
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advanced spectroscopic analysis and 15N-isotopic labelling study of nitrate and nitrite reduction to ammonia and nitrous oxide by E. coli†
George D.
Metcalfe
 a,
Thomas W.
Smith
ab and
Michael
Hippler
*a
a,
Thomas W.
Smith
ab and
Michael
Hippler
*a
aDepartment of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: M.Hippler@sheffield.ac.uk
bSchool of Chemical Engineering and Analytical Science, University of Manchester, Manchester M13 9PL, UK
First published on 15th October 2021
Abstract
Nitrate and nitrite reduction to ammonia and nitrous oxide by anaerobic E. coli batch cultures is investigated by advanced spectroscopic analytical techniques with 15N-isotopic labelling. Non-invasive, in situ analysis of the headspace is achieved using White cell FTIR and cavity-enhanced Raman (CERS) spectroscopies alongside liquid-phase Raman spectroscopy. For gas-phase analysis, White cell FTIR measures CO2, ethanol and N2O while CERS allows H2, N2 and O2 monitoring. The 6 m pathlength White cell affords trace gas detection of N2O with a noise equivalent detection limit of 60 nbar or 60 ppbv in 1 atm. Quantitative analysis is discussed for all four 14N/15N-isotopomers of N2O. Monobasic and dibasic phosphates, acetate, formate, glucose and NO3− concentrations are obtained by liquid-phase Raman spectroscopy, with a noise equivalent detection limit of 0.6 mM for NO3− at 300 s integration time. Concentrations of the phosphate anions are used to calculate the pH in situ using a modified Henderson–Hasselbalch equation. NO2− concentrations are determined by sampling for colorimetric analysis and NH4+ by basifying samples to release 14N/15N-isotopomers of NH3 for measurement in a second FTIR White cell. The reductions of 15NO3−, 15NO2−, and mixed 15NO3− and 14NO2− by anaerobic E. coli batch cultures are discussed. In a major pathway, NO3− is reduced to NH4+via NO2−, with the bulk of NO2− reduction occurring after NO3− depletion. Using isotopically labelled 15NO3−, 15NH4+ production is distinguished from background 14NH4+ in the growth medium. In a minor pathway, NO2− is reduced to N2O via the toxic radical NO. With excellent detection sensitivities, N2O serves as a monitor for trace NO2− reduction, even when cells are predominantly reducing NO3−. The analysis of N2O isotopomers reveals that for cultures supplemented with mixed 15NO3− and 14NO2− enzymatic activity to reduce 14NO2− occurs immediately, even before 15NO3− reduction begins. Optical density and pH measurements are discussed in the context of acetate, formate and CO2 production. H2 production is repressed by NO3−; but in experiments with NO2− supplementation only, CERS detects H2 produced by formate disproportionation after NO2− depletion.
1. Introduction
In the absence of oxygen (O2), Escherichia coli (E. coli) can utilise alternative terminal electron acceptors for anaerobic growth, such as nitrate (NO3−) and nitrite (NO2−). The sequence of reductions from NO3− to NO2− to ammonia (NH3, NH4+ at physiological pH) is generally referred to as Dissimilatory Nitrate Reduction to Ammonia (DNRA).1 The coupling of these reductions to the oxidation of organic substrates, such as formate, enables the generation of a proton gradient across the cytoplasmic membrane. DNRA is considerably more efficient for obtaining energy than the mixed acid fermentation pathways utilised when electron acceptors are unavailable. The expression of the respiratory NO3− and NO2− reductases is tightly controlled by FNR, an O2 sensitive transcription factor, and NarXL/NarQP, both of which are two-component NO3−/NO2− sensitive regulatory systems.2,3Although DNRA is the major NO3− reduction pathway in E. coli, the bacterium also generates minor amounts of the toxic radical nitric oxide (NO) from NO2− reduction. The low level of NO production by E. coli may be due to disproportionation of NO2− under acidic conditions or non-specific reduction by metalloproteins. The NADH-dependent cytoplasmic NO2− reductase (NirB),4 the membrane-bound periplasmic NO2− reductase (NrfA)5 and the major anaerobic NO3− reductase (NRA)6,7 have all been proposed to be significant sources of NO formation as a by-product of their roles in the DNRA pathway. Aerobically, flavohemoglobin (Hmp) detoxifies NO by oxidation back to NO3−; while anaerobically, NO is reduced further to nitrous oxide (N2O) reportedly by Hmp,8 flavorubredoxin (NorV)9 and hybrid cluster protein (Hcp).10 N2O is comparatively less toxic than NO and can rapidly diffuse out of the cell. E. coli is not a true denitrifier but N2O production by NO3− respiring E. coli cultures does share similarities with the denitrification pathway of NO3− to nitrogen (N2) via NO2−, NO and N2O. A summary of DNRA and NO generation and detoxification is shown in Fig. 1.
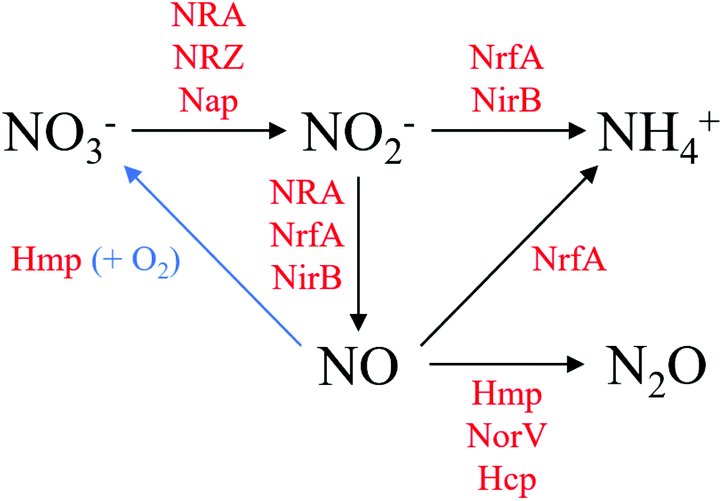 | ||
| Fig. 1 DNRA and NO generation and detoxification by E. coli. Enzymes are displayed in red: Hcp, hybrid cluster protein; Hmp, flavohemoglobin; Nap, periplasmic nitrate reductase, NirB, NADH-dependent nitrite reductase; NorV, flavorubredoxin; NRA, nitrate reductase A; NrfA, periplasmic nitrite reductase; NRZ, nitrate reductase Z. | ||
As a model organism, DNRA has been studied extensively in E. coli; however, comparatively less is known about the minor pathway leading to N2O and how its generation differs between NO3− and NO2− respiring cultures. To gain a better mechanistic understanding, monitoring the key compounds and parameters of these processes is essential. Accurate and reliable analytical techniques are crucial for understanding cell biochemistry and pathway elucidation. This represents a challenge for analytical chemistry, requiring a combination of advanced analytical techniques.
Mass spectrometry and chromatographic techniques are widely applicable to the detection and quantification of a broad range of metabolites.11 The tandem gas chromatography-mass spectrometry technique is considered the gold standard for the general analysis of volatile organic chemicals.12 Despite this, these techniques are not readily applicable to rapid, online analysis either due to the need for sampling or for downstream chemical/physical processing before analysis can occur. Electrochemical sensors are widely used for monitoring pH, conductivity, dissolved O2 and various other chemical species,13 including NO.14 Often such sensors are susceptible to cross-interferences from other species, changes in solution activity and long-term drift. For microbiological studies, the need for physical contact between the electrode and cell culture increases the risk of contamination, particularly in continuous cultures, and requires that the electrode is stable towards sterilisation.11
Spectroscopic techniques can be readily applied for monitoring bioprocesses in situ and online, with no sampling. Vibrational spectroscopic techniques, such as Fourier Transform Infrared (FTIR) and Raman spectroscopies, show high specificity for different molecules due to characteristic spectral bands, making them potentially very valuable for metabolic studies. Additionally, vibrational spectroscopies can distinguish different isotopologues and isotopomers, allowing online monitoring of isotope labelling experiments.15,16 Good sensitivities are observed in the condensed phase, but measuring headspace gases often suffers from low sensitivity, and special enhancement techniques are required such as Cavity Enhanced Raman Spectroscopy (CERS)15–22 or long-path absorption White cells in FTIR spectroscopy.23 Partial pressures in the headspace can be converted into concentrations in the solution via Henry's law. Quantum Cascade Laser (QCL) absorption spectroscopy has been applied to detect N2O and other trace gases;24–26 while sensitive, the limited tuning range of QCLs over a single IR absorption band limits the dynamic range due to band saturation effects. While FTIR spectroscopy has found some application in bioprocess monitoring, the broad absorption profile of water limits its application for monitoring metabolites at low concentrations in solution. In the gas-phase, the lack of an extended hydrogen network confines the absorption of water to certain spectral regions; molecules with absorption bands outside these regions can be readily detected, even in the presence of high levels of water vapour. Since Raman spectroscopy is comparatively insensitive to water, it is more readily applied to direct monitoring of the liquid-phase. However, fluorescence in complex media such as Lysogeny Broth (LB) can complicate the detection of the comparatively weak Raman light. Fluorescence can be avoided by moving to longer excitation wavelengths or by using media free of fluorescent components, such as M9 minimal media.23 Vibrational spectroscopic tools have been previously applied to monitoring NO3− metabolism in bacteria; CERS has been used to follow N2O and N2 production in denitrifying organisms, with the use of 15NO3− to produce 15N2 distinguishable from background 14N2.19,22 A robust CERS instrument has also been designed for field application to study the gas composition of soil samples.21
We report a combined approach for characterizing DNRA and N2O production in anaerobic E. coli batch cultures using mostly non-invasive spectroscopic techniques. Sampling of the bacterial culture was only done for NO2− colorimetry and FTIR detection of 14NH3 and 15NH3 isotopomers. Headspace gas analysis was provided by the complementary techniques of FTIR and CERS, with CERS being a technique recently introduced by us in this Journal.17 FTIR allowed detection of CO2, ethanol and N2O while CERS enabled monitoring of the homonuclear diatomic molecules N2, O2 and H2. Recently we introduced the capability of liquid culture analysis by Raman spectroscopy to monitor the microbial fermentation products of acetate and formate and the resulting in situ pH from phosphate signatures using a modified Henderson–Hasselbalch equation.23 Here, we report on improvements that also allowed NO3− and glucose analysis during DNRA. With the use of 15N-labelling, we report on mechanistic insights into NO3− and NO2− reduction to NH4+ and N2O through interpreting the different 14N/15N-isotopomers produced. The aims of this report are to introduce and characterise a unique combination of advanced spectroscopic techniques with great potential for bioanalytical applications, and to introduce an interesting biochemical application, a 15N-isotope labelling study on N2O production during DNRA by E. coli, with a focus on the differences observed between NO3− and NO2− reduction.
2. Experimental
Fig. 2 shows a scheme of our experimental setup. Since the previous iteration,23 it was modified to include CERS for H2, N2 and O2 detection with larger headspace and culture volumes to compensate for more frequent sampling. 250 mL of bacterial batch culture is contained in a round bottom flask with two side-arm ports and submerged in a 37 °C thermostated water bath. From the left side-arm, the bacterial suspension is circulated using a peristaltic pump (PP(l), 4.5 L h−1) for in situ OD600 (optical density at 600 nm in a 1 cm cuvette) and Raman spectroscopy measurements. From the central-neck, the headspace (1425 mL volume) is cycled by a second peristaltic pump (PP(g), 4.5 L h−1) for gas-phase FTIR and CERS analysis. The right side-arm has a rubber septum enabling sampling of the liquid culture for further analysis. The CERS cavity is equipped with a capacitance pressure gauge (PG), N2 inlet and vacuum line for purging O2 to give anaerobic growth conditions (1 atm N2) before starting experiments. | ||
| Fig. 2 Experimental setup for analysing the headspace by CERS and White cell FTIR spectroscopies and the liquid culture by Raman spectroscopy and in situ OD600 measurements. DM, dichroic mirror; LP, laser pointer; MO, microscope objective; PD, photodiode; PG, pressure gauge; PP(g), gas-phase peristaltic pump; PP(l), liquid-phase peristaltic pump; SM, supermirror; WC, White cell. | ||
Production of CO2, ethanol and N2O was quantified by gas-phase FTIR spectroscopy (Mattson Research Series, 0.4 cm−1 spectral resolution, MCT detector) with a home-built multiple-pass absorption White cell.23 The White cell pathlength was adjustable between 4–8 m, with 6 m used for this work. Spectra were recorded every 5 minutes. CO2 partial pressures were obtained by integrating the ν1 + 2ν2 + ν3 band (4920–5015 cm−1, ν0 = 4978 cm−1) of the Fermi triad and comparing with a reference spectrum from the PNNL database.27 N2O partial pressures were obtained by integrating the 2ν1 combination band from 2460–2580 cm−1 and comparing the integral with simulated spectra from HITRAN 2012.28 All four 14N/15N-isotopomers of N2O could be distinguished, which enabled the 15N-isotope labelling studies. A multiplier equivalent to ethanol partial pressure was obtained by a least-squares fit of 1 ppmv ethanol and water reference spectra in the 2800–3100 cm−1 region.23 Using Henry's law, all partial pressures could be converted into concentrations in solution. Using the ideal gas law, we estimated that 10% of the CO2 present in the sample was dissolved. Under our conditions, less than 1% of dissolved CO2 was expected to be converted to carbonic acid and carbonates. 7% of N2O and 99.7% of ethanol in the sample were also calculated to be dissolved.
The CERS setup has been described before with some modifications outlined below.15–17,20 A 40 mW 636 nm single-mode cw-diode laser (HL63133DG) is coupled via a short-pass filter, a Faraday isolator and a mode matching lens into a linear optical cavity composed of two highly reflective mirrors (Newport SuperMirrors, R > 99.99%). If the laser wavelength matches the cavity length, an optical resonance builds up optical power inside the cavity by up to 3 orders of magnitude, enhancing the Raman signals. After the cavity, a dichroic mirror separates leftover excitation light from Raman signals which are coupled into a round-to-linear glass fibre bundle (7 × ∅ 105 μm) and transferred to the monochromator (Andor Shamrock SR163, 1200 l mm−1 grating, DV420A-OE CCD). The 400–2500 cm−1 spectral range at 6 cm−1 resolution encompasses rotational S-branch lines of H2, the ν1/2ν2 Fermi resonance of CO2 and the vibrational fundamentals of O2 and N2. Part of the leftover excitation light is diverted back to the diode for optical feedback, locking the laser to the cavity. To normalize Raman signals, the N2 peak is used as an internal standard since N2 is not expected to change during bacterial activity. Raman intensities are converted to partial pressures using tabulated integrated peak areas.20 CO2 analysis by CERS was used to corroborate the FTIR analysis; however, CERS CO2 data was not displayed in this study due to FTIR CO2 detection being more sensitive. More details of the modified CERS setup are provided in the ESI.†
The bacterial suspension was circulated through a glass cuvette (1 cm path length) and the optical density OD600 was recorded in situ by measuring the scattering of red laser pointer light with a photodiode. The transmitted intensity was calibrated with start and end-point OD600 values externally measured using a UV-Vis spectrometer. The suspension was also circulated through a sealed borosilicate tube for recording liquid-phase Raman spectra using a home-built spectrometer.29,30 A 532.2 nm, 20 mW laser (Lasos, GL3dT) and monochromator (Shamrock SR-750-A, 1200 l mm−1 grating, DU420A-OE CCD) provided a spectral range from 830–1710 cm−1 at about 0.8 cm−1 resolution. Raman spectra were recorded every 5 minutes at 300 s integration time. No interfering fluorescence was noticeable in M9 minimal growth medium. The water bending vibration at 1630 cm−1 was used to normalise decreasing Raman intensities as the turbidity of the bacterial suspension increased.23 0.1 M reference spectra of individual glucose, KNO3, CH3CO2NH4, HCO2K, K2HPO4 and KH2PO4 solutions were recorded. As shown in Fig. 3, the 830–1200 cm−1 region contains characteristic Raman peaks for HPO42− (989 cm−1), H2PO4− (876 and 1076 cm−1), NO3− (1049 cm−1) and glucose (960–1180 cm−1).31 Using a least-squares fitting routine, Raman spectra of the bacterial suspension in this region were fitted to the reference spectra, as well as a linear baseline. The returned multipliers of the reference spectra were then converted into concentrations via calibration plots. Noise analysis of background sample measurements (pure water) provided noise equivalent (1σ) detection limits of 0.6 mM NO3− and 1.9 mM glucose at 300 s integration time. With additional averaging to an integration time of 0.5 h (as was done with all time-dependent data displayed in this study), the limits improve to 0.25 mM for nitrate and 0.8 mM for glucose. The concentrations of the phosphate anions were used to calculate the pH in situ using a modified Henderson–Hasselbalch equation.23,32 A least-squares fit determined acetate and formate concentrations in the 1310–1450 cm−1 region to the sum of acetate (1414 cm−1) and formate (1349 cm−1) models and a linear baseline, as shown in the ESI.† At 300 s integration time, the noise equivalent (1σ) detection limits of acetate and formate were 2.6 mM and 1.5 mM, respectively. These limits improve to 1.1 mM and 0.6 mM with additional averaging to 0.5 h integration time. Although NO2− has a peak at 1326 cm−1, the feature was too weak to be used in this study (1σ = 5.0 mM). Furthermore, NH3/NH4+ had no usable features within our spectral range.
 | ||
| Fig. 3 In black, an experimental Raman spectrum of M9 medium supplemented with 10 mM KNO3 and 30 mM glucose. In red, the sum of the fitted NO3−, glucose, HPO42− (47 mM) and H2PO4− (22 mM) models shown below the overlaid spectra. | ||
E. coli (strain K-12 MG1655) was transferred from glycerol stock (maintained at −80 °C) and streaked on LB-agar plates. Plates were left to grow overnight at 37 °C. Before a measurement, 50 mL of sterile LB medium was inoculated with a single colony and incubated anaerobically in a sealed 50 mL centrifuge tube for 16 h (37 °C, 200 rpm) to a typical OD600 of 1.2. From the starter culture, 20 mL was centrifuged, and the pellet resuspended into 20 mL of fresh M9 minimal medium. Our M9 medium formulation is given in the ESI;† but notably, it contains 30 mM glucose and 18 mM NH4Cl. The M9 medium was supplemented with 10 mM K15NO3 (10 mM, 98 atom % 15N, Sigma-Aldrich) and/or 5 mM KNO2 (either 14N or 15N). A further 230 mL of M9 medium was prepared in the round bottom flask with two side-arms. The flask was pre-warmed and maintained at 37 °C using a thermostated water bath under rapid stirring to enable efficient gas transfer. The 20 mL M9 medium containing E. coli was added to the 230 mL M9 medium in the flask, giving a typical starting OD600 of 0.1. The flask was then sealed and purged of O2 by alternating between evacuating the headspace and refilling with N2 at least five times. Experiments began once CERS measurements confirmed no O2 remained.
During experiments, 1 mL of the bacterial culture was sampled every 40 min and centrifuged. The supernatant was analysed using a colorimetric method to determine NO2− concentration based on the Griess test.33 Our M9 media began with a typical pH of 6.9 and ended between 5.0–5.5 due to organic acid excretion. With a pKa of 9.25, NH3 exists almost entirely as NH4+ at acidic pH. For 14N/15N-analysis of NH4+ samples, 2 mL 1 M NaOH was added to 0.6 mL of sample to release NH3. The gas was analysed by a second FTIR setup (Bruker Alpha FTIR, 0.8 cm−1 spectral resolution) with a home-built White cell (2.0 m pathlength). Spectra were recorded every 5 minutes with around 30 minutes needed before NH3 concentration peaked in the headspace. The basified solution was rapidly stirred and the 2 L headspace in the closed system was cycled between the sample flask and White cell using a peristaltic pump. The ν2 band is the strongest in the FTIR spectrum of NH3 and can be used for 14N/15N-analysis.34 At the end of bacterial activity, the suspension was centrifuged, washed and dried to record the dry biomass (typically around 200 mg when corrected for sampling). For comparison with the in situ spectroscopic pH measurements, the pH of start and end-point samples was recorded externally using a Mettler Toledo SevenMulti pH meter. See the ESI† for further experimental spectra and calibration plots for all aforementioned analytical techniques.
3. Results and discussion
3.1 FTIR spectroscopy of N2O and its 14N/15N-isotopomers
N2O has four 14N/15N-isotopomers, i.e., 14N2O, the structural isomers 14N15NO and 15N14NO, and 15N2O. N2O is amenable to 15N-isotope labelling studies due to the low natural abundance of the 15N-isotope (0.37%). In the 2000–3000 cm−1 spectral range, characteristic partially rotationally resolved bands of the N2O isotopomers are available for FTIR analysis. Apart from ca. 2250–2400 cm−1 which is saturated by CO2, this region is free from significant spectral interferences. The HITRAN molecular database contains line lists for the three most abundant 14N/15N-isotopomers, excluding 15N2O.28 A survey of HITRAN and our experimental spectra has shown that the following vibrational bands are available for quantitative analysis, including band position of 14N2O, integrated absorption cross-sections G and peak absorbances Apeak (defined as ln(I0/I)) under our experimental conditions for 1 μbar (1 ppmv) at 6 m path length: the ν3 fundamental near 2224 cm−1 with G = 5.55 × 10−17 cm and Apeak ≈ 0.023 for rotational lines in the P- and R-branches, the 2ν1 overtone near 2563 cm−1 with G = 1.33 × 10−18 cm and Apeak ≈ 6 × 10−4 for rotational features in the P- and R-branches, and the ν2 + ν3 combination near 2798 cm−1 with G = 9.0 × 10−20 cm and Apeak ≈ 2.6 × 10−4 of its Q-branch. Characteristic spectral shifts allow distinction of the isotopomers, while their G and Apeak values remain essentially the same. For accurate quantitative results, Apeak should not exceed unity. The dynamic range of the ν3 fundamental thus extends from trace levels up to ca. 45 μbar N2O, the 2ν1 overtone up to 1.7 mbar, and the ν2 + ν3 combination up to 3.8 mbar. This range can be extended by reducing the absorption pathlength of the White cell.Fig. 4 shows the ν3 fundamental with distinct P- and R-branch features, with 14N2O having its origin near 2224 cm−1. In a spectrum containing only 14N2O, a least-squares fit to the reference spectrum in the region denoted ‘D’ in Fig. 4 returns a multiplier which corresponds to N2O partial pressure. A simple integration over the ν3 band would not be suitable because part of the R-branch is buried in 13CO2 absorptions at higher wavenumbers. The region ‘D’ was selected because it has some of the strongest absorption features, it is very characteristic with partially resolved lines, and it is least affected by CO2. With this fitting routine, noise analysis of blank samples provides a noise equivalent detection limit of 60 nbar (60 ppbv at 1 bar total pressure) at 6 m pathlength and 128 accumulations which take 2 min to acquire. Detection limits can be improved by more averaging or increasing the path length. Note that this is sufficient to detect the 330 ppbv ambient levels of N2O for environmental analytical applications. The heavier isotopomers shift to lower wavenumbers, 2201 cm−1 for 15N14NO, 2178 cm−1 for 14N15NO, and 2155 cm−1 for 15N2O. Since the bands are overlapping, only a simultaneous fit to all four model spectra can yield individual isotopomer partial pressures. A fit in the entire 2100–2220 cm−1 region, however, has serious problems with cross-correlations. After a careful analysis, a simultaneous fit only including the regions ‘A’ to ‘D’ in Fig. 4 returned multipliers which are not noticeably affected by cross-correlations. Each region was chosen so that an individual isotopomer has a maximum weight with the other isotopomers having as little weight as possible. This procedure yields reliable isotopic partial pressures up to a dynamic range of about 45 μbar per isotopomer.
 | ||
| Fig. 4 ν 3 fundamental of N2O isotopomers with partially resolved rotational P- and R-branches. Absorbances scaled to correspond to 1 μbar (1 ppmv) at 6 m path length. (a) Experimental FTIR spectra of 15N2O (blue) and 14N2O (brown). (b) Isotopomers (structural isomers) 14N15NO (red) and 15N14NO (green) calculated from the HITRAN database. A to D denotes spectral ranges used in the fit. | ||
Fig. 5 shows the weaker absorption bands that are more suitable for N2O analysis above 45 μbar. In isotopically pure samples, the 2ν1 overtone near 2563 cm−1 can be integrated from 2505–2613 cm−1 to obtain 14N2O partial pressure after comparison with a reference spectrum (Fig. 5a). For 15N2O the shifted band near 2523 cm−1 can be integrated from 2460–2580 cm−1 (Fig. 5b). In samples with mixtures of isotopomers (Fig. 5c), the 2ν1 bands overlap and require a more sophisticated simultaneous fit similar to the one described above for the ν3 fundamental. Fortunately, this is not required as the ν2 + ν3 combination band (2798 cm−1 for 14N2O) has a sharp, characteristic Q-branch which remains well resolved and separated in isotopic mixtures. After comparison with reference spectra, simple integrations over the separate Q-branch peaks yield isotopic partial pressures in a mixture up to a dynamic range of about 3.8 mbar.
 | ||
| Fig. 5 Experimental FTIR spectra of N2O overtone and combination bands for (a) 1 mbar 14N2O, (b) 1 mbar 15N2O and (c) a mixture of 2.8 mbar 15N2O (49%), 1.1 mbar 14N15NO (20%), 1.2 mbar 15N14NO (22%) and 0.5 mbar 14N2O (9%). The isotopomer mixture was recorded at 30 h during the anaerobic respiration of E. coli supplemented with 10 mM 15NO3− and 5 mM 14N-nitrite (see section 3.4). | ||
3.2 Spectroscopic analysis of nitrate reduction by E. coli
Fig. 6 is a typical example of pH, OD600 and number of moles (n) of electron acceptors and other metabolites measured during the reduction of 10 mM 15NO3− by anaerobic E. coli. Concentrations (mM) in solution were converted to n (mmol) by multiplying by the culture volume (0.25 L), as were partial pressures using the ideal gas law (V = 1.425 × 10−3 m3, T = 310 K) and correcting for the dissolved percentage calculated via Henry's law. All biological experiments were repeated in triplicate, and all repeats showed essentially the same behaviour. The time-dependent data displayed in this study is for a single representative experiment selected from the repeats. Phase A (0–6.5 h) lasted until all NO3− was reduced to NO2−. Phase B (6.5–10 h) lasted until all NO2− was reduced to NH4+ and N2O. Phase C (>10 h) had no electron acceptors remaining so the bacteria utilised fermentative pathways solely. The 15N-label transferred to 15NH4+ and 15N2O with no trace of other N2O isotopomers formed. This was consistent with other studies that found the N-atoms in N2O both originate from NO3−/NO2− and not other sources such as N2 or NH4+.19,22,35 The externally measured start and end-point pH measurements showed good agreement with the time-dependent spectroscopically determined pH. | ||
| Fig. 6 Anaerobic E. coli growth in M9 medium supplemented with 10 mM 15NO3−. A to C denotes three distinct phases: NO3− reduction (A), NO2− reduction (B) and NO2− depletion (C). (a) Time-dependent number of moles (n) of 15NO3−, 15NO2−, 15NH4+ and 15N2O (×10). (b) n of glucose, acetate and formate. (c) n of CO2, ethanol (×5) and 14NH4+. (d) Spectroscopically determined pH (open circles), externally measured pH (solid squares) and OD600. | ||
After a brief lag phase, exponential growth began at 3 h with a rapid increase in the OD600. NO3− reduction to NO2− mirrored the growth curve with most of the NO2− produced excreted to prevent cytoplasmic toxification.36E. coli expresses three NO3− reductases: the respiratory NO3− reductases A and Z (NRA and NRZ) and the periplasmic NO3− reductase (Nap).37–39 NRA is the most active reductase at high NO3− levels (>2 mM).40 Nap is induced by low NO3− levels, while NRZ is expressed at low levels constitutively and may function under stress-associated conditions.40–42 NO2− peaked at 2.2 mmol, less than the initial 2.5 mmol NO3−, as some NO2− was reduced alongside NO3− during A. 0.3 mmol 15NH4+ and 1.6 μmol 15N2O was produced, accounting for the total N-balance. Only 1% of the 0.3 mmol NO2− reduced in A was converted to N2O instead of NH4+. E. coli expresses two NO2− reductases: the NADH-dependent cytoplasmic NO2− reductase (NirB) and the membrane-bound periplasmic NO2− reductase (NrfA). NirB likely produced NH4+ during A as it is active when NO3− is readily available, unlike NrfA.43 Evidence also suggests NirB can generate NO.4 Anaerobically, NO is detoxified by reduction to N2O, which is comparatively non-toxic and rapidly diffuses out of the cell. Flavorubredoxin (NorV),9 hybrid cluster protein (Hcp),10 NirB44 and NrfA45 have all been proposed to have NO detoxifying activity. Flavohemoglobin (Hmp) is primarily an NO oxidase but also acts as an NO reductase anaerobically.8
As E. coli does not possess any known N2O reductases, further reduction to N2 was not expected. However, there is some evidence that N2 can be produced from high amounts of N2O by a yet unknown mechanism.46 To investigate whether under our conditions N2 was produced, we repeated the experiment, but under an argon atmosphere instead of N2. No trace of N2 production was observed in the CERS spectra within our detection limit of ca. 0.2 mbar or 12 μmol N2.
Formate oxidation to CO2 by the NO3−-inducible formate dehydrogenase (FdhN) is a physiological source of electrons for NO3− reduction.38 Other sources include NADH, lactate and glycerol.37 1.7 of the 2.5 mmol NO3− reduced was coupled to FdhN activity as CO2 increased by such in A. The remaining 0.8 mmol NO3− was likely coupled to NADH oxidation.47 As no formate was excreted in A, all formate produced by pyruvate formate lyase (PFL) must have been oxidised to CO2. For each formate produced by PFL, one acetyl-CoA is formed which can be either directed into the anaerobic TCA cycle or converted to acetate (to produce ATP) or ethanol (to remove reducing equivalents). 1.7 mmol acetate and 0.05 mmol ethanol were excreted during A corresponding to 1.75 mmol formate, in good agreement with the 1.7 mmol CO2 produced. Acetate must be excreted to prevent cytoplasmic acidification and caused the extracellular pH to decrease from 7.1 to 6.7. The minor amount of ethanol produced was due to reducing equivalents being coupled directly into reduction of NO3−. Previous studies have found a similar repression of substrate-level NADH consuming pathways when electron acceptors are available.48 Glucose decreased by 1.1 mmol owing to the production of CO2, acetate, ethanol and biomass synthesis.
During phase B, NO2− was reimported into E. coli and reduced. From 6.5 to 10 h, 2.2 mmol 15NO2− was reduced almost linearly to 2.0 mmol 15NH4+ and 0.1 mmol 15N2O. 91% NO2− was reduced to NH4+ and 9% to N2O, a higher partitioning to N2O than observed in A (1%). A higher partitioning to N2O after NO3− was depleted is consistent with several studies of E. coli and Salmonella enterica that have implicated NRA as the enzyme that produces the majority of NO when NO2− is abundant and NO3− absent.6,7,49,50 NrfA, which is induced by NO2− but repressed by NO3−, may have also contributed towards the higher partitioning to N2O in B as it has been proposed as a source of NO.5,51 The radical NO has a distinct line-resolved absorption band centred at 1904 cm−1 (for 14NO) and a favourable partitioning into the headspace.52 However, no intermediate 15NO gas was observed to accumulate, owing to its rapid detoxification to 15N2O by E. coli. During B, a further 1.9 mmol CO2 was produced and the pH dropped from 6.7 to 5.7 due to the excretion of 5.7 mmol formate and a further 7.5 mmol acetate. Due to the 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of formate oxidation to CO2:NO2− reduction to NH4+, 0.6 mmol NO2− was coupled to formate by NrfA.53 The 5.7 mmol formate excreted during B would be plentiful to couple to the remaining 1.4 mmol NO2−. However, NrfA is most active at low NO2− levels while NirB is most active at high NO2− levels for detoxification of excess NO2−.36,43 Thus, 1.4 mmol NO2− was likely reduced by NirB.
:1 stoichiometry of formate oxidation to CO2:NO2− reduction to NH4+, 0.6 mmol NO2− was coupled to formate by NrfA.53 The 5.7 mmol formate excreted during B would be plentiful to couple to the remaining 1.4 mmol NO2−. However, NrfA is most active at low NO2− levels while NirB is most active at high NO2− levels for detoxification of excess NO2−.36,43 Thus, 1.4 mmol NO2− was likely reduced by NirB.
Phase C started with exponential growth ending as the OD600 peaked at 1.7, due to the depletion of glucose and NO2−. With no electron acceptors available, the bacteria funnelled reducing equivalents into ethanol as a further 0.7 mmol was made over the next 5 h. The remaining 5.7 mmol formate was slowly oxidised to CO2 at a rate of 0.03 mmol h−1. Under anaerobic conditions, the presence of formate induces formate hydrogenlyase (FHL) activity that disproportionates formate to CO2 and H2.54 O2 and NO3− repress FHL expression and instead induce the aerobic and the formate-NO3− respiratory chains. High formate concentrations can partially reverse the repression by NO3−, but not by O2.55,56 However, CERS measurements detected no H2 production during our 10 mM 15NO3− reduction experiments. During C, there was a slight decline in N2O observed due to the gas adsorbing to tubing and glass surfaces.
Experiments were terminated after 2 days with 5 mmol formate still remaining. The dry biomass was typically around 200 mg. As E. coli can be approximated to be 48% carbon and 14% nitrogen by mass,57ca. 8 mmol C and 2 mmol N in the biomass originated from the 7.5 mmol glucose (45 mmol C) and NH4+, respectively. 44 out of the 45 mmol C from glucose can be accounted for in the biomass, 5 mmol CO2, 5 mmol formate, 12 mmol acetate (24 mmol C) and 1 mmol ethanol (2 mmol C). During exponential growth, 14NH4+ decreased from 4.5 to 3.0 mmol accounting for 1.5 out of the 2 mmol N in the biomass. The remaining 0.5 mmol N likely was taken from the excreted 15NH4+. The 2.5 mmol 15N-label can be accounted for in the 2.0 mmol 15NH4+, 0.1 mmol 15N2O (0.2 mmol 15N) and ∼0.5 mmol 15NH4+ used for biosynthesis.
3.3 Spectroscopic analysis of nitrite reduction by E. coli
To study the response to NO2− alone, anaerobic E. coli was supplemented with 5 mM 15NO2−, as shown in Fig. 7. Phase A′ (0–9 h) corresponded to the reduction of NO2− to NH4+ with concurrent N2O production via NO. Phase B′ (9–15 h) was when the bacteria utilised fermentative pathways only, due to NO2− depletion. During the first 9 h of phase A′, 1.25 mmol 15NO2− was reduced almost exponentially to 1.15 mmol 15NH4+ (90%) and 0.06 mmol 15N2O (10%), mirroring the bacterial growth curve which increased to an OD600 of 0.7. The 10% partitioning to N2O here was consistent with the 9% observed during the NO2− reduction phase B in section 3.2. During A′, 1.9 mmol CO2, 3.7 mmol acetate, 2.0 mmol formate and 0.3 mmol ethanol were produced as glucose decreased from 7.5 to 5.2 mmol. Excretion of acetate and formate caused the pH to decrease from 6.9 to 6.3. The sum of acetate and ethanol (4.0 mmol) showed good agreement with the sum of CO2 and formate (3.9 mmol). Due to the 3:1 stoichiometry of formate oxidation:NO2− reduction and 1.9 mmol CO2 being produced, 0.63 out of the initial 1.15 mmol NO2− reduced to NH4+ was coupled to formate oxidation to CO2. The remaining 0.52 mmol NO2− was likely reduced via coupling to NADH oxidation by NirB.
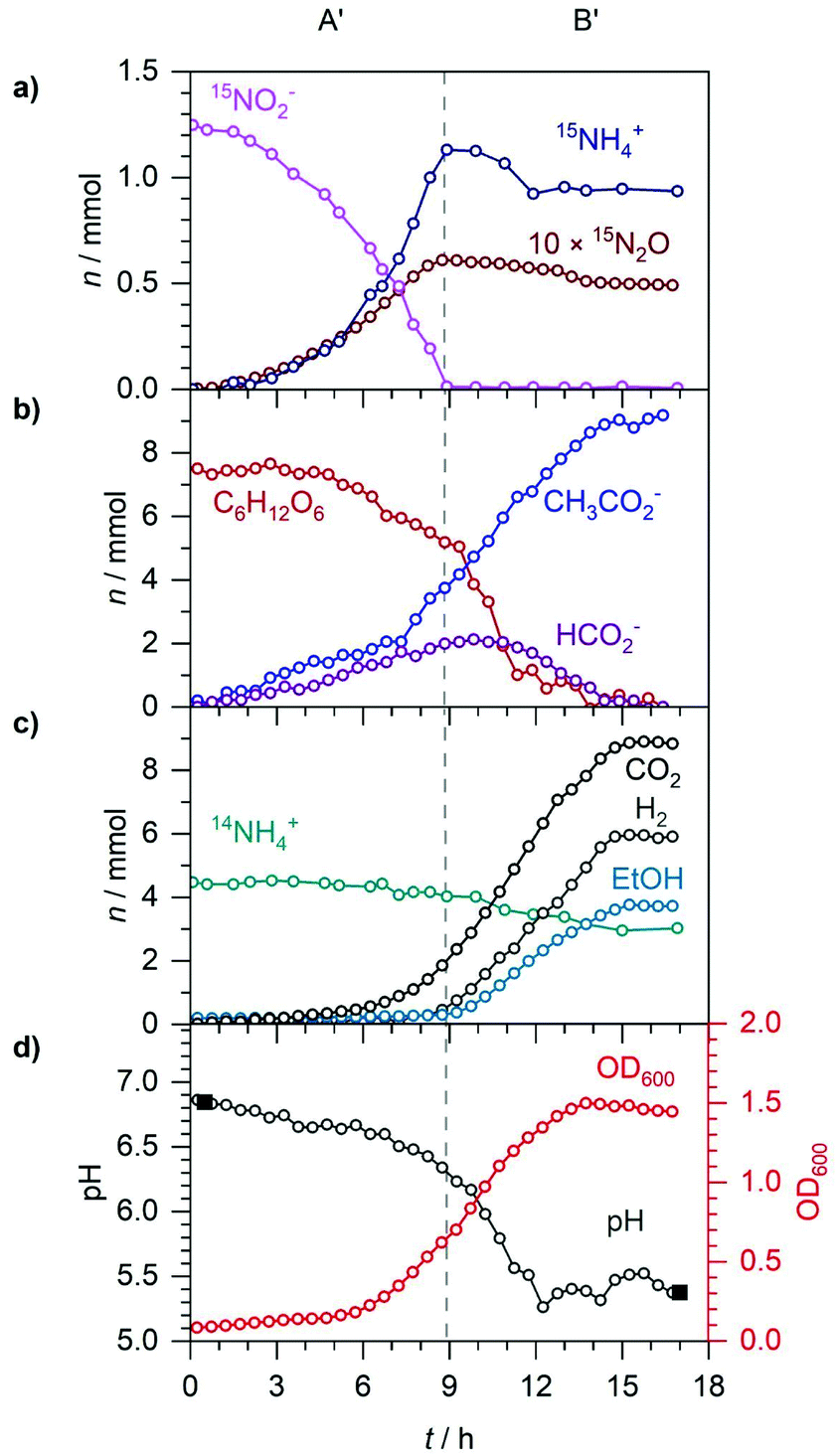 | ||
| Fig. 7 Anaerobic E. coli growth in M9 medium supplemented with 5 mM 15NO2−. A′ and B′ denote two distinct phases: NO2− reduction (A′) and NO2− depletion (B′). (a) Time-dependent number of moles (n) of 15NO2−, 15NH4+ and 15N2O (×10). (b) n of glucose, acetate and formate. (c) n of CO2, H2, ethanol and 14NH4+. (d) Spectroscopically determined pH (open circles), externally measured pH (solid squares) and OD600. | ||
Phase B′ began at 9 h when 15NO2− was depleted. E. coli could only utilise fermentative pathways in the absence of NO2−. The most notable difference between 10 mM 15NO3− reduction (discussed in section 3.2), and 5 mM 15NO3− reduction was H2 production that occurred after NO2− depletion in Fig. 7. No H2 production was observed during A′ as formate-dependent NO2− reduction likely made the intracellular formate unavailable for FHL induction. The presence of formate is required for FHL expression but it can be made unavailable by coupling to the reduction of electron acceptors. This inhibiting effect has been observed for NO3− and trimethylamine N-oxide respiring E. coli cultures and in both cases the effect could be partially relieved by adding exogenous formate.56,58 When NO2− was depleted, 5.2 mmol glucose remained meaning further formate could be produced during B′ which may have triggered the induction of FHL. From 9–15 h, 6.0 mmol H2 and a further 6.0 mmol CO2 were produced from the disproportionation of formate. At 10 h, there was a peak of 2.1 mmol formate excreted. During B′, a further 5.3 mmol acetate and 3.5 mmol ethanol were produced. By 12 h, the pH dropped to 5.4 and then remained stable as 1.6 mmol acetate was produced and balanced by the reimport and disproportionation of 1.5 mmol formate. By 14 h, the OD600 peaked at 1.5, just before the end of bacterial activity at 15 h due to the depletion of glucose and formate. 42.8 out of the 45 mmol C from glucose can be accounted for in the biomass (∼8 mmol C), 8.9 mmol CO2, 9 mmol acetate (18 mmol C) and 3.8 mmol ethanol (7.6 mmol C). During exponential growth, 14NH4+ decreased from 4.5 to 2.9 mmol as did 15NH4+ from a peak value of 1.15 to 0.9 mmol accounting for 1.85 mmol out of the ∼2 mmol N in the biomass.
3.4 Simultaneous nitrate and nitrite reduction
In section 3.2, when E. coli was supplemented with NO3−, there was a distinct hierarchy of metabolic pathways between phases A, B and C. NO3− reduction dominated in A, followed by NO2− reimport and reduction in B and finally fermentation in C. However, in A it was observed that some NO2− was simultaneously reduced alongside NO3− to NH4+ and N2O. To further investigate the overlap between the reductions of NO3− and NO2− in A, anaerobic E. coli was supplemented with 10 mM 15NO3− and 5 mM 14NO2− as shown in Fig. 8 and 9. Phase A (0–9 h) lasted until all 15NO3− was reduced to 15NO2−. Phase B (9–30 h) corresponded to the reduction of NO2− to NH4+ with concurrent N2O production via NO. At 15.5 h, the OD600 peaked and exponential growth of E. coli ended; thus, phase B1 (9–15.5 h) was NO2− reduction with glucose still present and phase B2 (15.5–30 h) was NO2− reduction during glucose depletion. Fig. 8 displays n of 15NO3−, NO2−, 14NH4+, 15NH4+ (×10) and N2O isotopomers in A. The complete characterization of bacterial growth is given in Fig. 9. | ||
| Fig. 8 N2O isotopomers produced in the NO3− reduction phase (A) of anaerobic E. coli growth in M9 medium supplemented with 10 mM 15NO3− and 5 mM 14NO2−. (a) Time-dependent number of moles (n) of 15NO3−, NO2−, 14NH4+ and 15NH4+ (×10). (b) n of N2O isotopomers produced. 14N15NO is omitted due to essentially having the same behaviour as 15N14NO. | ||
 | ||
| Fig. 9 Anaerobic E. coli growth in M9 medium supplemented with 10 mM 15NO3− and 5 mM 14NO2−. A to C denotes three distinct phases: NO3− reduction (A), NO2− reduction with glucose present (B1) and NO2− reduction with glucose depleted (B2). (a) Time-dependent number of moles (n) of 15NO3−, NO2− (both 14N and 15N), 15NH4+ and sum of all 14N/15N-isotopomers of N2O (×10). (b) n of glucose, acetate and formate. (c) n of CO2 and 14NH4+. (d) Spectroscopically determined pH (open circles), externally measured pH (solid squares) and OD600. | ||
In phase A, 2.5 mmol 15NO3− was reduced and ca. 2.25 mmol 15NO2− was excreted. NO2− colorimetry cannot distinguish between 14NO2− and 15NO2−, so NO2− was observed to increase from 1.25 to 3.5 mmol. During A, as in section 3.2, some NO2− was reduced alongside 15NO3− to 0.2 mmol 15NH4+ and N2O isotopomers. 14NO2− reduction to 14N2O occurred immediately, with 2.2 μmol 14N2O produced almost linearly by 9 h. This indicated that even before NO3− reduction began, some unknown enzymatic activity to reduce small quantities of NO2− to N2O was immediately active. For the first 3 h, 15NO3− and NO2− measurements were virtually constant suggesting a lag in the expression of NRA. This lag was best indicated by the highly sensitive positional isomers 14N15NO and 15N14NO which were not detected until 15NO2− was made available by 15NO3− reduction starting from 3 h. 15N2O production also began at 3 h, but much slower than the production of 14N2O and the positional isomers, due to 14NO2− initially being more readily available than 15NO2−. By the end of A, 1.5 μmol each of 14N15NO, 15N14NO and 15N2O were produced alongside the 2.2 μmol 14N2O, totalling 6.7 μmol. It is unknown if 14NO2− was also immediately reduced to 14NH4+, due to the large background of 4.5 mmol 14NH4+ in the growth medium. It can be assumed ca. 0.25 mmol NO2− was reduced during A based on the NO2− colorimetry measurements giving a partitioning of 5% NO2− reduced to N2O, instead of NH4+. This was a higher value than the 1% observed during A in section 3.2, indicating that the added 14NO2− led to more NO generation and detoxification to N2O. During A, glucose decreased from 7.5 to 5.6 mmol due to the production of 2.1 mmol CO2, 2.0 mmol acetate and biomass synthesis. The OD600 began increasing indicating exponential bacterial growth while acetate excretion caused the pH to decrease from 7.0 to 6.5. No formate was excreted during A, as the n of CO2 and acetate suggested all formate formed was oxidised to CO2. No ethanol was detected during the entire 30 h experiment, likely due to the abundance of electron acceptors to couple reducing equivalents to.
Phase B1 began with 15NO3− depletion and ended at 15.5 h when glucose was depleted, coinciding with the OD600 peaking at 1.2. The pH dropped further to 5.6 due to the excretion of 5.0 mmol formate and a further 6.0 mmol acetate. The sum of formate excreted and the further 1.3 mmol CO2 produced was in good agreement with the amount of acetate excreted.
Phase B2 lasted until NO2− depletion at 30 h. From NO2− reduction, 1.6 mmol 15NH4+ and 0.35 mmol N2O were produced overall. The final composition of N2O isotopomers was previously introduced in Fig. 5c. As the majority of N2O production occurred in B when the NO2− composition was ca. 66% 15NO2− and 33% 14NO2−, a near statistical mixture of N2O isotopomers was formed of 0.17 mmol 15N2O (49%), 0.08 mmol 15N14NO (22%), 0.07 mmol 14N15NO (20%) and 0.03 mmol 14N2O (9%). For comparison, a perfect statistical mixture would have produced 44.4% 15N2O, 22.2% 15N14NO, 22.2% 14N15NO and 11.1% 14N2O. It is unknown whether the slight preference for 15N14NO over 14N15NO is significant or due to experimental uncertainty. The partitioning of the 3.5 mmol NO2− to N2O in B was 20%, a much higher value than the 10% observed during A in section 3.2. This is consistent with previous studies that found that between 5–36% of NO3− is converted to N2O by E. coli, depending on growth conditions.35 During B2, CO2 increased by a further 0.5 mmol while the pH remained constant at 5.6 due to no significant change in acetate and formate. 33 out of the 45 mmol C from glucose can be accounted for in the biomass (∼8 mmol C), 4 mmol CO2, 8 mmol acetate (16 mmol C) and 5 mmol formate. The higher NO2− content may have had cytotoxic effects in E. coli resulting in other products that have not been accounted for in the C balance. During B2, the OD600 dropped from 1.2 to 0.8 suggesting cell death or changes in cellular size and morphology, possibly due to the cytotoxicity of NO2− and NO. The 2.5 mmol 15N label was accounted for in the 1.6 mmol 15NH4+, 0.17 mmol 15N2O (0.34 mmol 15N), 0.08 mmol 15N14NO, 0.07 mmol 14N15NO and ca. 0.5 mmol 15NH4+ assumed to have been used for biosynthesis. As ca. 2.0 mmol NH4+ was needed for biosynthesis, it was assumed ca. 1.5 mmol was taken from 14NH4+, which decreased overall from 4.5 to 4.0 mmol suggesting ca. 1.0 mmol 14NH4+ produced from the reduction of the 1.25 mmol 14NO2−. This was in good agreement with the 0.26 mmol 14NO2− reduced to N2O isotopomers with 0.03 mmol 14N2O (0.06 mmol 15N), 0.08 mmol 15N14NO and 0.07 mmol 14N15NO.
4. Conclusions
We have studied NO3− and NO2− reduction during DNRA by anaerobic E. coli batch cultures by a combination of advanced spectroscopic analytical techniques in conjunction with 15N-isotopic labelling. The online spectroscopic techniques described here are non-invasive, avoiding any contact with the bacterial suspension, and provide concentrations in real-time. We discussed in detail the spectroscopy, which spectral features are most useful for analysis, and data analysis and fitting routines for quantitative analysis. In situ analysis of the headspace is achieved using cavity-enhanced Raman (CERS) and long-path White cell FTIR spectroscopies alongside liquid-phase Raman spectroscopy. Gas phase CERS allows CO2, H2, N2 and O2 monitoring while White cell FTIR measures CO2, ethanol and N2O. The 6 m pathlength White cell affords trace gas detection of N2O with a noise equivalent detection limit of 60 nbar or 60 ppbv in 1 atm (1σ noise equivalent, 128 scans corresponding to 120 s acquisition). This extremely high sensitivity could be utilised in situations where N2O cannot be allowed to build up, e.g. in continuous culture studies. Quantitative analysis is discussed for all four 14N/15N-isotopomers, including the positional isomers 14N15NO and 15N14NO, a unique capability not available to other analytical techniques.15N-isotopic labelling of NO3− identifies the sources of N-atoms in products of E. coli metabolism, in particular, it provides insight into the mechanism of N2O production during mixed NO3− and NO2− reduction. This study is one of very few reporting quantitative analysis of N2O production by E. coli under various conditions. The reductions of 15NO3−, 15NO2−, and mixed 15NO3− and 14NO2− to NH4+ and N2O have been discussed. In a major pathway, NO3− is reduced to NH4+via NO2−, with the bulk of NO2− reduction occurring after NO3− depletion. By isotopically labelling 15NO3−, 15NH4+ production is distinguished from background 14NH4+ in the growth medium. In a minor pathway, NO2− is reduced to N2O via the toxic radical NO. With excellent detection sensitivities, N2O monitors trace NO2− reduction even when cells are predominantly reducing NO3−; the analysis of N2O isotopomers reveals that some enzymatic NO2− reduction activity occurs immediately for cultures supplemented with mixed 15NO3− and 14NO2−. Optical density and pH measurements are discussed in context of acetate, formate and CO2 production. H2 production is repressed by NO3−, but with NO2− only, CERS detects H2 produced by formate hydrogenlyase after NO2− depletion.
In future work, we want to extend our spectroscopic approach to monitor different bacterial pathways, in particular, the relationship between fermentative and other respiratory pathways and to study nitrifying and denitrifying bacteria. These spectroscopic techniques are capable of detecting key species in the nitrogen cycle and with the ability to sensitively distinguish N2O isotopomers they may be of great interest for helping better understand global N2O budgets. Spectroscopic monitoring of bioprocesses has excellent potential to supplement or replace traditional techniques in analytical chemistry.
Author contributions
All authors have contributed equally to the conception of the project, the experimental work, the analysis and to the writing of the manuscript.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
We are grateful to Dr Jim Reid and Profs Jeff Green and Robert Poole for help and discussions. We acknowledge the University of Sheffield and the EPSRC research council (DTP scholarship to GDM) for financial support of our research.References
- M. Tiso and A. N. Schechter, Nitrate reduction to nitrite, nitric oxide and ammonia by gut bacteria under physiological conditions, PLoS One, 2015, 10, e0127490, DOI:10.1371/journal.pone.0119712.
- V. Stewart, Nitrate- and nitrite-responsive sensors NarX and NarQ of proteobacteria, Biochem. Soc. Trans., 2003, 31, 1–10, DOI:10.1042/bst0310001.
- J. Green, M. D. Rolfe and L. J. Smith, Transcriptional regulation of bacterial virulence gene expression by molecular oxygen and nitric oxide, Virulence, 2014, 5, 794–809, DOI:10.4161/viru.27794.
- C. E. Vine and J. A. Cole, Nitrosative stress in Escherichia coli: reduction of nitric oxide, Biochem. Soc. Trans., 2011, 39, 213–215, DOI:10.1042/BST0390213.
- H. Corker and R. K. Poole, Nitric Oxide Formation by Escherichia coli: Dependence On Nitrite Reductase, The NO-Sensing Regulator Fnr, and Flavohemoglobin Hmp, J. Biol. Chem., 2003, 278, 31584–31592, DOI:10.1074/jbc.M303282200.
- M. S. Smith, Nitrous oxide production by Escherichia coli is correlated with nitrate reductase activity, Appl. Environ. Microbiol., 1983, 45, 1545–1547, DOI:10.1128/AEM.45.5.1545-1547.1983.
- R. Metheringham and J. A. Cole, A reassessment of the genetic determinants, the effect of growth conditions and the availability of an electron donor on the nitrosating activity of Escherichia coli K-12, Microbiol., 1997, 143, 2647–2656, DOI:10.1099/00221287-143-8-2647.
- S. O. Kim, Y. Orii, D. Lloyd, M. N. Hughes and R. K. Poole, Anoxic function for the Escherichia coli flavohaemoglobin (Hmp): reversible binding of nitric oxide and reduction to nitrous oxide, FEBS Lett., 1999, 445, 389–394, DOI:10.1016/S0014-5793(99)00157-X.
- A. M. Gardner, R. A. Helmick and P. R. Gardner, Flavorubredoxin, an Inducible Catalyst for Nitric Oxide Reduction and Detoxification in Escherichia coli, J. Biol. Chem., 2002, 277, 8172–8177, DOI:10.1074/jbc.M110471200.
- J. Wang, C. E. Vine, B. K. Balasiny, J. Rizk, C. L. Bradley, M. Tinajero-Trejo, R. K. Poole, L. L. Bergaust, L. R. Bakken and J. A. Coole, The role of the hybrid cluster protein, Hcp and its reductase, Hcr, in high affinity nitric oxide reduction that protects anaerobic cultures of Escherichia coli against nitrosative stress, Mol. Microbiol., 2016, 100, 877–892, DOI:10.1111/mmi.13356.
- N. M. Dixon and D. B. Kell, The control and measurement of ‘CO2’ during fermentations, J. Microbiol. Methods, 1989, 10, 155–179, DOI:10.1016/0167-7012(89)90048-1.
- X. Chen, R. Hu, L. Hu, Y. Huang, W. Shi, Q. Wei and Z. Li, Portable Analytical Techniques for Monitoring Volatile Organic Chemicals in Biomanufacturing Processes: Recent Advances and Limitations, Front. Chem., 2020, 8, 837, DOI:10.3389/fchem.2020.00837.
- S. Sachse, A. Bockisch, U. Enseleit, F. Gerlach, K. Ahlborn, T. Kuhnke, U. Rother, E. Kielhorn, P. Neubauer, S. Junne and W. Vonau, On the use of electrochemical multi-sensors in biologically charged media, J. Sens. Sens. Syst., 2015, 4, 295–303, DOI:10.5194/jsss-4-295-2015.
- M. D. Brown and M. H. Schoenfisch, Electrochemical Nitric Oxide Sensors: Principles of Design and Characterization, Chem. Rev., 2019, 119, 11551–11575, DOI:10.1021/acs.chemrev.8b00797.
- T. W. Smith and M. Hippler, Cavity-Enhanced Raman Spectroscopy in the Biosciences: In situ, Multicomponent, and Isotope Selective Gas Measurements To Study Hydrogen Production and Consumption by Escherichia coli, Anal. Chem., 2017, 89, 2147–2154, DOI:10.1021/acs.analchem.6b04924.
- G. D. Metcalfe, S. Alahmari, T. W. Smith and M. Hippler, Cavity-Enhanced Raman and Helmholtz Resonator Photoacoustic Spectroscopy to Monitor the Mixed Sugar Metabolism of E. coli, Anal. Chem., 2019, 91, 13096–13104, DOI:10.1021/acs.analchem.9b03284.
- R. Salter, J. Chu and M. Hippler, Cavity-enhanced Raman spectroscopy with optical feedback cw diode lasers for gas phase analysis and spectroscopy, Analyst, 2012, 137, 4669–4676, 10.1039/c2an35722d.
- R. Keiner, T. Frosch, T. Massad, S. Trumbore and J. Popp, Enhanced Raman multigas sensing - a novel tool for control and analysis of 13CO2 labeling experiments in environmental research, Analyst, 2014, 139, 3879–3884, 10.1039/C3AN01971C.
- R. Keiner, M. Hermann, K. Küsel, J. Popp and T. Frosch, Rapid monitoring of intermediate states and mass balance of nitrogen during denitrification by means of cavity enhanced Raman multi-gas sensing, Anal. Chim. Acta, 2015, 864, 39–47, DOI:10.1016/j.aca.2015.02.007.
- M. Hippler, Cavity-Enhanced Raman Spectroscopy of Natural Gas with Optical Feedback cw-Diode Lasers, Anal. Chem., 2015, 87, 7803–7809, DOI:10.1021/acs.analchem.5b01462.
- A. Sieburg, T. Jochum, S. Trumbore, J. Popp and T. Frosch, Onsite cavity enhanced Raman spectrometry for the investigation of gas exchange processes in the Earth's critical zone, Analyst, 2017, 142, 3360–3369, 10.1039/C7AN01149K.
- A. Blohm, S. Kumar, A. Knebl, M. Herrmann, K. Küsel, J. Popp and T. Frosch, Activity and electron donor preference of two denitrifying bacterial strains identified by Raman gas spectroscopy, Anal. Bioanal. Chem., 2021 DOI:10.1007/s00216-021-03541-y.
- G. D. Metcalfe, T. W. Smith and M. Hippler, On-line analysis and in situ pH monitoring of mixed acid fermentation by Escherichia coli using combined FTIR and Raman techniques, Anal. Bioanal. Chem., 2020, 412, 7303–7319, DOI:10.1007/s00216-020-02865-5.
- J. Mohn, B. Tuzson, A. Manninen, N. Yoshida, S. Toyoda, W. A. Brand and L. Emmenegger, Site selective real-time measurements of atmospheric N2O isotopomers by laser spectroscopy, Atmos. Meas. Tech., 2012, 5, 1601–1609, DOI:10.5194/amt-5-1601-2012.
- P. Wunderlin, M. F. Lehmann, H. Siegrist, B. Tuzson, A. Joss, L. Emmenegger and J. Mohn, Isotope Signatures of N2O in a Mixed Microbial Population System: Constraints of N2O Producing Pathways in Wastewater Treatment, Environ. Sci. Technol., 2013, 47, 1339–1348, DOI:10.1021/es303174x.
- H. Moser, W. Pölz, J. P. Waclawek, J. Ofner and B. Lendl, Implementation of a quantum cascade laser-based gas sensor prototype for sub-ppmv H2S measurements in a petrochemical process gas stream, Anal. Bioanal. Chem., 2017, 409, 729–739, DOI:10.1007/s00216-016-9923-z.
- S. W. Sharpe, T. J. Johnson, R. L. Sams, P. M. Chu, G. C. Rhoderick and P. A. Johnson, Gas-Phase Databases for Quantitative Infrared Spectroscopy, Appl. Spectrosc., 2004, 58, 1452–1461, DOI:10.1366/0003702042641281.
- L. S. Rothman, I. E. Gordon, Y. Babikov, A. Barbe, D. Chris Benner, P. F. Bernath, M. Birk, L. Bizzocchi, V. Boudon, L. R. Brown, A. Campargue, K. Chance, E. A. Cohen, L. H. Coudert, V. M. Devi, D. B. Drouin, A. Fayt, J.-M. Flaud, R. R. Gamache, J. J. Harrison, J.-M. Hartmann, C. Hill, J. T. Hodges, D. Jacquemart, A. Jolly, J. Lamouroux, R. J. Le Roy, G. Li, D. A. Long, O. M. Lyulin, C. J. Mackie, S. T. Massie, S. Mikhailenko, H. S. P. Müller, O. V. Naumenko, A. V. Nikitin, J. Orphal, V. Perevalov, A. Perrin, E. R. Polovtseva, C. Richard, M. A. H. Smith, E. Starikova, K. Sung, S. Tashkun, J. Tennyson, G. C. Toon, V. G. Tyuterev and G. Wagner, The HITRAN2012 molecular spectroscopic database, J. Quant. Spectrosc. Radiat. Transfer, 2013, 130, 4–50, DOI:10.1016/j.jqsrt.2013.07.002.
- C. Mohr, C. L. Spencer and M. Hippler, Inexpensive Raman Spectrometer for Undergraduate and Graduate Experiments and Research, J. Chem. Educ., 2010, 87, 326–330, DOI:10.1021/ed800081t.
- Y. Ryabenkova, N. Jadav, M. Conte, M. Hippler, N. Reeves-McLaren, P. D. Coates, P. Twigg and A. Paradkar, Mechanisms of Hydrogen-Bonded Complex Formation between Ibuprofen and Nanocrystalline Hydroxyapatite, Langmuir, 2017, 33, 2965–2976, DOI:10.1021/acs.langmuir.6b04510.
- M. D. Fontana, K. B. Mabrouk and T. H. Kauffmann, Raman spectroscopic sensors for inorganic salts, in Spectroscopic Properties of Inorganic and Organometallic Compounds: Techniques, Materials and Applications, Royal Society of Chemistry, Cambridge, U.K., 2013, vol. 44, pp. 40–67, 10.1039/9781849737791-00040.
- M. Hippler and G. D. Metcalfe, Using activities to correct the Henderson-Hasselbalch equation, Bunsenmagazin, 2020, 22, 102–105, DOI:10.26125/y7p7-an56.
- M. B. Shinn, Colorimetric Method for Determination of Nitrate, Ind. Eng. Chem., Anal. Ed., 1941, 13, 33–35, DOI:10.1021/i560089a010.
- I. L. Marr, A. Kindness and M. S. Cresser, Measurement of 14N: 15N ratios by Fourier transform infrared spectrometry, Analyst, 1987, 112, 1491–1494, 10.1039/AN9871201491.
- B. H. Bleakley and J. M. Tiedje, Nitrous Oxide Production by Organisms Other than Nitrifiers or Denitrifiers, Appl. Environ. Microbiol., 1982, 44, 1342–1348, DOI:10.1128/AEM.44.6.1342-1348.1982.
- T. M. Khlebodarova, N. A. Ree and V. A. Likhoshvai, On the control mechanisms of the nitrite level in Escherichia coli cells: the mathematical model, BMC Microbiol., 2016, 16, S7, DOI:10.1186/s12866-015-0619-x.
- V. Stewart, Nitrate respiration in relation to facultative metabolism in enterobacteria, Microbiol. Rev., 1988, 52, 190–232, DOI:10.1128/mr.52.2.190-232.1988.
- V. Bonnefoy and J. A. Demoss, Nitrate reductases in Escherichia coli, Antonie van Leeuwenhoek, 1994, 66, 47–56, DOI:10.1007/BF00871632.
- V. Stewart, Y. Lu and A. J. Darwin, Periplasmic nitrate reductase (NapABC enzyme) supports anaerobic respiration by Escherichia coli K-12, J. Bacteriol., 2002, 184, 1314–1323, DOI:10.1128/JB.184.5.1314-1323.2002.
- H. Wang, C. P. Tseng and R. P. Gunsalus, The napF and narG Nitrate Reductase Operons in Escherichia coli Are Differentially Expressed in Response to Submicromolar Concentrations of Nitrate but Not Nitrite, J. Bacteriol., 1999, 181, 5303–5308, DOI:10.1128/JB.181.17.5303-5308.1999.
- J. Cole, Nitrate reduction to ammonia by enteric bacteria: redundancy, or a strategy for survival during oxygen starvation?, FEMS Microbiol. Lett., 1996, 136, 1–11, DOI:10.1016/0378-1097(95)00480-7.
- L. Chang, L. I. Wei, J. P. Audia, R. A. Morton and H. E. Schellhorn, Expression of the Escherichia coli NRZ nitrate reductase is highly growth phase dependent and is controlled by RpoS, the alternative vegetative sigma factor, Mol. Microbiol., 2002, 34, 756–766, DOI:10.1046/j.1365-2958.1999.01637.x.
- H. Wang and R. P. Gunsalus, The nrfA and nirB Nitrite Reductase Operons in Escherichia coli Are Expressed Differently in Response to Nitrate than to Nitrite, Genet. Mol. Biol., 2000, 182, 5813–5822, DOI:10.1128/jb.182.20.5813-5822.2000.
- B. Weiss, Evidence for Mutagenesis by Nitric Oxide during Nitrate Metabolism in Escherichia coli, J. Bacteriol., 2006, 188, 829–833, DOI:10.1128/JB.188.3.829-833.2006.
- S. R. Poock, E. R. Leach, J. W. B. Moir, J. A. Cole and D. J. Richardson, Respiratory Detoxification of Nitric Oxide by the Cytochrome c Nitrite Reductase of Escherichia coli, J. Biol. Chem., 2002, 277, 23664–23669, DOI:10.1074/jbc.M200731200.
- M. Kaldorf, K. H. Linne von Berg, U. Meier, U. Servos and H. Bothe, The reduction of nitrous oxide to dinitrogen by Escherichia coli, Arch. Microbiol., 1993, 160, 432–439, DOI:10.1007/BF00245303.
- J. van der Plas, K. J. Hellingwerf, H. G. Seijen, J. R. Guest, J. H. Weiner and W. N. Konings, Identification and localization of enzymes of the fumarate reductase and nitrate respiration systems of Escherichia coli by crossed immunoelectrophoresis, J. Bacteriol., 1983, 153, 1027–1037, DOI:10.1128/jb.153.2.1027-1037.1983.
- W. J. Dobrogosz, Altered End-Product Patterns and Catabolite Repression in Escherichia coli, J. Bacteriol., 1966, 91, 2263–2269, DOI:10.1128/JB.91.6.2263-2269.1966.
- N. J. Gilberthorpe and R. K. Poole, Nitric oxide homeostasis in Salmonella typhimurium, J. Biol. Chem., 2008, 283, 11146–11154, DOI:10.1074/jbc.M708019200.
- C. E. Vine, S. K. Purewal and J. A. Cole, NsrR-dependent method for detecting nitric oxide accumulation in the Escherichia coli cytoplasm and enzymes involved in NO production, FEMS Microbiol. Lett., 2011, 325, 108–114, DOI:10.1111/j.1574-6968.2011.02385.x.
- L. Page, L. Griffiths and J. A. Cole, Different physiological roles of two independent pathways for nitrite reduction to ammonia by enteric bacteria, Arch. Microbiol., 1990, 154, 349–354, DOI:10.1007/BF00276530.
- R. H. Gillette and E. H. Eyster, The Fundamental Rotation-Vibration Band of Nitric Oxide, Phys. Rev., 1939, 56, 1113–1119, DOI:10.1103/PhysRev.56.1113.
- A. Abou-Jaoudé, M. Chippaux and M.-C. Pascal, Formate-Nitrite Reduction in Escherichia coli K-12. 1. Physiological Study of the System, Eur. J. Biochem., 1979, 95, 309–314, DOI:10.1111/j.1432-1033.1979.tb12966.x.
- J. S. McDowall, B. J. Murphy, M. Haumann, T. Palmer, F. A. Armstrong and F. Sargent, Bacterial formate hydrogenlyase complex, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3948–3956, DOI:10.1073/pnas.1407927111.
- A. Pecher, F. Zinoni, C. Jatisatienr, R. Wirth, H. Hennecke and A. Böck, On the redox control of synthesis of anaerobically induced enzymes in enterobacteriaceae, Arch. Microbiol., 1983, 136, 131–136, DOI:10.1007/BF00404787.
- R. Rossmann, G. Sawers and A. Böck, Mechanism of regulation of the formate-hydrogenlyase pathway by oxygen, nitrate and pH: definition of the formate regulon, Mol. Microbiol., 1991, 5, 2807–2814, DOI:10.1111/j.1365-2958.1991.tb01989.x.
- R. Grosz and G. Stephanopoulos, Statistical mechanical estimation of the free energy of formation of E. coli biomass for use with macroscopic bioreactor balances, Biotechnol. Bioeng., 1983, 25, 2149–2163, DOI:10.1002/bit.260250904.
- H. Abaibou, G. Giordano and M.-A. Mandrand-Berthelot, Supression of Escherichia coli formate hydrogenlyase activity by trimethylamine N-oxide is due to drainage of the inducer formate, Microbiol., 1997, 143, 2657–2664, DOI:10.1099/00221287-143-8-2657.
Footnote |
| † Electronic supplementary information (ESI) available: S.1. Key nitrate and nitrite reduction enzymes, S.2. M9 medium formulation, S.3. FTIR spectroscopy of CO2 and ethanol, S.4. cavity enhanced Raman spectroscopy (Experimental details, spectral fitting procedures and calibration plots), S.5. liquid phase Raman spectroscopy (Experimental details of the home-built Raman spectrometer, spectral fitting procedures and calibration plots) and S.6. analysis of bacterial culture samples (nitrite colorimetry, 14N/15N-ammonium analysis). See DOI: 10.1039/d1an01261d |
| This journal is © The Royal Society of Chemistry 2021 |