
Microsphere mediated exosome isolation and ultra-sensitive detection on a dielectrophoresis integrated microfluidic device
Wenjie
Zhao
 ab,
Lingqian
Zhang
a,
Yifei
Ye
ab,
Yuang
Li
ab,
Xiaofeng
Luan
a,
Jinlong
Liu
a,
Jie
Cheng
ab,
Yang
Zhao
a,
Mingxiao
Li
*a and
Chengjun
Huang
*ab
ab,
Lingqian
Zhang
a,
Yifei
Ye
ab,
Yuang
Li
ab,
Xiaofeng
Luan
a,
Jinlong
Liu
a,
Jie
Cheng
ab,
Yang
Zhao
a,
Mingxiao
Li
*a and
Chengjun
Huang
*ab
aInstitute of Microelectronics, Chinese Academy of Sciences, Beijing, People's Republic of China. E-mail: limingxiao@ime.ac.cn
bSchool of Future Technology, University of Chinese Academy of Sciences, Beijing, People's Republic of China. E-mail: huangchengjun@ime.ac.cn
First published on 8th September 2021
Abstract
Tumor-derived exosomes have been recognized as potential biomarkers for cancer diagnosis because they are actively involved in cancer progression and metastasis. However, progress in practical exosome analysis is still slow due to the limitation in exosome isolation and detection. The development of microfluidic devices has provided a promising analytical platform compared with traditional methods. In this study, we develop an exosome isolation and detection method based on a microfluidic device (ExoDEP-chip), which realized microsphere mediated dielectrophoretic isolation and immunoaffinity detection. Exosomes were firstly isolated by binding to antibodies pre-immobilized on the polystyrene (PS) microsphere surface and were further detected using fluorescently labeled antibodies by fluorescence microscopy. Single microspheres were then trapped into single microwells under the DEP force in the ExoDEP-chip. A wide range from 1.4 × 103 to 1.4 × 108 exosomes per mL with a detection limit of 193 exosomes per mL was obtained. Through monitoring five proteins (CD81, CEA, EpCAM, CD147, and AFP) of exosomes from three different cell lines (A549, HEK293, and HepG2), a significant difference in marker expression levels was observed in different cell lines. Therefore, this method has good prospects in exosome-based tumor marker detection and cancer diagnosis.
Introduction
Exosomes, a subtype of extracellular vesicles (EVs) with a diameter of 30–150 nm,1 are secreted by mammalian cells and are abundant in various body fluids (blood, urine, ascites, etc.).2–5 It was discovered that exosomes play an important role in many biological processes, including cell migration, cell communication,6 tumorigenesis, metastasis,7 and cardiovascular generation.8 Exosomes could reflect their parental cell and tissue origin by the shuttling signaling molecules they carry, i.e., proteins9 and nucleic acids.10 Thus, exosomes show great potential as new specific biomarkers for non-invasive early cancer diagnosis, and have been attracting more and more attention from researchers. In the reported studies, various techniques have been introduced for exosome isolation and detection, characterization of their size and morphology, etc. Reported methods for exosome isolation include ultracentrifugation (UC), polymer precipitation (PEG), and filtration.11 The UC method involves isolation of exosomes based on size and density under high centrifugal g-force (up to 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g), and is considered as the gold standard for exosome isolation. However, this method is time-consuming (i.e., ∼5 h for a sample) and requires expensive ultracentrifugation equipment.12 The PEG precipitation method isolates exosomes by competitively binding water molecules with exosomes or water-soluble compounds based on the hydrophobicity of exosomal membrane, which is relatively simple. But this method is limited by high cost and low specificity. The filtration method involves isolation of exosomes based on size, and is often combined with ultracentrifugation.13 Filtration is generally faster than centrifugation, but the detrimental clogging effects usually lead to low exosome yields. Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) have been widely used to observe the morphology and size of single exosomes. Meanwhile, nanoparticle tracking analysis (NTA) has been reported to measure the concentration and size distribution of exosomes. The enzyme-linked immunosorbent assay (ELISA) and western blot analysis are usually considered as standard methods for exosome detection. However, they are limited by the large sample requirement and multistep operation.6 The analysis of nucleic acids of exosomes can be performed by the polymerase chain reaction (PCR) or sequencing techniques. These detection methods require dedicated instruments or complex multi-step workflows. Therefore, the clinical utilization of exosomes is still lagging due to the challenges in rapid isolation and high sensitivity detection of exosomes.
000g), and is considered as the gold standard for exosome isolation. However, this method is time-consuming (i.e., ∼5 h for a sample) and requires expensive ultracentrifugation equipment.12 The PEG precipitation method isolates exosomes by competitively binding water molecules with exosomes or water-soluble compounds based on the hydrophobicity of exosomal membrane, which is relatively simple. But this method is limited by high cost and low specificity. The filtration method involves isolation of exosomes based on size, and is often combined with ultracentrifugation.13 Filtration is generally faster than centrifugation, but the detrimental clogging effects usually lead to low exosome yields. Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) have been widely used to observe the morphology and size of single exosomes. Meanwhile, nanoparticle tracking analysis (NTA) has been reported to measure the concentration and size distribution of exosomes. The enzyme-linked immunosorbent assay (ELISA) and western blot analysis are usually considered as standard methods for exosome detection. However, they are limited by the large sample requirement and multistep operation.6 The analysis of nucleic acids of exosomes can be performed by the polymerase chain reaction (PCR) or sequencing techniques. These detection methods require dedicated instruments or complex multi-step workflows. Therefore, the clinical utilization of exosomes is still lagging due to the challenges in rapid isolation and high sensitivity detection of exosomes.
To overcome the limits of these traditional methods, various microfluidic platforms have been developed to isolate and detect exosomes or exosome subpopulations14 from different sample types based on their physical15–18 or biochemical properties.19–23 Several microfluidic devices have been designed based on the different size of exosomes from other EVs. A microfluidic system based on viscoelasticity was presented to directly separate exosomes from cell culture media or serum.16 The viscoelastic forces exerted on EVs were controlled by adding a biocompatible polymer in the media. A DEP-based method is also an effective way for exosome isolation.24–27 An alternating current electrokinetic (ACE) microarray chip was designed to isolate and recover exosomes from undiluted human plasma samples based on the differences between the dielectric properties of the exosomes and the surrounding plasma.24 Size-based methods are generally simple and fast, which could provide relatively high throughput and label-free isolation of exosomes. A higher percentage of contaminants (similar EVs with different origins and proteins) is still the bottleneck of the method. The exosome isolation technique based on the affinity properties of exosomes, such as surface proteins, is considered as the method with higher specificity. Immunomagnetic bead-based exosome isolation and detection methods are common methods using microfluidic devices.28–30 An ExoSearch chip was designed to achieve on-chip isolation and enrichment of exosomes streamlined with multiplexed detection of marker combinations.28 Magnetic beads with bound exosomes were retained as tight aggregates in the microchamber by magnetic force for quantitative isolation and detection of exosomes. Compared with the UC method, the ExoSearch chip yielded a higher percentage of vesicles smaller than 150 nm (∼80% vs. ∼60%) with high specificity. Zhang et al. reported a 3D nanostructure via patterned colloidal self-assembly9,31 to detect eight markers on single addition of exosomes and achieved a low limit of detection. The nanostructure promoted microscale mass transfer and increased the surface area and probe density to enhance the efficiency and speed of exosome binding. In these devices, the interference during fluorescence observation caused by the aggregation of microbeads is inevitable. To reduce the influence of fluorescence interference on exosome detection, a bead-based microarray was proposed for exosome isolation and multiplexed tumor marker detection.22 The bead was trapped and queued among micropillars to avoid the optical interference. Three quantum dot (QD) probe-labeled lung cancer markers were used to conduct multiplexed detection of exosome surface protein markers. Engineered superparamagnetic materials (gold-loaded ferric oxide nanocubes32 (Au-NPFe2O3NCs) or carboxyl group-functionalized iron oxide nanoparticles33 (C-IONPs)) were also used to direct isolation and subsequent electrochemical detection of a specific population of exosomes. In Boriachek's work,32 Au-NPFe2O3NCs functionalized with the CD63 antibody were used as “dispersible nanocarriers” to capture exosomes. The limit of detection was low, 103 exosomes per mL. Recently, Tayebi et al. designed a microfluidic device with trapping arrays to permit multiplexed exosome capture by multiple surface modification.23 Fluorescence detection and quantification of a single Exobead, captured in individual trapping sites, reduced the optical interference of background noise and improved the accuracy of the statistical comparison of different exosomal biomarkers. These methods still face the limitation that passive trapping devices only capture microbeads of a specific size. Meanwhile, these platforms required complex fabrication or sophisticated sensing methods.
Here, we reported a microsphere-mediated exosome isolation and detection method based on a DEP integrated microfluidic device, called ExoDEP-chip. Antibody labeled polystyrene (PS) microspheres were used to capture exosomes on their surface. A DEP-based trapping chamber with a large number of microwells and a pair of interdigital DEP electrodes in each microwell was designed to trap the individual PS microspheres into the microwell. This method allows fluorescence quantification of single microspheres and reduces the optical interference. Compared with DEP-based devices mentioned before, this method achieved specific isolation of exosomes. Exosomes are captured on the surface of microspheres through the antigen–antibody affinity. Only microspheres need to be manipulated in the DEP trapping chamber, which greatly reduced the complexity of devices and operation. Compared with passive trapping devices, the ExoDEP-chip is more flexible. By adjusting the applied voltage, PS microspheres or cells with different diameters and dielectric properties can be selectively captured or released. The combination of microsphere-mediated exosome isolation and DEP-based immunoaffinity detection provides a specific and effective method for exosome isolation and detection. In addition, the platform is capable of multiplexed protein analysis for exosomes captured on microspheres. The obtained results demonstrate that our method achieved exosome isolation and ultra-sensitive detection and offers great potential in low-concentration exosome detection and cancer diagnosis.
Materials and methods
Materials
1-Ethyl-3-[3dimethylaminopropyl]carbodiimide (EDC), sulfo-N-hydroxysulfosuccinimide (NHS), IgG elution buffer, Tris buffer, bovine serum albumin (BSA), hexamethyldisilazane (HMDS), fluorescent carbocyanine dye (DiO), calcein, and glutaraldehyde solution were purchased from Sigma-Aldrich. PDMS was purchased from Dow Corning. Microsphere solution was purchased from BaseLine ChromTech (Tianjin, China). Biotin anti-human CD63 antibody, FITC anti-human CD81 antibody, FITC anti-human CD326 (EpCAM) antibody, anti-human CD66d/e (CEA), anti-human CD147 antibody, and anti-human AFP antibody were purchased from BioLegend. Exosome-free FBS was purchased from SunBio.Design and fabrication of the device
The ExoDEP-chip is illustrated in Fig. 1A with a cross-section view in Fig. 1B. The device consists of a multi-layer structure. On the bottom, Au interdigitated electrodes (IDEs) were fabricated on the top surface of the glass substrate. A large number of microwells, the material of which was Parylene, were covered on the Au IDEs. The polydimethylsiloxane (PDMS) microfluidic channel was finally covered on the top of the microwell array with an inlet and an outlet. | ||
| Fig. 1 The ExoDEP-chip. A. Schematic diagram of the ExoDEP-chip for exosome capture and detection. B. The cross-section view of the ExoDEP-chip. The schematic diagram and cross-section view are not drawn to scale. | ||
The mechanism of microsphere trapping is based on the dielectrophoresis (DEP) force, which has been widely applied for single-cell trapping34 and polystyrene particle manipulation.35 To evaluate the DEP trapping capability of the microspheres by the designed electrodes, the electric field inside the channel was simulated using COMSOL 5.4 (Burlington, MA). The AC/DC module physics was used to model the 2-D cross-section of the device including the channel floor, the fluid, the microwells, and the channel top.
The ExoDEP-chip was fabricated by standard microfabrication processes. In brief, Au IDEs with a thickness of 50 nm were fabricated on a cleaning glass substrate by the Au lift-off process. The width and spacing of the electrode were 15 μm and 10 μm, respectively. In the next step, a 6 μm-thick Parylene film was grown on the electrode by PECVD. The microwell structure was formed by removing excess Parylene by plasma etching and exposing the bottom electrodes. The diameter of microwells was 60 μm. The PDMS microfluidic channel was fabricated by standard soft lithography techniques. The PDMS layer was further bonded on the glass substrate after oxygen plasma activation. The assembled device was placed into an oven at 80 °C for 1 h to enhance bonding.
Preparation of exosome-capturing microspheres
Carboxylic cross-linked PS microspheres were activated through incubation with sulfo-NHS and EDC under acidic conditions (pH = 5) for 1 h at 25 °C, followed by mixing the activated microspheres with exosome-capturing antibodies (anti-CD63 antibody) to bind with the amine groups on the antibodies. After incubation for 1 h, the antibody-labeled microspheres were resuspended in 1× PBS with 5% BSA and stored at 4 °C for 2 h.Chip operation
Before the microfluidic experiment, exosome samples, antibody-labeled microspheres, and DiO dye were mixed and incubated at 37 °C for 30 min. A DEP buffer was freshly prepared by dissolving 8.5 wt% of sucrose and 0.3 wt% of glucose in DI water to adjust the osmotic pressure inside and outside of the exosomal membrane. The conductivity of the DEP buffer was 2 μS cm−1.36 After exosome isolation and DiO staining, the microspheres were resuspended into the DEP buffer to remove the excess dye.The experimental platform contained an injection system, signal generator, ExoDEP-chip, fluorescence microscope, and computer. The microsphere solution was injected into the ExoDEP-chip using a syringe pump (Harvard apparatus, Pump 11 Elite, USA) when the experiment began. The sinusoidal signal (NF Corporation, WF1974, Japan) was applied using the signal generator. The fluorescence microscope (OLYMPUS, BX51, Japan) was used to monitor the particle motion, and the computer was used to record images in the ExoDEP-chip through the microscope.
Prior to microsphere injection, a blocking buffer (1× PBS buffer with 0.1% BSA and 0.05% Tween-20) was introduced into the microchannel for 30 min to minimize non-specific adsorption of the ExoDEP-chip. Subsequently, the prepared exosome-captured microspheres were dispersed in the DEP buffer and introduced into the ExoDEP-chip with an external syringe pump. Meanwhile, an AC voltage with a frequency of 10 kHz and an amplitude of 20VP–P was applied on the IDEs. Under these conditions, a non-uniform electric field was generated inside the channel, and the microsphere would be attracted into the individual microwells under the positive DEP force. The whole injection process was performed at a flow rate of 1 μL min−1.
In the parallel set of experiments, the DiO dye was replaced by fluorescent-labeled detection antibodies (CD81, CEA, EpCAM, CD147, and AFP) to demonstrate the immunoaffinity detection capability of the device.
Cell culture and purified exosome sample preparation
Adenocarcinomic human alveolar basal epithelial cells (A549), HEK293 cells, and human hepatocellular cancer cells (HepG2) were purchased from China Infrastructure of Cell Line Resources (Beijing, China). A549 cells were cultured in RPMI 1640 supplemented with 10% exosome-free FBS and 1% penicillin/streptomycin and sub-cultured every 48 h using 0.25% trypsin-EDTA solution to obtain 80–90% confluency under 37 °C, 5% (v/v) CO2 in a humidified incubator. HEK293 cells and HepG2 cells were cultured in DMEM supplemented with 10% exosome-free FBS and 1% penicillin/streptomycin and sub-cultured every 48 h using 0.25% trypsin-EDTA solution to obtain 80–90% confluency under 37 °C, 5% (v/v) CO2 in a humidified incubator. Purified exosome samples were isolated from the A549 cell culture supernatant by standard ultracentrifugation at 4 °C. Briefly, the cell culture supernatant was collected (100 mL) and centrifuged at 2000g for 10 min to remove dead cells and cell debris. Then, the supernatant was ultra-centrifuged at 4 °C for 45 min at 10000g to remove microvesicles and again at 100000g for 2 h to pellet exosomes. Exosome pellets were then resuspended in 10 mL of PBS for a wash step and then collected again by ultracentrifugation at 4 °C for 60 min at 110000g in Beckman Coulter Quik-Seal Centrifuge Tubes. After aspiration of the PBS supernatant, the exosome pellet was resuspended in 100 μL. The particle size and concentration of purified exosomes were measured by NTA with ZetaView PMX 110 (Particle Metrix, Meerbusch, Germany) and the corresponding software ZetaView 8.04.02.
Sample processing for SEM
Microspheres were characterized using a field-emission SEM after exosome isolation. Microspheres with captured exosomes were fixed with 4% glutaraldehyde for 30 min and then dehydrated in a series of alcohol concentrations (50% for 10 min, 70% for 10 min, 90% for 10 min, 95% for 10 min, 100% twice for 10 min each). Then, the microspheres were further dehydrated with 50% HMDS in alcohol for 10 min and then transferred to 100% HMDS and uniformly spread onto a silica glass, followed by overnight air drying in the hood. After that, the silica glass was vacuumed and sputter-coated with gold at room temperature for 60 s. Finally, the morphology of exosomes on microspheres was examined under SEM.Data collection and analysis
Images were obtained using a CCD camera on a fluorescence microscope. The fluorescence signal intensity of a single PS microsphere was calculated using ImageJ software (National Institutes of Health, USA), and the average value of the fluorescence signal intensity of each microsphere was acquired.Results and discussion
Numerical simulation of DEP microsphere trapping
The mechanism of trapping microspheres into microwells is based on the DEP force. DEP is the motion of dielectrically polarized particles in a non-uniform electric field.35 The DEP force is given by| FDEP = πr3εmre[K(ω)]∇|E|2 | (1) |
 | (2) |
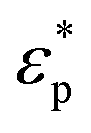 and
and  are the complex permittivities of the particle and medium (ε* = ε − (jσ/ω), where the subscripts p and m represent the particles and suspending medium, respectively), ε is the permittivity, and σ is the conductivity. By adjusting the frequency of the AC electric field, the value of re[K(ω)] can vary from negative to positive and vice versa. Particles moved to the field maximum (Re[K(ω)] > 0, positive DEP) or the field minimum (Re[K(ω)] < 0, negative DEP), which depends on the difference between the dielectric properties of the particles and suspending medium. The difference in size and dielectric properties between samples results in the different strengths and directions of the DEP force, which can lead to more effective separation. Thus, dielectrophoresis is a very sensitive sorting technology for target samples.
are the complex permittivities of the particle and medium (ε* = ε − (jσ/ω), where the subscripts p and m represent the particles and suspending medium, respectively), ε is the permittivity, and σ is the conductivity. By adjusting the frequency of the AC electric field, the value of re[K(ω)] can vary from negative to positive and vice versa. Particles moved to the field maximum (Re[K(ω)] > 0, positive DEP) or the field minimum (Re[K(ω)] < 0, negative DEP), which depends on the difference between the dielectric properties of the particles and suspending medium. The difference in size and dielectric properties between samples results in the different strengths and directions of the DEP force, which can lead to more effective separation. Thus, dielectrophoresis is a very sensitive sorting technology for target samples.
The electric field and the flow field distribution inside the channel were simulated using COMSOL 5.4. The electric field distribution is illustrated in Fig. 2A. The results showed that a non-uniform electric field was generated in the channel. The strength of the electric field inside the microwell was stronger than the strength of the electric field above the channel. The maximum of the electric field was at the edge of the electrode (∼107 V m−1).
 | ||
| Fig. 2 COMSOL simulation of the ExoDEP-chip. A. The electric field distribution in the microchannel. B. Electric field intensity in microwells with different microwell diameters (60 μm, 50 μm, 40 μm, 30 μm, and no microwell). C. The flow field distribution inside the microchannel. D. The flow velocity was at 7 μm above the substrate of different structures. | ||
In order to further determine the diameter of microwells, structures with different diameters (60 μm, 50 μm, 40 μm, 30 μm, and no microwell) of microwells were simulated and the electric field and flow field distribution inside the microwell were analyzed. The width and spacing of electrodes were 15 μm and 10 μm, respectively. The electric field intensity distribution curves at 1 μm above the electrodes inside the microwells (the red dotted line in Fig. 2A) with different microwell diameters are shown in Fig. 2B. The zero point of the x-axis was defined as the left side inside the microwell with 60 μm diameter, which is marked in Fig. 2A. It is shown in Fig. 2A that a pair of electrodes were in the center of the microwell and the Parylene wall was on the electrode beside them, when the diameter of the microwells was set as 60 μm. Due to the existence of Parylene, the geometric shape above the electrode changed and a stronger non-uniform electric field was generated near the edge of the microwell, which was higher than the maximum value of the electric field in the no-microwell structure. This can also be proved by the electric field intensity in Fig. 2B. The purple line shows the electric field intensity of the no-microwell structure, where the maximum of the electric field (∼1.8 × 106 V m−1) was at each edge of the electrode. The black line shows the electric field intensity of the microwell structure with 60 μm diameter, where the maximum of the electric field (∼2.4 × 107 V m−1) was at each edge of the microwell (near 0 μm and 60 μm in Fig. 2B). In the microwell structure with other diameters, this phenomenon was not found. According to the formula of DEP (eqn (1)), the sharp increase of electric field would significantly increase the magnitude of the DEP force. Therefore, this design of the structure was very beneficial for the trapping of microspheres.
The flow field distribution of the structure with 60 μm-diameter microwells and without microwells is shown in Fig. 2C. Different from the structure without microwells where the streamlines were always along the horizontal direction, in the structure with microwells, the direction of streamlines was slightly deflected downwards. It means that when the PS microspheres flowed over the microwell, the trajectory would deflect under the drag force. In this case, PS microspheres were more easily trapped into the microwells when the drag force was combined with the DEP force. Fig. 2D shows the flow velocity at 7 μm above the substrate (the black dotted line in Fig. 2C) of structures with different diameters. With the increase of the diameter, the maximum of flow rate increased, which means that the deflection effect caused by microwells was better in the structure with a larger diameter. Therefore, the structure with 60 μm-diameter microwells was chosen to fabricate the ExoDEP-chip.
Isolation evaluation of the ExoDEP-chip
To capture the microspheres in the chip uniformly, we designed an ExoDEP-chip comprising a PDMS slab bonded with a glass substrate that contained a large number of microwells fabricated on interdigitated Au electrodes. The photo of the ExoDEP-chip is shown in Fig. 3A. Fig. 3B shows the micrograph of the device. The width and distance of electrodes were 15 μm and 10 μm, respectively. The height of the microchannel was 30 μm. The microwell structure was made from 6 μm thick Parylene deposited on a glass substrate and the diameter was 60 μm, which was bigger than the diameter of microspheres (15 μm) to ensure that the microsphere can be completely attracted into the microwell. The microwells were aligned with the interdigitated electrodes in order to locate a pair of electrodes in each of the microwells. The wall's minimum thickness, defined as the minimum gap distance of two microwells, was designed to be 15 μm, equal to the electrode's width. Fig. 3C shows the SEM images of the microwell area. It can be seen from the images that microwells distributed above the electrode uniformly, and a pair of electrodes were located at the bottom of the microwell. | ||
| Fig. 3 Characterization of the ExoDEP-chip. A. The fabricated ExoDEP-chip used in this study. B. Microwell and electrode structure in the ExoDEP-chip. Scale bar, 60 μm. C. SEM images of microwells. Scale bar, 50 μm. Insert: The SEM image of the sidewall of Parylene. Scale bar, 10 μm. D. Experimental platform for exosome detection including the ExoDEP-chip, an injection system, a signal generator, a fluorescence microscope, and a computer. | ||
The experimental platform containing an injection system, signal generator, ExoDEP-chip, fluorescence microscope, and computer is shown in Fig. 3D. Before the experiment, the ExoDEP-chip was washed in PBS buffer with 0.5% BSA for 5 min. The microsphere solution was injected into the ExoDEP-chip using a syringe pump when the experiment began. The sinusoidal signal was applied by the signal generator. The fluorescence microscope was used to monitor the particle motion, and the computer was used to record images in the ExoDEP-chip through the microscope.
To investigate the performance of the ExoDEP-chip, we studied the ability of the ExoDEP-chip to separate PS microspheres and A549 cells. Before the DEP experiment, the ExoDEP-chip was washed with PBS buffer with 0.5% BSA for 5 min. To distinguish PS microspheres from A549 cells, A549 cells were stained with Calcein, which emitted green fluorescence, while PS microspheres were modified with the CEA antibody, which emitted red fluorescence. Fig. 4A shows the fluorescence image of a mixture of PS microspheres and A549 cells. The mixture solution was injected into the ExoDEP-chip at a rate of 1 μL min−1 and the frequency and peak-to-peak voltage were set as 50 kHz and 20 V, respectively. It has been reported that the DEP force acted on A549 cells was n-DEP in the range of 1 kHz to 50 kHz at a low conductivity37 while the DEP force acted on 13 μm PS particles (similar to the PS microspheres used in this work) was p-DEP in this frequency range.38 When the mixture solution flowed through the microwell area, PS microspheres were attracted to a high electric field area (inside the microwell) under p-DEP force, while A549 cells were pushed to the low electric field area under n-DEP force and flowed to the outlet with the fluid. As shown in Fig. 4B, PS microspheres (red plot) were trapped into the microwells, and there were almost no A549 cells (green plot) in this area. Fig. 4C shows the fluorescence image of the solution collected from the outlet after applying the voltage for 5 min. The percentage of A549 cells and PS microspheres before and after the DEP experiment was calculated as shown in Fig. 4D. The percentage of PS microspheres reduced from 24% to 3%. The capturing efficiency of PS microspheres was 90.5%, which confirmed that most of the PS microspheres were trapped in the electrode area during DEP processing. Thus, the ExoDEP-chip has the ability to separate PS microspheres from complex samples. Moreover, the ExoDEP-chip has the potential to separate different particles with different sizes and dielectric properties. The frequency and amplitude of the applied voltage can be adjusted as needed, which is more flexible in different applications.
 | ||
| Fig. 4 Separation of PS microspheres and A549 cells. A. Fluorescence image of the mixture of PS microspheres and A549 cells. B. Fluorescence image of the electrode area when the voltage was on. C. Fluorescence image of the solution collected from the outlet after the DEP experiment. D. Percentage of PS microspheres and A549 cells before and after the DEP experiment. Scale bar: 80 μm. | ||
Fluorescence detection of captured exosomes
Anti-CD63 labeled PS microspheres were used to capture exosomes from the A549 cell culture supernatant. Anti-CD63 antibody was chosen as the capture antibody for the immunocapture of exosomes because CD63 protein is a member of the transmembrane-4 superfamily and richly found on the surface of exosomes.39 The anti-CD63 labeled PS microspheres were mixed with the A549 supernatant to capture exosomes on their surface and stained with DiO dye with green fluorescence emission before the DEP experiment. The PS microspheres were then resuspended in DEP buffer. When the experiment began, the injection rate of the PS microsphere solution was set as 1 μL min−1. The frequency and the peak-to-peak value of the sinusoidal signal were set as 10 kHz and 20 V during the PS microsphere trapping experiment, respectively. A non-uniform electric field would be generated inside the channel. When PS microspheres pass above the channel, they will be affected by the DEP force and move to the high electric field area. As shown in Fig. 2, the maximum of the electric field is located at the inner edge of the microwell due to the change of geometry. Therefore, PS microspheres will be trapped into the microwells under DEP force. Fig. 5A shows the bright field and fluorescence images of the PS microspheres trapping experiment. The result showed that a single 15 μm PS microsphere was trapped into the microwell under DEP force and emitted green fluorescence, which was caused by the exosomes captured on the surface of PS microspheres stained with DiO dye. | ||
| Fig. 5 Analysis of the fluorescence results. A The bright field and fluorescence images of individual 15 μm microspheres trapped into the microwell under DEP force. Scale bar: 40 μm. B. Fluorescence images of anti-CD63 labeled PS microspheres mixed with the A549 supernatant, Exo-free FBS, and PBS, respectively. Scale bar: 30 μm. C. Comparison of the fluorescence intensity of anti-CD63 labeled microspheres mixed with the A549 supernatant, Exo-free FBS, and PBS. | ||
To further analyze the source of captured exosomes, anti-CD63 labeled microspheres were mixed with different samples (A549 supernatant, Exo-free medium, and PBS), followed by mixing with DiO dye to stain the lipid bilayer membrane of exosomes. After that, the PS microspheres were resuspended in DEP buffer and introduced into the ExoDEP-chip. A single microsphere was trapped into the microwell under DEP force, and the fluorescence microscopy images are shown in Fig. 5B. The average fluorescence intensity of single PS microspheres was calculated and is shown in Fig. 5C. Compared with Exo-free medium and PBS buffer as negative controls (lack of exosomes), the fluorescence intensity of PS microspheres mixed with the A549 cell culture supernatant showed at least 6-fold higher fluorescence. This proved that exosomes were captured on the surface of the microspheres and exosomes were secreted by A549 cells. Meanwhile, the very low fluorescence intensity of control microspheres could still be observed for the Exo-free microspheres, which was probably caused by the nonspecific adsorption of the dye.
On-chip quantitative detection of exosomes
Purified exosome samples isolated from the A549 cell culture supernatant by UC were used to investigate the relationship between the fluorescence intensity and the concentration of exosomes. The concentration of purified exosomes was 1.4 × 108 exosomes per mL according to the NTA results. The exosome samples with concentrations ranging from 1.4 × 103 to 1.4 × 108 exosomes per mL were prepared by diluting purified exosomes with PBS. The concentration of anti-CD63 labeled PS microspheres was 1 × 104 particles per mL. The mixture of anti-CD63 labeled PS microsphere and purified exosome samples was stained with the DiO dye. After that, the PS microspheres were resuspended in DEP buffer and introduced into the ExoDEP-chip. The average fluorescence intensity of single PS microspheres at different concentrations of exosomes was calculated as shown in Fig. 6A. The fluorescence intensity rapidly increased with the increase of exosome concentration, and finally reached the saturation level at a concentration of about 1.4 × 108 exosomes per mL. No signal increase was observed when the concentration was further increased to 1.8 × 108 exosomes per mL. A linear correlation between the fluorescence intensity and the logarithm of exosome concentration from 1.4 × 103 exosomes per mL to 1.4 × 108 exosomes per mL was obtained, as shown in the inset of Fig. 6A. The obtained calibration curve showed quantitative detection over a 4-log dynamic range, with a limit of detection (LOD) of 193 exosomes per mL for purified A549 exosomes, which was calculated by the equation:9,40 | (3) |
 | ||
| Fig. 6 On-chip quantitative detection of exosomes. A. Fluorescence intensity was measured after DiO staining as a function of the concentration of purified A549 exosomes. The concentration of anti-CD63 labeled PS microspheres was 1 × 104 particles per mL. Insert: The fluorescence intensity as a function of the logarithm of the A549 exosome concentration (R2 = 0.9707, LOD = 193 exosomes per mL). B. The reproducibility test for the Exo-DEP chip from the same A549 supernatant. Four devices were tested under the same conditions. C. The specificity of the ExoDEP chip. The A549 cell culture supernatant or PBS buffer was incubated with PS microspheres labeled with/without the anti-CD63 antibody. | ||
The reproducibility of the device was examined using four independently fabricated devices. When the four devices were used to detect the exosomes from the same A549 supernatant, the fluorescence intensity of the four devices was obtained as shown in Fig. 6B. The relative standard deviation (RSD) was 2.6%. In addition, a series of experiments were designed to characterize the specificity of the device. A549 cell culture supernatant or PBS buffer was incubated with PS microspheres labeled with/without the anti-CD63 antibody. Anti-CD63 labeled PS microspheres were incubated with the A549 cell culture supernatant (the positive experiment). Anti-CD63 labeled PS microspheres incubated with PBS and label-free PS microspheres incubated with the A549 supernatant were designed as the negative experiment. The fluorescence intensity in the three experiments is shown in Fig. 6C. Compared with the negative control, the positive experiment showed at least 6-fold higher fluorescence, indicating that the ExoDEP chip has reasonable specificity.
In previous studies, many microfluidic devices were used for exosome isolation and low concentration detection. LOD, specificity, and reproducibility are very important parameters for the performance and application potential evaluation. Table 1 lists the performance of several typical exosome isolation and detection devices. Adequate mixing of biological samples and detection/capture antibodies is an important factor to achieve a low LOD. In some devices which achieved exosome capture on-chip, low reagent consumption and rapid detection are achieved. However, it is difficult to achieve sufficient mixing if there is no complex structure to cause the disturbance of the liquid, because of this some exosomes in the sample cannot be detected. Using our method, the exosome isolation was realized out of chip; therefore, the contact between exosomes and the antibody-labeled PS microspheres can be increased through vibration or other methods. Meanwhile, we determined the fluorescence intensity of single microspheres and calculated their average value. The design of the microwell array allowed the PS microspheres to be evenly and dispersedly distributed in the electrode area. The fluorescence interference between microspheres was greatly reduced. Differing from magnetic bead-based methods, where magnetic beads were aggregated as a larger-scale sphere, PS microspheres were dispersedly distributed in a plane in our method. PS microspheres were scattered more sparsely due to the microwells, which avoided the fluorescence interference caused by the aggregation of microspheres. Meanwhile, more information neglected in the immunomagnetic-based method was obtained in this method, which was the reason that the LOD was lower than those of other fluorescence-based methods. The specificity of our device was at the same level as those of the other devices as shown in Table 1. The RSD of our device is 2.6% according to Fig. 6B, which is lower than those of the devices in Table 1. Therefore, our device has good specificity, reproducibility and stability.
| Method | LOD (exosomes per mL) | Specificity | Reproducibility (RSD) | Ref. |
|---|---|---|---|---|
| PS-ED chip | 9.5 × 104 | 5 fold | 5.4% | 41 |
| ExoSearch chip | 7.5 × 105 | 3–5 fold | <10% | 28 |
| isExoCD | 1 × 103 | 3.5 fold | — | 42 |
| AuNP-amplified SAW sensor | 1.1 × 103 | 7 fold | — | 43 |
| ExoProfile chip | 2.1 × 104 | 10 fold | — | 9 |
| Double-filtration and photonic crystals | 8.9 × 103 | 9 fold | 4.3% | 44 |
| Electrokinetic-based sensor | 1.75 × 105 | 3.5 fold | — | 21 |
| ExoPCD-chip | 4.39 × 103 | — | — | 19 |
| Microfluidic device based on ac-EHD induced nanoshearing | 2.76 × 107 | — | — | 45 |
| Au-NPFe2O3NC-based method | 1 × 103 | 25 fold | 5.5% | 32 |
| ExoDEP-chip | 1.93 × 102 | 6 fold | 2.6% | This work |
In order to further verify the accuracy of the curve, anti-CD63 labeled PS microspheres were mixed with the A549 cell culture supernatant and injected into the chip after DiO staining. The concentration of exosomes in the A549 cell culture supernatant was 3.1 × 107 exosomes per mL according to the NTA result, which is shown in Fig. 7A. The average fluorescence intensity of single microsphere was 129.091 in this experiment. As shown in Fig. 7B, the calculated exosome concentration in the A549 cell culture supernatant was 3.52 × 107 exosomesper mL according to the standard curve. The degree of difference between the concentration obtained by our method and the commonly used NTA result was 13.5%, which proved that the result of this method was relatively similar to the NTA result. This difference may due to the fact that the modified PS microspheres captured more vesicles smaller than 150 nm, which was confirmed from the SEM results (Fig. 8).
 | ||
| Fig. 7 Comparison of NTA and the standard curve. A. NTA result of the concentration of exosomes in the A549 cell culture supernatant. B. Calculated exosome concentration in the A549 cell culture supernatant from the standard curve. | ||
 | ||
| Fig. 8 Morphological characterization of exosomes captured on the microspheres. A. SEM image of anti-CD63 labelled microspheres captured with exosomes. B. SEM image of control microspheres. C. Diameter distribution of ultracentrifugation-isolated exosomes measured by NTA and exosomes captured by anti-CD63 labelled microspheres measured by SEM. Fitting curves were Gaussian fitting (R2 > 95%). | ||
Characterization of exosomes captured on PS microspheres
After DEP trapping and fluorescence detection of PS microspheres, the voltage was turned off and microspheres were released from the microwell area. PS microspheres flowed to the outlet with the fluid. The microchannel with DEP buffer was further rinsed three times with 1 min each time to ensure that most of the microspheres in the channel were flushed out and collected at the outlet. The solution at the outlet was transferred with a pipette. Morphological characterization of exosomes captured on the surface of the anti-CD63 labeled PS microspheres was conducted by SEM imaging. Fig. 8A and B show the SEM images of microspheres captured with exosomes from the A549 supernatant and BSA coated control microspheres, respectively. Compared with the control, a large number of exosomes were captured on the surface of the anti-CD63 labeled microspheres. The diameters of exosomes captured on the surface of microspheres were measured by ImageJ, as shown in Fig. 8C. Meanwhile, the diameter distribution of purified exosomes isolated from the A549 supernatant by ultracentrifugation was measured by NTA, which is shown in Fig. 8C. It can be found that the size distribution of exosomes isolated by two methods both conformed to Gaussian distribution. Exosomes captured on the anti-CD63 labeled microspheres exhibited a narrower range in size distribution. The FWHM of the two Gaussian curves was 78.46 nm (SEM) and 86.68 nm (NTA). The mean size of the captured exosomes by the microsphere-based method was less than 150 nm (118 nm). In addition, the samples treated by our method showed a higher percentage (83.5%) of vesicles smaller than 150 nm compared with the ultracentrifugation samples (62.5%), which verified that our method is capable of capturing exosome-sized vesicles.The captured exosomes can also be released from the PS microspheres for further analysis. PS microspheres were selected from the outlet and mixed with IgG elution buffer for 10 min to dissociate the antibody–antigen interaction and release the captured exosomes. Fig. 9A and B show the SEM image of microspheres before and after releasing of exosomes. Nanoparticles adsorbed on the surface of the microspheres were obviously decreased after release, which was also proved from the fluorescence intensity, as shown in Fig. 9C. After release, the fluorescence intensity of the microspheres decreased by 69.97% and the value was similar to the fluorescence intensity of the PBS control (Fig. 5C). It is noticed that the conductivity of the IgG buffer is about 10 mS cm−1, which is not suitable for releasing exosomes in the DEP trapping chamber. This problem can be solved by adding a reaction chamber after the DEP trapping chamber in the future.
 | ||
| Fig. 9 Characterization of the release of exosomes. A. SEM images of the microspheres before the release of exosomes. B. SEM images of the microspheres after the release of exosomes. C. Fluorescence intensity before and after the release of exosomes. | ||
Protein analysis of exosomes from different cells
Although significantly higher amounts of exosomes have been observed from tumor cells,46 it is hard to detect cancer cells effectively from the concentration of exosomes alone. Therefore, surface proteins of exosomes are usually used for further analysis. It is not accurate enough to diagnose the cancer type using one tumor marker because some cancer markers are expressed in a variety of cancers. In this work, we simultaneously monitored multiple protein markers (CD81, CEA, EpCAM, CD147, and AFP) on the surface of exosomes secreted from three different cells (A549, HEK293, HepG2) by our device. CD81 is a member of the transmembrane-4 superfamily and has been found to be abundant in most of the exosomes’ subpopulations.47 However, it is hard to classify exosomes relying on the four tetraspanin protein family because they are widely present on the surface of most exosomes. Therefore, a combination of specific tumor biomarkers was chosen to analyze the exosomes from different cell lines.9 EpCAM is known as a disease-specific biomarker and it is overexpressed on the surface of cancer cells and cancer cell-derived exosomes. CEA is a broad-spectrum tumor protein marker most commonly observed in adenocarcinoma, such as the A549 cell line.48 CD147 and AFP were reported as the biomarkers for hepatoma carcinoma.49,50A sandwich ELISA assay was used to detect the surface proteins of the isolated exosomes quantitatively. Fluorescently labeled detection antibodies enabled specific recognition of individual markers. Anti-CD81 antibody, CEA antibody, EpCAM antibody, CD147 antibody, and AFP antibody were used as fluorescence detection antibodies. Exosomes from A549, HEK293, and HepG2 cell culture supernatants were isolated using anti-CD63 labeled PS microspheres, and five detection antibodies were mixed with the PS microspheres and incubated for 30 min to detect the exosomes captured on the surface of the PS microspheres, respectively.
The fluorescence images of PS microspheres trapped in the microwells after DEP are shown in Fig. 10A, where significant differences are observed in the fluorescence intensity between three cell lines. The fluorescence intensity enhancement rate of the five detection antibodies from the three cell lines compared with the PBS control is shown in Fig. 10B. The expression levels of the five biomarkers showed a similar trend to that reported in the literature.22,51,52 There was little difference in the expression level of CD81 for the three types of samples since CD81 is widely present in most exosomes. However, the expression levels of CEA, EpCAM, CD147, and AFP showed significant differences in different cancer cell lines, which is suitable for further tumor classification. Therefore, cancer cell types could be identified by monitoring the expression level of the protein markers. Fig. 10C shows the principal component analysis (PCA) plot of the three cell supernatant samples. In the plot, the HepG2 cell supernatant, A549 cell supernatant, and HEK293 cell supernatant were well separated. Compared with single markers, the method based on multiple markers provided more accurate classification. The device can be designed as a multi-channel structure to achieve the detection of multiple markers simultaneously, which can reduce the detection time.
 | ||
| Fig. 10 Surface protein analysis of exosomes from different cell lines. A. Fluorescence images of exosomes from different cell lines using five different fluorescence detection antibodies. Scale bar: 40 μm. B. Average fluorescence intensity of five biomarkers measured in three cell lines. * indicates statistical significance (p < 0.05), ** indicates statistical significance (p < 0.01), and *** indicates statistical significance (p < 0.001). C. Principal component analysis of different cell samples. | ||
Conclusion
In this study, an ExoDEP-chip combined with antibody-labeled microspheres has been developed for exosome isolation and detection. The antibody-based exosome capture strategy could realize rapid and stable binding kinetic and versatile exosomal biomarker selection. The diameter range of the particles captured by anti-CD63 labeled microspheres showed a great agreement with that of the purified exosomes isolated by ultracentrifugation. The detection method showed a low LOD (193 exosomes per mL) and a large detection range. Single microspheres were trapped into single microwells in the ExoDEP-chip, which avoided the fluorescence interference caused by microsphere agglomeration during the fluorescence quantification, such as in magnetic bead-based methods, the overlapping areas between the beads in the cluster may hide the fluorescence information. Fluorescence may not be observed at all for some beads inside the cluster, which would seriously affect the exosomal quantitative analysis. In addition, the ExoDEP-chip achieved protein analysis for different cell lines. Significant differences are observed in multiple markers of exosomes for three cell lines, which is helpful for further cancer diagnosis.Overall, the proposed ExoDEP-chip is highly suitable for fluorescence-based exosome quantification and selective release of the trapped PS microspheres as needed. The method is convenient, sensitive, and accurate, which may provide a potential platform for exosome-based early cancer diagnosis and treatment.
Author contributions
The manuscript was written through the contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support from the National Key Research and Development Program of China (2020YFC2004502) and the Beijing Nova Program (Z201100006820083).References
- M. Colombo, G. Raposo and C. Thery, Annu. Rev. Cell Dev. Biol., 2014, 30, 255–289 CrossRef CAS PubMed.
- F. Andre, N. E. C. Schartz, M. Movassagh, C. Flament, P. Pautier, P. Morice, C. Pomel, C. Lhomme, B. Escudier, T. Le Chevalier, T. Tursz, S. Amigorena, G. Raposo, E. Angevin and L. Zitvogel, Lancet, 2002, 360, 295–305 CrossRef CAS.
- J. Skog, T. Würdinger, S. van Rijn, D. H. Meijer, L. Gainche, W. T. Curry, B. S. Carter, A. M. Krichevsky and X. O. Breakefield, Nat. Cell Biol., 2008, 10, 1470–1476 CrossRef CAS PubMed.
- T. Pisitkun, R.-F. Shen and M. A. Knepper, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13368 CrossRef CAS PubMed.
- H. Im, H. Shao, Y. I. Park, V. M. Peterson, C. M. Castro, R. Weissleder and H. Lee, Nat. Biotechnol., 2014, 32, 490–495 CrossRef CAS PubMed.
- A. V. Vlassov, S. Magdaleno, R. Setterquist and R. Conrad, Biochim. Biophys. Acta, 2012, 1820, 940–948 CrossRef CAS PubMed.
- K. Boriachek, M. N. Islam, A. Moller, C. Salomon, N. T. Nguyen, M. S. A. Hossain, Y. Yamauchi and M. J. A. Shiddiky, Small, 2018, 14, 1702153 CrossRef PubMed.
- S. W. Ferguson, J. Wang, C. J. Lee, M. Liu, S. Neelamegham, J. M. Canty and J. Nguyen, Sci. Rep., 2018, 8, 1419 CrossRef PubMed.
- P. Zhang, X. Zhou and Y. Zeng, Chem. Sci., 2019, 10, 5495–5504 RSC.
- G. Rabinowits, C. Gercel-Taylor, J. M. Day, D. D. Taylor and G. H. Kloecker, Clin. Lung Cancer, 2009, 10, 42–46 CrossRef CAS PubMed.
- S. Lin, Z. Yu, D. Chen, Z. Wang, J. Miao, Q. Li, D. Zhang, J. Song and D. Cui, Small, 2019, e1903916, DOI:10.1002/smll.201903916.
- M. He and Y. Zeng, J. Lab. Autom., 2016, 21, 599–608 CrossRef CAS PubMed.
- J. C. Contreras-Naranjo, H. J. Wu and V. M. Ugaz, Lab Chip, 2017, 17, 3558–3577 RSC.
- K. Singh, R. Nalabotala, K. M. Koo, S. Bose, R. Nayak and M. J. A. Shiddiky, Analyst, 2021, 146, 3731–3749 RSC.
- J. H. Moore, W. B. Varhue, Y.-H. Su, S. S. Linton, V. Farmehini, T. E. Fox, G. L. Matters, M. Kester and N. S. Swami, Anal. Chem., 2019, 91, 10424–10431 CrossRef CAS PubMed.
- C. Liu, J. Guo, F. Tian, N. Yang, F. Yan, Y. Ding, J. Wei, G. Hu, G. Nie and J. Sun, ACS Nano, 2017, 11, 6968–6976 CrossRef CAS PubMed.
- Y. O. Mengxi Wu, Z. Wanga, R. Zhang, Po.-H. Huang, C. Chen, H. Li, P. Li, D. Quinn, M. Daog, S. Sureshh, Y. Sadovskyc and T. J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2017, 10584–10589 Search PubMed.
- Z. Ramshani, C. Zhang, K. Richards, L. Chen, G. Xu, B. L. Stiles, R. Hill, S. Senapati, D. B. Go and H. C. Chang, Commun. Biol., 2019, 2, 189 CrossRef PubMed.
- H. Xu, C. Liao, P. Zuo, Z. Liu and B. C. Ye, Anal. Chem., 2018, 90, 13451–13458 CrossRef CAS PubMed.
- Y. S. Chen, Y. D. Ma, C. Chen, S. C. Shiesh and G. B. Lee, Lab Chip, 2019, 19, 3305–3315 RSC.
- S. Cavallaro, J. Horak, P. Haag, D. Gupta, C. Stiller, S. S. Sahu, A. Gorgens, H. K. Gatty, K. Viktorsson, S. El Andaloussi, R. Lewensohn, A. E. Karlstrom, J. Linnros and A. Dev, ACS Sens., 2019, 4, 1399–1408 CrossRef CAS PubMed.
- Y. Bai, Y. Lu, K. Wang, Z. Cheng, Y. Qu, S. Qiu, L. Zhou, Z. Wu, H. Liu, J. Zhao and H. Mao, Nano-Micro Lett., 2019, 11, 59 CrossRef CAS PubMed.
- M. Tayebi, Y. Zhou, P. Tripathi, R. Chandramohanadas and Y. Ai, Anal. Chem., 2020, 92, 10733–10742 CrossRef CAS PubMed.
- S. D. Ibsen, J. Wright, J. M. Lewis, S. Kim, S. Y. Ko, J. Ong, S. Manouchehri, A. Vyas, J. Akers, C. C. Chen, B. S. Carter, S. C. Esener and M. J. Heller, ACS Nano, 2017, 11, 6641–6651 CrossRef CAS PubMed.
- L. Shi, A. Rana and L. Esfandiari, Sci. Rep., 2018, 8, 6751 CrossRef PubMed.
- L. Shi, D. Kuhnell, V. J. Borra, S. M. Langevin, T. Nakamura and L. Esfandiari, Lab Chip, 2019, 19, 3726–3734 RSC.
- J. M. Lewis, A. D. Vyas, Y. Qiu, K. S. Messer, R. White and M. J. Heller, ACS Nano, 2018, 12, 3311–3320 CrossRef CAS PubMed.
- Z. Zhao, Y. Yang, Y. Zeng and M. He, Lab Chip, 2016, 16, 489–496 RSC.
- F. Niu, X. Chen, X. Niu, Y. Cai, Q. Zhang, T. Chen and H. Yang, Micromachines, 2020, 11, 503 CrossRef PubMed.
- S. Fang, H. Tian, X. Li, D. Jin, X. Li, J. Kong, C. Yang, X. Yang, Y. Lu, Y. Luo, B. Lin, W. Niu and T. Liu, PLoS One, 2017, 12, e0175050 CrossRef PubMed.
- P. Zhang, X. Zhou, M. He, Y. Shang, A. L. Tetlow, A. K. Godwin and Y. Zeng, Nat. Biomed. Eng., 2019, 3, 438–451 CrossRef CAS PubMed.
- K. Boriachek, M. K. Masud, C. Palma, H.-P. Phan, Y. Yamauchi, M. S. A. Hossain, N.-T. Nguyen, C. Salomon and M. J. A. Shiddiky, Anal. Chem., 2019, 91, 3827–3834 CrossRef CAS PubMed.
- F. Z. Farhana, M. Umer, A. Saeed, A. S. Pannu, M. Shahbazi, A. Jabur, H. J. Nam, K. Ostrikov, P. Sonar, S. H. Firoz and M. J. A. Shiddiky, ACS Appl. Nano Mater., 2021, 4, 1175–1186 CrossRef CAS.
- J. Y. Chan, A. B. Ahmad Kayani, M. A. Md Ali, C. K. Kok, B. Yeop Majlis, S. L. L. Hoe, M. Marzuki, A. S. Khoo, K. K. Ostrikov, M. Ataur Rahman and S. Sriram, Biomicrofluidics, 2018, 12, 011503 CrossRef PubMed.
- Q. Chen and Y. J. Yuan, RSC Adv., 2019, 9, 4963–4981 RSC.
- J. Zhang, S. Yan, G. Alici, N.-T. Nguyen, D. Di Carlo and W. Li, RSC Adv., 2014, 4, 62076–62085 RSC.
- S. I. Han, Y. D. Joo and K. H. Han, Analyst, 2013, 138, 1529–1537 RSC.
- J. Zhang, D. Yuan, Q. Zhao, S. Yan, S.-Y. Tang, S. H. Tan, J. Guo, H. Xia, N.-T. Nguyen and W. Li, Sens. Actuators, B, 2018, 267, 14–25 CrossRef CAS.
- M. S. Pols and J. Klumperman, Exp. Cell Res., 2009, 315, 1584–1592 CrossRef CAS PubMed.
- L. Kashefi-Kheyrabadi, J. Kim, S. Chakravarty, S. Park, H. Gwak, S. I. Kim, M. Mohammadniaei, M. H. Lee, K. A. Hyun and H. I. Jung, Biosens. Bioelectron., 2020, 169, 112622 CrossRef CAS PubMed.
- S. Zhou, T. Hu, F. Zhang, D. Tang, D. Li, J. Cao, W. Wei, Y. Wu and S. Liu, Anal. Chem., 2020, 92, 1574–1581 CrossRef CAS PubMed.
- C. Qian, Y. Xiao, J. Wang, Y. Li, S. Li, B. Wei, W. Du, X. Feng, P. Chen and B.-F. Liu, Sens. Actuators, B, 2021, 333, 129559 CrossRef CAS.
- C. Wang, C. Wang, D. Jin, Y. Yu, F. Yang, Y. Zhang, Q. Yao and G. J. Zhang, ACS Sens., 2020, 5, 362–369 CrossRef CAS PubMed.
- X. Dong, J. Chi, L. Zheng, B. Ma, Z. Li, S. Wang, C. Zhao and H. Liu, Lab Chip, 2019, 19, 2897–2904 RSC.
- R. Vaidyanathan, M. Naghibosadat, S. Rauf, D. Korbie, L. G. Carrascosa, M. J. Shiddiky and M. Trau, Anal. Chem., 2014, 86, 11125–11132 CrossRef CAS PubMed.
- J. C. Akers, D. Gonda, R. Kim, B. S. Carter and C. C. Chen, J. Neurooncol., 2013, 113, 1–11 CrossRef PubMed.
- E. R. Abels and X. O. Breakefield, Cell. Mol. Neurobiol., 2016, 36, 301–312 CrossRef CAS PubMed.
- M. Grunnet and J. B. Sorensen, Lung Cancer, 2012, 76, 138–143 CrossRef CAS PubMed.
- J. Hu, T. Gou, W. Wu, J. Sun, S. Zhang, S. Zhou, J. Yin and Y. Mu, Anal. Chim. Acta, 2019, 1076, 118–124 CrossRef CAS PubMed.
- M.-H. Lee, A. Ahluwalia, K.-M. Hsu, W.-T. Chin and H.-Y. Lin, RSC Adv., 2014, 4, 36990–36995 RSC.
- Y. Kabe, M. Suematsu, S. Sakamoto, M. Hirai, I. Koike, T. Hishiki, A. Matsuda, Y. Hasegawa, K. Tsujita, M. Ono, N. Minegishi, A. Hozawa, Y. Murakami, M. Kubo, M. Itonaga and H. Handa, Clin. Chem., 2018, 64, 1463–1473 CrossRef CAS PubMed.
- N. Sun, Y. T. Lee, R. Y. Zhang, R. Kao, P. C. Teng, Y. Yang, P. Yang, J. J. Wang, M. Smalley, P. J. Chen, M. Kim, S. J. Chou, L. Bao, J. Wang, X. Zhang, D. Qi, J. Palomique, N. Nissen, S. B. Han, S. Sadeghi, R. S. Finn, S. Saab, R. W. Busuttil, D. Markovic, D. Elashoff, H. H. Yu, H. Li, A. P. Heaney, E. Posadas, S. You, J. D. Yang, R. Pei, V. G. Agopian, H. R. Tseng and Y. Zhu, Nat. Commun., 2020, 11, 4489 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |