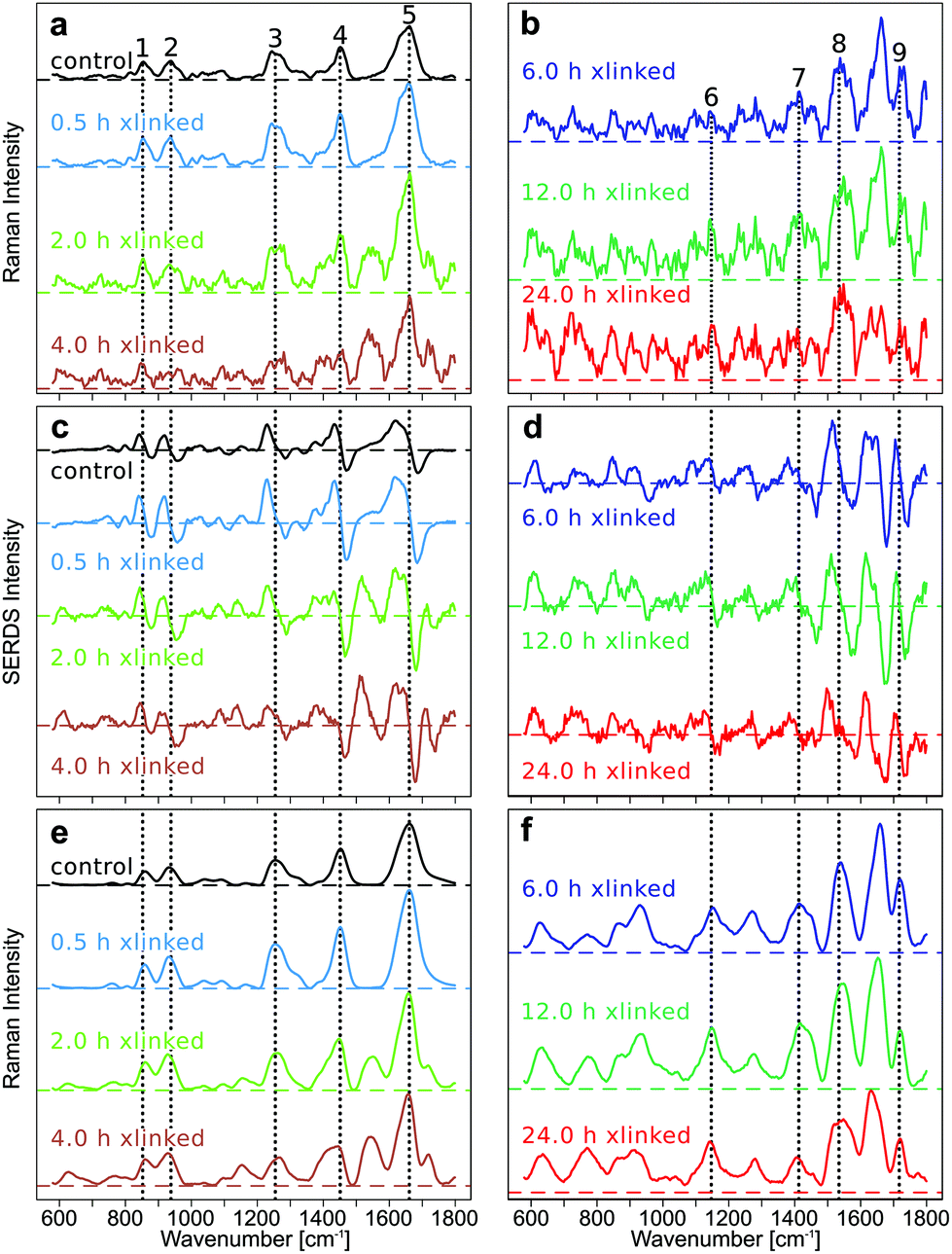 | ||
Fig. 1 Mean SNIP corrected Raman spectra of the different cross-linking time points (a and b), mean difference optimized SERDS spectra after SNIP correction (c and d), and mean reconstructed and SNIP corrected Raman spectra of SERDS spectra in 1c and 1d (e and f). Raman band positions of collagen (left) and genipin (right) are indicated by numbers and dotted lines: 1 = 852 cm−1 (Hydroxyproline); 2 = 938 cm−1 (C–Cα stretch); 3 = 1255 cm−1 (Amide III); 4 = 1452 cm−1 (CH2 collagen); 5 = 1662 cm−1 (Amide I); 6 = 1147 cm−1 and 7 = 1413 cm−1 (in-plane vibrations of ring system of GE cross-links); 8 = 1535 cm−1 (5-ring of GE cross-links); 9 = 1718 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). O). | ||
Results
The improvement in the quality of Raman spectra, in particular for crosslinking above 6 h, demonstrates the progress of the data processing and the importance of the method. Typical collagen bands are labelled from 1 to 5, and GE bands from 6 to 9 in Fig. 1.All raw spectra show a low fluorescent background for the early GE crosslinking time points and a high fluorescent background for longer GE crosslinking durations (Fig. S3–S6 in ESI†). The fluorescence background, which was estimated from the maxima of the mean spectra at the first excitation wavelength 784 nm, was approximately 7000 counts for the control sample and increased to 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (0.5 h), 60000 (2 h), 80000 (4 h), 100000 (6 h), 140000 (12 h) and 160000 counts (24 h) for GE-cross-linked samples. The increase in the fluorescent background is consistent with the spectral changes observed in Shaik et al.,5 and can attributed to the increased pigmentation of the samples upon GE cross-linking. The photobleaching is evident from reduced maxima of the mean spectra at the last excitation wavelength 786 nm which decreased for the control sample to approximately 6000 counts, and for GE cross-linked samples to approximately 9500 (0.5 h), 30000 (2 h), 50000 (4 h), 70000 (6 h), and 100000 (12 h, 24 h).
000 (0.5 h), 60000 (2 h), 80000 (4 h), 100000 (6 h), 140000 (12 h) and 160000 counts (24 h) for GE-cross-linked samples. The increase in the fluorescent background is consistent with the spectral changes observed in Shaik et al.,5 and can attributed to the increased pigmentation of the samples upon GE cross-linking. The photobleaching is evident from reduced maxima of the mean spectra at the last excitation wavelength 786 nm which decreased for the control sample to approximately 6000 counts, and for GE cross-linked samples to approximately 9500 (0.5 h), 30000 (2 h), 50000 (4 h), 70000 (6 h), and 100000 (12 h, 24 h).
The SERDS method was assessed by comparing the baseline corrected Raman spectra (Fig. 1a and b), the SERDS spectra (Fig. 1c and d) and the SNIP baseline corrected, reconstructed Raman spectra of the SERDS spectra (Fig. 1e and f). The Raman spectra measured at an excitation wavelength of 785 nm were baseline corrected using different computational baseline correction algorithms like EMSC, polynomial, rubberband, and SNIP. For simplicity, we only show the SNIP corrected mean spectra in Fig. 1a and b. The mean Raman spectra and their standard deviation of the other methods like EMSC (Fig. S7†), rubberband (Fig. S7†), polynomial (Fig. S8†), and SNIP (Fig. S8†) can be found in the ESI.†
The SERDS spectra (Fig. 1c and d) were calculated by subtracting the Raman spectra measured at different excitation wavelengths (λex1–λex2 = 784.0 nm–786.0 nm), after the preprocessed spectra of each wavelength were baseline corrected and then difference optimized. The difference optimization was applied for a better spectral overlap of the spectral pairs as explained below. The reconstructed spectra (Fig. 1e and f) were generated by integrating the SERDS spectra and then baseline correcting them with the SNIP method.
In Fig. 1a and b, the mean SNIP corrected Raman spectra of shorter and longer cross-link durations are shown. In the untreated control EP the typical type I collagen Raman bands are present at 852 cm−1 (Hydroxyproline), 938 cm−1 (C–Cα stretch), 1255 cm−1 (Amide III), 1452 cm−1 (CH2 collagen), and 1662 cm−1 (Amide I).6 New Raman bands emerge at 1147 cm−1 and 1413 cm−1, which can be assigned to the in-plane vibrations of the ring system of GE cross-links, at 1535 cm−1 (vibrations of the five membered ring of the GE cross-links) and 1718 cm−1 (CO) upon GE cross-linking.5 For clarity, collagen bands are only labeled in Fig. 1a, c and e, whereas GE bands were only labeled in Fig. 1b, d and f. Of course, GE bands are also evident in Fig. 1a, c and e, collagen bands in Fig. 1b, d and f. All emerging bands increase in intensity with longer cross-link durations relative to the collagen related bands. The quality of the baseline corrected Raman spectra decreases with longer cross-linking duration due to the high spectral background. This high spectral background results in an increase of the shot noise in the spectra, which impairs discrimination of the Raman bands, even in the baseline corrected spectra. Especially for the long cross-linking time points (12 h and 24 h) most Raman bands are hard to discriminate. For all other investigated baseline correction methods, similar results can be found (Fig. S7 and S8 in the ESI†). Changes in collagen and GE crosslinking bands were also observed in the mean SERDS spectra (Fig. 1c and d, mean and standard deviation in Fig. S9†), but the exact band positions are difficult to determine in the 1st derivative-like signatures. The successful background compensation is evident from the almost symmetric difference features around the zero line and the virtually absent slope. A progression from dominating collagen difference features to GE cross-linked features can be found in the SERDS spectra. Compared to the baseline corrected Raman spectra, spectral features are more pronounced in SERDS spectra, and structural changes can be better discerned for the longer GE cross-link durations. To determine the band positions from the SERDS signatures, Raman spectra were reconstructed from the SERDS spectra by integration and were afterwards baseline corrected using the SNIP algorithm (Fig. 1e and f, mean and standard deviation Fig. S9†). Whereas this reconstruction compromised the spectral resolution, the signal to noise ratio was improved. The reconstructed mean Raman spectra showed the spectral changes occurring during the cross-linking more clearly, the spectra could be better interpreted, in particular for longer time points, than for the baseline corrected Raman spectra, and all main GE bands were identified that are poorly resolved at 12 and 24 h crosslinking in Fig. 1b. The reason for this is a better compensation of instrument inherent backgrounds with the SERDS technique as further explained below.
Details of data processing for the generation of the SERDS spectra shown in Fig. 1 are presented in the following paragraphs focusing on the longer crosslinking durations (6 h, 12 h, and 24 h). Since these samples show the most intense background in Raman spectra, almost no collagen and GE bands are resolved, and the progress of the SERDS approach is most evident here.
By subtracting the preprocessed Raman spectra measured with λex2 = 786 nm (RS2) from the preprocessed Raman spectra acquired with λex1 = 784 nm (RS1) the SERDS spectra (DS12) were calculated. In Fig. 2a, the mean SERDS spectra of the longer cross-linking durations are shown without any previous normalization of the spectra. The difference intensities are consistent with the photobleaching described above and observed in Fig. S4 and S5.† It is evident from the intensity scales that the residual background increases with increased GE cross-linking duration. Photobleaching of the auto-fluorescence background is caused by the long laser exposure during the acquisition. To counter these photobleaching effects and therefore reduce the residual background in the SERDS spectra, an optimization or normalization has to be performed on the Raman spectra before subtraction as described in our previous work.36 The difference optimization will be summarized next.
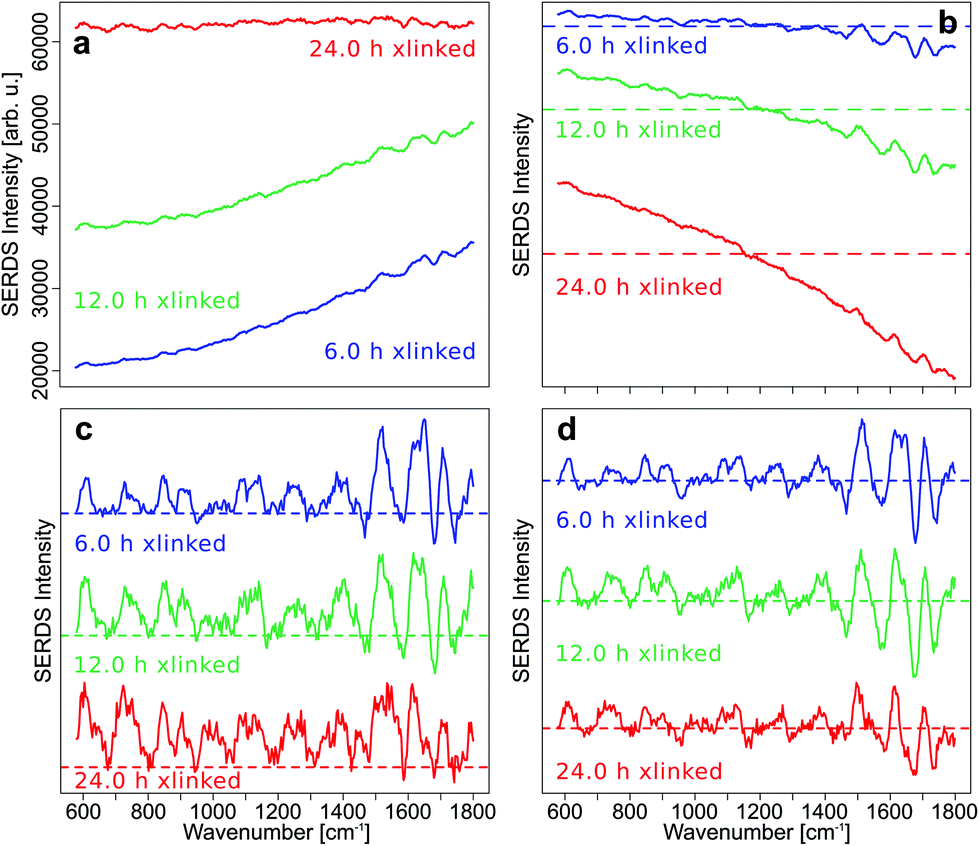 | ||
| Fig. 2 Mean SERDS spectra (a) without normalization or optimization, (b) with difference optimization, (c) with SNIP baseline correction before difference calculation and (d) with SNIP baseline correction and subsequent difference optimization before difference calculation for longer cross-linking durations. | ||
For each spectral pair, the subtrahend spectrum RS2 is multiplied with a factor k before subtraction from the minuend spectrum RS1 resulting in the SERDS spectrum DS12. The area under the curve (AUC) of the absolute values of the SERDS spectrum DS12 is calculated. Factor k is iteratively adjusted until the AUC reaches a minimum.
Even this simple difference optimization (Fig. 2b) step is not sufficient to compensate these strong photobleaching effects. In the difference optimized mean SERDS spectra, a strong tilt in the spectra can be observed, since the spectra are not aligned along the zero line, as it would be expected for a background free SERDS spectrum. They show a varying offset, which is positive for very low wavenumbers and decreases towards higher wavenumbers of the fingerprint region until the offset is negative. The reason for this is a higher auto-fluorescence intensity in the lower wavenumbers. Therefore, changes in background intensity due to photobleaching also effects the lower wavenumber region stronger. Since the simple difference optimization only uses one factor for the whole spectrum to compensate for variations in background, it cannot compensate a non-constant offset, that occurs during photobleaching.
For this reason, a baseline correction was performed using the SNIP algorithm before calculating the SERDS spectra from the preprocessed but not difference optimized Raman spectra (Fig. 2c). Since the SERDS spectra are mostly positive for longer cross-linking times, which makes it hard to discern the difference bands, the baseline corrected spectra were difference optimized after the baseline correction and then the SERDS spectra were calculated (Fig. 2d). This procedure corrected the SERDS spectra successfully for the residual offset. Since the combination of a baseline correction and a subsequent difference optimization before the calculation of the SERDS spectra resulted in an optimal photobleaching correction, these preprocessing steps were performed to calculate the SERDS spectra used for the assessment (Fig. 1c and d) and also for the reconstruction step (Fig. 1e and f).
To demonstrate the effect of intensity calibration on SERDS approach, the mean SNIP corrected Raman spectra with and without intensity calibration were compared with the mean difference optimized and SNIP corrected SERDS spectra with and without intensity calibration (Fig. 3).
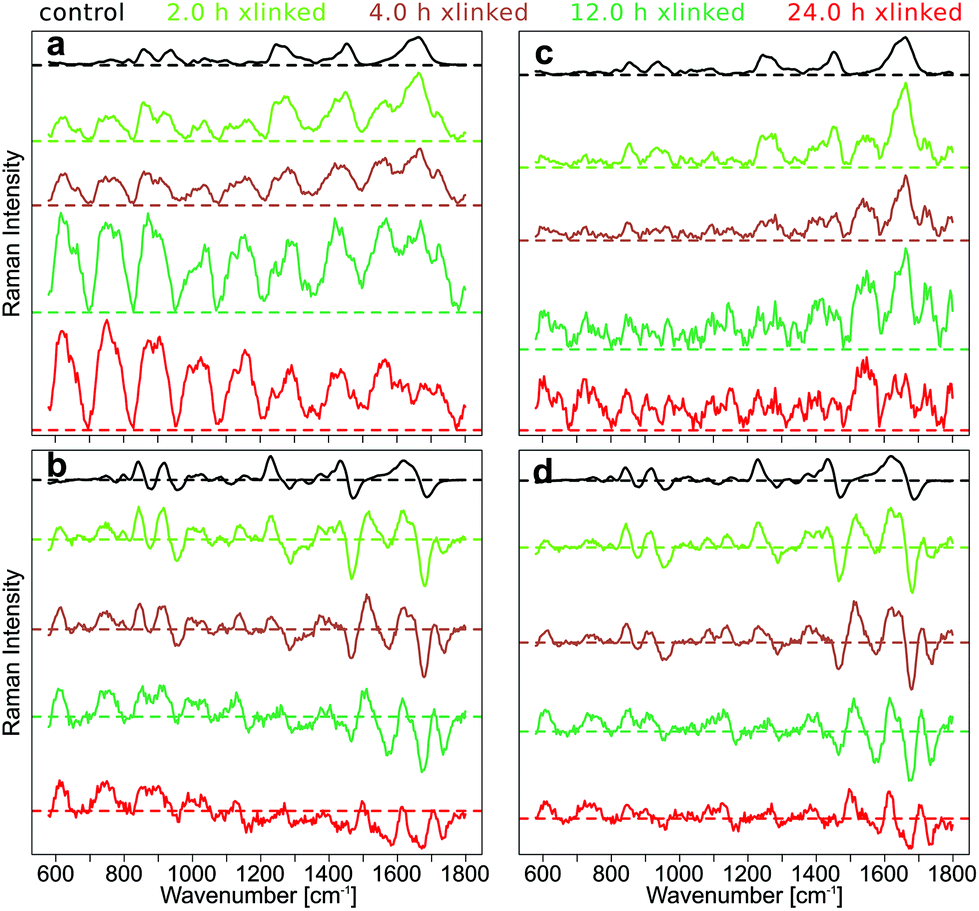 | ||
| Fig. 3 The influence of intensity calibration on SNIP baseline corrected and SERDS spectra of the cross-linked pericardium with different cross-linking times; (a) SNIP corrected mean Raman spectra and (b) SNIP corrected, difference optimized mean SERDS spectra without intensity calibration; (c) SNIP corrected mean Raman spectra and (d) SNIP corrected, difference optimized mean SERDS spectra with intensity calibration. | ||
The fluorescence background was quite low in the Raman spectra of control samples, and the SNIP baseline correction gave excellent spectra without an intensity calibration (Fig. 3a). However, the overall intensities of the raw Raman spectra increased with cross-linking time and introduced a wave-like artifact as spectral background after SNIP baseline correction which makes the interpretation of the Raman spectra very difficult and has frequently been observed in Raman publications.51 This background is an instrument inherent background which originates primarily from the optical filters used in the setup. Etaloning effects can be neglected in the front illuminated, open-electrode CCD detector used here, but may occur in back illuminated CCD detectors.52 The mean SERDS spectra after difference optimization and SNIP correction are shown without intensity calibration of the Raman spectra in Fig. 3b, and the wave-like artifact is absent here. Only for the spectra of the 24 h cross-linking samples, the mean spectrum is tilted by a small degree due to a decreasing positive offset towards higher wavenumbers in the spectral region below 1100 cm−1 and an increasing negative offset towards higher wavenumbers above 1100 cm−1. If the Raman spectra are intensity calibrated and then baseline corrected with the SNIP algorithm, the wave-like artifact can also be removed (Fig. 3c). However, Raman bands are hard to discern for the 12 h and 24 h cross-linking timepoints as already shown in Fig. 1b. An intensity calibration of the raw spectra further improves the SERDS result (Fig. 3d) which is most significant for the 24 h cross-linking timepoint. The mean SERDS spectra of all other timepoints are quite similar without (Fig. 3b) and with (Fig. 3d) intensity calibration. Overall, the intensity calibration step is usually not necessary for the SERDS method, since constant, instrument inherent background contributions are suppressed by this difference method.
Discussion
Photobleaching is challenging for SERDS measurements of samples with extremely high fluorescent backgrounds. Since a simple difference optimization did not sufficiently suppress the strong photobleaching effect, a baseline correction was included during data processing. An even better way to compensate for photobleaching is the accumulation of very short measurement intervals (50 ms or less), where the excitation wavelength is changed after each very short acquisition. Upon multiple iterations, this acquisition technique distributes the photobleaching more evenly among the spectra measured with different excitation wavelengths and hence efficiently compensates photobleaching.29,53 Since this was not feasible with the used laser source, an advanced difference optimization approach was incorporated in the preprocessing step to correct a baseline before the difference optimization and calculation of the difference spectra. Then, SERDS outperforms common baseline correction methods offering a better correction and spectral interpretation, in particular for very high backgrounds. Reconstruction of Raman spectra from the SERDS spectra reduces the spectral resolution, but the spectral features are more clearly visible. The dependency of the spectral resolution on Raman-based cell classification was recently studied.54 The accuracy was found to be almost unchanged for spectral resolutions between 8 and 48 cm−1. Consequently, the diminished resolution does not seem to be relevant for cell and tissue applications with relatively broad bands. Since two Raman spectra are necessary for the calculation of the SERDS spectra, the Raman spectra for the comparison with the SERDS technique should have twice the integration time of the SERDS spectra. This was partly compensated in this study by almost twice laser intensities at 785 nm.We demonstrated that an intensity calibration is not necessary only for Raman spectra with low spectral background and intense signals. However, for extremely high backgrounds the system transfer function had a strong influence and virtually masked all Raman bands in a wave-like pattern after baseline correction. For SERDS, intensity calibration is not as important since the system transfer function is an instrument inherent, excitation wavelength-independent background contribution, that is automatically suppressed by the SERDS approach.
We conclude, that SERDS can be used to study GE cross-linked EP at higher cross-linking durations despite its intense fluorescence. The here presented data processing will be used in a forthcoming publication to determine the biochemical signature of GE cross-linked EP, quantify GE cross-links and correlate the biochemical changes derived from SERDS with other optical modalities. Better quality spectra with higher signal to noise ratios are expected to improve also the contrast in Raman images. Furthermore, this SERDS strategy can be transferred to Raman studies on biological and non-biological samples with intense background at 785 nm and also shorter excitation wavelengths which benefit from more intense scattering intensities and higher quantum efficiencies of CCD detectors.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Conceptualization: FK, CK, JP. Investigation and methodology: FK, TS. Data curation: FK. Supervision: CK, JP. Funding acquisition: JP. Writing – original draft: FK. Writing, review and edit: all.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Dr. Claudia Beleites, Prof. Dr. Iwan W. Schie and Dr. Clara Stiebing for the fruitful discussions and their helpful comments. We also thank Oliver Bloch (Auto Tissue, Berlin, Germany) for providing us with the decellularized equine pericardium. Florian Korinth acknowledged funding by the Leibniz Association within the project “HYPERAM” (SAW-2016-IPHT-2).References
- E. Cordero, J. Rüger, D. Marti, A. S. Mondol, T. Hasselager, K. Mogensen, G. G. Hermann, J. Popp and I. W. Schie, Bladder tissue characterization using probe–based Raman spectroscopy: Evaluation of tissue heterogeneity and influence on the model prediction, J. Biophotonics, 2020, 13, 1–15, DOI:10.1002/jbio.201960025.
- J. Desroches, M. Jermyn, M. Pinto, F. Picot, M. A. Tremblay, S. Obaid, E. Marple, K. Urmey, D. Trudel and G. Soulez, et al. A new method using Raman spectroscopy for in vivo targeted brain cancer tissue biopsy, Sci. Rep., 2018, 8, 1–10, DOI:10.1038/s41598-018-20233-3.
- R. Galli, M. Meinhardt, E. Koch, G. Schackert, G. Steiner, M. Kirsch and O. Uckermann, Rapid Label-Free Analysis of Brain Tumor Biopsies by Near Infrared Raman and Fluorescence Spectroscopy — A Study of 209 Patients, Front. Oncol., 2019, 9, 1–13, DOI:10.3389/fonc.2019.01165.
- M. S. Bergholt, W. Zheng, K. Lin, J. Wang, H. Xu, J. L. Ren, K. Y. Ho, M. Teh, K. G. Yeoh and Z. Huang, Characterizing variability of in vivo Raman spectroscopic properties of different anatomical sites of normal colorectal tissue towards cancer diagnosis at colonoscopy, Anal. Chem., 2015, 87, 960–966, DOI:10.1021/ac503287u.
- T. A. Shaik, A. Alfonso-Garcia, M. Richter, F. Korinth, C. Krafft, L. Marcu and J. Popp, FLIm and Raman Spectroscopy for Investigating Biochemical Changes of Bovine Pericardium upon Genipin Cross-Linking, Molecules, 2020, 25, 3857, DOI:10.3390/molecules25173857.
- T. A. Shaik, J. L. Lagarto, E. Baria, M. Goktas, P. I. Onoja, K. G. Blank, F. S. Pavone, J. Popp, C. Krafft and R. Cicchi, Monitoring Changes in Biochemical and Biomechanical Properties of Collagenous Tissues Using Label-Free and Nondestructive Optical Imaging Techniques, Anal. Chem., 2021, 93, 3813–3821, DOI:10.1021/acs.analchem.0c04306.
- H.-W. Sung, R.-N. Huang, L. L. H. Huang and C.-C. Tsai, In vitro evaluation of cytotoxicity of a naturally occurring cross-linking reagent for biological tissue fixation, J. Biomater. Sci., Polym. Ed., 1999, 10, 63–78, DOI:10.1163/156856299X00289.
- M. F. Butler, Y. Ng and P. D. A. Pudney, Mechanism and kinetics of the crosslinking reaction between biopolymers containing primary amine groups and genipin, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3941–3953, DOI:10.1002/pola.10960.
- H. W. Sung, Y. Chang, C. T. Chiu, C. N. Chen and H. C. Liang, Crosslinking characteristics and mechanical properties of a bovine pericardium fixed with a naturally occurring crosslinking agent, J. Biomed. Mater. Res., 1999, 47, 116–126, DOI:10.1002/(SICI)1097-4636(199911)47:2<116::AID-JBM2>3.0.CO;2-J.
- R. Touyama, Y. Takeda, K. Inoue, I. Kawamura, M. Yatsuzuka, T. Ikumoto, T. Shingu, T. Yokoi and H. Inouye, Studies on the Blue Pigments Produced from Genipin and Methylamine. I. Structures of the Brownish-Red Pigments, Intermediates Leading to the Blue Pigments, Chem. Pharm. Bull., 1994, 42, 668–673, DOI:10.1248/cpb.42.668.
- S. Fujikawa, S. Nakamura and K. Koga, Genipin, a new type of protein crosslinking reagent from gardenia fruits, Agric. Biol. Chem., 1988, 52, 869–870, DOI:10.1271/bbb1961.52.869.
- D. Wei, S. Chen and Q. Liu, Review of Fluorescence Suppression Techniques in Raman Spectroscopy, Appl. Spectrosc. Rev., 2015, 50, 387–406, DOI:10.1080/05704928.2014.999936.
- F. Ariese, H. Meuzelaar, M. M. Kerssens, J. B. Buijs and C. Gooijer, Picosecond Raman spectroscopy with a fast intensified CCD camera for depth analysis of diffusely scattering media, Analyst, 2009, 134, 1192–1197, 10.1039/b821437a.
- M. Kögler and B. Heilala, Time-gated Raman spectroscopy – a review, Meas. Sci. Technol., 2020, 32, 1–17, DOI:10.1088/1361-6501/abb044.
- A. De Luca, K. Dholakia and M. Mazilu, Modulated Raman Spectroscopy for Enhanced Cancer Diagnosis at the Cellular Level, Sensors, 2015, 15, 13680–13704, DOI:10.3390/s150613680.
- S. Dochow, N. Bergner, C. Krafft, J. Clement, M. Mazilu, B. B. Praveen, P. C. Ashok, R. Marchington, K. Dholakia and J. Popp, Classification of Raman spectra of single cells with autofluorescence suppression by wavelength modulated excitation, Anal. Methods, 2013, 5, 4608–4614, 10.1039/c3ay40193f.
- D. Craig, M. Mazilu and K. Dholakia, Quantitative detection of pharmaceuticals using a combination of paper microfluidics and wavelength modulated Raman spectroscopy, PLoS One, 2015, 10, 1–10, DOI:10.1371/journal.pone.0123334.
- M. J. Wirth and S. H. Chou, Comparison of Time and Frequency Domain Methods for Rejecting Fluorescence from Raman Spectra, Anal. Chem., 1988, 60, 1882–1886, DOI:10.1021/ac00169a009.
- M. Morhác and V. Matousek, Peak clipping algorithms for background estimation in spectroscopic data, Appl. Spectrosc., 2008, 62, 91–106, DOI:10.1366/000370208783412762.
- M. Morháč, An algorithm for determination of peak regions and baseline elimination in spectroscopic data, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 600, 478–487, DOI:10.1016/j.nima.2008.11.132.
- E. W. Stark and H. Martens, Multiplicative signal correction method and apparatus, U.S. Pat., 005568400A, 1990 Search PubMed.
- N. K. Afseth and A. Kohler, Extended multiplicative signal correction in vibrational spectroscopy, a tutorial, Chemom. Intell. Lab. Syst., 2012, 117, 92–99, DOI:10.1016/j.chemolab.2012.03.004.
- H. Martens and E. Stark, Extended multiplicative signal correction and spectral interference subtraction: New preprocessing methods for near infrared spectroscopy, J. Pharm. Biomed. Anal., 1991, 9, 625–635, DOI:10.1016/0731-7085(91)80188-F.
- C. A. Lieber and A. Mahadevan-Jansen, Automated Method for Subtraction of Fluorescence from Biological Raman Spectra, Appl. Spectrosc., 2003, 57, 1363–1367, DOI:10.1366/000370203322554518.
- M. A. Kneen and H. J. Annegarn, Algorithm for fitting XRF, SEM and PIXE X-ray spectra backgrounds, Nucl. Instruments Methods Phys. Res. Sect. B, 1996, 109/110, 209–213, DOI:10.1016/0168-583X(95)00908-6.
- M. Pirzer and J. Sawatzki, Method and device for correcting a spectrum, U.S. Pat., 7359815, 2008 Search PubMed.
- A. P. Shreve, N. J. Cherepy and R. A. Mathies, Effective Rejection of Fluorescence Interference in Raman Spectroscopy Using a Shifted Excitation Difference Technique, Appl. Spectrosc., 1992, 46, 707–711, DOI:10.1366/0003702924125122.
- F. Korinth, A. S. Mondol, C. Stiebing, I. W. Schie, C. Krafft and J. Popp, New methodology to process shifted excitation Raman difference spectroscopy data: a case study of pollen classification, Sci. Rep., 2020, 10, 11215, DOI:10.1038/s41598-020-67897-4.
- F. Korinth, E. Schmälzlin, C. Stiebing, T. Urrutia, G. Micheva, C. Sandin, A. Müller, M. Maiwald, B. Sumpf and C. Krafft, et al. Wide field spectral imaging with shifted excitation raman difference spectroscopy using the nod and shuffle technique, Sensors, 2020, 20, 1–19, DOI:10.3390/s20236723.
- K. Noack, B. Eskofier, J. Kiefer, C. Dilk, G. Bilow, M. Schirmer, R. Buchholz and A. Leipertz, Combined shifted-excitation Raman difference spectroscopy and support vector regression for monitoring the algal production of complex polysaccharides, Analyst, 2013, 138, 5639–5646, 10.1039/c3an01158e.
- K. Sowoidnich and H.-D. Kronfeldt, Fluorescence Rejection by Shifted Excitation Raman Difference Spectroscopy at Multiple Wavelengths for the Investigation of Biological Samples, ISRN Spectrosc., 2012, 2012, 1–11, DOI:10.5402/2012/256326.
- Z. Han, B. D. Strycker, B. Commer, K. Wang, B. D. Shaw, M. O. Scully and A. V. Sokolov, Molecular origin of the Raman signal from Aspergillus nidulans conidia and observation of fluorescence vibrational structure at room temperature, Sci. Rep., 2020, 10(5428), 1–8, DOI:10.1038/s41598-020-62112-w.
- M. A. da Silva Martins, D. G. Ribeiro, E. A. P. dos Santos, A. A. Martin, A. Fontes and H. da Silva Martinho, Shifted-excitation Raman difference spectroscopy for in vitro and in vivo biological samples analysis, Biomed. Opt. Express, 2010, 1, 617–626, DOI:10.1364/BOE.1.000617.
- C. Knipfer, J. Motz, W. Adler, K. Brunner, M. T. Gebrekidan, R. Hankel, A. Agaimy, S. Will, A. Braeuer and F. W. Neukam, et al. Raman difference spectroscopy: a non-invasive method for identification of oral squamous cell carcinoma, Biomed. Opt. Express, 2014, 5, 3252, DOI:10.1364/BOE.5.003252.
- E. Schmälzlin, B. Moralejo, D. Bodenmüller, M. E. Darvin, G. Thiede and M. M. Roth, Ultrafast imaging Raman spectroscopy of large-area samples without stepwise scanning, J. Sensors Sens. Syst., 2016, 5, 261–271, DOI:10.5194/jsss-5-261-2016.
- E. Cordero, F. Korinth, C. Stiebing, C. Krafft, I. Schie and J. Popp, Evaluation of Shifted Excitation Raman Difference Spectroscopy and Comparison to Computational Background Correction Methods Applied to Biochemical Raman Spectra, Sensors, 2017, 17, 1724, DOI:10.3390/s17081724.
- K. Sowoidnich and H.-D. Kronfeldt, In situ species authentication of frozen-thawed meat and meat juice using shifted excitation Raman difference spectroscopy, Biophotonics: Photonic Solutions for Better Health Care VI, Proc. SPIE, 2018, 10685, 106850L, DOI:10.1117/12.2306659.
- M. T. Gebrekidan, R. Erber, A. Hartmann, P. A. Fasching, J. Emons, M. W. Beckmann and A. Braeuer, Breast Tumor Analysis Using Shifted-Excitation Raman Difference Spectroscopy (SERDS), Technol. Cancer Res. Treat., 2018, 17, 1533033818782532, DOI:10.1177/1533033818782532.
- M. T. Gebrekidan, C. Knipfer, F. Stelzle, J. Popp, S. Will and A. Braeuer, A shifted-excitation Raman difference spectroscopy (SERDS) evaluation strategy for the efficient isolation of Raman spectra from extreme fluorescence interference, J. Raman Spectrosc., 2016, 47, 198–209, DOI:10.1002/jrs.4775.
- J. Zhao, M. M. Carrabba and F. S. Allen, Automated Fluorescence Rejection Using Shifted Excitation Raman Difference Spectroscopy, Appl. Spectrosc., 2002, 56, 834–845, DOI:10.1366/000370202760171491.
- P. Matousek, M. Towrie and A. W. Parker, Simple Reconstruction Algorithm for Shifted Excitation Raman Difference Spectroscopy, Appl. Spectrosc., 2005, 59, 848–851, DOI:10.1366/0003702054280757.
- R. Willett, Multiscale Reconstruction for Photon-Limited Shifted Excitation Raman Spectroscopy, In Proceedings of the 2007 IEEE International Conference on Acoustics, Speech and Signal Processing - ICASSP '07, IEEE, 2007; pp. 833–836 Search PubMed.
- H. Schmidt, D. P. Kaiser and M. Maiwald, Method For Generating and for Detecting a Raman Spectrum, U.S. Pat., 8310672, 2012 Search PubMed.
- S. Guo, O. Chernavskaia, J. Popp and T. Bocklitz, Spectral reconstruction for shifted-excitation Raman difference spectroscopy (SERDS), Talanta, 2018, 186, 372–380, DOI:10.1016/j.talanta.2018.04.050.
- R Core Team R: A Language and Environment for Statistical Computing, 2018 Search PubMed.
- C. Beleites and V. Sergo, HyperSpec: a package to handle hyperspectral data sets in R, 2018 Search PubMed.
- C. Beleites, Cbmodels: Collection of “combined” models: PCA-LDA, PLS-LDA, PLS-LR as well as EMSC, 2015 Search PubMed.
- C. Beleites, Ramancal: Calibration routines for Raman spectrometers, 2013 Search PubMed.
- H. W. Borchers, Pracma: Practical Numerical Math Functions, 2018 Search PubMed.
- S. Gibb and K. Strimmer, MALDIquant: a versatile R package for the analysis of mass spectrometry data, Bioinformatics, 2012, 28, 2270–2271, DOI:10.1093/bioinformatics/bts447.
- S. Dochow, N. Bergner, C. Matthäus, B. B. Praveen, P. C. Ashok, M. Mazilu, C. Krafft, K. Dholakia and J. Popp, Etaloning, fluorescence and ambient light suppression by modulated wavelength Raman spectroscopy, Biomed. Spectrosc. Imaging, 2012, 1, 383–389, DOI:10.3233/BSI-120031.
- J. R. Walsh, W. Freudling, N. Pirzkal and A. Pasquali, Modelling the fringing of the ACS WFC and HRC chips, ST-ECF Instrum. Sci. Rep., ACS 2003-012, 2003, 1–23 Search PubMed.
- K. Sowoidnich, M. Towrie, M. Maiwald, B. Sumpf and P. Matousek, Shifted Excitation Raman Difference Spectroscopy with Charge-Shifting Charge-Coupled Device (CCD) Lock-In Detection, Appl. Spectrosc., 2019, 73, 1265–1276, DOI:10.1177/0003702819859352.
- I. W. Schie, C. Krafft and J. Popp, Cell Classification with Low-Resolution Raman Spectroscopy (LRRS), J. Biophotonics, 2016, 9, 994–1000, DOI:10.1002/jbio.201600095.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information about other baseline correction techniques. See DOI: 10.1039/d1an01376a |
| This journal is © The Royal Society of Chemistry 2021 |