
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bespoke reagent free amperometric chloride sensor for drinking water
Yanjun
Guo
and
Richard G.
Compton
 *
*
Department of Chemistry, Physical and Theoretical Chemistry Laboratory, Oxford University, South Parks Road, Oxford, OX1 3QZ, UK. E-mail: Richard.Compton@chem.ox.ac.uk
First published on 1st July 2021
Abstract
Chloride quantification is important in drinking water quality control. A bespoke, rapid and reagent free electrochemical method is reported for a simple and accurate chloride sensor specifically for mineral water without the need for added electrolyte. The voltammetry used embraces first the reduction of oxygen to clean and activate the electrode surface and ensure reproducibility without the requirement for any mechanical polishing, followed by silver chloride formation and stripping. A linear correlation was found with silver chloride stripping peak currents and chloride concentrations within the range of 0.4 mM to 3.2 mM on a silver macro disc electrode. The chloride concentrations in two different mineral water samples were measured giving excellent agreement with independent analysis.
Introduction
Chloride is one of the major ionic components of natural water4 the concentration of which is important to both human health5 as well as, in the case of drinking water, taste. The WHO recommends 20 mg to 100 mg chloride should be consumed via drinking water per person per day6 and high chloride mineral water (chloride >200 mg L−1) is widely used in intestinal, bile duct and liver treatments.7 The recommended upper limit of chloride levels in potable water is given by WHO as 250 mg L−1 (ca. 7 mM), but the actual concentration of chloride in branded mineral waters varies significantly from 0.17 mM to 37.7 mM (Table 1). If the drinking water has too high a chloride level, an unpleasant salty taste is detectable and the corrosion of pipes delivering the water is likely to occur, releasing in some cases unwanted toxic lead and copper cations into the drinking water supply.6 Determination of chloride in drinking water is essential.| mg L−1 | mM | |
|---|---|---|
| WHO limit | 2503 | 7 |
| Mineral waters | ||
| Highland | 6.1 | 0.17 |
| Evian | 10 | 0.28 |
| Tesco | 14 | 0.39 |
| Aqua Pura | 15 | 0.42 |
| Volvic | 15 | 0.42 |
| Nestle Pure Life | 18 | 0.51 |
| Buxton | 37 | 1.04 |
| Natural mineral waters | ||
| Voluvesi | 7.3 | 0.2 |
| Saaremaa Vesi | 13.4 | 0.4 |
| Saku | 27.1 | 0.8 |
| Nabeghlavi | 42–95 | 1.2–2.7 |
| Narzan Kislovodsk | 100–150 | 2.8–4.2 |
| Smironovskaya | 250–300 | 7.1–8.5 |
| Borjomi (sparkling) | 380 | 10.7 |
| Jernuk | 300–400 | 8.5–11.3 |
| Varska | 444 | 12.5 |
| Vichy | 601.5 | 17.0 |
| Varska original | 1338 | 37.7 |
Current chloride detection and quantification techniques for application in drinking water include titration, electrochemistry and chromatography,6,8,9 where ion chromatography is specifically recommended by CEN and ISO10 for a large number of samples. Each sample is added to a carbonate–bicarbonate eluant. After ion-exchange the separated chloride ions are directed into a suppressor where the eluant is neutralised. Chloride is identified by retention time standards and quantified via conductivity measurements.8 Apart from facile analysis for multiple samples, ion chromatography also avoids toxic reagents such as silver nitrate or mercury salts as used in titrimetric analysis and a low detection limit of 4 μg L−1 chloride can be achieved if required using ion chromatography. However, one drawback to the method are the costs of consumables, notably the expensive columns and eluants required.
Meanwhile, electrochemical methods have been applied in chloride detection including potentiometry via the chloride ion-selective electrode (Cl-ISE) based on a silver chloride membrane and the coulometric chloridometer. The Cl-ISE can detect chloride in the range 10 μM to 100 μM accurately, but the addition of nitrate reagent is required to maintain stable performance in very dilute solutions.11 In the coulometric method applied to chloride its concentration is determined by monitoring the conductivity of the analyte solution whilst it is titrated with in situ generated silver ions.12 However, the equipment requires trained personnel to operate and the addition of reagents is required. Ideally a direct measure of chloride in drinking water is required which simply and exclusively only needs the introduction of a probe into the solution.
Amperometric quantification via voltammetry opens up a possible alternative inexpensive and robust, reagent free method for halides.13–16 In particular, silver electrodes are analytically useful for chloride in seawater and sweat,17,18 utilizing the voltammetric response of metallic silver oxidation in chloride media. Notice that these literature studies were done with excess supporting electrolyte or high ionic strength media (‘full support’ conditions) in contrast to the low ionic strength solutions found in drinking water (‘low support’ conditions). The challenge in low support conditions is the distortion of voltammetric signals due to significant attraction/repulsion from the charged electrode surface (migration effect).19 Recently, the feasibility of making quantitative electrochemical reactions in aqueous media without supporting electrolyte has been demonstrated using microelectrodes by Li et al.19 with ultra-low conductivity H2O (60 nS cm−1). Silver deposition and stripping voltammetry has also been studied in low support aqueous solutions on gold microelectrodes,20,21 suggesting that the detection of chloride with silver electrodes in low support aqueous systems may be possible. This paper explores the possibility of simple voltammetric analysis directly in bottled mineral and tap water without the addition of any reagents, including the supporting electrolyte commonly added to such experiments.
Herein we present a chloride sensor for bespoke use in drinking water employing a silver macro electrode and utilizing the formation and stripping of silver chloride (eqn (1)) without any added supporting electrolyte or the need for microelectrode instrumentation. In addition, to avoid the need for the cleaning of the electrode the reduction of oxygen naturally present in the water via air saturation (eqn (2) and (3)) is used as an ‘in situ’ activation process18 to activate the electrode surface in a reproducible manner prior to the analysis.22
| AgCl(s) + e− ⇌ Ag + Cl−; E° = 0.2223V | (1) |
| O2 + e− ⇌ O2˙−; E° = −0.284 V | (2) |
| O2˙− + H2O + e− ⇌ HO2− + OH−; E° = −0.0649 V (in alkaline solutions) | (3) |
Generically the work suggests the feasibility of electro-analysis beyond the constraints imposed by the usual demands of operating with a fully supported solution phase together with rigorous outgassing of the solution which, whilst essential for quantitative physical electrochemistry is seen to be unnecessary in the context of at least some bespoke electroanalytical contexts even when using macro-electrodes.
Experimental
(i) Chemical reagents
Potassium chloride (>99.5%, KCl), silver nitrate (>99%, AgNO3) were received from Sigma Aldrich. Potassium nitrate (≥99%, KNO3) was obtained from Honeywell Fluka. Ultrapure water (spectrophotometric grade, H2O) was purchased from Alfa Aesar. Tesco Ashbeck water (2000 mL), Volvic Natural Mineral Water (1500 mL) and Borjomi Sparkling mineral water (500 mL) were used as received with the exception that the Borjomi water was analysed after the release of dissolved gas. Then, the Borjomi water was 10 times diluted with Millipore water. The spherical citrate-AgNPs ca.100 nm diameter (Nanoxact, 0.02 mg mL−1 silver, 2 mM sodium citrate) were purchased from Nanocomposix, USA. For voltammetry analysing silver chloride peaks with silver nanoparticles, solutions were rigorously degassed with nitrogen gas (N2, 99.998%, BOC Gases plc, Guildford, UK) at a sufficient flowrate to ensure nitrogen saturation and exclusion of oxygen. For voltammetry analysing both silver chloride peaks and ORR signals at a bulk silver electrode, solutions (5.0 mL) were stirred (400 rpm) under conditions open to the air for 15 min to ensure saturation of air for each measurement. Solutions were prepared using Millipore water with a resistivity of 18.2 MΩ cm at 25 °C unless stated otherwise.(ii) Electrochemical analysis
All electrochemical measurements were performed in a conventional three-electrode cell in a Faraday cage and a μAutolab II potentiostat manufactured by Metrohm-Autolab BV, Utrecht, Netherlands under 25 °C. The experimental temperature was maintained constant at 25 ± 0.5 °C by a thermostated water bath and the cell consisting of a platinum wire counter electrode, a homemade Ag/AgNO3 reference electrode (10 mM AgNO3 and 90 mM KNO3) and a working electrode (WE). The reference electrode consisted of a silver wire submerged in regularly recharged 10 mM AgNO3, 90 mM KNO3 solution and was calibrated against a Ag/AgCl [saturated KCl] (+0.20 V vs. NHE) reference electrode via cross referencing with the oxygen reduction peak in 0.1 M KNO3 air-saturated solution. The peaks were found to reproducibly differ by +0.48 V. Thus, this homemade reference has a potential of +0.68 V vs. NHE, in agreement with the previously reported potential of Ag/AgNO3 reference electrode in 10 mM AgNO3 and 90 mM NaNO3 with a value of +0.69 V vs. NHE.17A glassy carbon macro disc electrode (GC, radius 1.49 mm, BAS, Technical, UK) or a silver macro-disc electrode (Ag, homemade, radius 1.13 mm) calibrated as reported in previous papers18,23 were used as working electrodes. Both electrodes were polished using a sequence of 1.0, 0.3 and 0.05 μm alumina lapping compounds (Bucher, Germany).
(iii) Chloride detection on a silver macro-electrode
Cyclic voltammetry was conducted in KCl solutions made with Tesco water (Tesco–KCl solutions) or Millipore water (pure KCl solutions) as a solvent to realise a final chloride concentration range of 0.4 mM–0.32 mM. Each scan was undertaken in air-saturated solutions at 0.10 V s−1 from −0.50 V to −0.90 V then anodically to −0.05 V before returning to −0.50 V and repeated three times.(iv) Chloride detection with silver nanoparticles on a GC electrode
To allow comparison of the silver chloride stripping features between a bulk silver electrode and a silver nanoparticle modified electrode, a suspension of the as-supplied commercial AgNPs with a volume of 15.0 mL was washed with ultrapure water twice. The solution was then centrifuged at 400 rpm for 10 min and 10.0 mL of the supernatant was removed and replaced with a further 10.0 mL ultrapure water for two repetitions. Finally, 14.5 mL of the supernatant was removed, so that 0.5 mL suspension was obtained at 30 times the stock concentration. Before drop-casting onto the GC electrode, the washed AgNPs sample was dispersed by sonication for 15 min. Specifically, after polishing the glassy carbon macro-electrode, 5.0 μL of washed AgNPs was applied to the electrode surface and dried at 50 °C for 10 min. The electrode was then used to detect chloride levels in Tesco–KCl solutions and compared to that in a blank scan with 1 mM KNO3. Each scan was undertaken in nitrogen saturated solutions at 0.10 V s−1 from −0.70 V to +0.50 V then returning to −0.70 V and repeated three times.Results and discussion
We explore, develop and validate the proposed detection method in four steps. First, silver chloride formation and stripping were studied in combination with the ORR reaction under both ‘self-support’ and ‘low support’ solutions. The former relates to the total absence of any deliberately added electrolyte whereas the latter experiments had a low level of intentionally introduced supporting electrolyte for comparative purposes. Experimentally the silver macro-disc electrode was oxidized in the solution of known potassium chloride concentration with variable potassium nitrate levels to control the ionic strength. Voltammetric peaks associated with silver chloride formation and stripping, along with other observed peaks and their waveshapes were analysed and the feasibility of detection in drinking water discussed in the light of the ionic strength of branded water.Second, the detection of chloride using a silver electrode was undertaken in pure potassium chloride solutions and Tesco minerial water of chloride concentrations in the range 0.4 mM to 3.2 mM comparable to that in found in typical mineral waters (see Table 1). Linear correlations between the voltammetric peak current of silver chloride formation and chloride concentration were obtained.
Third, detections of chloride concentration in Tesco brand mineral water with varying added chloride concentrations were performed using a silver nanoparticles modified glassy carbon electrode via stripping voltammetry and the results compared with those from the silver macro disc electrode.
Finally, we performed chloride detections of two real samples using the calibration curve obtained in Tesco water with the silver macro disc electrode to validate the analytical procedure for the measurement of the chloride content of authentic samples of mineral waters under self-support conditions and without the addition of electrolyte or other reagents.
(i) Voltammetry under ‘self-support’ and ‘low support’ conditions
The voltammetric responses under ‘self-support’ and ‘low support’ conditions were analysed with a silver macro-disc electrode (Fig. 1) in air-saturated solutions containing 0.4 mM KCl with 0 mM, 1.0 mM or 4.0 mM KNO3. Each scan started from −0.50 V, swept negatively at 0.01 V s−1 towards −0.90 V to encompass the oxygen reduction reaction (ORR) to remove absorbed chloride ions before detections and ensure reproducibility of measurements to avoid any need for mechanical polishing of the electrode surface as described previously.18 Then the potential cycle reversed to +0.05 V and returned to −0.50 V.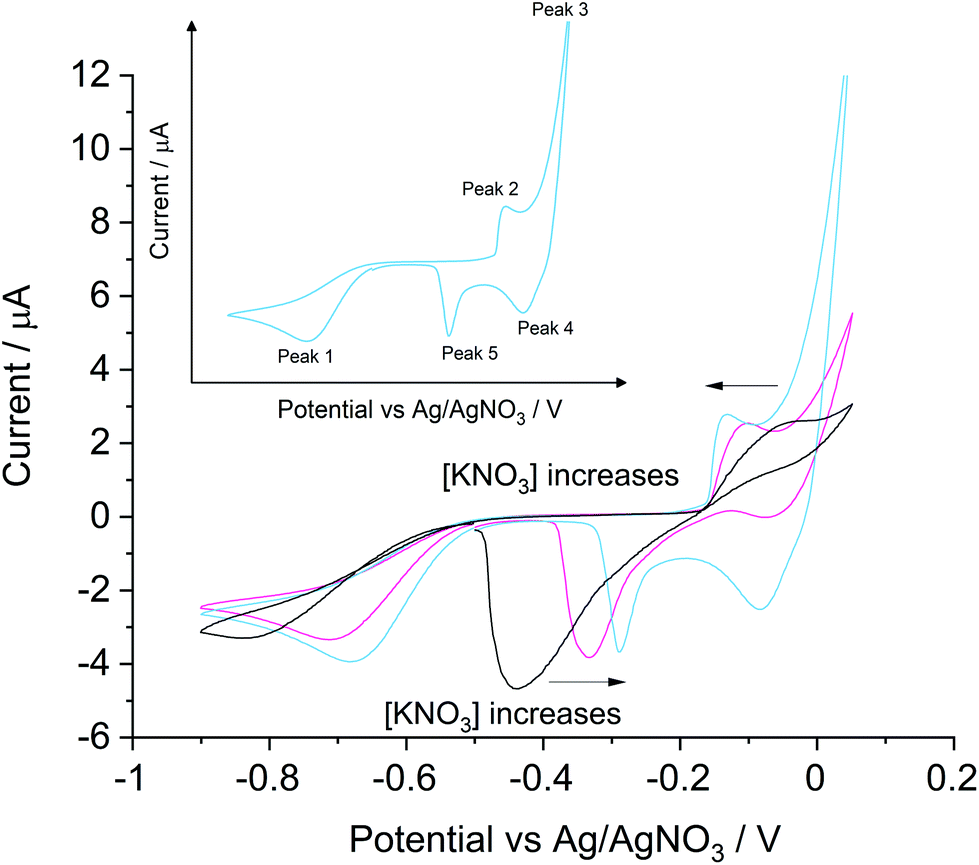 | ||
| Fig. 1 The oxidative stripping of silver macro-disc in air-saturated 0.4 mM KCl in KNO3 solutions at a scan rate of 0.01 V s−1, 25 °C. The black line refers to data measured in the absence of KNO3. The pink and blue lines refer to data with the addition of 1 mM and 4 mM KNO3 respectively. Voltammetric peaks of interests are labelled as peak X (X = 1, 2, 3, 4 or 5) in the inset using data from the solution of 0.4 mM KCl, 4 mM KNO3. | ||
Five peaks can be observed in Fig. 1, as labelled schematically in the inset, depending on the concentration of KNO3 in the solution. First, the large diffusional peak 1 in the anodic scan corresponds to the two-electron transfer reduction of oxygen24 (eqn (2) and (3)). Peak 1 was observed at −0.84 V vs. Ag/AgNO3 under ‘self-support’, which gradually shifted to a more positive potential at −0.68 V with its peak height more than doubled when 4.0 mM KNO3 was added. The oxygen reduction on silver with 0.1 M NaClO4 at 0.01 V s−1 was reported at −0.3 V vs. SCE. by Neumann et al.24 which is equivalent to −0.74 V vs. Ag/AgNO3. The distortion of the ORR peak under the ‘low support’ condition as compared to that in the ‘full support’ case is expected and the low conductivity of the medium and additionally may reflect the sensitivity of the formation of peroxide to the more extended and dilute double layer.
In peak 2, silver was oxidized to form silver chloride with a corresponding stripping feature (peak 5) seen on the reverse, cathodic scan. Significantly broadened peaks were observed under ‘self-support’ conditions regarding silver chloride formation and stripping at −0.04 V and −0.45 V respectively. In the KCl solution with 4.0 mM KNO3, peak 2 and peak 5 shifted to −0.13 V and −0.30 V respectively and both of their shapes narrowed. The peak potentials are consistent with the values reported by De Mele et al.25 in 0.09 M NaCl, 0.91 M NaClO4 solution at 0.02 V s−1. The formation and stripping of silver chloride on a silver electrode were seen at ca. 0.37 V and ca. 0.15 V vs. NHE respectively, which are −0.11 V and −0.33 V vs. the Ag/Ag+ reference electrode used in the present study.
Peak 3 and peak 4 were only clearly observed in the potential range studied in ‘low support’ experiments under conditions where the local chloride ions were significantly depleted via conversion into AgCl in peak 2. This suggests that peak 3 represents the onset of the silver oxidation signal leading to Ag+ and peak 4 represents the Ag+ reduction (eqn (4)). The onset of the silver oxidation is consistent with the formal potential of the Ag/Ag+ couple estimated to be 0.12 V vs. the reference electrode used in this study. Comparison of the voltammograms shown in Fig. 1 in the absence of added electrolyte and those with low levels of support indicates a more sustained silver oxidation as the level of electrolyte support increases. Notice that when the concentration of KNO3 increases from 0 to 4.0 mM, the continuous shift in peak 3 causes difficulties in the signal subtraction from the AgCl formation process, thus the stripping peak of AgCl is used in chloride determinations as reported below.22
| Ag+(aq) + e− ⇌ Ag; E° = 0.8 V vs. NHE | (4) |
Next, we question whether we can realistically expect to be able to apply the silver chloride stripping peak current under a certain ‘support level’ to quantify chloride ions in common drinking waters with varying ionic strength. The compositions of typical commercially available mineral waters were used to generate the conductivity data shown in Table 2. Estimations were made via the labelled compositions and the literature values for the equivalent molar conductivities.26 Overall support levels in each sample are listed along with the equivalent potassium nitrate concentrations with the same conductivity. Most branded mineral waters have ionic strengths corresponding to ‘low support’ conditions and can be approximated as solutions with concentrations in the range of 1 mM to 4 mM KNO3. As shown in Fig. 1, the AgCl stripping peak current is almost unchanged from solutions with 1 mM to 4 mM KNO3, suggesting, on the basis of Table 2, similar voltammetric parameters can permit direct measurements in most drinking waters, though dilutions to reach this range are required in (surprisingly) high salt mineral waters such as Borjomi and Varska waters.
| Conductivity/μS cm−1 | Equivalent KNO3 concentration/mM | |
|---|---|---|
a Conductivity values estimated from compositions labelled on the mineral water package via equation 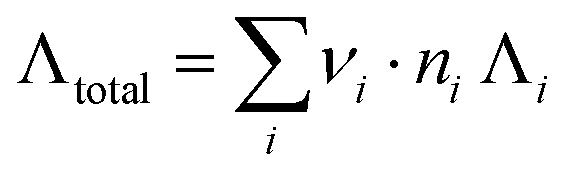 where νi, ni, Λi refer to the number of charges, the moles, and the equivalent ionic conductivity of species i respectively.
1 where νi, ni, Λi refer to the number of charges, the moles, and the equivalent ionic conductivity of species i respectively.
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The ionic conductivity values are obtained from the literature.26 Conductivity values in brackets are provided in manufacture annual reports or measured in other literatures.27 Equivalent KNO3 concentrations were calculated according to KNO3 solutions with the same conductivity value. The ionic conductivity values are obtained from the literature.26 Conductivity values in brackets are provided in manufacture annual reports or measured in other literatures.27 Equivalent KNO3 concentrations were calculated according to KNO3 solutions with the same conductivity value. |
||
| Mineral waters | ||
| Highland Spring | 313a (33627) | 2.16 (2.32) |
| Evian | 442a (600) | 3.05 (4.14) |
| Tesco | 159a | 1.10 |
| Aqua Pura | 218a | 1.50 |
| Volvic | 218a (220) | 1.50 (1.52) |
| Nestle Pure Life | 437a (160–960) | 3.01 (1.10–6.63) |
| Buxton | 582a | 4.02 |
| Natural mineral waters | ||
| Voluvesi | 5961 | 4.11 |
| Saaremaa Vesi | 5881 | 4.06 |
| Saku | 1751 | 1.21 |
| Borjomi Sparkling | 4114a | 52.27 |
| Varska | 21301 | 14.70 |
| Varska original | 43701 | 30.16 |
(ii) Chloride determination in pure KCl solutions and Tesco mineral water KCl solutions (Tesco–KCl solutions)
To develop the sensor for use in mineral water samples which contain additional ions such as sulfate and carbonate, Tesco mineral water was used to prepare a series of standard solutions spiked with different amounts of KCl so as the create a calibration curve based on the voltammetric method developed in the section (i) above. This was first applied in pure KCl solution and results were used for comparison with Tesco–KCl solutions at the same chloride level.The electroanalytical responses of the silver/silver chloride stripping signal measured at a scan rate of 0.01 V s−1 were explored in air-saturated pure KCl solutions and Tesco–KCl solutions with chloride concentration ranges from 0.4 mM to 3.2 mM. Fig. 2 shows the voltammograms which consist of oxygen reduction (to activate the electrode and ensure reproducibility of the analytical signal) and silver chloride deposition/reduction, scanned from −0.50 V to −0.90 V then scanned positively to −0.05 V and finally swept back to −0.50 V. Three peaks (peak 1, peak 2 and peak 5 as indicated in Fig. 2) were observed for each scan in all the concentration range studied.
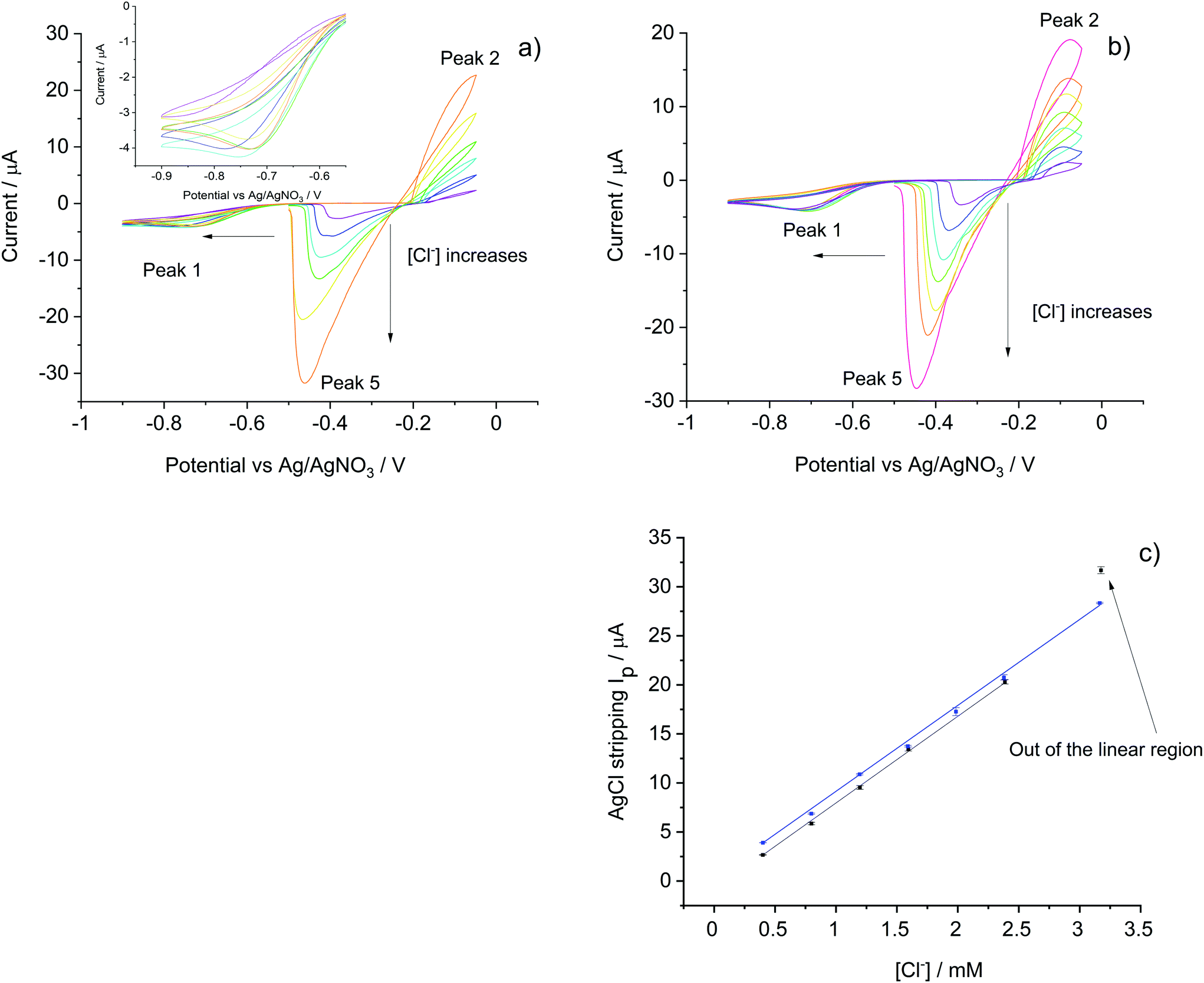 | ||
| Fig. 2 The oxidative stripping voltammetry of silver macro-disc electrode at 0.01 V s−1, 25 °C in air-saturated solutions. (a) Pure KCl solutions of 0.4 mM, 0.8 mM, 1.2 mM, 1.6 mM, 2.4 mM and 3.2 mM. Chloride concentrations increase as indicated by the arrow. (b) Tesco–KCl solutions of 0.4 mM, 0.8 mM, 1.2 mM, 1.6 mM, 2.0 mM, 2.4 mM and 3.2 mM supplementing the chloride present in Tesco water which contributes 0.4 mM. Chloride concentrations increased as indicated by the arrow. (c) The calibration curves determined with the silver chloride stripping peak currents. Data in blue shows results from Tesco–KCl solutions, data in black shows results from pure KCl solutions. Each data contains three repeats. The regression coefficient for pure KCl solution and Tesco–KCl solution are both 0.999. The high concentration point is discussed in the text. | ||
First, the ORR signals (peak 1) in pure KCl solutions were compared to those in Tesco–KCl solutions. In the former medium in the concentration range 0.4 mM to 0.8 mM KCl solution, the peroxide feature shifted from −0.85 V to ca. −0.75 V and the peak current increased from 3 μA to 4 μA, reflecting the increased ionic concentration. In contrast, the ORR signal in Tesco–KCl water was fixed at ca. −0.70 V plausibly due to the higher conductivity in the mineral water. As the ORR signal reflects more the ionic strength instead of the real chloride levels in the solution, it has little analytical value in the present study beyond and very importantly creating a reproducible surface for silver chloride formation and stripping.
Next, silver chloride formation signals (peak 2) were analysed. In pure KCl solutions due to the extreme low ionic strength, the peaks were broad and the current steadily increased within the potential range studies creating a voltammetric ‘loop’ to be was observed with the highest current at −0.05 V. In the Tesco–KCl solutions, clear silver chloride formation peaks were observed at −0.1 V. In both cases the formation peak currents increased as the chloride concentration increased.
On the cathodic scan, silver chloride stripping peaks (peak 5) were observed during the reverse scans for both pure KCl and Tesco–KCl solutions. As more silver chloride formed on the electrode surface, the peak shifted to more negative potentials and its height increased. Linear relationships were observed between AgCl stripping peak currents and chloride concentrations (Fig. 2c) for both solutions. The correlation ranges were 0.4 mM to 2.4 mM and 0.4 mM to 3.2 mM for pure KCl solutions and Tesco–KCl solutions respectively with the different intercepts reflecting the different amounts of AgCl formation within the potential window. The extreme data point of 3.2 mM KCl solution deviated from linearity likely due to the increased ionic strength. Most importantly, the calibration curve slope in pure KCl solutions is 8.99 μA mM−1, almost identical to that in Tesco–KCl solutions (8.76 μA mM−1), indicating that the detection sensitivity is not affected by mineral water interferences, validating the use of Tesco mineral water as the solvent in the following sections.
(iii) Chloride determinations in Tesco–KCl solutions using silver nanoparticle modified electrodes
Meanwhile, the analytical methodology developed above with bulk silver was compared to that using silver nanoparticles (AgNPs) seeking enhanced mass transport under low support systems. The oxidative stripping voltammetry was performed in nitrogen saturated Tesco–KCl solutions with chloride concentrations from 0.4 mM to 3.2 mM on AgNPs modified glassy carbon electrode (GC). The voltammetry started from −0.70 V, scanned positively at 0.1 V s−1 to 0.50 V and reversed to −0.70 V.Two broad peaks were observed in the oxidative scan in chloride solutions. As shown in Fig. 3, peak a was observed at −0.10 V, partially overlapping with peak b at a higher potential between 0.05 V and 0.25 V. With a constant amount of AgNPs modified on the electrode, peak a increased in magnitude as the chloride concentration increased in contrast to peak b which current was gradually decreased and potential shifted to a lower value. A further experiment was performed in 1 mM KNO3 to exclude the presence of chloride and which has a conductivity identical to that of the Tesco mineral water (see Table 2). As only peak b was observed in the blank data, we infer peak a correlates to the formation of AgCl whilst peak b is associated with the oxidation of the AgNPs to Ag+. Similar observations were reported by Toh et al.17 who performed the stripping of AgNPs modified GC in ‘full support’ conditions using 0.1 M NaNO3 solution with an addition of 2 mM to 40 mM KCl content. Two broad peaks were seen each at −0.2 V (peak a) and 0.2 V (peak b) vs. Ag/AgNO3 for the silver chloride formation and the silver cations formation respectively. Compared to results reported in ‘full support’ conditions, both peaks shifted to more positive potentials in this study probably due to the low ionic strength.
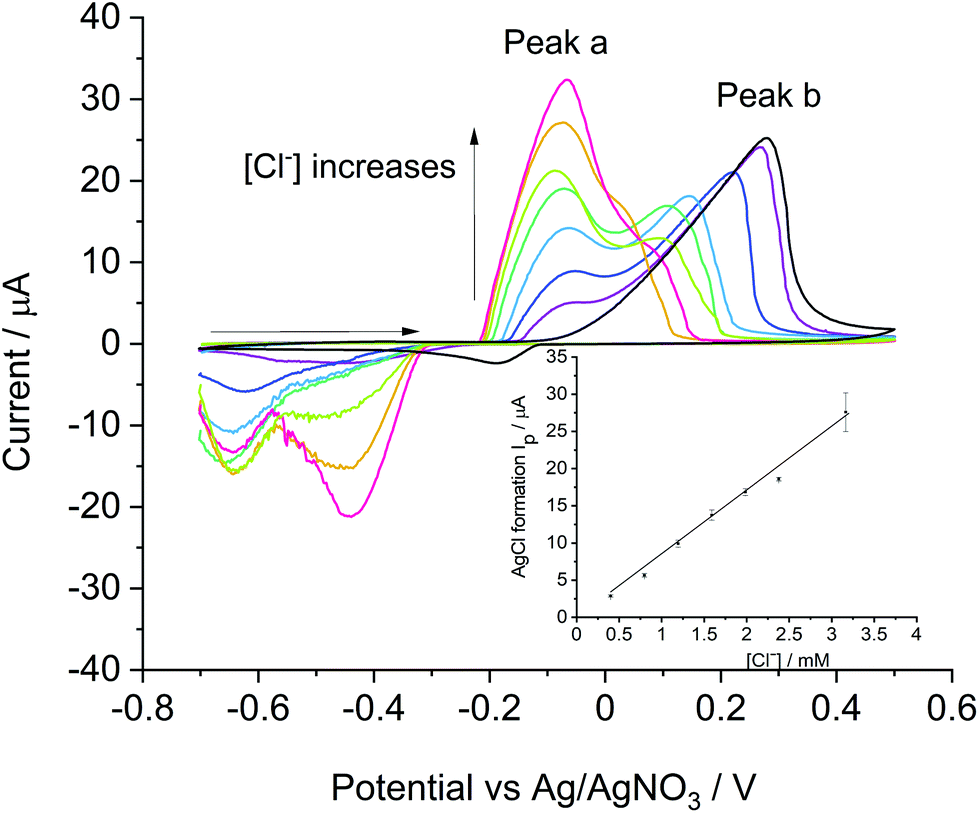 | ||
| Fig. 3 The oxidative stripping of AgNPs from a GC electrode at a scan rate of 0.01 V s−1, 25 °C in N2 saturated Tesco–KCl solutions. The blank experiment was performed in 1 mM KNO3 (black); Chloride detections were performed in Tesco–KCl solutions of 0.4 mM, 0.8 mM, 1.2 mM, 1.6 mM, 2.0 mM, 2.4 mM and 3.2 mM supplementing the chloride present in Tesco water which contributes a further 0.4 mM. Chloride concentrations increase as indicated by the arrow. The inset shows the calibration curve where chloride in Tesco water has also been counted. Each point contains three repeats. Regression coefficient R2 = 0.995. | ||
A linear correlation was observed between the current of peak a and the concentration of chloride ions with a slope of 8.57 μA mM−1, which is comparable to that observed with the silver macro disc electrode (8.76 μA mM−1). With sufficient silver deposited (27.8 nmol of Ag) to ensure the sensitivity with AgNPs,17 the similarity in slopes indicates that the reaction of AgCl formation is chloride concentration limited, and the use of a silver macro disc electrode can achieve the same sensitivity as with AgNPs; the analytical signal is controlled in both cases by the diffusion of the chloride to the geometric area of the electrode, either pure silver or the silver nanoparticle modified glassy carbon electrode. Accordingly, the use of the silver electrode was adopted preferentially for reasons of simplicity and speed. In this manner chloride concentrations in real samples were estimated on a bulk silver electrode using the calibration curve evaluated from Tesco–KCl solutions as reported in the next section.
(iv) Chloride determination in real samples
To evaluate the feasibility for practical chloride detections in mineral waters, two real samples were tested against the system. Sample 1 was Volvic water used directly as purchased, sample 2 was Borjomi sparkling water which has an unusually high mineral content (and taste!), hence prior to analysis this was diluted with deionized water by a factor of ten after all gases were released. The dilution was essential for the Borjomi water as first, its original chloride concentration was 10.7 mM, beyond the linear sensing range of 0.4 mM to 3.2 mM and second, its conductivity equivalent to 52.3 mM KNO3 which is far above ‘low support’ levels so differs from the conditions applied in the construction of the calibration curve. The test with the diluted Borjomi water extended the application of the developed chloride determination methodology from regular to the very highest mineral level drinks.A cyclic voltammogram was recorded from −0.50 V to −0.90 V before sweeping positively to −0.05 V and returned to −0.50 V at a scan rate of 0.01 V s−1. Each experiment was repeated three times. Fig. 4 shows the voltammograms recorded from the mineral water samples. Three peaks were observed. First, the ORR feature (peak 1) was seen at −0.75 V and −0.70 V for sample 1 and 2 respectively, reflecting the conductivity increased from 218 μS cm−1 to 411 μS cm−1 (recall Table 2). Second, peak 2 was observed at −0.12 V and −0.15 V corresponding to the silver chloride formation in sample 1 and sample 2, followed by the generation of Ag+ at −0.05 V. Finally, silver chloride was removed in the reversed scan, for each solution, a shoulder was observed at ca. −0.32 V before the main peak appeared at ca. −0.36 V. The shoulder in silver chloride stripping peak has been reported,18,28,29 possibly reflecting instantaneous AgCl nucleation or silver dissolution.
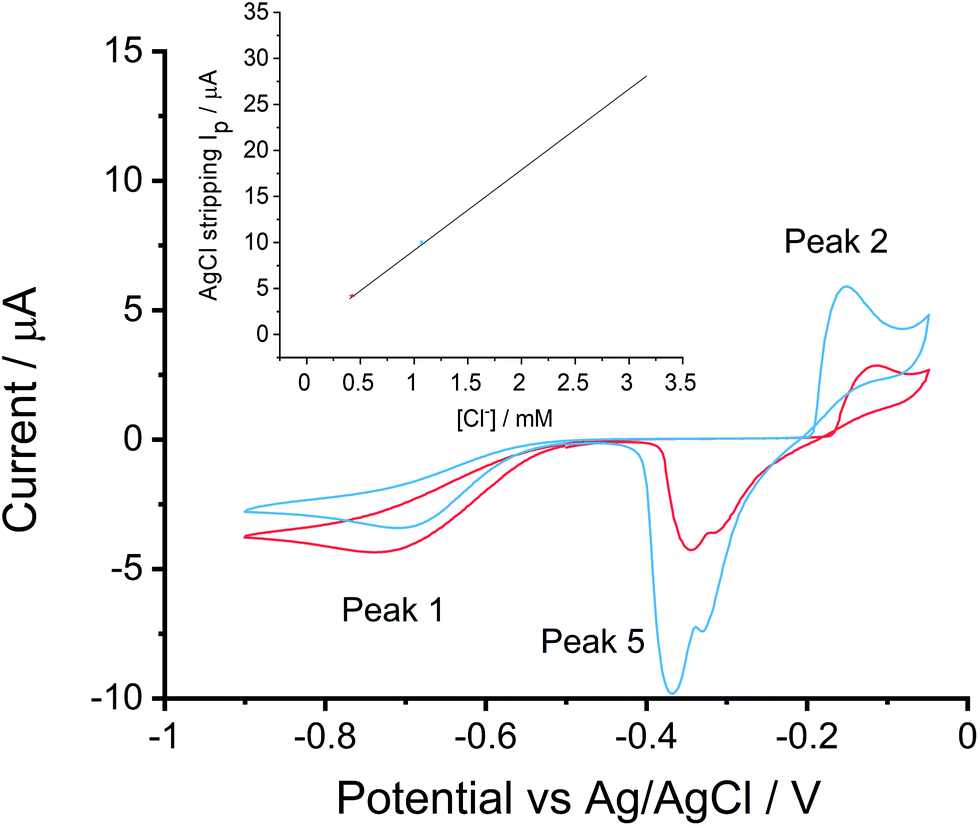 | ||
| Fig. 4 Chloride determinations in real mineral water samples at 0.01 V s−1. Red: Volvic water; blue: Borjomi sparkling water diluted with deionized water by a factor of ten. Inset shows the chloride concentration estimated with the calibration curve generated in Tesco–KCl solutions. Each point represents three repeats. | ||
By measuring the current in peak 3, we estimated the chloride concentration in samples. The experimentally measured values were 0.44 ± 0.01 mM and 1.10 ± 0.02 mM for Volvic water and 1/10 Borjomi mineral water respectively and the corresponding manufacturer reported chloride concentrations were 0.42 mM (Volvic) and 1.07 mM (Borjomi). The inset of Fig. 4 displays the samples at the expected chloride concentration, showing the measured chloride concentration in examined samples fitted well to the expected values. In this way, the developed method was validated in and applied to authentic mineral waters.
Conclusions
This paper presents a facile chloride detection method for mineral water samples and was specifically developed for low ionic strength solutions. The method utilizes the peak current of the AgCl stripping process on a silver macro disc electrode over a well-defined potential range as a measure of chloride concentrations while applying the ORR reaction to activate the electrode surface so that reproducible measurements can be realised without mechanical cleaning. The method was validated with chloride ions quantifications in Volvic mineral water and Borjomi sparkling mineral water where results were in agreements with mineral water labelled compositions. More generally quantitative electroanalytical measurements were realised using a macro-electrode under conditions of low electrolyte support so allowing reagent free analysis and the use of conventional voltammetry.Author contributions
Yanjun Guo: Data curation, formal analysis, investigation, methodology, validation, writing (original draft). Richard G. Compton: Conceptualization, resources, supervision, writing (review & editing).Conflicts of interest
There are no conflicts to declare.References
- L. Bityukova and V. Petersell, J. Geochem. Explor., 2010, 107, 238–244 CrossRef CAS.
- V. Gvakharia, A. Jgamadze, J. Gabechava, T. Adamia, D. Chkhaidze, T. Jebashvili, D. Sozashvili, B. Lebanidze and A. Maglakelidze, Ann. Agric. Sci., 2018, 16, 27–31 Search PubMed.
- E. Dinelli, A. Lima, S. Albanese, M. Birke, D. Cicchella, L. Giaccio, P. Valera and B. De Vivo, J. Geochem. Explor., 2012, 112, 368–389 CrossRef CAS.
- J. D. Hem, Study and interpretation of the chemical characteristics of natural water, Report 2254, Reston, VA, 1985 Search PubMed.
- L. McCallum, S. Lip and S. Padmanabhan, Pflügers Arch., 2015, 467, 595–603 CrossRef CAS PubMed.
- WHO, Guidelines for drinking-water quality, WHO, Geneva, 2011 Search PubMed.
- S. Quattrini, B. Pampaloni and M. L. Brandi, Clin. Cases Miner. Bone Metab., 2016, 13, 173–180 Search PubMed.
- L. S. Clesceri, A. E. Greenberg and A. D. Eaton, Standard Methods for the Examination of Water and Wastewate, APHA American Public Health Association, USA, 20th edn, 1998 Search PubMed.
- Department of the Environment, Chloride in waters, sewage, and effluents 1981, H.M.S.O., London, 1982 Search PubMed.
- P. Quevauviller and K. C. Thompson, Analytical Methods for Drinking Water: Advances in Sampling and Analysis, Wiley, West Sussex, 2006 Search PubMed.
- A. Bratovcic and A. Odobasic in Environmental Monitoring, Dr Ema Ekundayo, InTech, Croatia Search PubMed.
- W. Holiczer, Coulometric method of the chloride ion concentration measurement: mathematical model of the measuring transducer and its experimental verification, SPIE, 2001 Search PubMed.
- K. Arai, F. Kusu, N. Noguchi, K. Takamura and H. Osawa, Anal. Biochem., 1996, 240, 109–113 CrossRef CAS PubMed.
- J. Bujes-Garrido, D. Izquierdo-Bote, A. Heras, A. Colina and M. J. Arcos-Martínez, Anal. Chim. Acta, 2018, 1012, 42–48 CrossRef CAS PubMed.
- A. Gupta, M. Maynes and S. Silver, Appl. Environ. Microbiol., 1998, 64, 5042–5045 CrossRef CAS PubMed.
- X. Qin, H. Wang, Z. Miao, X. Wang, Y. Fang, Q. Chen and X. Shao, Talanta, 2011, 84, 673–678 CrossRef CAS PubMed.
- H. S. Toh, C. Batchelor-McAuley, K. Tschulik and R. G. Compton, Analyst, 2013, 138, 4292–4297 RSC.
- Y. Guo and R. G. Compton, Talanta, 2021, 232, 122502 CrossRef CAS PubMed.
- X. Li, C. Batchelor-McAuley, E. Laborda and R. G. Compton, Chem. – Eur. J., 2017, 23, 15222–15226 CrossRef CAS PubMed.
- K. Ngamchuea, C. Batchelor-McAuley and R. G. Compton, J. Electroanal. Chem., 2018, 830–831, 122–130 CrossRef CAS.
- Y. Guo, M. Yang, C. Batchelor-McAuley and R. G. Compton, J. Electroanal. Chem., 2019, 849, 113370 CrossRef CAS.
- A. J. Bard, R. Parsons and J. Jordan, International Union of Pure and Applied Chemistry, Standard potentials in aqueous solution, M. Dekker, New York, 1985 Search PubMed.
- Y. J. Guo, M. J. Yang, R. C. Xie and R. G. Compton, Chem. Sci., 2021, 12, 397–406 RSC.
- C. C. M. Neumann, E. Laborda, K. Tschulik, K. R. Ward and R. G. Compton, Nano Res., 2013, 6, 511–524 CrossRef CAS.
- M. F. L. de Mele, R. C. Salvarezza, V. D. Vasquez Moll, H. A. Videla and A. J. Arvia, J. Electrochem. Soc., 1986, 133, 746–752 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, Taylor & Francis, 86th edn, 2005 Search PubMed.
- A. Alsulaili, M. Al-Harbi and K. Al-Tawari, J. Eng. Res., 2015, 3, 2 CrossRef.
- F. Pargar, H. Kolev, D. A. Koleva and K. van Breugel, J. Mater. Sci., 2018, 53, 7527–7550 CrossRef CAS.
- V. I. Birss and C. K. Smith, Electrochim. Acta, 1987, 32, 259–268 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |